

Translate this page into:
Drug design of new anti-EBOV inhibitors: QSAR, homology modeling, molecular docking and molecular dynamics studies
⁎Corresponding author. i.hdoufane@uca.ac.ma (Ismail Hdoufane)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Ebola virus disease is a deadly pathogenic disease with a fatality rate of 25–90 % as recorded in previous outbreaks. The Ebola Virus glycoprotein (EBOV-GP) plays a crucial role in the entry of viruses into human cells, making it an interesting target for therapeutic discovery. Therefore, inhibiting this protein can directly limit the virus replication and disease progression at an early stage of infection. The present study focuses on the design of novel potent EBOV-GP inhibitors using multiple computational techniques. In this context, two QSAR models were built from a set of 86 amodiaquine derivatives as anti-EBOV-GP using Monte Carlo and genetic algorithm multiple linear regression methods. Both models confirmed their predictive performance with satisfactory statistical parameters of the validation (R2 = 0.9129; Q2 = 0.8848 for the CORAL model and R2 = 0.8848; Q2 = 0.8148 for the GA-MLR model). From the outputs of the CORAL model, the structural fragments responsible for increasing and decreasing the inhibition activity were extracted and interpreted. This molecular information was used to design 26 new potentially safe and active candidate drugs. Molecular docking and dynamics simulations have affirmed the efficacy of the designed compounds. Specifically, compounds D2 (pIC50_coral = 7.12; pIC50_GA-MLR = 7.07), D3 (pIC50_coral = 7.83; pIC50_GA-MLR = 7.10), and D5 (pIC50_coral = 7.26; pIC50_GA-MLR = 7.55) displayed notable predicted inhibitory activity, according to both models. These compounds also exhibited conformational and structural stability, as well as a favorable binding profile. Furthermore, these potential drug candidates were found to be non-toxicity and have acceptable pharmacological properties.
Keywords
Ebola virus
Glycoprotein
QSAR
Homology modeling
Molecular docking
Molecular dynamics

1 Introduction
Ebola virus disease (EVD) is a severe and rapidly progressing hemorrhagic fever caused by the Ebola virus (EBOV) (hong Ma et al., 2023). Since the virus was first identified in the Democratic Republic of Congo in 1976, there have been several outbreaks in which it has been responsible for intermittent but highly destructive epidemics, mainly affecting regions in Central and West Africa (Dash et al., 2017; Garcia-Rubia et al., 2023). The most devastating of these outbreaks occurred in West Africa between 2013 and 2016 with over 28,000 infections and more than 11,000 deaths (hong Ma et al., 2023). The repeated and severe outbreaks have emphasized the significant risk that EBOV poses to human health. In the face of this disease, current treatment strategies focus on complementary treatment, as there is no specific approved antiviral therapy for widespread use. The high mortality rate associated with EBOV emphasizes the urgent need to develop drugs that can inhibit different stages of the EBOV replication cycle.
EBOV belongs to the Filoviridae family and is characterized by its filamentous structure and an 18 kb single-stranded RNA genome encoding seven structural proteins, including viral structural proteins (VP35, VP40, VP30 and VP24), nucleoprotein (NP), RNA-dependent RNA polymerase (L) and glycoprotein (GP) (Peng et al., 2022; Mali and Chaudhari, 2019; Volchkova et al., 1998; Volchkov and Klenk, 2018; Lee and Saphire, 2009). The GP gene of EBOV transcribes into three different GP forms. Full-length GP chains express the attachment protein (GP1) and the entry/fusion protein (GP2) from messenger RNAs (mRNAs) containing an additional adenosine that is not present in the template (Lee et al., 2008). The soluble GP (sGP), on the other hand, is synthesized from the primary RNA transcript. The small soluble GP (ssGP) is produced by the insertion of two additional adenosine residues (Volchkova et al., 1998). These GPs, which are located on the surface of the virion, play a crucial role in promoting membrane fusion and penetration into host cells (Lee and Saphire, 2009). Therefore, inhibition of this protein can prevent the virus from entering and infecting host cells, which is an important strategy for antiviral therapies and vaccine development.
In silico investigations represent a very promising methodology in drug discovery to develop new potent and safe anti-EBOV drugs in terms of time and cost. This shift towards these methods is evident in the growing importance of Computer-Aided Drug Design (CADD) in the field. CADD encompasses various computational approaches, including structure-based drug design (SBDD) and ligand-based drug design (LBDD) (Mallipeddi et al., 2014). Additionally, CADD involves the estimation of pharmacokinetics, therapeutic, and toxicological properties in drug discovery journey with computational approaches before the experimentations (Lipinski et al., 2012). Despite the advantages of CADD in modern drug design, there are still limitations, as many identified candidate molecules do not show the desired activity in biological systems. In fact, only 40 % of drug candidates identified via CADD make it through clinical trials and receive approval for clinical use. To overcome these limitations, using the most accurate tools available and combining multiple CADD techniques can increase the reliability of the predictions (Baig et al., 2018).
In the quest for new treatments for EVD, this study focused on the development of quantitative structure–activity relationships (QSAR) to design new inhibitors of EBOV-GP. The initial QSAR model employed the Monte Carlo optimization using simplified molecular input line representations (SMILES) to explore the molecular fragments associated with enhancing/reducing of the biological activity. Using this molecular information, we designed new molecules with potential inhibition against EBOV-GP. Subsequently, a Genetic Algorithm Multiple Linear Regression (GA-MLR) based QSAR model was applied to the newly designed molecules to confirm their potential inhibition activity. To thoroughly evaluate these compounds, we conducted molecular docking and molecular dynamics simulations (MD) together with Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) assessments to ensure a comprehensive evaluation of their interaction with the EBOV-GP protein, their stability, and their pharmacokinetic profiles.
2 Materials and methods
2.1 Data collection and preparation
A dataset of 86 amodiaquine derivatives with EBOV-GP inhibitory activity were collected from the published work by Sakurai et al. (Sakurai et al., 2018). After drawing the chemical structures of these derivatives using ChemDraw software (PerkinElmerInformatics, Inc, ChemDraw, 2021), their geometries were optimized in the gas phase using the AM1 semi-empirical method. The optimization procedure was performed using the Gaussian 09 software package (Gaussian 09, n.d.). The optimized molecular structures were converted into isomeric SMILES notations for the modeling process of the CORAL model using the Openbabel program (O’Boyle et al., 2011). For the GA-MLR model, the molecular descriptors of the optimized 3D structures were calculated using AlvaDesc (Mauri and Bertola, 2022). To avoid multicollinearity in the QSAR model, we reduced the number of generated variables by removing descriptors with a correlation coefficient greater than 0.95 and retaining only one of the pairs. As a result, a total of 832 descriptors were selected from the original set of 4149 descriptors. The experimental data value of each molecule (half-maximal inhibitory concentration: IC50) was converted into negative logarithmic scale (pIC50). These converted data were then utilized as variables for building QSAR models. The molecular structures and corresponding IC50 data are listed in Table S1 and their SMILES notation and converted pIC50 can be found in Table S2.
2.2 CORAL QSAR model
CORAL QSAR modeling software (Toropova and Toropov, 2014) utilizes SMILES attributes for endpoint prediction, based on optimal descriptors, namely correlation weights (CWs), along with the balance-of-correlation approach. Four splits were randomly made from the entire dataset with each split in turn divided into four partitions: Training (Tr: 35 %), Invisible Training (Inv-Tr: 35 %), Calibration (Calib: 15 %), and Validation (Valid: 15 %). The specific functions of each set in the development of QSAR models are described in detail in the literature (Toropova and Toropov, 2014; Oubahmane et al., 2023). Table 1 illustrates the distribution and percentage of the identity of the compounds in the four splits.
Split
Set
Split 1 (%)
Split 2 (%)
Split 3 (%)
Split 4 (%)
Split 1
Total
100
29.1
25.6
32.6
Tr
100
36.7
26.7
40.0
Inv-Tr
100
33.3
33.3
36.7
Calib
100
15.4
15.4
30.8
Valid
100
15.4
15.4
7.7
Split 2
Total
100
33.7
24.4
Tr
100
40.0
33.3
Inv-Tr
100
33.3
23.3
Calib
100
30.8
23.1
Valid
100
23.1
7.7
Split 3
Total
100
33.7
Tr
100
33.3
Inv-Tr
100
43.3
Calib
100
23.1
Valid
100
23.1
Split 4
Total
100
Tr
100
Inv-Tr
100
Calib
100
Valid
100
The development of QSAR models based on SMILES notation uses the equation of the optimal descriptor of correlation weights (Eq.1).
Following the calculation of all CWs, the linear regression technique was then employed to develop QSAR models, as indicated in equation (Eq.2).
In order to optimize the QSAR modeling, based on the Monte Carlo method, two different target functions, namely TF1 (without the IIC) and TF2 (with value WIIC = 0.3) were used to generate the models. TF1 was calculated using the equilibrium of the correlation method (according to equation (Eq.3)), and the Index of Ideality of Correlation (IIC) (Liman et al., 2022; Toropov et al., 2019; Toropova and Toropov, 2017) was combined with TF1 to formulate the modified target function TF2 (Eq.4).
The IIC, given in equation (Eq.5), was proposed as a measure for evaluating the predictive ability of the developed QSAR models. In particular, it improves the precision of the model, which is determined by the determination coefficient (R2) and the mean absolute error (MAE). The WIIC coefficient, which represents the IIC weight, can change the extent of the influence of the IIC on the Monte Carlo optimization process. The optimal value of WIIC is influenced by two aspects: the diversity of the molecules and the characteristics of the endpoint.
Where;
Δk measures the quality prediction of the kth molecule.
To construct robust QSAR models, the optimal threshold (T*) and the number of epochs (N*) had to be determined by evaluating the most favorable statistical metrics for the calibration set. In the search for the ideal T* and N*, ranges from 1 to 10 were used for the threshold and 1 to 30 for the number of epochs in the optimization process, using three optimization probes. For this study, the WIIC was set to 0 for TF1 and adjusted to 0.3 for TF2.
2.3 GA-MLR QSAR model
The selection of the most relevant descriptors, from the entire computed ones, is a crucial step in QSAR modeling. This selection was achieved by stepwise MLR technique via XLSTAT (Addinsoft, 2020). A total of 832 molecular descriptors were filtered out to get a final selection of five relevant molecular descriptors. Using these descriptors, the MLR method with the ordinary least squares (OLS) algorithm, implemented in the QSARINS software (Addinsoft, 2020; Gramatica et al., 2013) was applied to establish a linear correlation between the pIC50 endpoints of the molecules and their corresponding molecular descriptors. The dataset was divided into a training set with 60 molecules and a test set consisting of 26 molecules, with a distribution ratio of 70 % and 30 % accordingly. The GA-MLR models were developed using the following parameters: all subsets up to a maximum of 5, a maximum generation of 10 000, and a mutation probability of 0.05. The remaining settings were configured with their default values.
2.4 QSAR model validation
The validation process is a crucial stage in QSAR establishment to assess the accuracy of the model and to make reliable predictions about the activity of new molecules. This process is considered the most important aspect in testing the robustness, predictability, and reliability of a QSAR model. Four steps are typically used to validate the generated model, including (a) internal validation or cross-validation using the training set, (b) Y-randomization, (c) independent validation using the test set, and (d) applicability domain (AD) evaluation. The validation procedures and criteria for the CORAL-based QSAR and GA-MLR QSAR models are well discussed in our earlier works (Gramatica et al., 2014; Hdoufane et al., 2019, 2022; Oubahmane et al., 2023; Toropova and Toropov, 2014).
2.5 EBOV-GP structure preparation
The experimental 3D structure of EBOV-GP (PDB ID: 5JQ7) has some missing residues that are essential for its functional mechanism in three regions (LYS191-SER210, THR284-GLY286 and LYS294-ARG302). To fill these missing residues and facilitate subsequent molecular docking and MD simulations, we used the SWISS-MODEL web server for homology modeling to reconstruct the full structure of EBOV-GP (Schwede et al., 2003). The server applies a rigorous protocol that includes the use of BLAST and HHblits to find structural templates from its extensive library, the SWISS-MODEL Template Library (SMTL), which is sourced from the PDB. This ensures that high quality templates are provided, as determined by the Global Model Quality Estimate (GMQE) and the Quaternary Structure Quality Estimate (QSQE). After selecting the most suitable template, which in this case was the same PDB ID due to its high similarity to the target structure, the incomplete EBOV-GP structure was aligned with the template. Subsequently, the missing loop regions were carefully filled in using OpenStructure and the ProMod3 modeling algorithm (Studer et al., 2021). The accuracy and reliability of the final EBOV-GP model was checked using the Qualitative Model Energy ANalysis (QMEAN) assessment function to ensure that the final model was of high quality and suitable for the intended advanced computational analyses (Benkert et al., 2011).
2.6 Molecular docking
Molecular docking analysis was performed to investigate the ligand–protein interactions of the designed compounds and the reference lead compound (78) within the binding site of the modeled EBOV-GP protein. Prior to the docking process, the co-crystallized ligand (Toremifene: TOR) was used to generate the grid-box coordinates of the binding site of the EBOV-GP protein. The grid box coordinates are as follows: xyz coordinates of −45, 16.75, −9, and grid box dimensions of 24 Å3. The protein was then prepared by removing water molecules and the co-crystallized ligand and adding polar hydrogens and Gasteiger charges using AutodockTools (Morris et al., 2009). The protein and compounds were saved in PDBQT format using the same software. Molecular docking analysis were performed using AutoDock Vina software (Trott and Olson, 2010) with exhaustiveness of 20. The docking results were analyzed by the Discovery Studio visualizer and Pymol softwares (BIOVIA Discovery Studio Visualizer, n.d.; Yuan et al., 2017).
2.7 Molecular dynamics simulations
The apo form of the EBOV-GP protein and the designed compounds complexed with the EBOV-GP protein were subjected to all-atom molecular dynamic simulations using GROMACS 2021.3 software (Van Der Spoel et al., 2005). Prior to the MD simulations, the CHARMM-GUI web server (Jo et al., 2008) was utilized to generate initial input parameters, implementing the CHARMM36 force field (Huang et al., 2017). Each complex was solvated in TIP3P water within a rectangular grid box (Jorgensen et al., 1983) and counterions (Na+, Cl-) were added to maintain a 0.15M salt concentration through Monte Carlo ion displacement. The size and shape of the systems were considered when the periodic boundary conditions (PBC) were imposed. Non-bound interactions were addressed using Lennard-Jones and Coulomb potentials with a cut-off distance of 12 Å, and the Verlet cut-off strategy was used to buffer the neighbor search list. The Particle-Mesh Ewald (PME) method (Darden et al., 1993) was used to address the long-range electrostatic interactions. The novel Linear Constraint Solver (LINCS) algorithm (Hess et al., 1997) constrained all covalent bonds, including hydrogen atoms. Energy minimization of each system was carried out using the steepest descent algorithm with a maximum of 50,000 steps and a maximum force of 10.0 KJ/mol. The systems were equilibrated using a two-step process: first, a canonical (NVT) ensemble was employed to equilibrate the temperature at 303 K for 500 ps, followed by an isothermal-isobaric (NPT) ensemble for another 500 ps to equilibrate the pressure, both set at 303 K and 1.01325 bar respectively. The Nose-Hoover thermostat (Hess et al., 1997; Nosé, 1984) and Parrinello-Rahman barostat (Parrinello and Rahman, 1981) were used to regulate the temperature and pressure respectively.
The production run was conducted for 100 ns to analyze the structural stability of the designed molecules within the binding site of EBOV-GP protein. The structural stability of the designed molecules within the EBOV-GP was assessed using the root mean square deviation (RMSD), the radius of gyration (Rg), the root mean square flexibility (RMSF), and the solvent accessibility area (SASA) based on the dynamics trajectory results. The data obtained were visualized using Xmgrace software (GRACE - GRaphing, Advanced Computation and Exploration of data, (n.d.). https://plasma-gate.weizmann.ac.il/Grace/ (accessed March 4, 2024).
2.8 Drug likeness and ADMET prediction
The evaluation of ADMET has a great importance in the initial phases of pharmaceutical exploration. It is expected that a therapeutic drug of better quality would demonstrate exceptional activity at the target receptor, as well as better ADMET profile at therapeutic dosage. Therefore, it is fundamental to assess the pharmacokinetic properties of hit compounds to minimize the risk of subsequent medication failure. The qualities related to drug-likeness are important to understand the distribution of a molecule in an organism, which in turn affects its pharmacological effectiveness. ADMET predictions for the designed compounds were performed via the AdmetLab2, ProTox-II, and Osiris Property Explorer servers (Xiong et al., 2021; Banerjee et al., 2018; Property explorer, 2024).
3 Results and discussion
3.1 CORAL-based QSAR model
A total of eight QSAR models were created by performing four random splits. The two different target functions, namely TF1 and TF2, were used to generate these models. The obtained statistical parameters from the analysis of these QSAR models suggest that the inclusion of WIIC = 0.3 significantly increases the influence of IIC in the Monte Carlo optimization process. Table 2 presents the statistical parameters for all splits, indicating the robustness and validity of the models according to the criteria established by Tropsha et al. (Golbraikh and Tropsha, 2002) and Ojha et al. (Ojha et al., 2011). Fig. 1 displays the correlation between the experimental and predicted pIC50 values across the four splits. The QSAR model derived from split 3 was chosen as the optimal model based on its superior validation metrics (
= 0.9129;
). The mathematical model equation for split 3 is given below (Eq.9):
Split
TF
Set
n
R2
CCC
IIC
Q2
Q2F1
Q2F2
Q2F3
S
MAE
F
Avg Rm2
Δ Rm2
Equation
1
TF1
Tr
30
0.9472
0.9729
0.7442
0.9395
0.106
0.085
502
pIC50 = 3.853 (±0.015) + 0.129 (±0.001)
DCW(3,30)
Inv-Tr
30
0.9289
0.7204
0.5278
0.9211
0.328
0.261
366
Calib
13
0.8191
0.8338
0.7272
0.7058
0.7926
0.7414
0.7161
0.275
0.233
50
0.5578
0.2363
Valid
13
0.7919
0.8259
0.3625
0.7251
0.250
0.5721
0.2334
TF2
Tr
30
0.8196
0.9009
0.6923
0.7922
0.197
0.148
127
pIC50 = 3.498 (±0.040) +
0.110 (±0.002)
DCW(3,30)
Inv-Tr
30
0.8209
0.8609
0.4263
0.7952
0.246
0.188
128
Calib
13
0.8623
0.8863
0.9285
0.7672
0.8538
0.8176
0.7998
0.231
0.181
69
0.6406
0.1787
Val
13
0.8807
0.9333
0.7306
0.8463
0.158
0.8260
0.0745
2
TF1
Tr
30
0.9684
0.9839
0.4920
0.9628
0.082
0.051
857
pIC50 = 3.210 (±0.018) + 0.142 (±0.001)
DCW(1,30)
Inv-Tr
30
0.9476
0.7806
0.4236
0.9433
0.298
0.237
506
Calib
13
0.6199
0.7704
0.5066
0.4494
0.6947
0.6193
0.5820
0.333
0.258
18
0.4752
0.2594
Valid
13
0.5718
0.7471
0.6509
0.2983
0.311
0.4290
0.1397
TF2
Tr
30
0.8484
0.9180
0.6141
0.8243
0.180
0.139
157
pIC50 = 4.152 (±0.027) + 0.079 (±0.001)
DCW(3,30)
Inv-Tr
30
0.8352
0.8236
0.3810
0.8190
0.267
0.206
142
Calib
13
0.8731
0.8782
0.9344
0.7877
0.8524
0.8159
0.7978
0.232
0.188
76
0.5765
0.2120
Valid
13
0.8115
0.8836
0.8700
0.7540
0.207
0.6947
0.1769
3
TF1
Tr
30
0.9223
0.9596
0.7344
0.9099
0.153
0.102
332
pIC50 = 2.322 (±0.038) + 0.173 (±0.002)
DCW(4,30)
Inv-Tr
30
0.9219
0.9432
0.4511
0.9130
0.167
0.127
330
Calib
13
0.5525
0.6907
0.2647
0.3255
0.1773
0.1755
0.3708
0.428
0.309
14
0.3950
0.2702
Valid
13
0.7982
0.8892
0.7386
0.7127
0.252
0.7116
0.1171
TF2
Tr
30
0.8618
0.9258
0.8123
0.8287
0.204
0.157
175
pIC50 = 2.043 (±0.078) + 0.061 (±0.001)
DCW(1,30)
Inv-Tr
30
0.7631
0.8716
0.7205
0.7376
0.261
0.203
90
Calib
13
0.8369
0.9116
0.9148
0.7713
0.8296
0.8293
0.8697
0.195
0.143
56
0.8369
0.9116
Valid
13
0.9129
0.9512
0.5608
0.8848
0.147
0.8288
0.0885
4
TF1
Tr
30
0.9103
0.9530
0.7296
0.9008
0.149
0.099
284
pIC50 = 3.214 (±0.020) + 0.158 (±0.001)
DCW(6,30)
Inv-Tr
30
0.9353
0.9664
0.8614
0.9267
0.154
0.113
405
Calib
13
0.3941
0.6212
0.5782
0.1323
0.1718
0.1364
0.3877
0.435
0.335
7
0.2448
0.1002
Valid
13
0.6518
0.7316
0.5470
0.3660
0.405
0.3837
0.3548
TF2
Tr
30
0.7843
0.8791
0.3795
0.7608
0.231
0.163
102
pIC50 = 2.543 (±0.044) + 0.089 (±0.001)
DCW(1,30)
Inv-Tr
30
0.8346
0.8992
0.5306
0.8139
0.260
0.201
141
Calib
13
0.7857
0.8661
0.8863
0.6641
0.7449
0.7340
0.8114
0.241
0.176
40
0.6958
0.0742
Valid
13
0.7846
0.8787
0.3273
0.7097
0.201
0.6899
0.1885
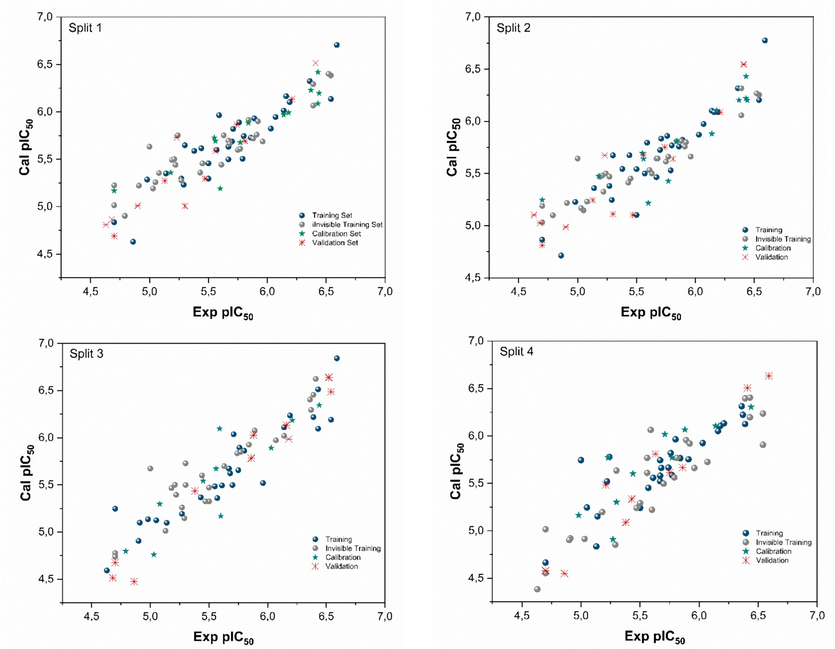
Experimental vs. calculated pIC50 values for the four Splits.
3.2 GA-MLR QSAR model
The GA-MLR model was built based on five selected descriptors from a comprehensive set of descriptors, namely IC2, SpMin7_Bh(s), P_VSA_Charge_7, CATS2D_05_DD, and T(O..O). The description of each descriptor was given in Table S5. These descriptors were found to significantly explain the structure activity relationship (SAR) between the pIC50 activity and the studied compounds and were subsequently used to construct the QSAR model. The generated model using the GA-MLR approach (Eq.10) and its corresponding statistical parameters are shown below:
NTr = 60, R2 Tr = 0.8490, Q2LOO = 0.8148
Next = 26, R2ext = 0.8661, MAEext = 0.2135.
Q2F1 = 0.8681, Q2F2 = 0.8599, Q2F3 = 0.8840, CCCext = 0.9229, F = 77.6036, s = 0.3138.
NTr and Next indicate the total number of samples used for the training and the test sets, respectively. CCCext is the external concordance correlation coefficient, while F refers to F-statistic. Additionally, the standard deviation is represented by “s” and the external validation criteria are expressed through , and (Chirico and Gramatica, 2012; Hess et al., 1997; Nosé, 1984).
The evaluation of the indicated parameters in the established GA-MLR model satisfies the validation requirements outlined in the OECD guidelines. Moreover, the scatter plot shown in Fig. 2a displays a good correlation between the experimental and predicted pIC50 endpoints. To confirm the reliability of the built model, the AD was evaluated using the leverage method, and presented through the Williams plot in Fig. 2b. The graphic includes dashed lines to indicate the cutoff value of ±3 s.d., and the warning line for the outliers (h*) was found to be 0.29. The findings from the Williams plot provide definitive evidence that the AD covers all compounds, except for compound 65. This observation can be attributed to the low activity of compound 65, as evidenced by its high value (IC50 = 20 μM), which is 69-fold higher than that of the lead compound (IC50 = 0.29 μM).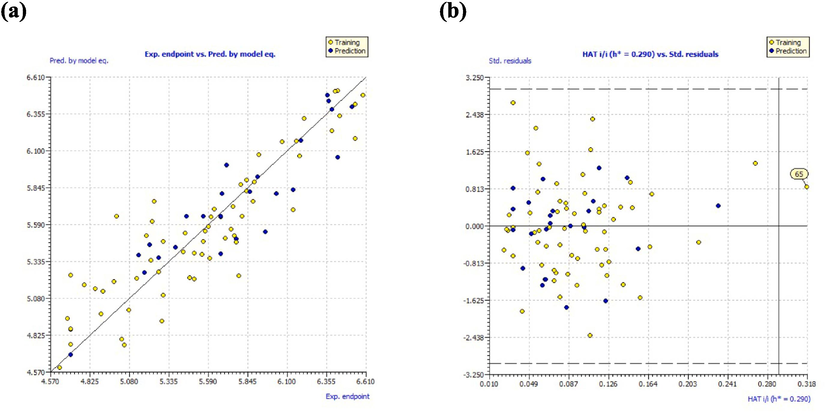
(a) Experimental vs. predicted pIC50 values; (b) William’s plot.
3.3 Mechanistic interpretation
The integration of mechanistic interpretations is a pivotal component within the framework of the OECD. These interpretations are helpful for identifying and evaluating the chemical features that contribute to the increase or decrease of a defined endpoint value. To provide a comprehensive understanding of the mechanistic interpretation of the CORAL model, numerous iterations of Monte Carlo optimization were used. The chemical features, obtained from SMILES descriptors, display positive (CWs) and recognize factors that support an increase in the pIC50 value. In contrast, negative CWs identify factors that encourage a reduction in the pIC50 value. While SMILES qualities that include both positive and negative CWs values remain undefined. Table S6 presents a complete summary of the main factors that contribute to the increase or decrease of pIC50 values, associated with their corresponding correlation weights (CWs). These results are derived from the three separate runs of the established QSAR model for split 3.
Based on the SAR study of Sakurai et al. (Sakurai et al., 2018) and on the insights obtained from Table S6, the factors increasing the inhibitory activity were incorporated into the lead compound 78 along with compounds 80 and 83. These two molecules exhibit low cytotoxicity and high pIC50 values and are closely aligned with the lead compound. While the promoters associated with a decrease were omitted. The influential promoters were systematically investigated at three distinct sites (R1, R2, R3) within the lead compound to strategically design novel inhibitors targeting the EBOV-GP, as depicted in Fig. 3. Therefore, 26 novel molecules were designed (Table S7). Table S8 gives the calculated activities of the newly designed molecules by the two QSAR models. The CORAL model predicted a pIC50 range [7.1239–7.9595] for all the designed compounds. The GA-MLR QSAR model confirmed the inhibitory potential of the newly developed compounds.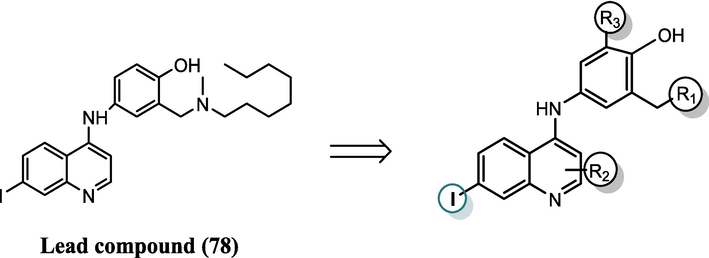
Scaffold chemical structure derived from the lead compound with highlighted substitution sites (R1, R2, R3) maintaining crucial Iodine for inhibition activity.
3.4 Modeling missing residues as loop regions
The loops are involved in many biological functions and constitute key components of protein structure. The loop modeling of the EBOV-GP (PDB ID: 5JQ7), successfully filled the missing residues in three critical regions (LYS191-SER210, THR284-GLY286 and LYS294-ARG302). These regions are essential for the functional mechanism of the EBOV-GP as depicted in Fig. 4. The process involved aligning the incomplete structure of EBOV-GP with its corresponding higher-resolution template, and the missing residues were completed using the ProMod3 modeling algorithm. The accuracy and reliability of the reconstructed EBOV-GP was confirmed by QMEAN and GMQE values of 0.69 and 0.85, respectively. Importantly, the loop model of the missing residues in region 1 is positioned and oriented upwards to allow the ligands to access the binding site. This structural arrangement is in agreement with the findings of previous research (Ren et al., 2018), suggesting that the modeled loop does not obstruct the binding pocket and thus facilitates potential inhibitor interactions. These findings indicate that the completed EBOV-GP structure is well constructed, confirming its suitability for subsequent molecular docking and MD simulations.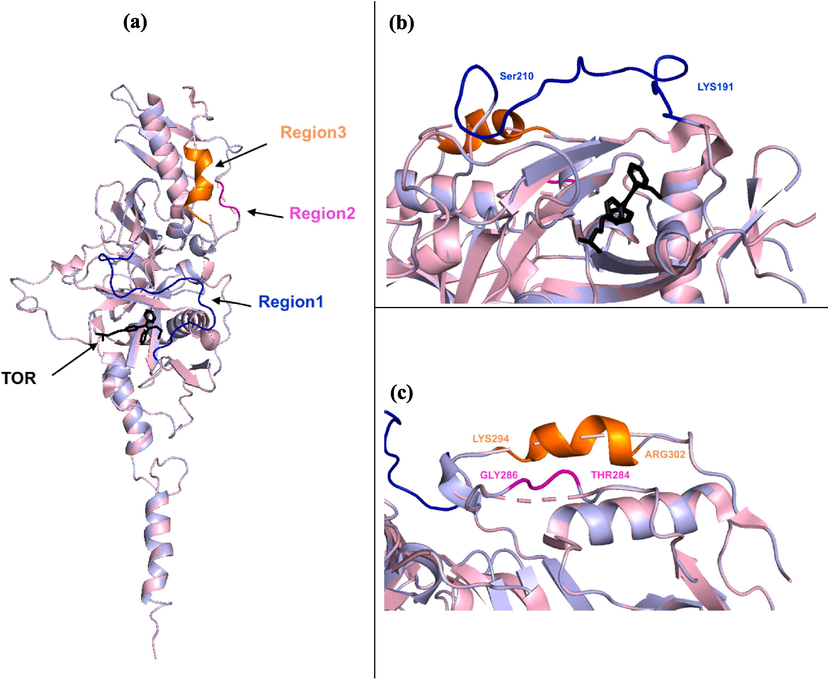
(a) The alignment of the uncompleted structure of EBOV-GP (PDB ID: 5JQ7, light pink) and the modeled EBOV-GP (light blue) containing the co-crystalized ligand (TOR). The modeled structure of the missing residues in the three regions: (b) Region 1 (LYS191-SER210, blue), (c) Region 2 (THR284-GLY286, magenta) and Region 3 (LYS294-ARG302, orange).
3.5 Molecular docking
To validate the efficacy of our design approach, we employed molecular docking methodology to explore the ligand–protein interactions involving the newly designed compounds, the reference lead compound, and the co-crystallized ligand (TOR) with the modelled EBOV-GP protein. A crucial step in this validation process was the re-docking analysis of the TOR ligand to assess the accuracy of the docking protocol. The RMSD between the x-ray and predicted poses of TOR was determined to be 0.664 Å (Figure S1), confirming the appropriateness of the docking protocol in reproducing native poses within the acceptable range of less than 2 Å. Subsequently, a comprehensive docking study was conducted for all designed compounds against the modelled EBOV-GP, and the resulting binding affinity values are detailed in Table S8. Notably, ten designed molecules exhibited higher binding affinity values compared to both the co-crystallized ligand (TOR: −8.4 Kcal/mol) and the lead compound (78: −7.4 Kcal/mol). The chemical structures of the top five newly designed compounds (D1-D5) ranked based on their binding affinities are presented in Fig. 5. While, Fig. 6 illustrates their 2D and 3D representations within the binding site of EBOV-GP. Their binding affinity values and ligand–protein interactions details are provided in Table 3 and Table S9, respectively. Analysis of the docking results elucidates that compound D1 exhibited a high binding affinity (−9.9 kcal.mol−1), forming stable interactions with the key residues of the active site of EBOV-GP. The interactions formed by compound D1 were three hydrogen bonds (with GLY B:45, TYR B:16, and VAL A:176), one Pi-Sulfur interaction with MET B:47, and three Alkyl and Pi-Alkyl interaction involving (ALA A:74), (LEU B:14), and (VAL A:39) residues. Compound D2 exhibited stability through three hydrogen bonds (two conventional with (PRO A: 160) and (TYR B:16), one carbon hydrogen bound with (LEU B:46), one pi–sulfur interaction with (MET B:47), one pi-pi Stacked interaction with (TYP B: 16), and seven alkyl and pi-alkyl interactions. Similarly, compound D3 formed interactions with (ARG A:37) and (VAL A:176) via conventional hydrogen bonds and with (GLY B:45) via carbon hydrogen bond. Furthermore, it engages in Pi-Donor hydrogen bonds with (TYR B:16), contributing to its structural stability. Additionally, D3 exhibits hydrophobic interactions with (ALA A:74), (LYS A:163), (LYS A:164), (VAL A:39), and (LEU B:14), further enhancing its binding affinity and molecular interactions. Compound D4 was stabilized by the formation of one carbon hydrogen bond with (LYS A:163) and eight hydrophobic interactions with (ALA A:74), (VAL A:39), (LEU A:41), (LEU B:14), (MET B:47) and (PHE A:167). Compound D5 formed one conventional hydrogen bond with (THR B:18) and seven hydrophobic interactions with (ALA A:74), (LEU B:14), (LYS A:163), (MET B:47), (VAL A:39), (THR B: 18), and (TYR B:16).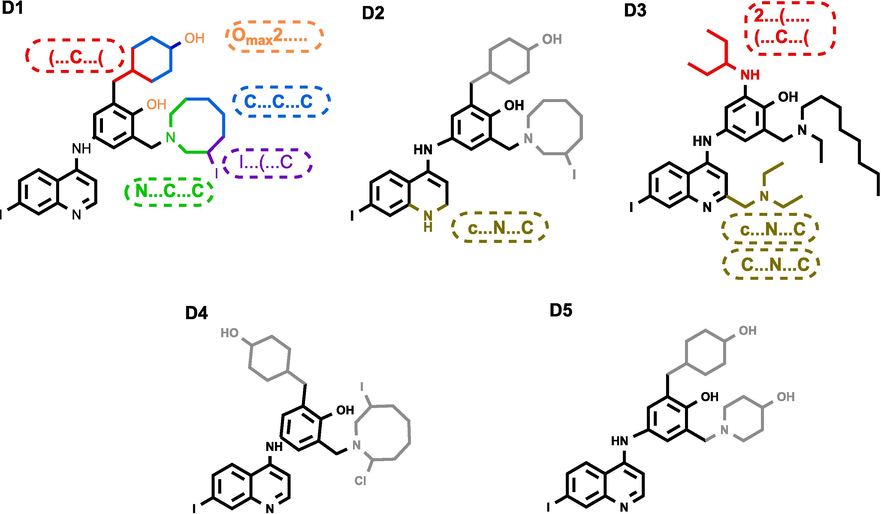
Chemical structures of the top five newly designed compounds.
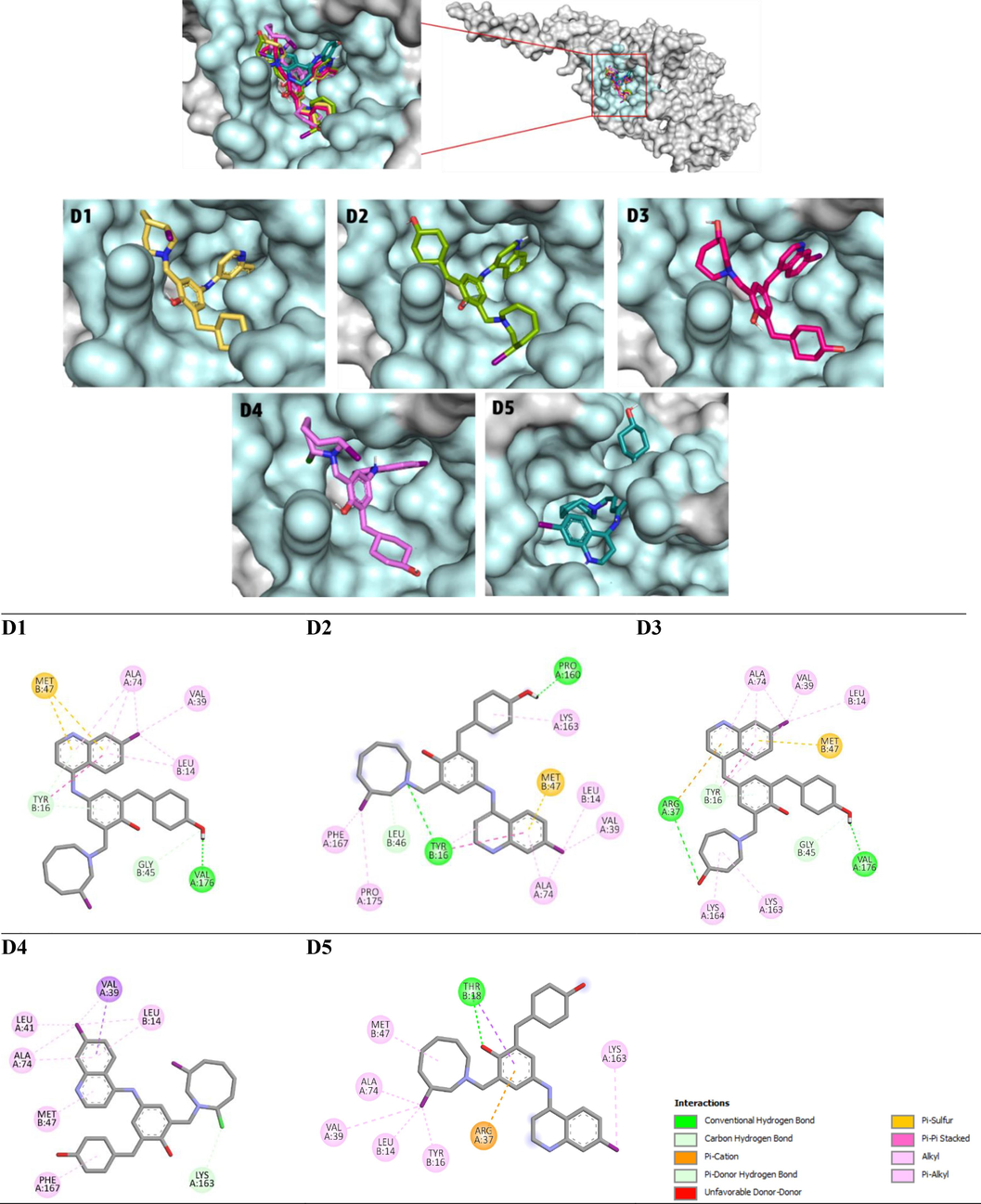
2D and 3D representation of the interactions of ligands D1-D5 within EBOV-GP binding pocket.
Ligand
CORAL pIC50
GA-MLR pIC50
Docking Score (kcal/mol)
Lead compound
6.84
6.77
−7.4
D1
7.63
7.04
−9.9
D2
7.12
7.07
−9.8
D3
7.83
7.10
−9.6
D4
7.68
7.04
−9.5
D5
7.26
7.55
−9.4
3.6 Molecular dynamics simulations
Molecular dynamics simulations were employed to assess the stability of interactions between proteins and ligands, as well as to quantify the binding affinity of ligands. MD simulations were run for the top five compounds (D1-D5). The study included the computation of diverse parameters (Table 4) such as RMSD, RMSF, Rg, and SASA metrics. The best three stable systems (D2, D3, and D5) that showed good stability with Apo form of the EBOV-GP and the EBOV-GP-lead compound complex are presented in the Table 4.
System
RMSD (nm)
RMSF (nm)
Rg (nm)
SASA (nm2)
Apo
0.399 ± 0.075
0.145 ± 0.117
2.268 ± 0.021
198.266 ± 31.119
Lead compound
0.532 ± 0.111
0.164 ± 0.149
2.293 ± 0.013
205.813 ± 32.156
D2
0.374 ± 0.081
0.181 ± 0.167
2.312 ± 0.018
206.417 ± 32.365
D3
0.416 ± 0.075
0.162 ± 0.146
2.291 ± 0.015
203.612 ± 31.916
D5
0.414 ± 0.078
0.178 ± 0.173
2.277 ± 0.014
204.020 ± 32.163
The RMSD serves as a measure to quantify the degree of deviation in a protein's backbone from its initial to final conformation during a simulation, providing valuable insights into its structural stability. A protein with consistent stability will exhibit minimal backbone deviation, while a less stable protein will show greater variation. Analysis of the MD results, as depicted in both the Fig. 7 and Table 4, reveals variations in the RMSD values for the Apo form of EBOV-GP and its complexes with different compounds. Interpreting these findings, the compound with the lowest average RMSD, namely compound D2, suggests a higher degree of stability compared to the complex protein-lead compound. Similarly, compound D3 and compound D5 also exhibit greater stability, as evidenced by their lower RMSD values. This implies that these compounds potentially contribute to maintaining the structural integrity of the protein during the simulation.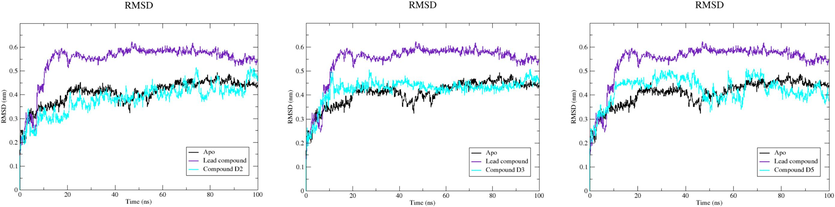
Time-dependent RMSD of c-α backbone of EBOV-GP Apo, lead compound, compound D2, compound D3, and compound D5.
To further investigate the ligand stability within the binding site of EBOV-GP, the RMSD values of the ligands were monitored (Fig. 8). Monitoring these trajectories would provide valuable information about the conformation and orientation of the simulated ligands within the binding site of EBOV-GP throughout the simulation period. The RMSD plot shows that compound D3 exhibits the most consistent and stable behavior, showing the least variation throughout the 100 ns MD simulation. In contrast, compounds D2 and D5 show higher RMSD values than the lead compound, indicating greater mobility within the binding site before reaching a semblance of stability later at the end of the simulation.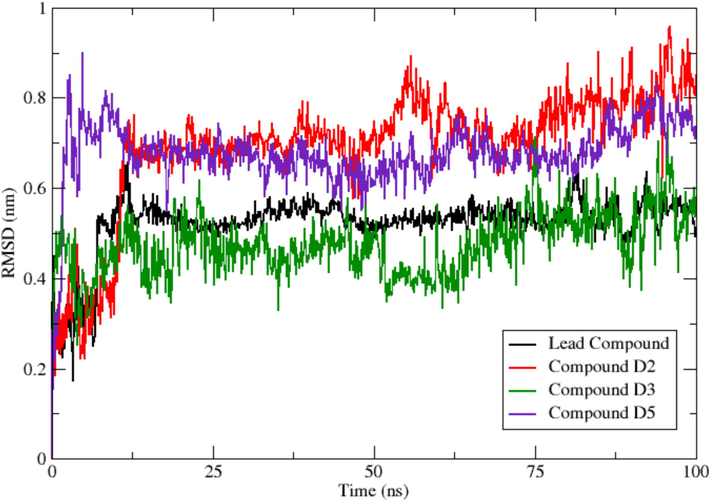
RMSD of the ligands (reference compound 78 and designed compounds D2, D3 and D4).
Analyzing the RMSF is essential for identifying flexible areas within protein–ligand complexes. Higher RMSF values in regions with lower structural organization, such as loops and turns, contrast with lower fluctuations in more ordered segments like alpha helices and beta sheets. It is crucial to note that the RMSF assessment covered C-alpha atoms across the entire simulation for both the complexes and the EBOV-GP Apo form. In evaluating the flexibility of the top five compounds binding to the EBOV-GP, RMSF values from the lead compound complex served as a reference baseline. Remarkably, compound D3 demonstrated the lowest RMSF (Table 4 and Fig. 9), corroborating the findings observed in the RMSD analysis. This underscores the consistent behavior of compound D3, not only in terms of structural stability (as indicated by the RMSD) but also in terms of exhibiting minimal fluctuations in flexible regions, further highlighting its potential as a stable and structurally sound ligand for EBOV-GP.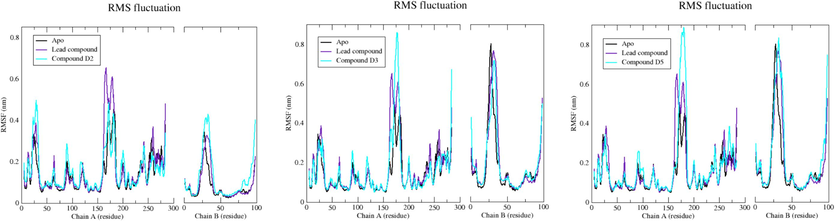
Plot of RMSF for c-α atoms of EBOV-GP Apo, lead compound, compound D2, compound D3, and compound D5.
Radius of gyration, serving as an indicator of protein structure compactness, is employed to evaluate changes induced by ligand binding through a comparison of Rg values before and after binding. A lower radius of gyration signifies a more compact protein structure. Fig. 10 presents the Rg plot for the Apo state, lead compound, compound D2, compound D3, and compound D5 unveiled average Rg values of 2.268, 2.293, 2.312, 2.291, and 2.277 nm, respectively. Particularly noteworthy is that compound D5 exhibited Rg values slightly lower than those of the EBOV-GP bound to the lead compound. This suggests that compound D5 may lead to a protein structure that is more densely packed compared to the lead compound, implying potential implications for the binding process and overall stability of the protein–ligand complex.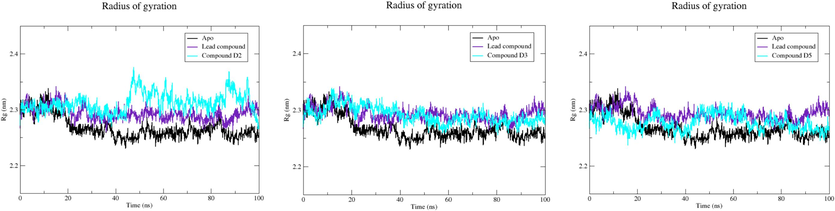
Plot of Rg vs. time for EBOV-GP Apo, lead compound, compound D2, compound D3, and compound D5.
The SASA parameter refers to the surface area of a protein that engages with solvent molecules, measuring the fraction of a protein's surface available to such molecules.
The SASA values for the EBOV-GP Apo state, lead compound, compound D2, compound D3, and compound D5 are 198.266, 205.813, 206.417, 203.612, and 204.020 nm2, respectively. The Apo structure inherently represents solvent accessibility, and the marginal increase in SASA with the lead compound indicates alterations upon ligand binding. Notably, compound D2, as showed in Fig. 11, exhibits higher SASA values, suggesting modifications in the protein's surface accessibility, likely attributed to the binding of distinct ligands. These SASA variations offer insights into the protein's dynamic nature, highlighting structural adaptations and potential functional implications associated with specific ligand interactions.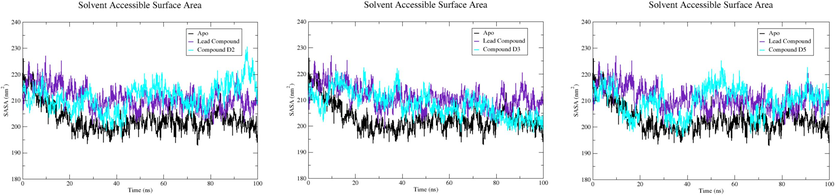
The SASA profile of EBOV-GP Apo, lead compound, compound D2, compound D3, and compound D5.
3.7 ADMET
In drug discovery and development, the ADMET emerges as a pivotal phase and plays a vital role in circumventing potential complications during clinical trials. A successful drug candidate must not only exhibit efficacy against the targeted therapeutic endpoint but also demonstrate favorable drug-like qualities and pharmacokinetic characteristics. In the present study, in silico ADMET analysis results of our newly compounds are summarized in Table 5. All the five compounds exhibit a reassuring absence of risks of tumorigenicity, irritancy, mutagenicity, reproductive effects, carcinogenicity, or hepatotoxicity, underscoring their potential safety and efficacy in the drug development pipeline. The human intestinal absorption (HIA) of an oral drug is a critical determinant of its apparent efficacy. The scale of the five compounds ranges from excellent (0–0.3) to poor (0.7–1.0), as it is represented in red in Table 5. Caco-2 cell permeability, expressed as a log cm/s value, is a critical index for evaluating the potential in vivo drug permeability of a compound, with a predicted value >-5.15 log cm/s indicating adequate permeability across intestinal cell membranes via various transport processes. With the exception of compounds D3 and D5, all compounds described have shown good predicted permeability (Barrett et al., 2022). In addition, solubility is a critical factor that influences the ADME of a drug candidate. The solubility is often expressed as the logarithm of its solubility in water (logS). A more negative logS value indicates lower solubility, while a less negative (or more positive) logS value indicates higher solubility. Compared to the lead compound, all the new compounds have higher solubility than the starting lead compound (−5.671), as indicated by their less negative logS values. This suggests an improvement in solubility, which is beneficial for drug development since higher solubility typically correlates with better bioavailability. D5, with the lowest permeability, had the highest solubility value (−4.409) as the permeability is inversely correlated with the solubility (Barrett et al., 2022). For drugs targeting the central nervous system (CNS), traversing the blood–brain barrier (BBB) is imperative to reach their molecular targets, while drugs with peripheral targets may require minimal or no BBB penetration to avoid CNS side effects. The evaluation criteria for BBB penetration are categorized as excellent (0–0.3), medium (0.3–0.7), and poor (0.7–1.0). As detailed in the table below all the proposed structures had excellent to medium values of BBB. Another essential parameter in ADMET analysis is the half-life of a drug (T1/2), a composite measure of both clearance and volume of distribution. It is a better assessment with reliable estimates of these two properties. Excellent categorization was attributed to all molecules as they are in the range 0–0.3. Additionally, these drug-like molecules are easy to synthesis according to their excellent Synthetic Accessibility scores (SA score ≤ 6). This score was estimated based on a combination of fragment contributions and a complexity penalty (Ertl and Schuffenhauer, 2009). Furthermore, neither inhibitory nor substrate interactions with the human cytochrome P450 family CYP450- 2C9 were observed, suggesting a favorable safety profile with minimal impact on drug metabolism mediated by CYP450-2C9, which is a positive characteristic in drug development. Regarding toxicity, all compounds pose no risk except compound D5, which may have an irritant effect. N: No risk.
Cmpd.
Lead compound
D1
D2
D3
D4
D5
Physicochemical and ADME properties
TPSA (Å2)
48.39
68.62
67.76
88.85
58.97
63.66
HBA
4
5
5
6
5
6
HBD
2
3
4
4
3
3
BBB
0.182
0.121
0.343
0.250
0.543
0.085
HIA
0.004
0.010
0.029
0.784
0.028
0.004
Solubility (log(S))
−5.671
−4.969
−4.953
−4.677
−4.694
−4.409
Caco-2 Permeability
−4.730
−5.267
−5.585
−5.105
−5.071
−5.614
CYP2C9 inhibitor/ CYP2C9 substrate
N/N
N/N
N/N
N/N
N/N
N/N
T1/2
0.068
0,02
0,03
0,057
0,031
0,026
Medicinal Chemistry
SA score
2.47
3.557
3.942
3.356
3.936
3.132
Drug score
Reference
0.100
0.200
0.220
0.310
0.050
Toxicity properties
AMES Toxicity
N
N
N
N
N
N
Tumorigenic
N
N
N
N
N
N
Irritant
N
N
N
N
N
Y
Reproductive affective
N
N
N
N
N
N
Carcinogenicity
N
N
N
N
N
N
Hepatotoxicity
N
N
N
N
N
N
4 Conclusion
This study was focused on designing novel and potent inhibitors for the Ebola virus, specifically targeting EBOV-GP, the crucial protein involved in attachment and fusion. Utilizing a series of amodiaquine derivatives, two QSAR models, CORAL and GA-MLR were developed to gain insights into the design of potent inhibitors for EBOV-GP. The first model, the CORAL QSAR model, was employed to identify the key features involved in increasing and decreasing the inhibition activity against EBOV-GP. Using these features, 26 new molecules were designed with better inhibition activity than the lead compound. The second model, a GA-MLR model, served to confirm the predicted inhibition activity of the designed compounds. Computational validation through molecular docking and dynamics simulations confirmed the efficacy of the designed compounds. Before conducting the molecular docking study, homology modeling was used to fill the missing residues in the EBOV-GP structure. The modeled and completed EBOV-GP structure was docked against the designed compounds where three designed compounds (D2, D3, and D5) showed remarkable binding affinity profiles. Furthermore, molecular dynamics simulations corroborated the docking results, revealing their stability when they are complexed with EBOV-GP protein. Moreover, these drug candidates demonstrated non-toxic and acceptable pharmacological properties. The combined computational methods employed in this study facilitated the identification of new drug candidates, paving the way for further evaluation of their efficacy and safety. Further studies may explore in vitro experiments to validate and reinforce our computational modeling results. Nevertheless, the computational methods, showcased in this study, underscore their potential utility in expediting drug discovery and guiding future research endeavors against the Ebola virus.
Funding
This research was supported by Princess Nourah bint Abdulrahman University. Researcher supporting project number (PNURSP2024R342), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
CRediT authorship contribution statement
Nouhaila Ait Lahcen: Writing – original draft, Investigation, Data curation, Conceptualization. Wissal Liman: Writing – original draft, Data curation. Mehdi Oubahmane: Writing – original draft, Methodology, Formal analysis, Conceptualization. Ismail Hdoufane: Writing – review & editing, Formal analysis, Conceptualization. Youssef Habibi: Writing – review & editing, Supervision. Ashwag S. Alanazi: Validation, Resources, Funding acquisition. Mohammed M. Alanazi: Writing – review & editing, Validation, Funding acquisition. Christelle Delaite: Validation, Resources. Mohamed Maatallah: Writing – review & editing, Supervision. Driss Cherqaoui: Writing – review & editing, Visualization, Validation, Supervision.
Acknowledgments
The authors extend their appreciation to Princess Nourah bint Abdulrahman University researcher supporting project number (PNURSP2024R342), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia for supporting this work.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Addinsoft (2020) XLSTAT Statistical and Data Analysis Solution. New York, (n.d). Scientific Research Publishing, (n.d.). https://www.xlstat.com (accessed March 4, 2024).
- Computer aided drug design and its application to the development of potential drugs for neurodegenerative disorders. Curr. Neuropharmacol.. 2018;16:740.
- [CrossRef] [Google Scholar]
- ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res.. 2018;46:W257-W263.
- [CrossRef] [Google Scholar]
- Discovery solubility measurement and assessment of small molecules with drug development in mind. Drug Discov. Today. 2022;27:1315-1325.
- [CrossRef] [Google Scholar]
- Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011;27:343-350.
- [CrossRef] [Google Scholar]
- BIOVIA Discovery Studio Visualizer, (n.d.). https://discover.3ds.com/discovery-studio-visualizer-download (accessed March 4, 2024).
- Real external predictivity of QSAR models. Part 2. New intercomparable thresholds for different validation criteria and the need for scatter plot inspection. J. Chem. Inf. Model.. 2012;52:2044-2058.
- [CrossRef] [Google Scholar]
- Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys.. 1993;98:10089-10092.
- [CrossRef] [Google Scholar]
- In silico-based vaccine design against Ebola virus glycoprotein. Adv. Appl. Bioinforma. Chem.. 2017;10:11-28.
- [CrossRef] [Google Scholar]
- Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J Cheminform. 2009;1:1-11.
- [CrossRef] [Google Scholar]
- N′-phenylacetohydrazide derivatives as potent ebola virus entry inhibitors with an improved pharmacokinetic profile. J. Med. Chem.. 2023;66:5465-5483.
- [CrossRef] [Google Scholar]
- Gaussian 09, Revision A.02, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janes and 56 Authors., (n.d.). https://gaussian.com/ (accessed March 4, 2024).
- GRACE - GRaphing, Advanced Computation and Exploration of data, (n.d.). https://plasma-gate.weizmann.ac.il/Grace/ (accessed March 4, 2024).
- QSARINS: A new software for the development, analysis, and validation of QSAR MLR models. J. Comput. Chem.. 2013;34:2121-2132.
- [CrossRef] [Google Scholar]
- QSARINS-chem: Insubria datasets and new QSAR/QSPR models for environmental pollutants in QSARINS. J. Comput. Chem.. 2014;35:1036-1044.
- [CrossRef] [Google Scholar]
- QSAR and molecular docking studies of indole-based analogs as HIV-1 attachment inhibitors. J. Mol. Struct.. 2019;1193:429-443.
- [CrossRef] [Google Scholar]
- In silico design and analysis of NS4B inhibitors against hepatitis C virus. J. Biomol. Struct. Dyn.. 2022;40:1915-1929.
- [CrossRef] [Google Scholar]
- LINCS: A Linear Constraint Solver for Molecular Simulations. John Wiley & Sons Inc; 1997.
- hong Ma, Y., Hong, X., Wu, F., feng Xu, X., Li, R., Zhong, J., qi Zhou, Y., wen Liu, S., Zhan, J., Xu, W., 2023. Inhibiting the transcription and replication of Ebola viruses by disrupting the nucleoprotein and VP30 protein interaction with small molecules, Acta Pharmacologica Sinica 2023 44:7 44 (2023) 1487–1499. Doi: 10.1038/s41401-023-01055-0.
- CHARMM36: an improved force field for folded and intrinsically disordered proteins. Biophys. J .. 2017;112:175a-a176.
- [CrossRef] [Google Scholar]
- CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem.. 2008;29:1859-1865.
- [CrossRef] [Google Scholar]
- Comparison of simple potential functions for simulating liquid water. J. Chem. Phys.. 1983;79:926-935.
- [CrossRef] [Google Scholar]
- Lee, J.E., Fusco, M.L., Hessell, A.J., Oswald, W.B., Burton, D.R., Saphire, E.O., 2008. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature, 454,7201 454 (2008) 177–182. Doi: 10.1038/nature07082.
- Ebolavirus glycoprotein structure and mechanism of entry. Future Virol. 2009;4:621-635.
- [CrossRef] [Google Scholar]
- Monte Carlo Method and GA-MLR-Based QSAR Modeling of NS5A Inhibitors against the Hepatitis C Virus. Molecules. 2022;27:2729.
- [CrossRef] [Google Scholar]
- Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev.. 2012;64:4-17.
- [CrossRef] [Google Scholar]
- Molecular modelling studies on adamantane-based Ebola virus GP-1 inhibitors using docking, pharmacophore and 3D-QSAR. SAR QSAR Environ. Res.. 2019;30:161-180.
- [CrossRef] [Google Scholar]
- Recent advances in computer-aided drug design as applied to anti-influenza drug discovery. Curr. Top. Med. Chem.. 2014;14:1875-1889.
- [CrossRef] [Google Scholar]
- Alvascience: A New Software Suite for the QSAR Workflow Applied to the Blood-Brain Barrier Permeability. Int. J. Mol. Sci.. 2022;23:12882.
- [CrossRef] [Google Scholar]
- AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem.. 2009;30:2785.
- [CrossRef] [Google Scholar]
- A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys.. 1984;81:511-519.
- [CrossRef] [Google Scholar]
- Further exploring rm2 metrics for validation of QSPR models. Chemom. Intel. Lab. Syst.. 2011;107:194-205.
- [CrossRef] [Google Scholar]
- Design of potent inhibitors targeting the main protease of SARS-CoV-2 Using QSAR modeling, molecular docking, and molecular dynamics simulations. Pharmaceuticals. 2023;16:608.
- [CrossRef] [Google Scholar]
- Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys.. 1981;52:7182-7190.
- [CrossRef] [Google Scholar]
- Peng, W., Rayaprolu, V., Parvate, A.D., Pronker, M.F., Hui, S., Parekh, D., Shaffer, K., Yu, X., Saphire, E.O., Snijder, J., 2022. Glycan shield of the ebolavirus envelope glycoprotein GP, Communications Biology 2022 5:1 5 (2022) 1–11. Doi: 10.1038/s42003-022-03767-1.
- PerkinElmerInformatics, Inc, ChemDraw, (2021), One month ChemOffice+ Cloud trial license.
- Property explorer, https://www.organicchemistry.org/prog/peo/ (accessed February 29, 2024).
- Target identification and mode of action of four chemically divergent drugs against ebolavirus infection. J. Med. Chem.. 2018;61:724-733.
- [CrossRef] [Google Scholar]
- Novel amodiaquine derivatives potently inhibit Ebola virus infection. Antiviral Res.. 2018;160:175-182.
- [CrossRef] [Google Scholar]
- SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res.. 2003;31:3381-3385.
- [CrossRef] [Google Scholar]
- ProMod3—A versatile homology modelling toolbox. PLoS Comput. Biol.. 2021;17:e1008667.
- [Google Scholar]
- The study of the index of ideality of correlation as a new criterion of predictive potential of QSPR/QSAR-models. Sci. Total Environ.. 2019;659:1387-1394.
- [CrossRef] [Google Scholar]
- CORAL software: Prediction of carcinogenicity of drugs by means of the Monte Carlo method. Eur. J. Pharm. Sci.. 2014;52:21-25.
- [CrossRef] [Google Scholar]
- The index of ideality of correlation: A criterion of predictability of QSAR models for skin permeability? Sci. Total Environ.. 2017;586:466-472.
- [CrossRef] [Google Scholar]
- AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem.. 2010;31:455-461.
- [CrossRef] [Google Scholar]
- Volchkov, V., Klenk, H.D., 2018. Proteolytic Processing of Filovirus Glycoproteins, Activation of Viruses by Host Proteases (2018) 99. Doi: 10.1007/978-3-319-75474-1_5.
- The nonstructural small glycoprotein sGP of ebola virus is secreted as an antiparallel-orientated homodimer. Virology. 1998;250:408-414.
- [CrossRef] [Google Scholar]
- ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res.. 2021;49:W5-W14.
- [CrossRef] [Google Scholar]
- Using PyMOL as a platform for computational drug design. Wiley Interdiscip. Rev.: Comput. Mol. Sci.. 2017;7:e1298.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2024.105870.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary Data 1
Supplementary Data 1




