

Translate this page into:
Establishment of a multiplex-PCR detection method and development of a detection kit for five animal-derived components in edible meat
⁎Corresponding author. yuanguangxin2007@163.com (Guangxin Yuan)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
A multiplex PCR detection kit was developed. Preparation of DNA cloning solutions of five meat positive controls by molecular cloning technique.
Abstract
A multiplex polymerase chain reaction (PCR) detection method for the simultaneous detection of animal-derived components from deer, cow, sheep, pig and horse in edible meat was established, and a multiplex PCR detection kit for the rapid detection of animal-derived components was developed. According to the mitochondrial cytochrome b (Cyt b) gene of bovine species, sheep species, pig species and horse species and the mitochondrial cytochrome c oxidase subunit I (COX 1) gene of sika deer and red deer as the target gene sequences of primers, the specific primers of five different species were designed, the PCR system was optimized, and the multiplex PCR identification method of five animal-derived components was established. The minimum detection amount was determined by sensitivity test. The results showed that five meat specific amplification bands could be found at the same time in the same reaction system, including 173 bp fragment for venison, 148 bp for beef, 261 bp for pork, 100 bp for mutton and 424 bp for horse, indicating that the method is specific and stable. The minimum detection limit by this method was 1 ng/μL, showing a high sensitivity. According to the different sites in different areas of animal mitochondrial genes, a multiplex PCR detection method was established and a detection kit was developed, and the rapid, sensitive, stable and high-throughput detection of five animal-derived components and adulterated animal components in edible meat can be realized by using the kit.
Keywords
Multiplex PCR
Mitochondrial DNA
Animal-derived components
Detection kit

1 Introduction
Meat products have become an indispensable part of human diet, and meat foods with a high content of nutrients are the source for people to obtain high-quality proteins (Vlachos et al.,2016). In order to ensure people's “safety on the tip of the tongue”, the authenticity evaluation of components in foods has become the main focus of protecting consumers (Qin, 2018). Nowadays, the main fraud encountered by meat industries is that someones break the law to sell fake products to earn more money illegally, such as selling high-price animal meats mixed with cheap animal meats (rat meat as mutton), rat meat foods containing a variety of harmful substances (bacteria and heavy metals), and mutton products mixed with pork to religious consumers (Amaral, 2017), endangering people's health and damaging the people's rights and interests. Therefore, strengthening the research on the authenticity analysis method of meat products is of great significance to control the safety of food circulating in the market.The early analysis methods commonly used in the recognition of meats were mainly based on the sensory and immunological characteristics of meats to distinguish them (Hu et al., 2018; Li et al.,2013; Shi et al., 2019). However, most proteins are unstable to heat, and proteins in some foods processed at a high temperature will be denatured, so the application of the conventional methods in testing hot processed foods will be limited (Chen et al., 2000; Montowsk et al., 2015). DNA molecule is more stable than proteins, and now DNA research methods based on PCR technology have become a hot spot, which can analyze heat-processed foods quickly, sensitively, specifically and repeatedly. Common PCR technologies, such as species-specific PCR (Ai et al., 2018; Chen et al.,2020; Ghovvati et al., 2009; Kesmen et al.,2007; Qin, 2019, restriction fragment length polymorphism PCR (RFLP-PCR) (Li et al.,2011), multiplex PCR (Li et al., 2019a, 2019b) and fluorescence quantitative PCR (qPCR) (Chen et al.,2017; Liliana et al.,2017; Tang et al.,2018), are widely used for the detection of meat foods. The DNA analysis technology based on PCR is widely used in the identification of animal-derived components, but it is difficult for a PCR reaction only targeting a single gene to detect multiple animal-derived components due to the diversity of adulterants (Safdar et al.,2014). The development and application of multiple DNA detection kit can avoid the limitations in the low efficiency, high cost and high professional requirements for technicians.
DNA fragments of different species can be detected at the same time by adding primer pairs of multiple species to a single reaction system in multiplex PCR reaction, so as to reduce the detection cost and improve the detection efficiency (Alikord et al.,2017). In this study, mitochondrial Cyt b gene and COX 1 gene were used to establish a PCR method that could detect five animal-derived components in meat foods at the same time, and a multiple DNA detection kit was developed. By using the kit to simplify the operation, the efficient and high-throughput simultaneous detection of animal-derived components in meat foods could be realized, which was expected to provide a reliable and stable biological detection technology to identify the authenticity of meat products for the judicial identification of meat foods, ensuring the people’s dietary safety and maintaining the social stability.
2 Materials and methods
2.1 Samples and reagents
The fresh products of beef, pork, horse meat, mutton, venison, donkey meat, mink meat and mouse meat were provided by Changchun Food and Drug Inspection Center (Changchu, Jilin, China). Chicken, duck, goose, dog meat and rabbit meat were purchased in a local supermarket in Jilin City (Jilin, Jilin, China). GelRed Nucleic Acid Gel Stain, DL100bp DNA Marker, 2 × Taq PCR Master Mix, Amp (ampicillin), X-Gal (5-bromo-4-chloro-3-indolyl-β-d-thiogalactoside), IPTG (isopropyl-β-d-thiogalactopyranoside), DH5α competent cells, DNA gel recovery kit, pGM-T vector linking kit and plasmid DNA small quantity extraction kit were purchased from Tiangen Biotech (Beijing) Co., ltd. (Beijing, China).
2.2 DNA extraction of meat samples
One hundred mg of meat sample each were placed in a 1.5-mL centrifuge tube and cut into pieces, and 500 μL of P1 cell lyssis buffer [10 mL of 10 % SDS, 1 mL of 1 mol/L Tris HCl (pH 7.5), 2 mL of 0.5 mol/L EDTA (pH 8.0), 0.58 g NaCl, 1 mL of 20 mg/mL PK and 5 μL RNase were added with sterilized ddH2O to set the constant volume to 100 mL] were added into the tube, and the meat-lysis buffer solution was mixed evenly by shaking and heated in a water bath at 55 ℃ for 1 h. Then, the solution was loaded onto a DNA purification column. The column was centrifuged at 12,000 r/min for 3 min, then the waste liquid was discarded, and 600 μL of P2 rinsing solution [26 μL of 5 mol/L KAc, 18 μL of 1 mol/L Tris HCl (pH 7.5), 3 μL of 0.5 mol/L EDTA (pH 8.0), 480 μL of 0.5 mol/L absolute ethanol and 73 μL sterilized ddH2O] was loaded onto the column. The column was centrifuged at 12,000 r/min for 1 min, and the waste liquid was discarded and the column was washed with? once again. The waste liquid was discarded and the column was centrifuged at 12,000 r/min for 2 min, then the DNA purification column was transferred into another centrifuge tube, and 100 μL of P3 sterilized ddH2O were added into the tube, left standing at room temperature for 10–20 min. Then, the column was centrifuged at 12,000 r/min for 2 min, and the centrifugate, namely the test sample DNA solution, was kept at −20 ℃ for standby.
2.3 Primer design
The sequence homology of a specific gene on the mitochondrial DNA of five different species was aligned and analyzed with a MEGA7 software. Targeting at the Cyt b gene sequences of cow (NC_006853.1), pig (NC_000845.1), sheep (NC_001941.1), horse (NC_001941.1), and the Cox 1 gene sequences of sika deer (NC_007704.2) and red deer (NC_018595.1), the primers for various species were designed using a bioinformatics software to find specific SNP sites as the target sites. The synthesis of the designed specific primer sequences (Table 1) was entrusted to Huada Gene Technology Co., ltd (Beijing, China).
Species
Primer
Sequence (5′-3′)
Primer region
Size of products (bp)
Deer
Forward
AATATCAAACCCCTCTATTC
COX 1
173
Reverse
GAACAAGTGTTGATATAGAATA
Cattle
Forward
CCCATACATCGGCACAAGTT
Cyt b
148
Reverse
TTCGTGGAGGAATAATAGGTGG
Pig
Forward
GAATGAATCTGAGGGGACT
Cyt b
261
Reverse
GTCTGGTGAGAATAGTACAAGG
Sheep
Forward
TTCCACCCTTATTACACCG
Cyt b
100
Reverse
GGTCTCCGAGTAAGTCAG
Horse
Forward
TCACAGCCCTGGTAGTCGT
Cyt b
424
Reverse
GCCACTAAGAGTCAGAACAC
2.4 Primer specificity detection and screening
Meat DNA was extracted by the method in Section 2.2, and mixed primers (deer-derived, bovine-derived, sheep-derived, pig-derived and horse-derived primers) were used to detect venison, beef, mutton, pork and horse meat samples. The PCR amplification was carried out at 52 ℃, and the specificity of primers was identified based on whether the experimental results could indicate the occurrence of a non-specific amplification.
2.5 Multiplex PCR reaction and condition optimization
The total volume of multiplex PCR reaction was 50 μL, including 25 µL of 2 × Taq Mix, 0.5 ∼ 5 μL of the upstream (10 µmol/L) and downstream (10 µmol/L) primers of different species, 2 μL of 50 ng/μL DNA solution, and the insufficient reaction solution volume was made up with sterilized ddH2O.
The PCR reaction tube was placed in a PCR instrument, and the procedures were set as 94 ℃ for 2 min, with 30 cycles (94 ℃ for 30 s, 52 ℃ for 30 s, 72 ℃ for 30 s), and extension at 72 ℃ for 5 min. Based on these conditions, the conditions of the reaction system at different primer addition concentrations were optimized.
GelRed nucleic acid stain was added to 2 % agarose gel (the final concentration was 0.075 μL/mL). After PCR, 5 μL of the reaction solution was loaded onto the gel, and the 100 bp DNA ladder loading amount was 5 μL, in which 1 × TBE buffer was used. The electrophoresis was performed at 80 V for 1 h, and the bands were examined and photographed with an UV analyzer.
2.6 Cloning and preparation of standard control solution
The DNA of venison, beef, mutton, pork and horse meat were subjected to PCR reaction. The agarose gel electrophoresis on 5 μL of the PCR products was carried out, and the target gene gel was recovered. The recovered products were connected to pGM-T vectors at 16 ℃ overnight according to the instructions of DNA gel recovery kit. The products were transferred to DH5α competent cells in an ice bath for 30 min, then the cells were incubated and left standing in a hot water bath at 42 ℃ for 90 s, placed on ice for 2 min, and seeded into plates and incubated at 37 ℃ overnight. The positive monoclonal antibodies were screened by blue-white spot screening, and the plasmid DNA was extracted by following the procedures proposed in the small plasmid DNA extraction kit after multiplication culture. The amplified plasmid by PCR was analyzed by electrophoresis to determine whether it was consistent with the expected fragment, and sequenced to determine the accuracy of its sequence.
2.7 Development of kit
In order to make the multiplex PCR detection method established in this study easy to be popularized, a multiplex PCR detection kit was developed based on this method. Using the kit to simplify the operation made it possible to detect animal-derived components in meat foods simultaneously in a high efficient and high-throughput way. One kit could be used for 20 tests, and the kit was divided into 8 parts, including DNA extraction and PCR amplification (Table 2).
Number
Size
Effect
Storage
Usage
P1
15 mL
Lysing cells to release DNA
−20 °C
melting completely
P2
30 mL
Isolating DNA
−20 °C
melting completely
P3
5 mL
Dissolving DNA
−20 °C
melting completely
Positive control liquid
50 μL
Positive control
−20 °C
melting completely
Negative control liquid
50 μL
Negative control
−20 °C
melting completely
PCR reaction tube
48 μL × 20
PCR reaction system
−20 °C
melting completely
Chromatographic column
20 ↑
Purifying DNA
RT
−20 °C
2.8 Kit evaluation
2.8.1 Specificity
The self-developed multiplex PCR detection kit was used for evaluating its specificity, in which venison, beef, mutton, pork, horse, chicken, duck, donkey, mouse, mink and dog DNAs were added into the PCR reaction tube, and the PCR amplification was conducted at 52 ℃ for observing the specificity of the kit.
2.8.2 Reproducibility
The same samples (venison, beef, mutton, pork and horse meat) were detected by three technical analysts in three laboratories using the kit, and the reproducibility of the reagents was evaluated according to whether the experimental results were consistent or not.
2.8.3 Stability
The stability of the reagents in a kit selected randomly was observed by three technical analysts in three laboratories at three months, six, nine and 12 months, respectively.
2.8.4 Sensitivity
Using the self-developed multiplex PCR kit, the PCR amplification was carried out at 52 ℃ with different concentrations of mutton, beef, venison, pork, horse DNA and mixed sample DNA as templates to investigate the detection ability and the minimum content detection limit of the kit for five meat DNA and mixed sample DNA.
2.9 Multiplex PCR results of mixed meat samples
Mutton, beef, venison, pork and horse meat (20 mg each) were mixed. The kit was used according to the instructions. The genomic DNA in the mixed meat samples was extracted as templates and amplified by PCR at 52 ℃ for investigating the detection ability of the kit for mixed meat samples.
2.10 Data processing
The gel electrophoresis patterns of the PCR-amplified products were all photographed by an ultraviolet imaging analyzer, and then processed by Photoshop CS6.
3 Results
3.1 Analysis of mtDNA
DNA was extracted by using the self-developed kit and repeatedly measured 3 times. SPSS statistical V17.0 software was used for the statistical analysis, and the results showed that the average value of AD260 nm/AD280 nm was about 1.95 ± 0.10, and the purity was 80~2.00, indicating that there was nearly no protein contamination in the DNA samples extracted by this method, with a high purity (Table 3).
Different animal-derived components
A260/A280
DNA concentration (ng/μL)
Beef
1.955 ± 0.05
692.80 ± 2.15
Mutton
1.957 ± 0.04
503.20 ± 4.22
Pork
1.976 ± 0.08
574.35 ± 2.60
Horse meat
1.899 ± 0.10
622.05 ± 1.25
venison
1.877 ± 0.05
803.15 ± 0.80
3.2 Primer specificity
A single species DNA was added into the mixed primers for the primer specificity experiment. Mixed primers (deer-, bovine-, sheep-, pig- and horse-derived primers) were used to detect venison, beef, mutton, pork and horse meat samples at 52 ℃ by PCR amplification. The results of agarose gel electrophoresis showed that there was a specific amplification band at the target positions of venison, beef, mutton, pork and horse meat samples (173 bp for venison, 148 bp for beef, 100 bp for mutton, 261 bp for pork and 424 bp for horse meat), there was no band in non-target species and blank control (Fig. 1), indicating that the primers of each species were specific.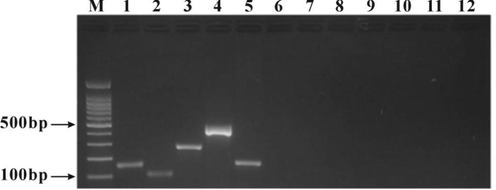
Primer-specific PCR electrophoresis. M: 100 bp Ladder, 1: beef DNA, 2: mutton DNA, 3: pork DNA, 4: horse meat DNA, 5: venison DNA, 6: chicken DNA, 7: duck DNA, 8: donkey meat DNA, 9: rat meat DNA, 10: mink meat DNA, 11: dog meat DNA, 12: blank control.
3.3 Primer concentration optimization
The ratio between primers has a serious impact on the multiplex PCR reaction, and adding the same amount of primers may cause a mutual inhibition between primers, leading to the loss of the band of a species. Specific bands of different species can be found by changing the concentration of various primers in the reaction system of a PCR amplification. In the basis of pig primer at the concentration of 1 μM in the reaction system, the deer primer concentration was increased in gradient (1, 1.1, 1.2, 1.3 μM) for observing the impact of primer concentration on the mutiplex PCR reaction, and the optimal concentration of upstream and downstream primers for deer was determined as 1.3 μM based on the results (Fig. 2A). On this basis, the optimal concentration of sheep primers was investigated by decreasing the concentration in gradient (1, 0.8, 0.6, 0.4, 0.2 μM), and the results (Fig. 2B) indicated that the optimal sheep primer concentration was 0.8 μM and the complete and clear characteristic bands could be found at this concentration. Considering the interaction between primers, the horse primer concentration was reduced in gradient (0.1, 0.09, 0.08, 0.07 μM), and the optimal concentration of horse primers was determined as 0.07 μM (Fig. 2C). On this basis, the optimal cow primer concentration was studied by increasing the concentration in gradient (0.8, 0.6, 0.4, 0.2 μM), and the results showed that the multiplex PCR bands were clear and there was no nonspecific amplification when the cow primer concentration was 0.2 μM (Fig. 2D). Finally, the optimal primer concentrations of cow, sheep, pig, horse and deer were determined as 0.2, 0.8, 1, 0.07 and 1.3 μM, respectively (Fig. 3).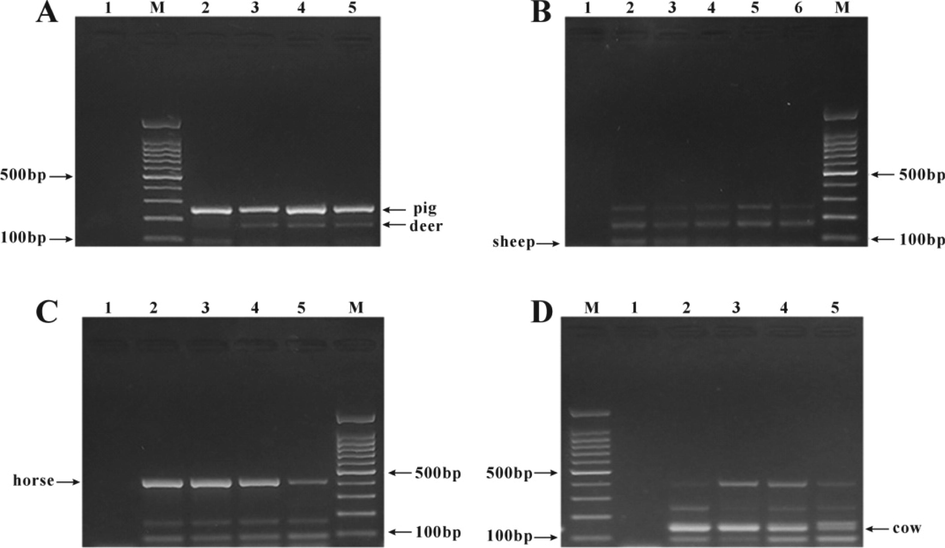
Screening of the optimum concentrations of five meat primers in the multiplex PCR reaction system. A. Screening of the best concentration of deer primers: M: 100 bp DNA ladder, 1: blank, 2: 1 μM, 3: 1.1 μM, 4: 1.2 μM, 5: 1.3 μM; B. Screening of the optimum concentration of sheep primers: M: 100 bp DNA ladder; 1: blank control; 2: 1 μM; 3: 0.8 μM; 4: 0.6 μM; 5: 0.4 μM; 6: 0.2 μM. C. Screening of the optimum concentration of horse primers. M: 100 bp DNA ladder; 1: blank control; 2: 0.1 μM; 3: 0.09 μM; 4: 0.08 μM; 5: 0.07 μM. D. Screening of the optimum concentration of bovine primers. M: 100 bp DNA ladder; 1: blank control; 2: 0.8 μM; 3: 0.6 μM; 4: 0.4 μM; 5: primer concentration 0.2 μM.
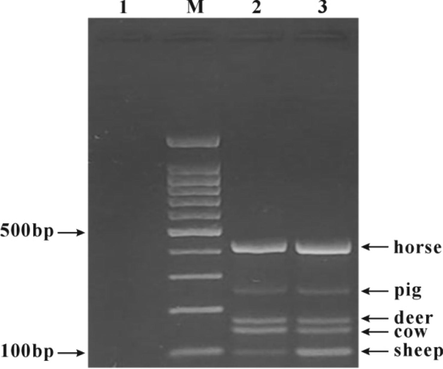
Electrophoresis of multiplex PCR products of five meat primers. M: 100 bp DNA ladder; 1: blank control; 2 and 3: mixed primers (the primer concentrations of cow, sheep, pig, horse and deer were 0.2 μM, 0.8 μM, 1 μM, 0.07 μM and 1.3 μM, respectively).
3.4 Standard control solution electrophoresis and plasmid DNA sequencing alignment
The white colonies in the blue-white screening plate were amplified and cultured, and the plasmids were extracted. The purified and recovered plasmids as the positive control liquid were identified by PCR reaction and gel electrophoresis. The results showed that after the plasmid was amplified, there was a consistent target band at the corresponding position in the gel electrophoresis map of each standard (Fig. 4), suggesting that the monoclone of the target gene was completely correct, and there was no change in some bases or their arrangement orders in the DNA sequence. The sequencing results were input into NCBI (https://www.ncbi.nlm.nih.gov/) for using BLAST to test the consistency of DNA sequences, and the results showed that the consistency with the DNA sequence of the specific fingerprint fragment was more than 99 % (Fig. S1 A-E). The diluted plasmid DNA as the standard control solution was put into the DNA detection kit.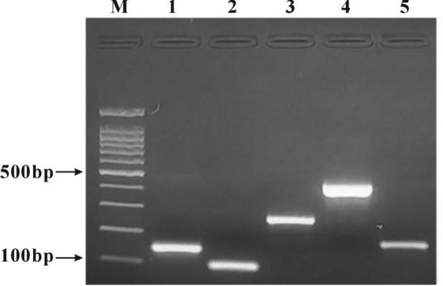
Electrophoresis of five positive control PCR products. M: 100 bp DNA ladder; 1: Cow; 2: Sheep; 3: Pig; 4: Horse; 5: Deer.
3.5 Evaluation of kit parameters
3.5.1 Specificity
The self-developed multiplex PCR detection kit was used. Deer, beef, mutton, pork, horse, chicken, duck, donkey, rat, mink and dog DNAs were added into the multiplex PCR reaction system, and the PCR amplification was carried out at 52 ℃ for observing the specificity of the kit. The results of agarose gel electrophoresis of PCR products showed that there was a specific amplification band of venison, beef, mutton, pork and horse meat samples each at the target position, while there was no amplification band of the other species and blank control (Fig. 5), indicating that the reagents in the kit was highly specific.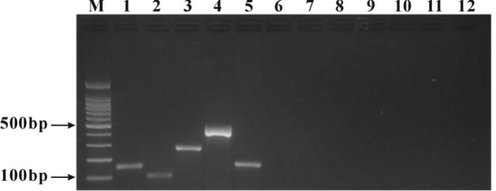
Specificity test of the self-developed multiplex PCR detection kit. M: 100 bp DNA ladder; 1: Beef DNA; 2: Mutton DNA; 3: Pork DNA; 4: Horse DNA; 5: Venison DNA; 6: Chicken DNA; 7: Duck DNA; 8: Donkey DNA; 9: Rat DNA; 10: Mink DNA; 11: Dog DNA; 12 Blank control.
3.5.2 Reproducibility
The same samples were tested by three technical analysts from three different laboratories for the study on the kit reproducibility. The results showed that there were specific amplification bands of the tested species samples and positive control at the expected positions, and there was no amplification band of the blank control, consistent with the above results, indicating that the reproducibility of the reagents was good (Fig. 6).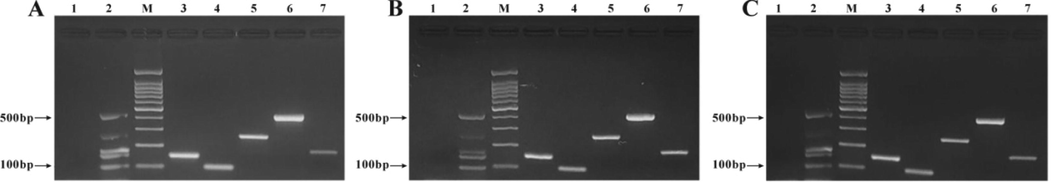
Reproducibility test of the self-developed multiplex PCR detection kit. A. Laboratory: molecular biology laboratory, School of Medical Technology, Beihua University; Experimenter: Duan Siqi, master degree, B. Laboratory: Jilin Province Traditional Chinese Medicine DNA Fingerprint Detection Technology Innovation Center; Experimenter: Jiang Haiying, graduate student. C. Laboratory: Jilin Leining Food and Drug Testing Technology Service Co., ltd.; Experimenter: Li Yingnuo, master degree. M: 100 bp DNA ladder; 1: Blank control; 2: Kit positive control; 3: Beef DNA; 4: Mutton DNA; 5: Pork DNA; 6: Horse DNA; 7: Venison DNA.
3.5.3 Stability
A self-developed multiplex PCR detection kit randomly selected was repeatedly frozenthawed for 3 months, 6 months, 9 months and 12 months, respectively, and then used to detect samples at the different time points. The results detected by the kit were consistent, showing a good stability of the reagents in the kit (Fig. 7).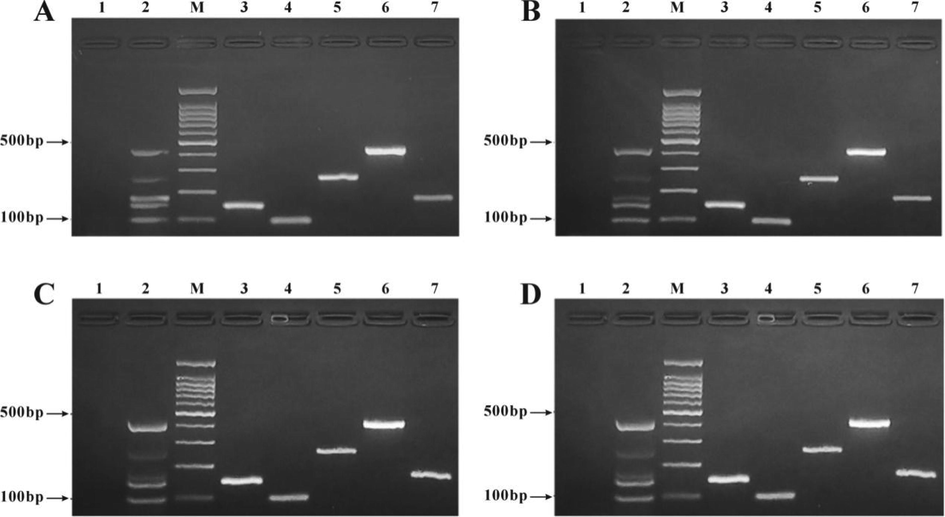
Stability test of the self-developed multiplex PCR detection kit. A: 3 Months; B: 6 Months; C: 9 Months; D; 12 Months; M: 100 bp DNA ladder; 1: Kit blank control, 2: Kit positive control; 3: Beef DNA; 4: Mutton DNA;, 5: Pork DNA; 6: Horse DNA; 7: Venison DNA.
3.5.4 Sensitivity
One hundred ng/μL deer DNA, beef DNA, mutton DNA, pork DNA and horse DNA each were diluted 10 times in turn, respectively, for investigating the sensitivity of the method. The results showed that when a single species was detected by the multiplex PCR system, the specific amplification bands of the target species could still be found at the minimum concentration of 100 pg/μL (Fig. 8A–E). The five meats were mixed in equal proportion (The concentrations were 200 ng/μL, 100 ng/μL, 50 ng/μL, 20 ng/μL, 10 ng/μL, 1 ng/μL and 0.5 ng/μL, respectively), for the investigation of the sensitivity of the method. The results showed that a clear target band could still be seen at the corresponding position in the electrophoretic map when the DNA content in the mixed sample was 1 ng/μL (Fig. 8F).
Sensitivity test of the self-developed multiplex PCR detection kit. A. Deer meat DNA detection. M: 100 bp DNA Ladder; 1: 100 ng/μL; 2: 10 ng/μL; 3: 1 ng/μL; 4: 100 pg/μL; 5: 10 pg/μL; 6: 1 pg/μL; 7: 0.1 pg/μL; 8: kit blank control. B. Beef DNA detection. M: 100 bp DNA Ladder; 1: 100 ng/μL; 2: 10 ng/μL; 3: 1 ng/μL; 4: 100 pg/μL; 5: 10 pg/μL; 6: 1 pg/μL; 7: 0.1 pg/μL; 8: Blank control. C. Mutton DNA detection. M: 100 bp DNA Ladder; 1: 100 ng/μL; 2: 10 ng/μL; 3: 1 ng/μL; 4: 100 pg/μL; 5: 10 pg/μL; 6: 1 pg/μL; 7: 0.1 pg/μL; 8: Blank control. D. Pork DNA detection. M: 100 bp DNA Ladder; 1: 100 ng/μL; 2: 10 ng/μL; 3: 1 ng/μL; 4: 100 pg/μL; 5: 10 pg/μL; 6: 1 pg/μL; 7: 0.1 pg/μL; 8: Blank control. E. Horse meat DNA detection. M: 100 bp DNA Ladder; 1: 100 ng/μL; 2: 10 ng/μL; 3: 1 ng/μL; 4: 100 pg/μL; 5: 10 pg/μL; 6: 1 pg/μL; 7: 0.1 pg/μL; 8: Blank control. F. Detection of mixed meat DNA in equal proportion. M: 100 bp DNA ladder; 1: Blank control; 2: 200 ng/μL; 3: 100 ng/μL, 4: 50 ng/μL; 5: 20 ng/μL; 6: 10 ng/μL; 7: 1 ng/μL; 8: 0.5 ng/μL.
3.6 Multiplex PCR detection of mixed meat samples
The results of agarose gel electrophoresis of PCR products showed that there were 5 clear and bright bands of the mixed meat samples at 100 bp, 148 bp, 173 bp, 261 bp and 424 bp, indicating that the kit could be used to accurately detect sheep-, cow-, deer-, pig- and horse-derived components in the mixed meat samples (Fig. 9).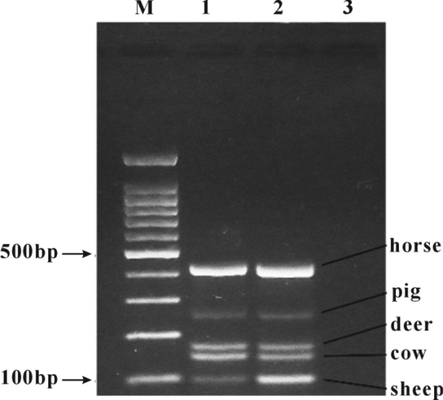
Multiplex PCR detection of mixed meat samples. M: 100 bp DNA ladder; 1: Positive control; 2: Mixed meat samples in equal proportion; 3: Blank control.
4 Discussion
DNA detection method has the advantages of specificity, accuracy and intuition. At present, many reports have used this method to identify animal samples (Afifa et al., 2021; Li et al., 2019a, 2019b; Martins et al., 2020; Zyrianova & Zaripov, 2022). However, these studies still have some shortcomings, such as poor stability, cumbersome operation, great difficulty in promotion, and lack of reference materials, which limit their application. In this experiment, self-developed reagents were used to simplify the operation steps, and the results showed that the effect of extracting DNA was stable, and the quality of DNA template could be guaranteed, so as to reduce the possibility of false negative or false positive caused by the cross-contamination of samples, which was considered to be important to ensure the smooth progress of follow-up experiments.
In a multiplex PCR experiment, the design of primers is a crucial step, with a direct impact on the successful detection of target species (Galal-Khallaf, 2021; Prusakova et al., 2018; Wang et al., 2018; Xu et al., 2018). In this experiment, the homology of the whole mitochondrial genome sequences was aligned for the sequence analysis to find the specific difference regions among species. The Cox 1 gene of sika deer and red deer as well as the Cyt b gene sequences of cow, sheep, pig and horse species were selected as the target regions of primer design. According to the sequence differences, the primers were designed by using a bioinformatics software. The annealing temperature was adjusted within the range of 50 ∼ 55 ℃, and the 3 'ends of forward and reverse primers were located at the SNP site to reduce the possibility of reaction between different primers to form a primer dimer and the amplification interference to the target gene.
In this study, the specificity of primers was verified by whether a single primer could amplify the DNA of non target species, and the experimental results showed that the primers, with a specificity, could be used to further explore the multiplex PCR reaction system. The concentration of five pairs of primers was optimized. In a reaction system, the same amount of primers (1:1:1) was added, and there was a mutual inhibition between primers, suggesting that there would be no band in a species. Then, through repeated screening, the primer amount was adjusted, and the primer concentrations of cow, sheep, pig, horse and deer were determined as 0.2 μM, 0.8 μM, 1 μM, 0.07 μM and 1.3 μM, respectively, so the specific amplification bands of the five species could be clearly found.
Based on the established multiplex PCR reaction system, a multiplex DNA detection kit was developed. The specific DNA sequences of cow, sheep, pig, horse and deer were cloned by using molecular cloning technology, and the isolated and purified cloned plasmid was extracted as the standard reference solution of the kit. The diluted plasmid DNA was amplified using the kit PCR reaction system, and the electrophoresis results were used to judge whether it contained the expected target gene DNA fragment. In addition, the plasmid DNA was sequenced and analyzed, and the sequencing results were compared with the target sequence of the species. The sequence alignment results showed that the recombinant plasmid prepared in the experiment could be used as the standard reference solution of the kit.
5 Conclusion
A multiplex PCR method for the simultaneous detection of animal-derived components from deer, cow, sheep, pig and horse in edible meat was established, and a rapid, sensitive, stable and high-throughput detection of five animal-derived components and adulterated animal components in edible meat could be realized. Based on this method, a multiplex PCR detection kit was developed. By simplifying the operation steps and standardizing the operation specifications, the kit may be easily used and popularized in the supervision of meat foods.
Funding
This work was financially supported by High Technical Industry of Development and Reform Commission of Jilin Province (2018C048-2), Local Standard Formulation Project of Chinese Medicinal Materials of Jilin Province (2016066), Jilin Science and Technology Development Plan Project of Jilin Province (20190303093SF, 20190301014NY, 20200403047SF) and Jilin Science and Technology Innovation Center Construction Project (20190902018TC).
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Not applicable in this study.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Detection of species adulteration in meat products and Mozzarella-type cheeses using duplex PCR of mitochondrial cyt b gene: a food safety concern in Bangladesh. Food Chem.. 2021;16(2):100017
- [CrossRef] [Google Scholar]
- Development on DNA detection kit of fox and its application in common meat products. J. Hygiene Res.. 2018;47(06):979-983. https://doi.org/CNKI:SUN:WSYJ.0.2018-06-027
- [Google Scholar]
- Multiplex-PCR as a rapid and sensitive method for identification of meat species in halal-meat products. J. Recent Pat Food Nutr. Agric.. 2017;8(3):175-182.
- [CrossRef] [Google Scholar]
- Quantitative detection of pork meat by EvaGreen real-time PCR to assess the authenticity of processed meat products. J. Food Control.. 2017;72(7):53-61.
- [CrossRef] [Google Scholar]
- Application of quantitative real-time PCR in meat and meat products. J. Sci. Technol. Food Ind.. 2017;17:324-328.
- [CrossRef] [Google Scholar]
- Detection of pork in heat-processed meat products by monoclonal antibody-based ELISA. J. J AOAC Int.. 2000;83(1):79-85.
- [CrossRef] [Google Scholar]
- Polymerase chain reaction with lateral flow sensor assay for the identification of horse meat in raw and processed meat products. J. Food Chem.. 2020;345(1):128840
- [CrossRef] [Google Scholar]
- Multiplex PCR and 12S rRNA gene sequencing for detection of meat adulteration: A case study in the Egyptian markets. Gene. 2021;5(764):145062
- [CrossRef] [Google Scholar]
- Fraud identification in industrial meat products by multiplex PCR assay. J. Food Control.. 2009;20(8):696-699.
- [CrossRef] [Google Scholar]
- A review of recent progress in detection of heterologous genes in meat products. J. Food Science.. 2018;39(15):275-282.
- [CrossRef] [Google Scholar]
- PCR assay for the identification of animal species in cooked sausages. J. Meat Sci.. 2007;77(4):649-653.
- [CrossRef] [Google Scholar]
- Identification and detection of Cordyceps sinensis with LAMP. J. Chinese Traditional Herbal Drugs.. 2011;42(8):1605-1608. https://doi.org/CNKI:SUN:ZCYO.0.2011-08-038
- [Google Scholar]
- Quick identification of five species of meat by PCR assay. J. Food Sci.. 2013;34(8):249-252. https://doi.org/CNKI:SUN:SPKX.0.2013-08-055
- [Google Scholar]
- Detection of goat meat adulteration by real-time PCR based on a reference primer. Food Chem.. 2019;277(30):554-557.
- [CrossRef] [Google Scholar]
- Rapid detection of common adulterated components in beef by multiple allele-specific polymerase chain reaction. J. Sci. Technol. Food Ind.. 2019;40(24):82-87.
- [CrossRef] [Google Scholar]
- EvaGreen real-time PCR to determine horse meat adulteration in processed foods. J. LWT - Food Sci. Technol.. 2017;75(8):408-416.
- [CrossRef] [Google Scholar]
- A mathematical modeling approach to the quantification of lactic acid bacteria in vacuum-packaged samples of cooked meat: Combining the TaqMan-based quantitative PCR method with the plate-count method. Int. J. Food Microbiol.. 2020;318(2):108466
- [CrossRef] [Google Scholar]
- Authentication of processed meat products by peptidomic analysis using rapid mass spectrometry. J. Food Chem.. 2015;187(4):297-304.
- [CrossRef] [Google Scholar]
- A simple and sensitive two-tube multiplex PCR assay for simultaneous detection of ten meat species. Meat Sci.. 2018;137:34-40.
- [CrossRef] [Google Scholar]
- S tudy on rapid identification technology of common meat products based on PCR principle D. Hefei: Hefei University of Technology; 2018.
- Self-signal-on fluorescent colorimetric protocol for rapid authentication of horsemeat adulterated beef samples with functional designed probes. J. Int. J. Food Sci. Technol.. 2019;5:1752-1759.
- [CrossRef] [Google Scholar]
- A highly sensitive and specific tetraplex PCR assay for soybean, poultry, horse and pork species identification in sausages: Development and validation. J. Meat Sci.. 2014;98(2):296-300.
- [CrossRef] [Google Scholar]
- Recent progress in techniques for adulteration of meat and meat products. J. Food Sci.. 2019;40(23):319-326.
- [CrossRef] [Google Scholar]
- Detection of chicken-derived components in livestock and poultry meat by fluorescent PCR assay. J. Modern Food Sci. Technol.. 2018;34(7):230-234.
- [CrossRef] [Google Scholar]
- An updated review of meat authenticity methods and applications. J. Crit. Rev. Food Sci. Nutr.. 2016;56(7):1061-1096.
- [CrossRef] [Google Scholar]
- Development of nested multiplex polymerase chain reaction (PCR) assay for the detection of Toxocara canis, Toxocara cati and Ascaris suum contamination in meat and organ meats. Parasitol Int.. 2018;67(5):622-626.
- [CrossRef] [Google Scholar]
- Multiplex TaqMan locked nucleic acid real-time PCR for the differential identification of various meat and meat products. Meat Sci.. 2018;137:41-46.
- [CrossRef] [Google Scholar]
- 18S ribosomal DNA-based PCR test for avian and mammalian DNA identification in meat products. Vet Anim Sci.. 2022;15:100234
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.104593.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




