

Translate this page into:
Experimental, DFT and Theoretical Corrosion Study for 4-(((4-ethyl-5-(thiophen-2-yl)-4H-1,2,4-triazole-3-yl)thio)methyl)-7,8-dimethyl-2H-chromen-2-one
⁎Corresponding author. rebaz.anwar@koyauniversity.org (Rebaz Anwar Omar)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
In this work, 4-(((4-ethyl-5-(thiophen-2-yl)-4H-1,2,4-triazole-3-yl)thio)methyl)-7,8-dimethyl-2H-chromen-2-one was synthesized by acetone-mediated condensation of 4-ethyle-5-(thiophen-2-yl)-4H-1,2,4-triazole-3-thiol and 4-(chloromethyl)-7,8-dimethyl-2H-chromen-2-one. The molecule results (3) were experimentally characterized using FT-IR, 1H-, and 13C NMR spectroscopy. Density Functional Theory (B3LYP/cc-pVDZ) was used to investigate the ideal molecule structure, vibrational frequencies, and 1H with 13C NMR (theoretically) chemical shifts. Theoretical and experimental spectroscopy results were compared and agreed with each other, which indicated the validity of the used developed molecular structure. The Dipole moment, hardness, softnes, electronegativity, electrophilicity index, nucleophilicity index, and chemical potential as electronic structural parameters linked to corrosion inhibition efficacy were investigated for the prepared compound. Furthermore, the fraction of transferred electrons was calculated to determine the interaction between the iron surface and organic molecules. The results indicated a favorable relationship between organic-based corrosion inhibitors and quantum chemical parameters processes. The corrosion inhibitors' behavior can be predicted without the need for experimental investigation.
Keywords
Anti-corrosion
DFT
FT-IR
NMR spectroscopy
Substituted 1,2,4-Triazole

1 Introduction
In recent decades, there has been a lot of interest in the synthesis of 1,2,4-triazole compounds having antibacterial (Ikizler et al., 1999; Yuksek et al., 2013; Zobo Mfomo et al., 2017; Raviprabha and Bhat, 2021), anti-inflammatory (Tozkoparan et al., 2000), analgesic (Turan-Zitouni et al., 1999), antitumorial (Demirbaş et al., 2002), antihypertensive (Purohit et al., 2011), anticonvulsant, and antiviral (Kritsanida et al., 2002) properties. For synthesizing 1,2,4-triazole, cyclization of thiosemicarbazide-structured compounds has proven to be a good method (Demirbas et al., 2004). The identical thiosemicarbazides were cyclized in the presence of NaOH to produce 1,2,4-triazole derivatives (Varvaresou et al., 1998; Küçükgüzel et al., 1999; Küçükgüzel et al., 2000; Karakuş and Rollas, 2002; Raviprabha and Bhat, 2019). Various medications for cancer treatment have been developed in recent years, with certain 1,2,4-triazole derivatives with shiff base structure being synthesized as anticancer agents (Bayrak et al., 2009; Omar et al., 2021).
Coumarin compounds having reactive oxygen, heterocycles, and a benzopyran backbone are important natural compounds (Borges et al., 2005). benzopyrans have a unique structure, its derivatives can easily influence a wide range of enzymes and receptors in organisms with weak binding interactions, illustrating the drug's broad potential (Nicolaou et al., 2000; Newman, 2008; Cragg and Newman, 2013; Jashari et al., 2014; Koparir et al., 2022). Coumarins were discovered in tonka bean for the first time, and have since been found in over 150 species belonging to more than 30 families. Despite their presence in all sections of the plant, the fruits have the largest quantities of coumarins (Venugopala et al., 2013). The coumarin family is one of the most widely utilized natural chemicals on the planet. Coumarin is a privileged framework for a wide range of medical qualities, and it is frequently used as a scaffold for developing innovative and potent analogs with physicochemical properties and a diverse, simple synthetic transformation into a variety of activated coumarins (Yang et al., 2011; Peng et al., 2013). Because of the substituents present in their parent benzopyran moiety, coumarin-containing compounds have a wide range of biological and pharmacological activity (Musa et al., 2011; Borah et al., 2012). As a result, their synthesis has become a popular issue and focus of interest (El Ansary, 1992). Coumarins are associated with a wide range of biological processes. Cancer, burns, brucellosis, and cardiovascular and rheumatic disorders have all been treated with them (Kontogiorgis and Hadjipavlou-Litina, 2005; Basri et al., 2020).
Among the modeling methodologies, the functional density theory (DFT) formalism can be highlighted, as it allows for the calculation of physicochemical properties of the researched molecules at a microscopic level with great precision and low processing cost (Raviprabha and Bhat, 2019; Marinho et al., 2021). For example, this quantum approach can be used to determine the molecule's frontier orbitals, nucleophilic and/or electrophilic sites, kinetics, and thermodynamic characteristics (Braga et al., 2016; Madkour et al., 2018; Raviprabha and Bhat, 2020; Koparir et al., 2021; Omer et al., 2021). Thus, DFT is a crucial step in determining the pharmacological potential and finding the key pharmacophore for drug development. The Density Functional Theory (DFT) approach can be used to calculate organic molecule geometric optimization. In addition, infrared (IR) spectra can be simulated, and Frontier Molecular Orbitals (FMO) can be generated for electronic characterization and to derive quantum reactivity descriptors in order to comprehend the global reaction of the molecule's behavior as a nucleophile or electrophile (Braga et al., 2016). Pan et al (Pan et al., 2005) studied the amentoflavone molecule using the DFT method with B3LYP at 6-31G(d) basis set, structural optimization was done in the gas phase (vacuum), and the Molecular Electrostatic Potential, Frontier Molecular Orbitals, and the Mulliken charge distribution were used to characterize the molecule. However, in order to generate conformational relevant data for the development of a pharmacological tool, it is necessary to perform DFT molecular modeling calculations taking into account the dielectric constants of the various solvents, because the investigated molecules will have to be dissolved before being applied to humans (Marinho et al., 2021).
The main objective of this study is to generate new molecular structures in laboratories that can predict and develop many of the molecules' actions while also having more active properties. The use of computers to do various forms of “theoretical molecular modeling” approaches has increased in popularity, and it has become a significant tool in determining a wide range of molecule behaviors. The structure and functional properties of these molecules, as well as theoretical calculations of the 4-(((4-ethyl-5-(thiophen-2-yl)-4H-1,2,4-triazol-3-yl)thio)methyl)-7,8-dimethyl-2H-chromen-2-one compound (ETZC), have yet to be identified. The ETZC molecule has been studied both experimentally and theoretically using quantum chemical simulations and a variety of spectrum methods. The theoretical calculations were computed using the DFT/cc-pVDZ and the results were compared to experimental data. The data from the theoretical simulations were used for the global reactive descriptor and anti-corrosion activity.
2 Material and methods
2.1 Experimental
The melting points of the compound were determined using the Gallenkamp melting point apparatus. The IR spectra for the compound were measured using a PerkinElmer Spectrum One FT-IR Spectrophotometer. The 1H and 13C NMR spectra were recorded on a Bruker AC-400 NMR spectrometer at 400 MHz for 1H and 100 MHz for 13C NMR. Tetramethylsilane (1H and 13C NMR) chemical shifts were evaluated, and Dimethyl sulfoxide (DMSO) was utilized as an NMR solvent. Compounds (1) and (2) in Fig. 2 were found during synthesis at Firat University Organic Lab, while the remaining Chemicals were acquired from Aldrich and used without further purification.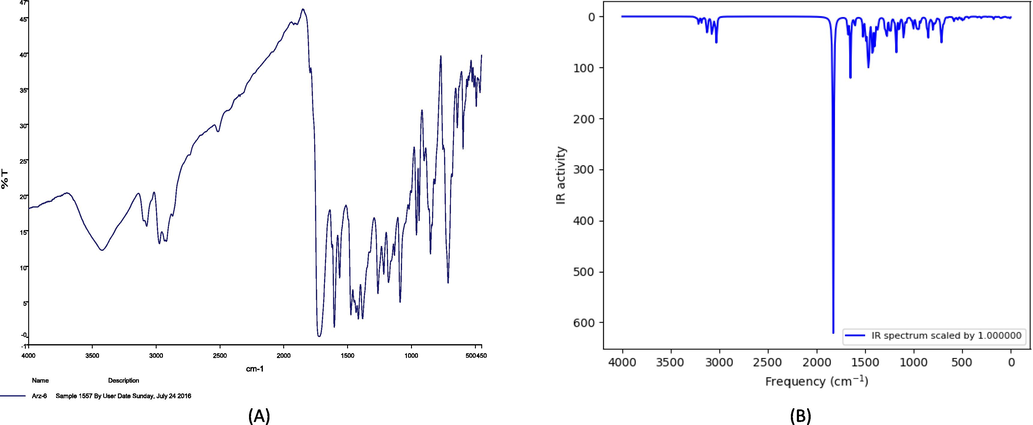
IR spectrum for the ETZC. (A) Experimental (B) Theoretical results.
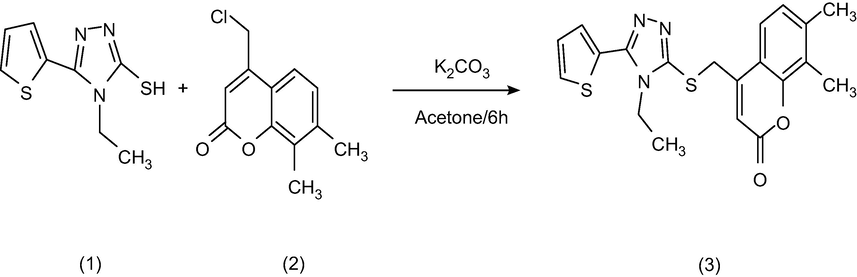
The synthesis of the compound (ETZC).
2.2 Computational analysis
The computational chemistry calculations were carried out using Density Functional Theory (DFT), which is one of the most effective methods for analyzing the stability and reactivity of chemical species. The Gaussian 09 program (Gaussian09 2009) and the Gauss View 5 program (Dennington et al., 2009) were used to draw the input molecules and perform DFT quantum calculations for the ground state electrical and reactive characteristics of the compound. This molecule have different orbital energies, a restricted Kohn-Sham functional was used, along with Becke's (Lee et al., 1988; Becke, 1993) three-parameter functional (B3) (Becke, 2003) for the exchange component and the Lee-Yang-Parr gradient corrected correlation functional (LYP) (Ditchfield et al., 1971) for the geometrical structure. The optimization was also done in the gas phase using the Gaussian 09 computer package to better understand the structural properties of the molecule due to the dimer's structure that allows for an angle between the two monomer structures (Cances et al., 1997). A vibrational frequency calculation was performed to get theoretical infrared spectra in the gas phase using the same degree of theory to confirm the validity of the optimization (Grimme, 2012; Rebaz et al., 2021). To assess the reactivity of the title molecule, the energy values of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) were used at the B3LYP/cc-pVDZ level of theory. (Ahmed and Rebaz, 2021; Ahmed and Rebaz, 2021; Kumar et al., 2015; Pereira et al., 2016). A larger HOMO orbital energy value suggests a reasonable probability for the chemical species to give electronic density, while a lower LUMO orbital energy value indicates a predisposition to receive electronic density (Fukui, 1982). As a result, the energy gap (n Egap), which is defined as the difference between the energies for the LUMO-HOMO orbitals (Eq. (1)), shows a direct relationship between the frontier orbitals and chemical reactivity: large values of (n Egap) indicate lower reactivity, while short values of (n Egap) indicate higher reactivity (Obot et al., 2015; Ahmed and Rebaz, 2020; Omer et al., 2020; Rebaz et al., 2020).
The ionization energy (I) and electron affinity (A) of a chemical compound are related to the energy of the HOMO orbital (Eq. (2)) and the energy of the LUMO orbital (Eq. (3)), respectively, according to the DFT theorem presented by Koopmans (Koopmans, 1934).
In DFT theory chemical variables like electronegativity (χ), chemical potential (μ), and global chemical hardness (η) can be described as the derivative of the molecule electronic energy (E) in relation to the number of total electrons (N) at a constant external potential (υ) (Parr et al., 1978; Parr and Pearson, 1983; Chermette, 1999). The following are the mathematical expressions:
According to Iczkowski and Margrave's (Madkour et al., 2018), electronegativity and chemical potential can be described as a function of the ionization energy and electron affinity (Eq. (6)). Janak's theorem and the valence state parabola model can be used to calculate global chemical hardness (Eq. (7)) (Janak, 1978; Von Szentpály, 1991).
The inverse of global chemical hardness is described as global softness (S) (Yang and Parr, 1985) (Eq. (8)).
The global electrophilicity index (ω) was proposed by Parr et al. (Raissi et al., 2021), which is defined as the susceptibility of a chemical species (atom, molecule, or ion) to accept electronic density. The higher the value of ω the better the electrophilicity of the species. As a result, lower values of ω describe a good nucleophile. Nucleophilicity (ε) is the inverse of the global electrophilicity index (Toro-Labbé, 2006; Marinho et al., 2021).
2.3 Synthesis of ETZC
In 30 ml of dry acetone, the potassium carbonate (K2CO3) (0.02 mol) was dissolved, added 4-(chloromethyl)-7,8-dimethyl-2H-chromen-2-one (0.02 mol). At room temperature, 4-Ethyl-5-(thiophene-2-yl)-4H-1,2,4-triazole-3-thiol (0.02 mol) was added dropwise to this solution for 6 h. The solid product was filtrated and dried with recrystallized by ethyl alcohol. FT-IR, 1H NMR, and 13C NMR techniques were used to illuminate the structure of the product. Fig. 2 depicts the general reaction of the product.
Yield is 75%; Melting point 175–177 °C; FT-IR (KBr, cm−1, ʋ): 2920–3074 (ʋAr-H), 1723(ʋC⚌O), 1623–1382 (ʋC⚌C), 1346–1474 (ʋC—O), 753 (ʋC—S); 1H NMR (400 MHz, DMSO‑d6, δ, ppm): 1.17 (t, J = 7.2 Hz, 3H, N-CH2- CH3), 2.38 (s, 3H, Ar-CH3), 3.29 (s, 3H, Ar-CH3), 4.08 (q, J = 7.1 Hz, 2H, N-CH2-CH3), 4.61 (s, 2H, S-CH2), 6.35 (s,1H, H-C—C⚌O), 7.27 (m, 2H, Ar-H), 7.55 (t, 1H, thiophene H), 7.68 (d, J = 3.6 Hz, 1H, thiophene-H), 7.81 (d, J = 5.1 Hz, 1H, thiophene-H); 13C NMR (100 MHz, DMSO‑d6, δ, ppm): 11.71, 15.31, 20.37, 33.83, 40.23, 114.29, 115.79, 122.51, 124.46, 126.07, 128.07, 128.80, 129.60, 142.15, 149.43, 150.32, 151.75, 151.93, 160.16, 160.16. Molecular Weight (C20H19N3O2S2): 397 g/mol.
3 Results and discussion
3.1 Infrared (IR) spectroscopy
Calculated harmonic frequencies are frequently greater than experimental values, leading to an overestimation of vibrational frequencies. The expected vibrations roughly reflect the experimental data after scaling, and the error distributions closely resemble the experimental frequencies. When a proper scaling factor is used to correct the calculated frequencies to compensate for the approximate treatment of electron correlation, anharmonicity effects, and basis set deficiencies, DFT calculations on harmonic frequencies have provided excellent vibrational frequencies for organic compounds (Omer and Anwer, 2021; Dennington et al., 2003). To correct the theoretical error, the theoretical harmonic frequencies were computed with B3LYP/cc-pVDZ and scaled by the classical factor of 0.979 according to the literature (Foresman and Frish, 1996). As demonstrated in Fig. 1, the computed spectra are more regular than the real FT-IR, and several bands reported in the predicted IR spectra were not found in the experimental spectra. This is because several other modes of infrared intensities are greatly altered as a result of multiple vibrations in the condensed phase. The vibrations of the C⚌C—C, and C—H rings are extensively utilized to quickly identify the presence of aromatic and thiophene rings in a structure. When comparing the aromatic C—H stretch to the aliphatic C—H stretch, the aromatic C—H stretch shows as a spate of weak to mediocre bands above 3000 cm−1 (Coates, 2000).
In this study using the B3LYP/ccp-VDZ method, the C—H stretching vibration FTIR spectra for thiophene was measured at 2974–3423 cm−1, which was theoretically in the range of 3030.32–3250.46 cm−1. C—H stretching vibration values for ethane groups would be lower than aromatic C—H ring stretching vibration values. According to the literature (Furić et al., 1992; Sajan et al., 2004) the symmetrical and asymmetrical CH2 stretching vibrations have ranges of 2800–2900 cm−1 and 2900–3000 cm−1, respectively. In the experimental IR spectra, the Ar-H stretching mode of the title molecule was found at frequencies between 2920 and 3074 cm−1. Wavenumbers in the range of 3026.26 3078.97 cm−1 were assigned to Ar-H stretching vibrations using the B3LYP/cc-pVDZ method, as shown in Table 1. The carbonyl characteristic frequency is a significant in the region of 1650–1850 cm-l that can be employed in a variety of applications (Omer et al., 2022). The C⚌O stretching vibration was detected at 1695, 1705, and 1712 cm−1 in the previous literature (Cansız et al., 2012; Koparir et al., 2022). In this study C⚌O stretching vibration has a band at 1723 cm−1 in the experimental IR spectra. The band was given a value of 1827.79 cm−1 in the computation, which is 104.79 cm−1 higher. Experiments have been performed to determine the C⚌N stretching modes of triazole derivatives at 1541 cm−1 (Koparir et al., 2013), 1592 cm−1 (Rebaz et al., 2020), and 1600 cm−1 (Panicker et al., 2015). Using the B3LYP method, the stretching vibrations of C⚌N in triazole were measured at 1458.93 cm−1, whereas the experimental value was 1453 cm−1. The stretching modes of C⚌C and C—O in the title compound were measured experimentally to be 1382–1623 and 1133 cm−1, respectively, which agrees with the theoretical value 1604.61–1524.79 and 1150.66 cm−1. In the title compound, the measured value for C—S-C stretching modes was 753 cm−1, which corresponded to the theoretical value of 773.57 cm−1. The vibration bands of the triazole ring of the molecule do not differ significantly from those of previously identified triazole derivatives (Cansız et al., 2012; Cansız et al., 2012; Koparir et al., 2013; Orek et al., 2018). With a correlation coefficient of 0.9978, the correlation graph between experimental and theoretical IR vibrational frequencies of the molecule is presented in (S1). The experimental and theoretical frequencies are found to be very similar. TV: Starching vibration, UN: anti, S: Symmetrical, RO: Rocking, SCI: Scissoring.
Assignments With TED
FT-IR (cm−1)
With KBrUnscaled Frequencies B3LYP/cc-pVDZ
Assignments With TED
FT-IR (cm−1) With KBr
Unscaled Frequencies B3LYP/cc-pVDZ
ST-CH in thiophene
3423
3250.46
UNSTV-CH3 in ring
1474
1450.90
STV-C28H31
3235.56
STV-C19-N18
1449
1420.16
STV-C1H7
3217.56
STV-C19=N17, C14-N18
1382
1394.37
UNST-CH in thiophene
3212.91
RO-ethylene group
1374.77
STV-C8H6
3208.86
STV-C=C in ring
1365.36
STV-C37H38
3182.11
STV-C in thiophene
1364.89
STV-C39H48
3165.37
STV-C4-O9
1262
1293.64
UNSTV-ethylene group
3131.27
RO-C11H12H13
1278.11
UNSTV-C43H44H45H46
3123.67
STV-CH in thiophene
1237.26
UNSTV-C23H24H25H26
3121.52
SCI-CH in ring
1216
1200.97
UNSTV-ethylene group
3110.01
STV-C5-O9
1180
1180.65
USTV-CH3 in ring
3081.75
RO-C11H12H13
1173.91
USTV-CH3 in ring
3074
3078.97
STV-ethylene group
1133
1150.66
STV-C11H12H13
3071.04
STV-N16-N17
1111.83
STV-C20H21H22
3062.17
RO-CH3 in ethylene
1099.13
STV-C23H24H25H26
3042.23
SCI-CH in thiophene
1088
1089.56
STV-C39H40H41H42
2974
3030.32
STV-C28-C30
1077.02
STV-C43H44H45H46
2920
3026.26
UNSTV-CH3 in ring group
1060.14, 1027.79,
STV-C5=O10
1723
1827.79
STV-C14-N18
1019
1022.03
STVC1=C2
1623
1678.7
STV-C20-C23
960
976.78
STV-C=C in ring
1604
1651.18
STV-C5-O9
940
953.41
STV-C=C in thiophene
1611.10
RO-C11H12H13
927.34
STV- C=C in ring
1604.61, 1524.79
UNSTV-CH in thiophene
900
913.48
STV-C30=C32 in thiophene
1521.68
STV-C32-S29
851
853.11
SCI-C20H21H22
1493.90
STV-C11-S15
816
798.06
SCI-C23H24H25
1478.66
STV-C11-S15
753
773.57
RO-C39H40H41H42
1471.30
STV-N16-C14-N18
712
722.85
STV-C19=N17, C14=N16
1467.90
STV-C11-S15
716.10
RO-CH3 in ring
1465.72
STV-CH in thiophene
710.74
STV-C23H24H25H26
1461.52
STV-N16-N17
693.79
STV-C19=N17
1453
1458.93
STV-C19-N18
685.47
3.2 1H and 13C nuclear magnetic resonance studies
NMR spectroscopy (nuclear magnetic resonance spectroscopy) is a technology that can be used to determine the structure of organic molecules. Quantum chemical computations have been proven to be sufficient for predicting NMR spectra and investigating the link between molecule structure and chemical shifts (Dennington et al., 2009; Daoud et al., 2017; Ahmed and Rebaz, 2019; Rebaz et al., 2022). As a result, using a combination of experimental and theoretical approaches to understand and predict molecular structure is tremendously beneficial. The 1H and 13C NMR spectra of ETZC (Fig. 2) in DMSO‑d6 were determined experimentally. The B3LYP/cc-pVDZ approach with the Gaussian 09 program was used to estimate theoretical 1H- and 13C NMR spectra, and the standard gauge-including atomic orbital (GIAO) strategy was used to optimize the geometry of the title chemical. The 1H NMR chemical shifts in gas phase ranged from 0.00 (Reference) to 9.00 ppm, whereas the experimental chemical shifts in DMSO‑d6 ranged from 0.00 to 8.00 ppm (see Fig. 3).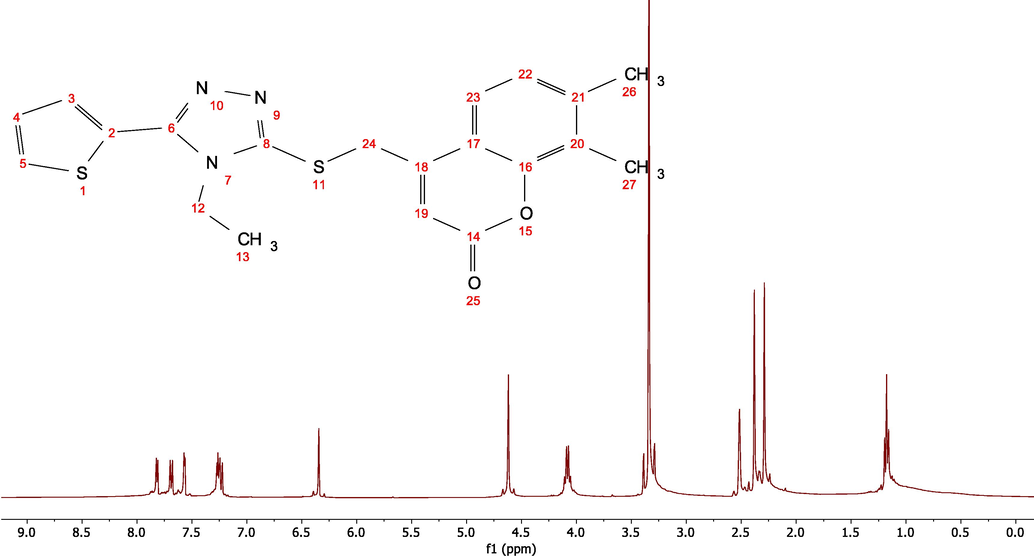
Experimental 1H NMR spectrum for the ETZC in DMSO‑d6.
The experimental 1H NMR spectra ascribed the hydrogen (H34) of the thiophene group to the doublet peak at 7.81 ppm. The theoretical proton resonance signal of this hydrogen (H34) was obtained at 7.66 ppm in the gas phase using the B3LYP/cc-pVDZ method. The aromatic proton's chemical shift value (H6 and H38) was determined to be 7.27 ppm experimentally and 7.72 and 7.46 ppm theoretically. The other doublet pack for thiophene hydrogen (H31) was determined to be 7.68 ppm experimentally, and 7.36 ppm computationally. In the gas phase, the triplet pack proton for hydrogen (H33) of thiophene was assigned at 7.55 ppm and computed as 7.35 ppm. In experiments, the single pack for (s,1H, H—C—C⚌O) was determined to be 6.35 ppm, but in computational analysis, it was found to be 6.34 ppm. The obtained experimentally single peak chemical shift for (s, 2H, S—CH2) was 4.61 ppm, but the computed values were 5.14 and 4.83 ppm, respectively. In the experiment, the quartet pack for (q, 2H, N—CH2—CH3) linked to the triazole ring was measured at 4.08 ppm, while computed observed at 4.47 and 4.28 ppm. The two single packs for (s, 3H, Ar—CH3) and (s, 3H, Ar—CH3) were assigned experimentally at 3.29 and 2.38 ppm, respectively, while the hydrogen packs were observed theoretically at a range of 2.30–3.87 ppm, see Table 2.
H number
Experimental Results
Theoretical Results
H34
7.81
7.66
H31
7.68
7.36
H6
7.27
7.72
H33
7.55
7.35
H38
7.27
7.46
H7
6.35
6.34
H13
4.61
5.14
H12
4.61
4.83
H22
4.08
4.47
H21
4.08
4.28
H40
3.29
3.87
H45
3.29
2.82
H44
3.29
2.81
H46
2.38
2.55
H41
2.38
2.31
H42
2.38
2.3
H25
1.17
2.22
H24
1.17
1.95
H26
1.17
1.74
The compoundv13C NMR resonance signals were detected in DMSO‑d6 between 11.71 and 160.16 ppm (Fig. 4) and in the gas phase between 20.55 and 160.78 ppm using the B3LYP/cc-pVDZ method with the Gaussian 09 program. Using 13C NMR, it was discovered that the chemical shift values of the C19, C4, and C14 atoms, which are close to the electronegative oxygen, as well as the sulfur atoms, are greater than those of the other carbons in the molecule. The 13C NMR resonance signals in DMSO‑d6 were found to be 160.16 ppm for both (C19 and C5) and 151.93 ppm for (C4), respectively, whereas their predicted values in the gas phase were 160.78 and 160.61 ppm for both (C19 and C5) and 160.11 ppm for (C4). Coumarin carbon shifts (C1, C2, C3, C8, C37, C36, and C35) were measured experimentally at 114.29, 150.32, 115.79, 124.46, 128.07, 149.43 and 128.80 ppm, while mesitylene carbon shifts were discovered at 120.24, 156.30, 125.97, 127.51, 131.73, 150.26 and 134.69 ppm. The carbon signals for thiophene (C7, C28, C30, and C32) were observed at 142.15, 122.51, 126.07, and 129.60 ppm, while the theoretical signals were observed at 147.21, 126.52, 131.19, and 139.71 ppm, respectively. The carbon chemical shift S-C11 was measured in the lab at 40.23 ppm and in theory at 49.51 ppm. The carbon chemical shifts for ethane, which is representative of the C20 and C23, were obtained experimentally at 33.83 and 15.31 ppm, respectively, whereas in the gas phase the carbon chemical shifts were detected at 49.00 and 24.28 ppm. The experimentally chemical shifts for H3C-Ar-CH3 Cs, which are representative for the C43 and C39, are 20.37 and 11.71 ppm, respectively, while theoretically found in the gas phase at 30.28 and 20.55 ppm. The experimental and theoretical 13C NMR chemical shift results are shown in Table 3. The proposed structure was validated by the experimental and theoretical 1H- and 13C NMR chemical shift values for the title molecule. The correlation between experimental and theoretical chemical shift values for 1H- and 13C NMR studies, with correlation coefficients of 0.9849 and 0.9984, respectively show in S2 and S3.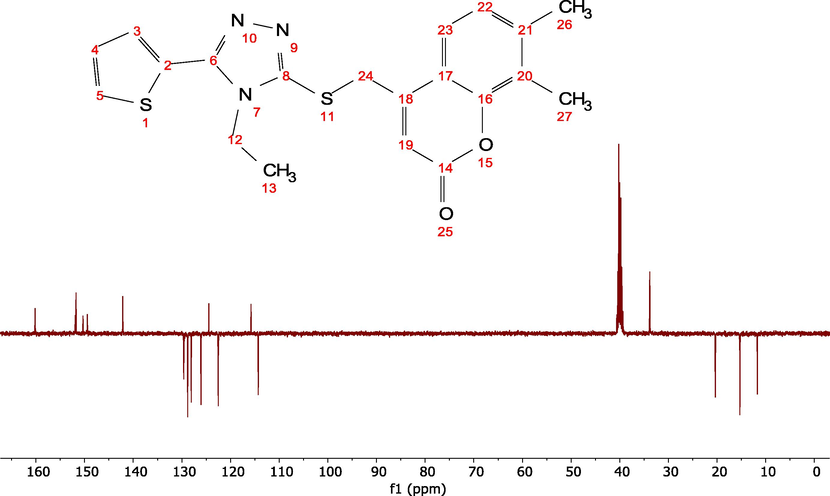
The experimental 13C NMR spectrum for the ETZC in DMSO‑d6.
C number
Experimental Results
Theoretical Results
C19
160.16
160.78
C5
160.16
160.61
C4
151.93
160.11
C14
151.75
157.62
C2
150.32
156.3
C36
149.43
150.26
C27
142.15
147.21
C32
129.60
139.71
C35
128.80
134.69
C37
128.07
131.73
C30
126.07
131.19
C8
124.46
127.51
C28
122.51
126.52
C3
115.79
125.97
C1
114.29
120.24
C11
40.23
49.51
C20
33.83
49
C43
20.37
30.4
C23
15.31
24.28
C39
11.71
20.55
3.3 Geometrical analysis
The molecular structure of chemical 3 was optimized using the DFT/cc-pVDZ method, and the molecule's structural characteristics in the gaseous phase were found to be optimal. The computed structural properties linked to the molecule's stable state are shown in Table 4, and the molecular structure of ETZC is shown in Fig. 5. Compound 3 is made up of the rings Thiophene (Thp: C27—C28— C30—C32—S29), Triazole (Trz: N18—C14—N16—N17—C19), and Coumarin (Com: C1—C2—C3—C8—C37—C36—C35—C4—O9—C5). The C⚌C bond lengths of Thp rings range from 1.371–1.382 A0, while from the Com ring the bond length for C⚌C is 1.357 A0. Because the oxygen atom is more electronegative than the carbon atom, the electrons in the C⚌O bond are removed from oxygen to the carbon atom, as a result of this fact, The C5⚌O10 bond shortens 1.207 A0 while the C5—C1 bond lengthens 1.456 A0. In the literature (Zhang et al., 2013; Ezhilarasu and Balasubramanian, 2016; Çelik et al., 2021), the C⚌O and C—C bond lengths were found as single-crystal structure molecules with similar results. When the literature's C⚌O and C—C bond length values are compared to the optimized compound estimated values, they appear to be in agreement. The bond angles of O9—C4—C3 and O10—C5—C1 are 121.082 and 126.079 A0, respectively. The C—C—O bond angles are 126.07 and 125.79 A0, according to the previous literature (Ezhilarasu and Balasubramanian, 2016). The C—C—O bond angles in the literature are slightly different from the computed angle values because the calculated values refer to the gaseous state of the molecule, whilst the supplied values refer to the solid phase of the molecule, also the intermolecular interactions in the solid phase calculation happened. The bond lengths between N18–N19 atoms in the Trz ring are 1.372 A0. In the literature, 1.354 A0 (Gilandoust et al., 2016) and 1.354 A0 (Kumar et al., 2015) have been reported as experimental values for molecules containing 1,2,3 triazole rings in the solid phase. The dihedral angles between the Thp, Trz, and Com rings in the molecule were discovered to be Trz/Com = 4.500, Thp/Com = 103.324, and Php/Trz = −178.554 in the molecule. The structural properties of the molecule appear to be in agreement with experimental structure parameters published in the literature. The phase difference of the molecules accounts for the small variations between calculated and experimental values in the literature. Finally, the methodologies and basis set suitability for optimization are determined by the agreement of the estimated values with experimental values in the literature.
Symbol
Bond Length
Symbol
Bond angle
Symbol
Dihedral angle
C2-C1
1.357
C3-C2-C1
118.945
C4-C3-C2-C1
−0.070
C3-C2
1.459
C4-C3-C2
118.104
C5-C1-C2-C3
0.016
C4-C3
1.411
C5-C1-C2
122.840
C8-C3-C2-C1
179.900
C5-C1
1.456
C8-C3-C2
124.071
O9-C4-C3-C2
0.025
C8-C3
1.408
O9-C4-C3
121.082
O10-C5-C1-C2
−179.964
O9-C4
1.368
O10-C5-C1
126.079
C11-C2-C1-C5
−179.836
O10-C5
1.207
C11-C2-C1
123.678
C14-C11-C2-C1
−2.263
C11-C2
1.506
C14-C11-C2
164.731
S15-C11-C2-C1
−1.336
C14-C11
4.768
S15-C11-C2
113.702
N16-C14-C11-C2
−173.209
S15-C11
1.841
N16-C14-C11
58.971
N17-N16-C14-C11
2.191
N16-C14
1.321
N17-N16-C14
108.231
N18-C14-C11-C2
4.500
N17-N16
1.372
N18-C14-C11
50.930
C19-N17-N16-C14
−0.068
N18-C14
1.387
C19-N17-N16
107.013
C20-N18-C14-C11
174.510
C19-N17
1.316
C20-N18-C14
129.957
C23-C20-N18-C14
96.000
C20-N18
1.463
C23-C20-N18
113.334
C27-C14-C11-C2
62.642
C23-C20
1.528
C27-C14-C11
176.185
C28-C27-C14-C11
−78.472
C27-C14
1.454
C28-C27-C14
131.917
S29-C27-C14-C11
103.324
C28-C27
1.382
S29-C27-C14
117.434
C30-C28-C27-C14
−178.554
S29-C27
1.754
C30-C28-C27
113.200
C32-C30-C28-C27
0.155
C30-C28
1.425
C32-C30-C28
112.645
C35-C4-C3-C2
179.986
C32-C30
1.371
C35-C4-C3
122.521
C36-C35-C4-C3
−0.020
C35-C4
1.407
C36-C35-C4
118.202
C37-C8-C3-C2
−179.970
C36-C35
1.408
C37-C8-C3
120.408
C39-C35-C4-C3
−179.987
C37-C8
1.387
C39-C35-C4
120.622
C43-C36-C35-C4
−179.936
C39-C35
1.508
C43-C36-C35
120.216
C43-C36
1.507
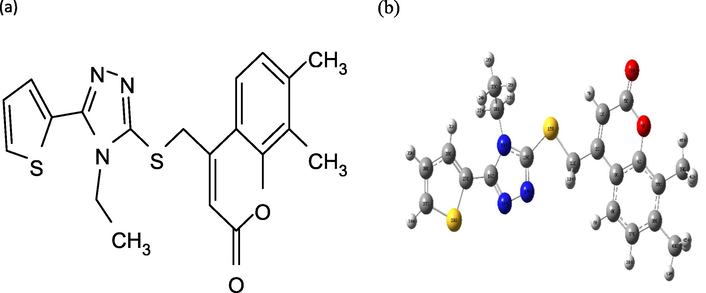
Structure of the ETZC A) Drawing by ChemBioDraw Ultra B) Gaussian Optimized by B3LYP/cc-pVDZ.
3.4 The energy of HOMO and LUMO
The chemical stability of a molecule is determined by the bandgap between the highest occupied molecular orbital (HOMO) and the lowest occupied molecular orbital (LUMO). The molecular orbitals influence optical and electrical properties, as well as the electronic absorption spectra (Tarı et al., 2015). The HOMO and LUMO energies of the molecule were estimated in the gas phase using the B3LYP/cc-pVDZ method, and 3D plots of the HOMO and LUMO were shown in Fig. 6. The estimated HOMO and LUMO energies for the title molecule were −0.2155 Hz (-5.866 eV) and −0.06817 Hz (−1.870 eV), respectively. The HOMO of the investigated molecule is concentrated on the sulfur atom, triazole ring, and thiophene ring, whereas the LUMO is concentrated on the coumarin ring. The energy gap (ΔE) between the HOMO and the LUMO was calculated to be 0.1468 Hartree (3.996 eV), as shown in Fig. 6. As a result, the density of states (DOS) in the gas phase was calculated using the Gauss Sum 3.0 software. Fig. 7 depicts the density of states diagram for the title compound.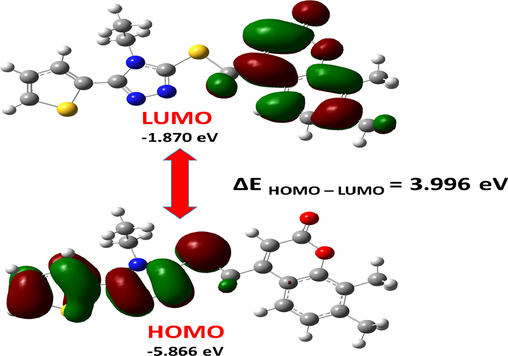
Energy levels and views on the electronic isosurfaces of the HOMO and LUMO of the ground state of the ETZC, obtained by using the B3LYP/cc-pVDZ method.
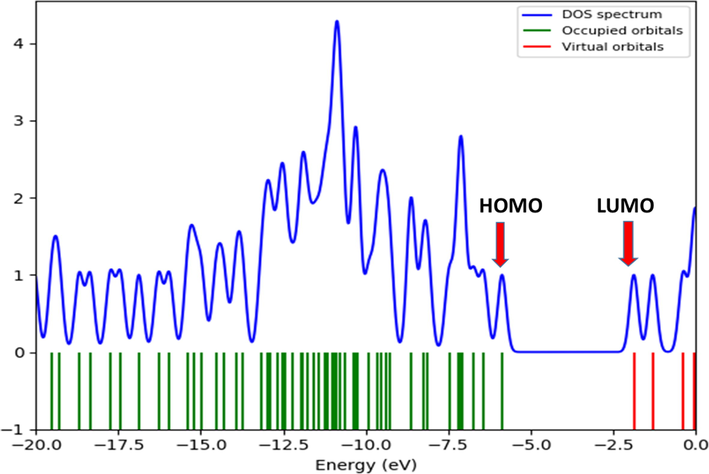
Diagrams depicting the density states for the ETZC.
3.5 Mulliken charges distributions
Because atomic charges have such a large impact on the electrical properties and reactivity of molecules, estimating Mulliken atomic charges is critical in quantum chemical calculations for molecular systems (Mathammal et al., 2016). The B3LYP/cc-pVDZ level was used to compute the Mulliken atomic charge distributions on the title compound in the gas phase, as shown in Fig. 8. In most cases, the positive and negative charges on the atoms of the molecule were roughly equal. The Mulliken atomic charges distribution on the electronegative atoms N16, N17, and N18 were calculated to be −0.188720, −0.200962, −0.239788. Also, it has been observed that the C9 and C10 carbon atoms in the coumarin ring have a negative charge of −0.2322 and −0.206369 a.u., respectively.
Millikan atoms charge distribution on the title compound (ETZC).
3.6 Inhibitor study
The electronic structural properties and total energy of the title molecule are shown in Table 5. Table 5 and Fig. 6 show that the energies of HOMO and LUMO for the structures studied are −5.866 and −1.870 eV, respectively. The ability to donate electrons is related to HOMO, which is important for corrosion study. The mechanism of charge transfers over the metal surface, which allows for adsorption related to the HOMO levels rise, the inhibitory effect of inhibitor compounds rises as well (Chen et al., 2014). The high EHOMO value of our molecule increases its inhibitory activity. The LUMO property refers to the ability to accept an electron. The low value of ELUMO indicates that the inhibitor (compound) will give the metal surface a negative charge. The LUMO energy value of our molecule is lower. Inhibitors with high HOMO energy values had lower LUMO energy values, indicating that they were active. The title compound has the highest corrosion inhibition activity based on HOMO-LUMO capacity.
Parameters
Equations
Results
Total Energy (a.u)
−1884.743
μ (D)
2.703
ELUMO (eV)
−1.870
EHOMO (eV)
−5.866
ΔE (eV)
3.996
I
I = -EHOMO
5.866
A
A = -ELUMO
1.870
χ (eV)
χ = (I + A) / 2
3.868
ɳ (eV)
ɳ = (I – A) / 2
1.998
σ (eV)
σ = I/ɳ
2.935
Pi (eV)
Pi = -χ
−3.868
ω (eV)
ω = Pi2/2ɳ
3.744
ε (eV)
ε = Pi. ɳ
7.729
ΔN
ΔN = (χmetal - χinhibitor) / 2. (ɳmetal - ɳinhibitor)
0.783
The difference in energy between EHOMO and ELUMO has important implications for theoretical inhibitory efficiency and static molecular reactivity. In inhibitor studies, comparing ΔE is useful because the shorter the energy distances, the more effective the inhibition. When compared to the ELUMO used for corrosion inhibitors research, the EHOMO is more related to the discovered ΔE value. Because the inhibitor derivatives have a high HOMO energy and a low ΔE, they can be used as anti-corrosion compounds (Chen et al., 2014; Beytur et al., 2019). The title chemical would have a significant inhibitory effect based on its maximum HOMO energy and lowest ΔE value (see Figs. 6 & 7).
The Mulliken atomic charge distribution is a popular technique for estimating inhibitor adsorption sites. Many scientists believe that when negatively charged heteroatoms are present, they have a greater ability to adsorb on metal surfaces via the donor–acceptor process (El Adnani et al., 2013; Omer and Anwer, 2020). Fig. 8 depicts the charge distribution on the atoms of our compound inhibitors. When compared to a positive charge, the number of negative charges accounts for one-quarter of the total atoms in our compound, this is an excellent example of our title chemical's anti-corrosion properties.
When evaluating an inhibitor's stability, reactivity, and inhibitor efficacy, Hardness (ɳ) and softness (σ) are important parameters to consider. Organic molecules that are Lewis-based and soft outperform hard molecules in terms of anti-corrosion activity. Organic molecules that are Lewis-based and soft have better anti-corrosion activity than hard molecules (Alexander and Moccari, 1993). Our compound inhibitors have a high softness and a low hardness due to their high EHOMO and low ΔE. Table 5 demonstrates that the title chemical has the highest inhibitory activity, with values of ɳ and σ. In order to evaluate inhibitor activity, the factors electronegativity (χ) and chemical potential (Pi) are also investigated. The calculated (χ) values indicate that the metal and the inhibitor have a coordinated covalent bond. This study looked at the corrosion inhibition behavior of compounds designed to be iron metal inhibitors. Table 5 demonstrates that the theoretical (χ) value for the title compound is less than the theoretical (χ) value for the iron bulk metal. The iron metal was able to form bonds by absorbing electrons from the inhibitor molecule. Because of its higher value of (χ), the title chemical inhibitor will be the most effective corrosion inhibitor in comparison to the literature (Martinez, 2003; Issa et al., 2008). The dipole moment (μ) is another property mentioned in Table 5. No previous research has discovered a direct relationship between dipole moment and inhibitory action. The inhibitory activity of the molecules increases as the value of (μ) increases in the same study, whereas inhibition impact decreases as the value of (μ) decreases in the other literature(Musa et al., 2012; Chen et al., 2014; Beytur et al., 2019). Our compound has a dipole moment of 2.703 D.
The electrophilicity index (ω) and the nucleophilicity index (ε) are two important parameters to consider when evaluating corrosion inhibitors. Inhibitor molecules' ability to absorb electrons is measured by their (ω), whereas their ability to donate electrons is measured by their (ε). The inhibition activity increases as the value of (ε) increases, and decreases as the value of (ω) decrease (Erkan et al., 2014; Karakus and Sayin, 2015). The value of (ε) has increased in our molecule study, while the value of (ω) has decreased. According to ε and ω measurements, our molecule has the most potent inhibitory activity. According to the ΔN findings (Table 5), the title molecule has a good inhibitor and can transfer more electrons to iron metal surfaces.
4 Conclusion
The title compound was synthesized and tested using FT-IR, 1H-, and 13C NMR spectroscopic techniques. The Gaussian 09 program was used at the B3LYP/cc-pVDZ level of the theory to construct the compound optimal shape, IR vibration frequencies, 1H- and 13C NMR chemical shifts, Mulliken atomic charges and HOMO-LUMO energies. Theoretical FT-IR, 1H-, and 13C NMR spectroscopy results were presented and compared to experimental data. The theoretical and experimental results were found to be compatible. The proposed molecule structure is supported by both theoretical and experimental evidence. The DFT method was used to calculate the HOMO and LUMO energies of the molecule, and the energy gap between HOMO and LUMO was found to be 3.996 eV, indicating that the molecule has an inhibitory function. The charge transfers from thiophene and triazole to the coumarin ring were viewable in the HOMO and LUMO 3D graphs. The Mulliken population method was used to compute the charge distributions of the molecules. The bigger positive charge was distributed on the atoms of a molecule in dissimilarity to a negative charge. When considering a compound inhibitor's atomic charges, the electronegative atoms are expected to have a significant impact on inhibitory efficacy. According to the molecular charge distribution analysis, the most negative potential sites were found on the N16, N17, N18, O9, and O10, while the most positive potential sites were found around the hydrogen atoms. The calculated values of ƞ, ω, σ, ε, Pi and χ confirm that the title chemical is the most effective inhibitor. The value of ΔN will increase the effect of corrosion will increase due to better absorption on the surface of the metal. As a result, this research should help with the design, synthesis, and corrosive activity of novel materials like the compound.
Acknowledgements
This work is a part of the Ph.D. thesis for the corresponding author at Firat University in turkey.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1H-Pyrrole, Furan, and Thiophene Molecule Corrosion Inhibitor Behaviors. J. Phys. Chem. Funct. Mater.. 2021;4:1-4.
- [Google Scholar]
- The Role of the Various Solvent Polarities on Piperine Reactivity and Stability. J. Phys. Chem. Funct. Mater.. 2021;4:10-16.
- [Google Scholar]
- A theoretical study on Dopamine molecule. J. Phys. Chem. Funct. Mater.. 2019;2:66-72.
- [Google Scholar]
- Spectroscopic properties of Vitamin C: A theoretical work. Cumhuriyet Sci. J... 2020;41:916-928.
- [Google Scholar]
- Evaluation of corrosion inhibitors for component cooling water systems. Corrosion.. 1993;49
- [Google Scholar]
- Exploration of Chromone-Based Thiosemicarbazone Derivatives: SC-XRD/DFT, Spectral (IR, UV–Vis) Characterization, and Quantum Chemical Analysis. ACS Omega. 2020;5:30176-30188.
- [Google Scholar]
- Synthesis of some new 1, 2, 4-triazoles, their Mannich and Schiff bases and evaluation of their antimicrobial activities. Eur. J. Med. Chem.. 2009;44:1057-1066.
- [Google Scholar]
- A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys.. 1993;98:1372-1377.
- [Google Scholar]
- A real-space model of nondynamical correlation. J. Chem. Phys.. 2003;119:2972-2977.
- [Google Scholar]
- Synthesis, characterization and theoretical determination of corrosion inhibitor activities of some new 4, 5-dihydro-1H-1, 2, 4-Triazol-5-one derivatives. Heliyon.. 2019;5:e01809
- [Google Scholar]
- Synthesis of some tetrazole fused pyrido [2, 3-c] coumarin derivatives from a one-pot three-component reaction via intramolecular 1, 3-dipolar cycloaddition reaction of azide to nitriles. Tetrahedron Lett.. 2012;53:5034-5037.
- [Google Scholar]
- Simple coumarins and analogues in medicinal chemistry: occurrence, synthesis and biological activity. Curr. Med. Chem.. 2005;12:887-916.
- [Google Scholar]
- Molecular electrostatic potential surface, HOMO–LUMO, and computational analysis of synthetic drug Rilpivirine. Int. J. Sci. Eng. Res.. 2016;7:315-319.
- [Google Scholar]
- A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys.. 1997;107:3032-3041.
- [Google Scholar]
- 6-Phenyl-3-(4-pyridyl)-1, 2, 4-triazolo-[3, 4-b][1, 3, 4] thiadiazole: Synthesis, experimental, theoretical characterization and biological activities. Spectrochim. Acta Part A Mol. Biomol. Spectrosc.. 2012;97:606-615.
- [Google Scholar]
- 4-Allyl-5-pyridin-4-yl-2, 4-dihydro-3H-1, 2, 4-triazole-3-thione: Synthesis, experimental and theoretical characterization. Spectrochim. Acta Part A Mol. Biomol. Spectrosc.. 2012;91:136-145.
- [Google Scholar]
- 1, 2, 3-triazole derivative: Synthesis, characterization, DFT, molecular docking study and antibacterial-antileishmanial activities. J. Indian Chem. Soc.. 2021;98:100105
- [Google Scholar]
- Quantum chemical study of some benzimidazole and its derivatives as corrosion ınhibitors of steel in HCl solution. Int. J. Electrochem. Sci.. 2014;9:5400-5408.
- [Google Scholar]
- Chemical reactivity indexes in density functional theory. J. Comput. Chem.. 1999;20:129-154.
- [Google Scholar]
- Natural products: a continuing source of novel drug leads. Biochimica et Biophysica Acta (BBA)-General Subjects.. 2013;1830:3670-3695.
- [Google Scholar]
- Experimental and theoretical study of a novel synthesized thiophene compound. J. Mol. Struct.. 2017;1137:50-59.
- [Google Scholar]
- Synthesis and antimicrobial activities of some new 1-(5-phenylamino-[1, 3, 4] thiadiazol-2-yl) methyl-5-oxo-[1, 2, 4] triazole and 1-(4-phenyl-5-thioxo-[1, 2, 4] triazol-3-yl) methyl-5-oxo-[1, 2, 4] triazole derivatives. Eur. J. Med. Chem.. 2004;39:793-804.
- [Google Scholar]
- Synthesis of 3-alkyl (Aryl)-4-alkylidenamino-4, 5-dihydro-1H-1, 2, 4-triazol-5-ones and 3-alkyl-4-alkylamino-4, 5-dihydro-1H-1, 2, 4-triazol-5-ones as antitumor agents. Bioorg. Med. Chem.. 2002;10:3717-3723.
- [Google Scholar]
- Dennington, R., Keith, T., Millam, J., 2009. GaussView, version 5.
- Dennington, R., Keith, T., Millam, J., et al., 2003. GaussView, Version 3.09, Semichem. Inc., Shawnee Mission, KS. 2, 1.
- Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys.. 1971;54:724-728.
- [Google Scholar]
- DFT theoretical study of 7-R-3methylquinoxalin-2 (1H)-thiones (RH; CH3; Cl) as corrosion inhibitors in hydrochloric acid. Corros. Sci.. 2013;68:223-230.
- [Google Scholar]
- Synthesis and biological activity of some new coumarins. Egypt. J. Pharm. Sci.. 1992;33:639-650.
- [Google Scholar]
- Theoretical studies on eight oxovanadium (IV) complexes with salicylaldehyde and aniline ligands. Hacettepe J Biol Chem.. 2014;42:337-342.
- [Google Scholar]
- Exploring chemistry. Pittsburg, USA: Gaussian Inc.; 1996.
- Raman spectroscopic study of H2O and D2O water solutions of glycine. J. Mol. Struct.. 1992;267:39-44.
- [Google Scholar]
- Gaussian09, R.A., 2009. 1, mj frisch, gw trucks, hb schlegel, ge scuseria, ma robb, jr cheeseman, g. Scalmani, v. Barone, b. Mennucci, ga petersson et al., gaussian. Inc., Wallingford CT. 121, pp. 150–166.
- 1-(2′-Ethoxy-4′-fluoro-[1, 1′-biphenyl]-4-yl)-4-phenyl-1H-1, 2, 3-triazole. IUCrData.. 2016;1:x161712
- [Google Scholar]
- Supramolecular binding thermodynamics by dispersion-corrected density functional theory. Chem.–A Eur. J.. 2012;18:9955-9964.
- [Google Scholar]
- Antimicrobial activities of some 4H–1, 2, 4-triazoles. Indian J. Pharmaceut. Sci.. 1999;61:271.
- [Google Scholar]
- Quantum chemical studies on the inhibition of corrosion of copper surface by substituted uracils. Appl. Surf. Sci.. 2008;255:2433-2441.
- [Google Scholar]
- Synthesis and cellular characterization of novel isoxazolo-and thiazolohydrazinylidene-chroman-2, 4-diones on cancer and non-cancer cell growth and death. Bioorg. Med. Chem.. 2014;22:2655-2661.
- [Google Scholar]
- The investigation of corrosion inhibition efficiency on some benzaldehyde thiosemicarbazones and their thiole tautomers: Computational study. J. Taiwan Inst. Chem. Eng.. 2015;48:95-102.
- [Google Scholar]
- Synthesis and antituberculosis activity of new N-phenyl-N′-[4-(5-alkyl/arylamino-1, 3, 4-thiadiazole-2-yl) phenyl] thioureas. Il Farmaco.. 2002;57:577-581.
- [Google Scholar]
- Synthesis and antiinflammatory activity of coumarin derivatives. J. Med. Chem.. 2005;48:6400-6408.
- [Google Scholar]
- Über die Zuordnung von Wellenfunktionen und Eigenwerten zu den einzelnen Elektronen eines Atoms. Physica. 1934;1:104-113.
- [Google Scholar]
- Synthesis, experimental, theoretical characterization and biological activities of 4-ethyl-5-(2-hydroxyphenyl)-2H-1, 2, 4-triazole-3 (4H)-thione. Spectrochim. Acta Part A Mol. Biomol. Spectrosc.. 2013;105:522-531.
- [Google Scholar]
- Synthesis, Characterization, and theoretical inhibitor study for (1E, 1'E)-2, 2'-thiobis (1-(3-mesityl-3-methylcyclobutyl) ethan-1-one) dioxime. El-Cezeri.. 2021;8:1495-1510.
- [Google Scholar]
- Synthesis, molecular characterization, biological and computational studies of new molecule contain 1, 2, 4-triazole, and Coumarin bearing 6, 8-dimethyl. Biointerface Res. Appl. Chem.. 2022;12:809-823.
- [Google Scholar]
- Synthesis and antiviral activity evaluation of some new 6-substituted 3-(1-adamantyl)-1, 2, 4-triazolo [3, 4-b][1, 3, 4] thiadiazoles. Il Farmaco.. 2002;57:253-257.
- [Google Scholar]
- Synthesis, characterization and antimicrobial evaluation of ethyl 2-arylhydrazono-3-oxobutyrates. Eur. J. Med. Chem.. 1999;34:153-160.
- [Google Scholar]
- Synthesis, characterization and pharmacological properties of some 4-arylhydrazono-2-pyrazoline-5-one derivatives obtained from heterocyclic amines. Eur. J. Med. Chem.. 2000;35:761-771.
- [Google Scholar]
- Crystal structure of 1-(4-fluorophenyl)-4-(4-methoxyphenyl)-1H-1, 2, 3-triazole. Acta Crystallogr. Section E: Crystallographic Commun.. 2015;71:o534
- [Google Scholar]
- XRD, vibrational spectra and quantum chemical studies of an anticancer drug: 6-Mercaptopurine. Spectrochim. Acta Part A Mol. Biomol. Spectrosc.. 2015;146:204-213.
- [Google Scholar]
- Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B.. 1988;37:785.
- [Google Scholar]
- Quantum chemical calculations, molecular dynamic (MD) simulations and experimental studies of using some azo dyes as corrosion inhibitors for iron. Part 2: Bis–azo dye derivatives. J. Mol. Struct.. 2018;1163:397-417.
- [Google Scholar]
- Quantum computational investigations and molecular docking studies on amentoflavone. Heliyon.. 2021;7:e06079
- [Google Scholar]
- Inhibitory mechanism of mimosa tannin using molecular modeling and substitutional adsorption isotherms. Mater. Chem. Phys.. 2003;77:97-102.
- [Google Scholar]
- Molecular structure, vibrational, UV, NMR, HOMO-LUMO, MEP, NLO, NBO analysis of 3, 5 di tert butyl 4 hydroxy benzoic acid. J. Mol. Struct.. 2016;1120:1-14.
- [Google Scholar]
- Molecular dynamic and quantum chemical calculations for phthalazine derivatives as corrosion inhibitors of mild steel in 1 M HCl. Corros. Sci.. 2012;56:176-183.
- [Google Scholar]
- Cytotoxic activity of new acetoxycoumarin derivatives in cancer cell lines. Anticancer Res.. 2011;31:2017-2022.
- [Google Scholar]
- Natural products as leads to potential drugs: an old process or the new hope for drug discovery? J. Med. Chem.. 2008;51:2589-2599.
- [Google Scholar]
- Natural product-like combinatorial libraries based on privileged structures. 1. General principles and solid-phase synthesis of benzopyrans. J. Am. Chem. Soc.. 2000;122:9939-9953.
- [Google Scholar]
- Density functional theory (DFT) as a powerful tool for designing new organic corrosion inhibitors. Part 1: an overview. Corros. Sci.. 2015;99:1-30.
- [Google Scholar]
- Population Analysis and UV-Vis spectra of Dopamine Molecule Using Gaussian 09. J. Phys. Chem. Functional Mater.. 2021;3:48-58.
- [Google Scholar]
- Population Analysis and UV-Vis spectra of Dopamine Molecule Using Gaussian 09. J. Phys. Chem. Funct. Mater.. 2020;3:48-58.
- [Google Scholar]
- Theoretical analysis of the reactivity of chloroquine and hydroxychloroquine. Indian J. Chemi.-Section A (IJCA).. 2020;59:1828-1834.
- [Google Scholar]
- Computational and spectroscopy study of melatonin. Indian J. Chem.-Section B (IJC-B). 2021;60:732-741.
- [Google Scholar]
- Characterization and Inhibitor Activity of Two Newly Synthesized Thiazole. J. Bio-and Tribo-Corrosion.. 2022;8:1-12.
- [Google Scholar]
- Synthesis, Structure Investigation, Spectral Properties and in Vitro Antioxidant Evaluation of New 1-(3-Methyl-3-Mesityl)-Cyclobutyl-2-(5-Thiophen-4-Ethyl-2h-[1, 2, 4] Triazol-3-Ylsulfanyl)-Ethanone. Fresenius Environ. Bull.. 2018;27:2992-3005.
- [Google Scholar]
- Amentoflavone and its derivatives as novel natural inhibitors of human Cathepsin B. Bioorg. Med. Chem.. 2005;13:5819-5825.
- [Google Scholar]
- FT-IR, HOMO–LUMO, NBO, MEP analysis and molecular docking study of 3-Methyl-4-{(E)-[4-(methylsulfanyl)-benzylidene] amino} 1H–1, 2, 4-triazole-5 (4H)-thione. Spectrochim. Acta Part A Mol. Biomol. Spectrosc.. 2015;151:198-207.
- [Google Scholar]
- Electronegativity: the density functional viewpoint. J. Chem. Phys.. 1978;68:3801-3807.
- [Google Scholar]
- Absolute hardness: companion parameter to absolute electronegativity. J. Am. Chem. Soc.. 1983;105:7512-7516.
- [Google Scholar]
- Current developments of coumarin compounds in medicinal chemistry. Curr. Pharm. Des.. 2013;19:3884-3930.
- [Google Scholar]
- New perspectives on the role of frontier molecular orbitals in the study of chemical reactivity: a review. Revista Virtual De Quimica. 2016:425-453.
- [Google Scholar]
- Synthesis and antimicrobial activity of some new 1, 3, 4-thiadiazoles and 1, 3, 4-thiadiazines containing 1, 2, 4-triazolo nucleus. Acta Chim. Slov.. 2011;58:53-59.
- [Google Scholar]
- Relationships between experimental and theoretical scales of electrophilicity of 7-L-4-nitrobenzofurazans. J. Mol. Struct.. 2021;1224:128843
- [Google Scholar]
- 5-(3-Pryridyl)-4H-1, 2, 4-triazole-3-thiol as potential corrosion inhibitor for AA6061 aluminium alloy in 0.1 M hydrochloric acid solution. Surf. Eng. Appl. Electrochem.. 2019;55:723-733.
- [Google Scholar]
- Inhibition effects of ethyl-2-amino-4-methyl-1, 3-thiazole-5-carboxylate on the corrosion of AA6061 alloy in hydrochloric acid media. J. Fail. Anal. Prev.. 2019;19:1464-1474.
- [Google Scholar]
- Electrochemical and Quantum Chemical Studies of 5-[(4-Chlorophenoxy) Methyl]-4H-1, 2, 4-Triazole-3-Thiol on the Corrosion Inhibition of 6061 Al Alloy in Hydrochloric Acid. J. Fail. Anal. Prev.. 2020;20:1598-1608.
- [Google Scholar]
- Corrosion inhibition effect of ethyl 1-(4-chlorophenyl)-5-methyl-1H-1, 2, 3-triazole-4-carboxylate on aluminium alloy in hydrochloric acid. Prot. Met. Phys. Chem. 2021;57:181-189.
- [Google Scholar]
- Computational determination the reactivity of salbutamol and propranolol drugs. Turkish Comput. Theoret. Chem.. 2020;4:67-75.
- [Google Scholar]
- Theoretical Determination of Corrosion Inhibitor Activities of Naphthalene and Tetralin. Gazi Univ. J. Sci. 2022 1–1
- [Google Scholar]
- Structure reactivity analysis for Phenylalanine and Tyrosine. Cumhuriyet Sci. J.. 2021;42:576-585.
- [Google Scholar]
- NIR-FT Raman and infrared spectra and ab initio computations of glycinium oxalate. Spectrochim. Acta Part A Mol. Biomol. Spectrosc.. 2004;60:173-180.
- [Google Scholar]
- Crystal structure, spectroscopic studies and quantum mechanical calculations of 2-[((3-iodo-4-methyl) phenylimino) methyl]-5-nitrothiophene. Spectrochim. Acta Part A Mol. Biomol. Spectrosc.. 2015;141:119-127.
- [Google Scholar]
- Theoretical aspects of chemical reactivity. Elsevier; 2006.
- 6-benzylidenethiazolo [3, 2-b]-1, 2, 4-triazole-5 (6h)-onessubstituted with ibuprofen: Synthesis, characterizationand evaluation of anti-inflammatory activity. Eur. J. Med. Chem.. 2000;35:743-750.
- [Google Scholar]
- Synthesis and analgesic activity of some triazoles and triazolothiadiazines. Il Farmaco.. 1999;54:218-223.
- [Google Scholar]
- Synthesis, lipophilicity and biological evaluation of indole-containing derivatives of 1, 3, 4-thiadiazole and 1, 2, 4-triazole. Il Farmaco.. 1998;53:320-326.
- [Google Scholar]
- Review on natural coumarin lead compounds for their pharmacological activity. BioMed Res. Int.. 2013;2013
- [Google Scholar]
- Studies on electronegativity equalization: part 1. Consistent diatomic partial charges. J. Mol. Struct. (Thoechem). 1991;233:71-81.
- [Google Scholar]
- Synthesis and biological evaluation of hydroxylated 3-phenylcoumarins as antioxidants and antiproliferative agents. Bioorg. Med. Chem. Lett.. 2011;21:6420-6425.
- [Google Scholar]
- Hardness, softness, and the fukui function in the electronic theory of metals and catalysis. Proc. Natl. Acad. Sci.. 1985;82:6723-6726.
- [Google Scholar]
- Synthesis and In Vitro Antioxidant Evaluation of New 1, 3, 5-Tri-{2-methoxy-4-[(4, 5-dihydro-1. J. Chem.. 2013;2013
- [Google Scholar]
- 4-Methoxy-3-(methoxymethyl) benzaldehyde. Acta Crystall Section E: Struct. Reports Online.. 2013;69 o112–o112
- [Google Scholar]
- Thermodynamics-antioxidant activity relationships of some 4-benzylidenamino-4, 5-dihydro-1h-1, 2, 4-triazol-5-one derivatives: Theoretical evaluation. Int. J. Food Prop.. 2017;20:1935-1948.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2022.104088.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1
Supplementary data 2
Supplementary data 2
Supplementary data 3
Supplementary data 3




