

Translate this page into:
Exploring the electronic and pharmacological properties of 1,2,3-triazole derivatives: Synthesis, DFT modeling, and AKT1 binding affinity
*Corresponding author: E-mail address: y.riadi@psau.edu.sa (Y. Riadi)
-
Received: ,
Accepted: ,
Abstract
This study presents a comprehensive multi-step synthesis followed by the characterization of two salicyldehyde-based 1,2,3-triazoles using a variety of analytical techniques. The structural integrity of the 1,2,3-triazole-O-tosyl derivatives 8a-b was confirmed through extensive NMR (1H and 13C) and high-resolution mass spectrometry (HRMS) analyses. To investigate the behavior of each of the synthesized compounds 8a-b in molecular nano-electronic systems, each one was connected to gold electrodes and their geometric structure was optimized using the theoretical level of density functional theory (DFT)/B3LYP/6-311+G (d,p) in different electric field intensities. According to the obtained results, although the energy gap and as a result the electron potential barrier decreased for both structures with the increase of the electric field intensity, the response of compound 8b to the application of the electric field was more impressive. Also, the I-V curve for each structure was calculated using Landauer’s theory and the results confirmed the higher conductivity of compound 8b. The analysis of the electron potential barrier under applied electric fields shows that the 8b structure can change to the ON state at EF>120×10-4 (a.u.) and act as a molecular switch of the field effect. Finally, findings from Quantum Theory of Atoms in Molecule (QTAIM) studies predicted lower Joule/Pelletier-like coefficients in compound 8b. To assess the pharmacological potential, in silico studies including molecular docking and absorption–distribution–metabolism–excretion–toxicity (ADMET) predictions were conducted. These investigations revealed the strong binding affinity of compound 8b to AKT1, with a binding energy score of -7.61 kJ/mol, highlighting its promising interactions for therapeutic applications.
Keywords
1,2,3-triazole
AKT1
DFT
Molecular docking
Nanoelectronic
NMR

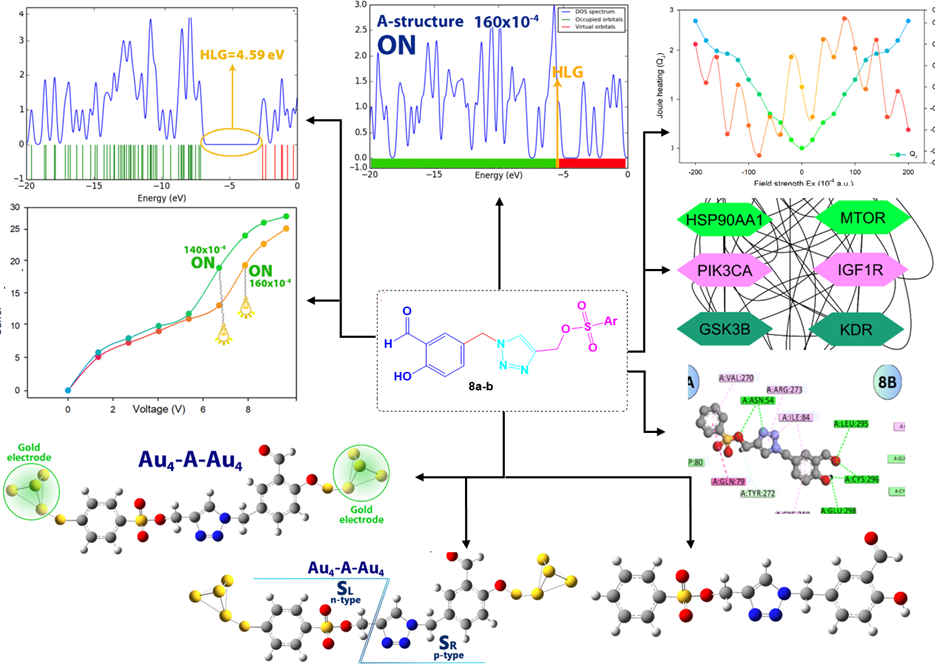
1. Introduction
Cancer-associated inflammation is a key factor in tumor development [1], first identified by Virchow in the 19th century and supported by genetic and epidemiological studies [2]. Both precancerous and malignant tissues often contain immune cells and inflammatory mediators, such as cytokines and chemokines, which play essential roles in tumor progression [3,4]. Chronic inflammation, seen in conditions like inflammatory bowel disease, accounts for approximately 20% of cancer-related deaths [2]. Due to the genetic stability of inflammation-associated cells, they are promising targets for anticancer therapies with lower resistance risks [5]. Despite significant advancements in cancer treatment, it remains the second leading cause of mortality worldwide. This is largely attributed to the challenges faced by chemotherapy, particularly issues like drug resistance and the difficulty of targeting cancer cells specifically without harming healthy tissue [6].
1,2,3-Triazoles, a class of nitrogen-containing five-membered heterocycles, are recognized for their critical role in a wide range of biologically active compounds. 1,4-disubstituted 1,2,3-triazoles have gained substantial attention due to their broad spectrum of biological activities, including antiviral [7], antiepileptic [8], and anticancer effects [9]. Additionally, various 1,2,3-triazole derivatives have demonstrated significant anticancer properties [10]. The ease of synthesizing these compounds via click chemistry has fueled their extensive exploration in medicinal chemistry, particularly for the discovery of new therapeutic agents [11].
Hybrid compounds combine distinct pharmacophores to reduce side effects and drug resistance [12]. Many are in clinical trials, targeting drug-resistant diseases, demonstrating hybridization’s potential in drug development [13]. Incorporating a 1,2,3-triazole scaffold with anticancer pharmacophores shows promise for improving therapeutic outcomes and reducing toxicity in oncology. Hybrid compounds incorporating 1,2,3-triazoles can be broadly categorized into two primary groups. Type I hybrids, where the 1,2,3-triazole moiety is connected to various anticancer pharmacophores via different linkers. Type II hybrids, which employ the 1,2,3-triazole as a bridging element between two distinct anticancer pharmacophores (Figure 1). This review focuses on Type II hybrids, highlighting their potential as anticancer agents.
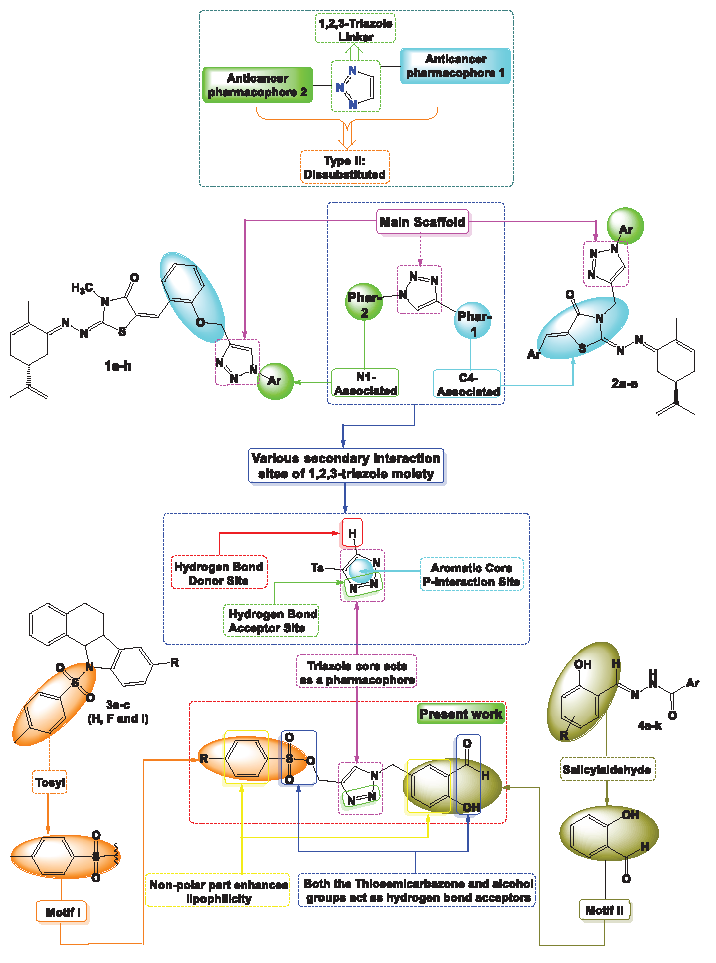
- Overview of the approach employed in the design of target compounds 8a-b.
In the same context of hybridization and according to the literature, Ait Itto et al. synthesized a series of 1,2,3-triazoles 1a-h using (R)-Carvone and salicylaldehyde to develop anticancer agents targeting various cancer cell lines. Cytotoxicity tests showed enhanced activity with IC50 values of 15.04 ± 0.71 µM, indicating the 1,2,3-triazole structure’s key role in biological interactions [14]. The same group explored the impact of heterocyclic frameworks, finding that the 1,2,3-triazole ring 2a-e significantly increased anticancer activity, while other rings reduced efficacy [15,16]. Buarque’s group synthesized tosyl-derived compounds 3a-c with antineoplastic properties against MDR leukemia and breast cancer [17,18], and further investigated their toxicity [19]. Doytchinova et al. examined salicylaldehyde derivatives 4a-k, demonstrating favorable drug-like characteristics and anticancer activity, with an in-silico analysis showing that pyridinyl-salicylaldehyde derivatives were the most effective for enhancing activity and selectivity [20] (Figure 1).
Computational methods have become indispensable in biology, offering valuable insights into molecular interactions within living systems. These techniques are widely employed in both basic and applied research, playing a significant role in the identification of new drugs and materials. Key applications include molecular docking, molecular dynamics simulations, ADMET assessments, and network pharmacology. The objective of this study is to investigate the interactions of novel 1,2,3-triazole derivatives, synthesized from salicylaldehyde, with proteins involved in cancer cell apoptosis, building on previous in-silico studies that identified heterocyclic compounds as potential inhibitors. This research aims to assess the binding affinities of these newly synthesized triazoles and evaluate their potential as modulators in cancer therapy. Additionally, the study explores the optical and electronic properties of these compounds.
2. Materials and Methods
2.1. Experimental part
The Products 8a-b were characterized utilizing a Bruker Avance III 300 MHz NMR spectrometer, with CDCl3 serving as the deuterated solvent. Molecular weights were assessed through HRMS data acquired via a Q-TOF mass spectrometer. It is important to mention that all intermediate compounds have been previously detailed in this publication [21].
2.2. Synthesis of 1,2,3-triazole-O-tosyl (8a-b)
An equimolar quantity (10 mmol) of 5-azidomethyl-2-hydroxybenzaldehyde 7 and propargyl-O-tosyl was dissolved in a mixture of 30 mL of ethanol and water (1:1). Subsequently, CuSO4·5H2O (10 mol %) and sodium ascorbate (20 mol %) were added to the solution. The reaction mixture was stirred for 4 hrs at room temperature (RT). The crude product obtained was then purified using chromatography.
1-(3-Formyl-4-hydroxybenzyl)-1H-1,2,3-triazol-4-yl)methyl benzenesulfonate 8a. (Yield : 75% ; Huil). 1H-δ : 4.71 (s, 2H, CH2), 5.48 (s, 2H, CH2), 7.00-7.30 (m, 6H, CHAr), 7.54 (s, 1H, CH), 7.55-7.67 (m, 2H, CHAr), 9.95 (s, 1H, CHO), 10.96 (s, 1H, OH) ppm. 13C-δ : 52.49, 56.19, 117.15, 119.87, 121.54, 123.15, 125.49, 129.21, 131.20, 132.86, 133.50, 136.17, 148.17, 161.17, 195.88 ppm. HRMS ([M+H]+) for 8a: C17H15N3O5S : 374.0733.
1-(3-Formyl-4-hydroxybenzyl)-1H-1,2,3-triazol-4-yl)methyl 4-methylbenzenesulfonate 8b. (Yield : 78 % ; Huil). 1H-δ : 2.44 (s, 3H, CH3), 4.73 (s, 2H, CH2), 5.50 (s, 2H, CH2), 7.00-7.30 (m, 3H, CHAr), 7.56 (s, 1H, CH), 7.35-7.70 (m, 4H, CHAr), 9.92 (s, 1H, CHO), 10.87 (s, 1H, OH) ppm. 13C-δ : 20.82, 51.13, 54.15, 118.22, 119.87, 121.86, 125.11, 126.86, 129.17, 130.12, 131.49, 136.14, 137.82, 148.19, 159.87, 195.20 ppm. HRMS ([M+H]+) for 8b: C18H17N3O5S : 388.0956
2.3. Computational methods
At first, each of the studied structures was drawn using the GaussView software. Then, the geometrical optimizations of each structure were performed using Gaussian software and DFT at the B3LYP/6-311+G theoretical level and in the water phase. The absence of imaginary frequency in each of the designed structures guarantees their stability. The B3LYP functional was chosen for this study due to its established accuracy in predicting molecular geometries and energies, making it a reliable choice for optimizing the studied structures. To check the stability of each of the studied structures, the cohesive energy was calculated using Eq. (1).
Here Etot, Ei, and ni represent the total energy of the designed molecules, the energy of each atom, and the number of atoms, respectively, with n denoting the total atom count.
Examining the energy gap is very important to understand the electronic properties of a molecule. Therefore, using Eq. (2), the energy gap of each of the designed structures was calculated.
EHOMO: energy level of the highest occupied molecular orbital and ELUMO: energy level of the lowest unoccupied molecular orbital.
In addition, the designed structures’ total hardness (η), chemical potential (μ), and total softness (S), were computed accordingly using Eqs. (3-5).
The polarizability (α) and first hyperpolarizability , which correspond to linear and nonlinear optical (NLO) properties, respectively, were calculated using Eqs. (6-8).
In which
Landauer’s formula (LT, Eqs. 9-11) was applied to predict the current-voltage (I-V) curve for A- and B-structured molecular systems. The temperature-independent direct-tunneling electric conduction (G) of a single-molecule nanoelectronics system is evaluated using this formula.
2.4. Network pharmacology
The potential target genes of Triazole Benzene Sulfonate (TBS) derivatives 8a-b in breast cancer were identified through a systematic approach. First, the Swiss Target Prediction database was used to predict Triazole Benzene Sulfonate (TBS) targets in Homo sapiens based on their Simplified molecular input line entry system (SMILES) notation [22]. This was followed by gene analysis [23], and filtering of breast cancer-related genes using the DisGeNET database [24]. The intersection of predicted targets and cancer-related genes was identified via Venn diagrams [25]. Pathway and functional enrichment analysis were performed using DAVID, focusing on Biological Process (BP), Molecular Function (MF), and Cellular Component (CC) with a p-value threshold of ≤ 0.05. The top ten Gene Ontology (GO) enrichments and kyoto encyclopedia of genes and genomes (KEGG) pathways were selected for further analysis. A network was built using Cytoscape 3.10.0, where nodes represented compounds and target genes, and edges denoted interactions. Key targets were identified based on network connectivity. The protein-protein interaction (PPI) network was constructed using STRING and further analyzed with the CytoHubba plugin to highlight core regulatory genes and key targets, consistent with previous findings [26].
2.5. Physicochemical properties
The pharmacological efficacy of a drug is intricately connected to its physicochemical characteristics, which are crucial in influencing its behavior in biological systems. Important attributes, including the partition coefficient (Log P), molecular weight, the number of hydrogen bond donors and acceptors, and the topological polar surface area (TPSA), are fundamental for predicting a compound’s absorption, distribution, and bioavailability. Online tools like Molinspiration software can be employed to compute these properties, facilitating the assessment of molecular features. Lipinski’s Rule of 5 offers a valuable guideline, indicating that compounds with a Log P of 5 or lower, a molecular weight of 500 or less, a maximum of 10 hydrogen bond acceptors, and no more than 5 hydrogen bond donors are more likely to have favorable bioavailability. Deviations from these criteria often suggest potential challenges regarding drug absorption and overall effectiveness.
2.6. Bioactivity
The bioactivity scores for the synthesized compounds were evaluated using the Molinspiration software, an essential resource in drug discovery and development processes. These scores offer valuable insights into a compound’s potential effectiveness against specific biological targets. A score exceeding 0.00 suggests that the compound may function as an agonist, indicating favorable interactions with the target. On the other hand, a negative score points to antagonistic or inhibitory effects. Compounds that score between -0.50 and 0.00 are regarded as moderately active, implying they may engage with the target to a degree, albeit less effectively than those with positive scores. In contrast, compounds with scores lower than -0.50 are deemed inactive, reflecting minimal significant interaction with the intended target.
2.7. Toxicity
Recent advancements in computational techniques have markedly improved the capacity to predict the toxicity of chemical products, offering essential safety data through in silico methods. These approaches facilitate a comprehensive evaluation of different toxicity parameters, such as hepatotoxicity, carcinogenicity, immunotoxicity, and mutagenicity. Utilizing the Pro Tox II server enables efficient prediction and analysis of toxicity profiles for newly developed compounds, thereby enhancing the safety of drug development and minimizing the reliance on extensive in vitro or in vivo testing [27].
2.8. Molecular docking
Molecular docking was conducted using AutoDock 4.2 software [28]. The ligand was imported into UCSF Chimera via its SMILES notation, converted into a 3D structure, and energy minimized to create PDB files. The AKT1 protein (PDB ID: 4EJN) was retrieved from the research collaboratory for structural bioinformatics (RCSB) Protein Data Bank and prepared by removing unnecessary residues and water molecules, adding polar hydrogens, and applying Kollmann charges. The PDB files were converted into PDBQT format, and a grid box was set for the docking procedure. Docking was performed with Lamarckian genetic algorithm parameters, and results were saved in the ‘.dlg’ format. Interaction analysis was performed using BIOVIA Discovery Studio to visualize ligand-protein conformations in 2D and 3D [29].
3. Results and Discussion
3.1. Synthesis
The hybrid derivative 8a-b, which consisted of a 1,2,3-triazole, salicylaldehyde, and a tosyl fragment, was synthesized through a four-step procedure outlined in Scheme 1. The process commenced with the chlorination of salicylaldehyde utilizing formaldehyde and hydrochloric acid, following the methodology described in reference [17]. In the second step, sodium azide was introduced into acetone under reflux conditions to replace the chlorine atom, yielding 5-azidomethyl-2-hydroxybenzaldehyde 7 with a notable yield of 88%. Simultaneously, a propargyl-O-tosyl support was created from an equimolar mixture of tosyl chloride derivatives and propargyl alcohol in dichloromethane, using triethylamine as a catalyst. The following step involved a 1,3-dipolar cycloaddition between compound 7 and propargyl-O-tosyl, facilitated by CuSO4 and sodium ascorbate as reducing agents, leading to the formation of a 1,4-disubstituted 1,2,3-triazole core (Compounds 8a-b) with an excellent yield, as illustrated in Scheme 1.
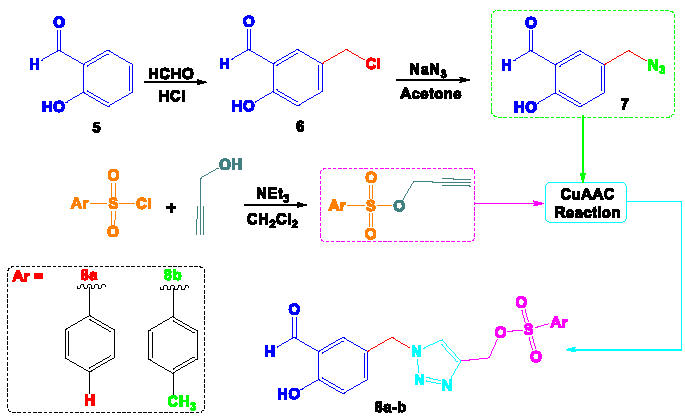
- Synthetic strategy for obtaining target compounds 8a-b.
The structures of all newly synthesized products 8a-b were unequivocally verified using HRMS, their spectra show the pseudo-molecular ion at 374.0733 and 388.0956 respectively, consistent with formula (8a: C17H15N3O5S and 8b: C18H17N3O5S). A comparative assessment of the NMR spectra for compound hybrids 8a-b demonstrated a notable similarity. This consistent spectral profile across the series serves as strong evidence for the successful O-alkylation and synthesis of the desired 1,2,3-triazoles 8a-b. To exemplify the distinct spectral variations linked to these reactions, we examined the NMR (1H & 13C) spectra of compound hybrid 8b. The appearance of new signals at (1H-δ: 7.56 ppm; 13C-δ: 125.11 ppm) indicates the formation of H-C=N within the newly created triazole ring. Furthermore, the CH2 connecting the 1,2,3-triazole structure to the paramethoxythymol unit and the tosyl group is evidenced respectively by signals at (1H-δ: 5.50 ppm; 13C-δ: 54.15 ppm) and (1H-δ: 4.73 ppm; 13C-δ: 51.13 ppm). Additionally, the aldehyde and alcohol functionalities of salicylaldehyde were identified at (1H-δ: 10.87 ppm; 13C-δ: 195.20 ppm) and (1H-δ: 9.92 ppm), respectively. The aromatic protons resonated between δ 7.00 and 7.75 ppm in the 1H-NMR spectrum, while the corresponding aromatic carbons were observed in the 13C-NMR spectrum at δ 118 to 137 ppm.
3.2. Geometry optimization
The geometrical structures of the studied molecules were optimized at the DFT/B3LYP/6-311+G(d,p) theoretical level to determine their most stable configurations (Figure 2). This optimization process helps in identifying the lowest energy conformation for each molecule, which is critical for understanding their physical and chemical properties. To further assess the stability of these structures, imaginary frequency calculations, and cohesive energy evaluations were performed. Imaginary frequencies are used in vibrational analysis to confirm that a structure represents a true minimum on the potential energy surface. A structure without any imaginary frequencies is considered stable, as the presence of such frequencies would indicate that the structure is in a transition state rather than a stable molecule. Cohesive energy, on the other hand, measures the amount of energy required to disassemble a molecule into its constituent atoms. Higher cohesive energy values suggest stronger bonding within the molecule and therefore greater structural stability.
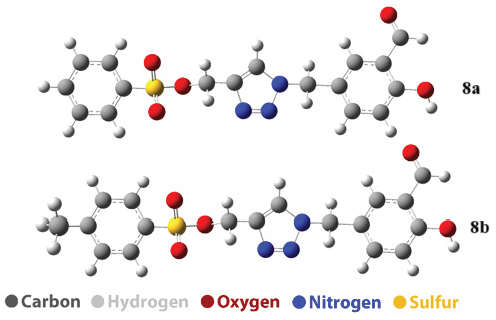
- The Optimized structure of 8a and 8b structure.
Table 1 presents the values of cohesive energy and the first vibrational frequency for the studied structures. Compound 8a has a first vibration frequency of 9.415 cm⁻1 and a cohesive energy of 89.12 kcal/mol, while compound 8b exhibits a higher first vibration frequency of 16.073 cm⁻1 and a higher cohesive energy of 91.86 kcal/mol. The increase in cohesive energy after the addition of methane (8b) indicates that the incorporation of methane strengthens the internal bonds and leads to a stronger structure. These results indicate that compound 8b is more stable than compound 8a.
| Compound | First vibration frequency | Cohesive energy |
|---|---|---|
| 8a | 9.415 | 89.12 |
| 8b | 16.073 | 91.86 |
3.3. Theoretical NMR
13C-NMR spectrum for both compounds 8a-b has been shown in Figure 1S. The NMR spectrum of compound 8a reveals multiple carbon environments with chemical shifts ranging from 117.720 to 176.115 ppm. Notable signals include those at 176.115 ppm (C39), indicating a carbonyl or highly deshielded carbon, and 165.215 ppm (C3) and 164.268 ppm (C13), which likely correspond to sp2 carbons adjacent to electronegative groups. Additional shifts, such as 152.855 ppm (C19) and 149.283 ppm (C14), suggest aromatic carbons possibly attached to electron-withdrawing groups. Carbons at 132.533 ppm (C1), 132.267 ppm (C5), 131.616 ppm (C15), and 130.659 ppm (C21) are likely part of aromatic systems. The lower shift at 117.720 ppm (C25) might correspond to a less deshielded aromatic carbon, while other aromatic carbons, such as those at 136.718 ppm (C6), 135.055 ppm (C20), and 139.698 ppm (C22), show variations in deshielding, indicating diverse functionalization or substitution patterns. The signal at 126.351 ppm corresponds to C4 and the signal at 126.251 ppm corresponds to (C23). The NMR spectrum data for compound 8b shows a range of chemical shifts associated with different carbon atoms. The chemical shifts observed are as follows: 132.783 ppm for atom 1, 126.951 ppm for atom 2, 162.522 ppm for atom 3, 126.242 ppm for atom 4, and 132.703 ppm for atom 5. For atom 6, the chemical shift is 150.450 ppm, while for atom 13 it is 164.112 ppm, and for atom 14, it is 149.302 ppm. Atom 15 exhibits a chemical shift of 131.589 ppm, and atom 19 has a shift of 152.882 ppm. The chemical shift for atom 20 is 135.070 ppm, and for atom 21, it is 130.692 ppm. Atom 22 shows a chemical shift of 139.698 ppm, while atom 23 shows 126.355 ppm. Atom 25 exhibits a shift of 117.561 ppm, and atom 27 has a chemical shift of 162.849 ppm. The highest chemical shift is observed at 176.112 ppm for atom 39, while atom 41 shows the lowest chemical shift in the dataset at 120.109 ppm.
The 1H-NM spectrum of each of the studied structures has been shown in Figure 2S. The 1H-NMR spectrum of compound 8a displays several distinct chemical shifts, indicating the presence of various proton environments within the molecule. At 6.741 ppm, the protons associated with atoms 7(H), 10(H), and 41(H) are observed, suggesting aromatic or vinylic hydrogen atoms. Another signal at 7.294 ppm corresponds to protons on atoms 8(H) and 24(H), while a nearby signal at 7.238 ppm is attributed to atom 9(H), further indicating the presence of aromatic hydrogens. A signal at 6.971 ppm corresponds to the 26(H) atom and another shift at 5.552 ppm corresponds to the 28(H) atom, possibly indicating hydrogens adjacent to the OH functional group. A deshielded proton is observed at 10.705 ppm in the 29 (H) atom, which indicates the bond of the proton to an oxygen (hydroxyl group). The spectrum also shows shifts at 4.651 ppm (atom 32(H)) and 4.932 ppm (atom 33(H)), which may be related to hydrogens near electronegative groups or in aliphatic regions. Another aromatic signal appears at 6.829 ppm for atom 34(H). The chemical shift at 4.112 ppm, assigned to atom 35 (H), and at 5.013 ppm, corresponding to atom 36 (H), is related to hydrogens adjacent to nitrogen groups. Finally, a shift at 3.422 ppm is attributed to the 37(H) atom, indicating the presence of the hydrogen of the hydroxyl group, which is less shielded than the aromatic regions. This detailed 1H-NMR analysis highlights the diversity of the proton environments in compound 8a, pointing to a complex molecular structure with aromatic, aliphatic, and functional group influences.
The 1H-NMR spectrum of compound 8b displays the following chemical shifts. A signal at 6.460 ppm corresponds to proton 7. Protons 8 and 9 show a signal at 7.207 ppm, while proton 10 has a shift at 6.566 ppm. Proton 24 exhibits a peak at 7.302 ppm, and proton 26 appears at 7.021 ppm. Proton 28 is detected at 5.561 ppm, and proton 29 shows a deshielded signal at 10.709 ppm, indicating the presence of an acidic proton. Proton 32 gives a signal at 4.642 ppm, while proton 33 appears at 4.925 ppm. Proton 34 has a shift at 6.841 ppm, and proton 35 is observed at 4.109 ppm. The signal for proton 36 is found at 5.015 ppm, and proton 37 appears at 3.411 ppm. A downfield shift at 2.037 ppm corresponds to proton 42, and proton 43 shows a signal at 1.758 ppm. Finally, proton 44 displays a signal at 1.386 ppm.
3.4. Uv spectra
The UV analysis data for compounds 8a-b reveals some similarities and differences in their optical properties. Both structures exhibit a maximum absorption wavelength (λmax) of approximately 364 nm, with A having λmax at 364.764 nm and 8b at 364.560 nm. The corresponding excitation energies (Eex) are nearly identical, with A at 3.3990 eV and B at 3.4009 eV. For both structures, the primary electronic transition is assigned as H-1 to L+1 with a high contribution (95%), and the oscillator strength (ƒ) for this transition is zero in each case (Figure 3 and Table 2). The second absorption peak for A is observed at 312.371 nm, with an excitation energy of 3.9691 eV and an oscillator strength of 0.0119. This transition is primarily assigned to H-6 to least unoccupied molecular orbital (LUMO) with a 59% contribution. In contrast, 8b’s second absorption peak occurs at a similar wavelength of 311.797 nm, with a slightly higher excitation energy of 3.9764 eV and a higher oscillator strength of 0.0131, assigned to H-7 to LUMO (70%).
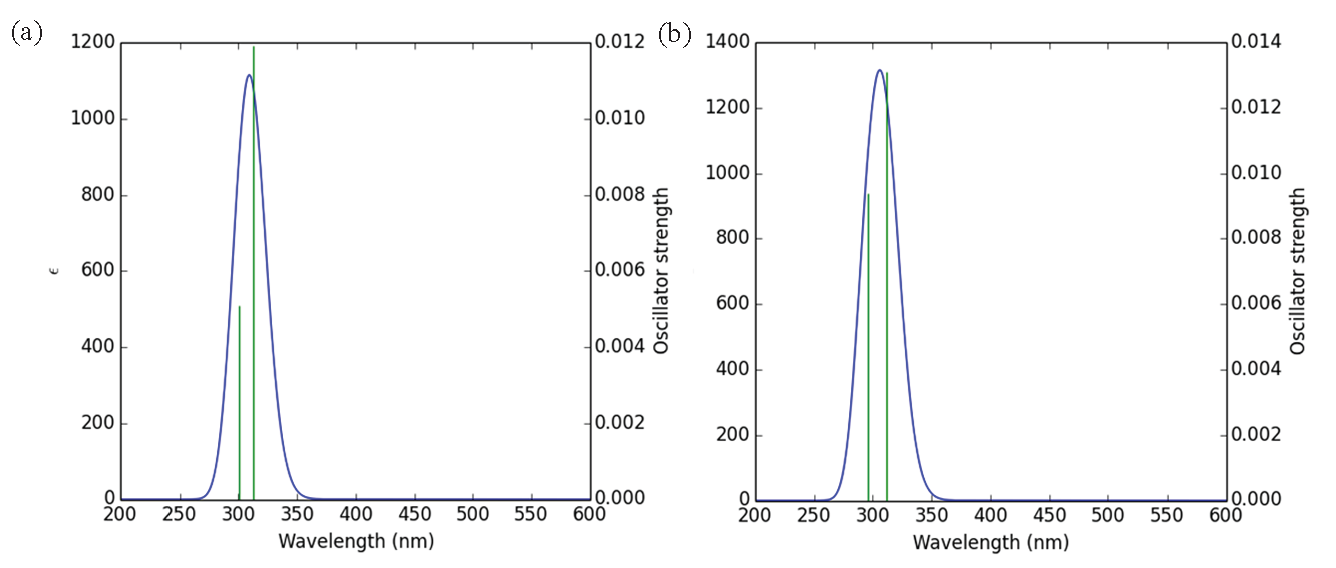
- UV spectrum of studied structures (a) derivative 8a and (b) derivative 8b.
| Structure | ʎmax (nm) | Eex (eV) | ƒ | Assignment |
|---|---|---|---|---|
| 8a | 364.764 | 3.3990 | 0.0 | H-1->L+1 (95%) |
| 312.371 | 3.9691 | 0.0119 | H-6->LUMO (59%) | |
| 300.412 | 4.1271 | 0.0051 | HOMO->LUMO (98%) | |
| 8b | 364.560 | 3.4009 | 0.0 | H-1->L+1 (95%) |
| 311.797 | 3.9764 | 0.0131 | H-7->LUMO (70%) | |
| 296.001 | 4.1886 | 0.0094 | HOMO->LUMO (96%) |
λmax: Maximum absorption wavelengths, ƒ: Oscillator strength, Eex: Excitation energies.
For the third absorption peak, 8a shows a wavelength of 300.412 nm with an excitation energy of 4.1271 eV and an oscillator strength of 0.0051, corresponding to the highest occupied molecular orbital (HOMO) to LUMO transition (98%). Compound 8b, on the other hand, has its third peak at a slightly lower wavelength of 296.001 nm, with a higher excitation energy of 4.1886 eV and an oscillator strength of 0.0094 for the same HOMO to LUMO transition (96%). Overall, while the maximum absorption wavelengths and the primary transitions are similar for both compounds, 8b displays slightly higher excitation energies and oscillator strengths for the subsequent transitions, indicating a more intense absorption in the UV region compared to 8a.
3.5. Polarizability, hyperpolarizability and dipole moment
The comparison of dipole moment (µ), polarizability (α), and first hyperpolarizability (β) for compounds 8a-b indicates that product 8b exhibits slightly higher values in all three parameters. Compound 8a has a dipole moment of 7.1645, while compound 8b shows a higher value of 7.5637, suggesting that 8b has a slightly stronger separation of charge (Table 3). Regarding polarizability (α), product 8a has a value of 21.03, while compound 8b’s value is higher at 22.46, indicating that 8b is more polarizable and may respond more easily to external electric fields. Similarly, the first hyperpolarizability (β) is also higher for compound 8b, with a value of 41.50 compared to 39.12 for compound 8a. This suggests that 8b has a stronger nonlinear optical response, making it potentially more suitable for applications requiring high optical activity.
| Compound | µ | α | β |
|---|---|---|---|
| 8a | 7.1645 | 21.03 | 39.12 |
| 8b | 7.5637 | 22.46 | 41.50 |
µ: Dipole moment, α: Polarizability, β: First hyperpolarizability
3.6. Frontier molecular orbital and quantum parameter
The comparison of the data presented in Table 4 highlights some key differences between compounds 8a-b regarding their electronic properties. Product 8a has a LUMO energy of -2.66 eV and a HOMO energy of -7.17 eV, resulting in a HOMO-LUMO gap (HLG) of 4.51 eV. Its chemical hardness (η) is 2.25 eV, chemical potential (μ) is -4.91 eV, and chemical softness (S) is 0.22 eV⁻1. In contrast, Structure B shows a slightly higher LUMO energy of -2.58 eV and a HOMO energy of -7.14 eV, yielding a slightly larger HLG of 4.59 eV. The density of states (DOS) diagram is a great way to show the energy gap in molecules. For this purpose, the DOS plot for each of the studied structures has been shown in Figure 4. The results of the DOS plot clearly confirm the further reduction of the energy gap in compound 8b. The chemical hardness of compound 8b is also marginally higher at 2.29 eV, while its chemical potential is slightly less negative at -4.86 eV. Additionally, compound 8b has a lower chemical softness value of 0.21 eV⁻1 compared to product 8a. These differences suggest that compound 8b is somewhat harder and has a slightly larger energy gap, indicating a potentially more stable configuration with a lower reactivity compared to compound 8a.
| Compound | LUMO (eV) | HOMO (eV) | HLG (eV) | η (eV) | μ (eV) | S (eV) |
|---|---|---|---|---|---|---|
| 8a | -2.66 | -7.17 | 4.51 | 2.25 | -4.91 | 0.22 |
| 8b | -2.58 | -7.14 | 4.59 | 2.29 | -4.86 | 0.21 |
η: Chemical hardness, μ: Chemical optional, S: Chemical softness.
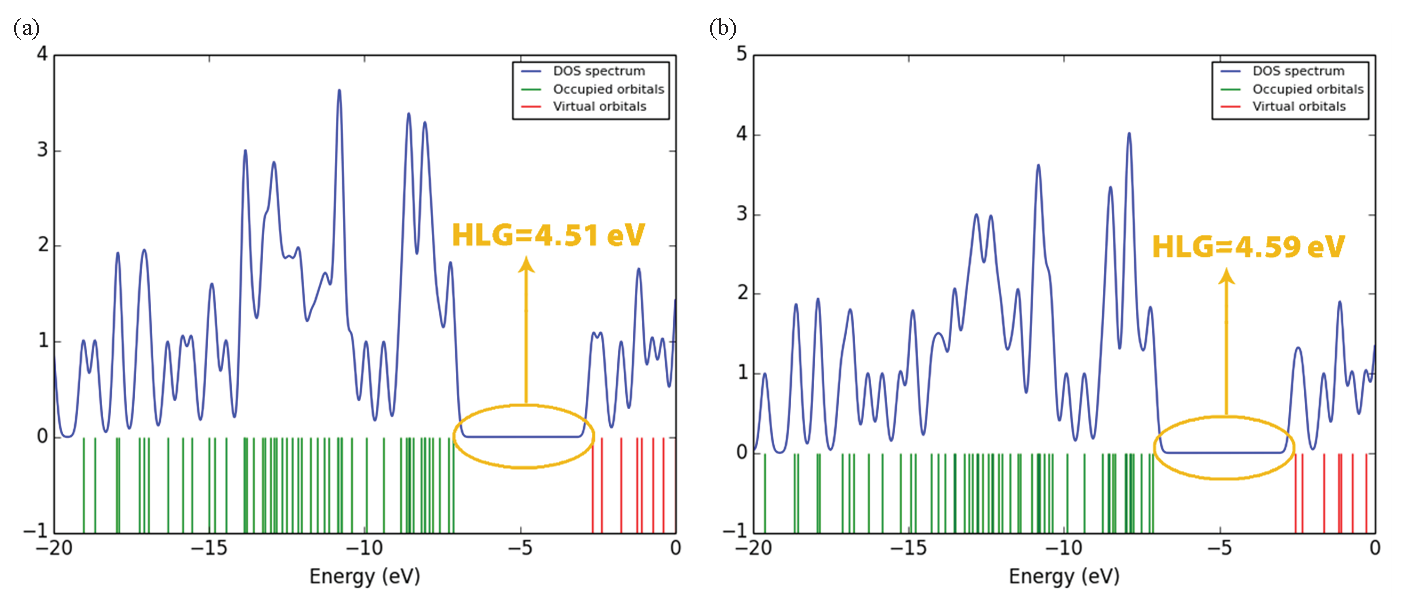
- DOS diagram: (a) for compound 8a and (b) for compound 8b.
3.7. Field effect
In this section, the effect of the electric field on each of the studied structures was investigated. For this purpose, each structure was connected to gold electrodes (Figure 5).
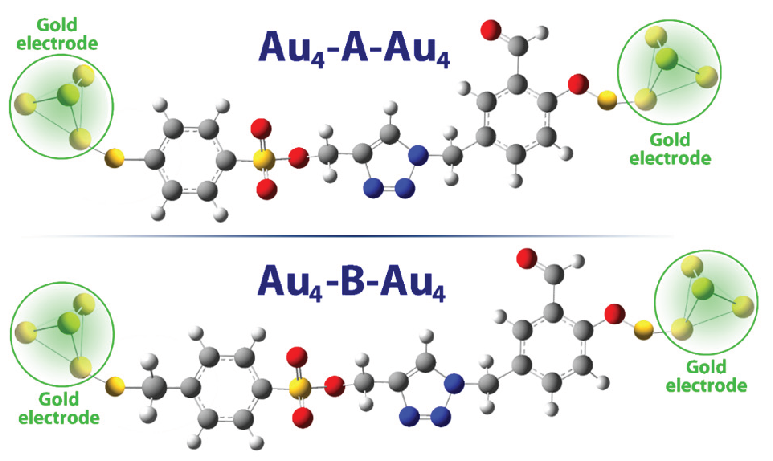
- Structures studied after attachment to gold electrodes (Au-A-Au and Au-B-Au).
3.8. Effect of the electric field on frontier orbitals
The comparison of the HLG values in 8a-b compounds under varying electric field intensities reveals a consistent decrease in the energy gap as the field strength increases. At zero field intensity, the HLG for 8a and 8b is 4.51 eV and 4.59 eV, respectively, showing that compound 8b initially has a slightly larger energy gap. As the electric field intensity increases to 20×10-4 a.u., both structures exhibit a reduction in their HLG values, with compound 8a decreasing to 4.38 eV and 8b to 4.26 eV. This trend continues with higher intensities; at 40×10⁻⁴ a.u., the HLG for 8a drops to 4.22 eV while 8b shows a more significant reduction to 3.97 eV (Table 5).
| Electrical field (10-4 a.u) | Compound 8a | Compound 8b | ||||
|---|---|---|---|---|---|---|
| LUMO (eV) | HOMO (eV) | HLG (eV) | LUMO (eV) | HOMO (eV) | HLG (eV) | |
| 0 | -2.66 | -7.17 | 4.51 | -2.58 | -7.14 | 4.59 |
| 20 | -3.13 | -7.51 | 4.38 | -2.21 | -7.47 | 4.26 |
| 40 | -3.16 | -7.38 | 4.22 | -3.17 | -7.14 | 3.97 |
| 60 | -3.57 | -7.04 | 3.47 | -3.59 | -6.79 | 3.20 |
| 80 | -3.97 | -6.7 | 2.75 | -4.0 | -6.44 | 2.44 |
| 100 | -4.37 | -6.35 | 1.98 | -4.42 | -6.09 | 1.67 |
| 120 | -4.78 | -6.21 | 1.43 | -4.83 | -5.74 | 0.91 |
| 140 | -4.78 | -6.01 | 1.23 | -5.03 | -5.61 | 0.58 |
| 160 | -5.3 | -5.63 | 0.33 | -5.18 | -5.5 | 0.32 |
| 180 | -5.33 | -5.64 | 0.30 | -5.25 | -5.52 | 0.27 |
| 200 | -5.38 | -5.64 | 0.26 | -5.31 | -5.53 | 0.22 |
With further increases in field intensity, both structures experience a marked decline in their HLG values. For instance, at 80 × 10⁻⁴ a.u., compounds 8a-b have HLG values of 2.75 eV and 2.44 eV, respectively, indicating that 8b’s gap reduces more quickly. This pattern persists at even higher intensities; at 120×10⁻⁴ a.u., A’s HLG is 1.43 eV compared to 0.91 eV for 8b, demonstrating that 8b’s structure is more sensitive to the electric field. At the maximum field intensity of 200 × 10⁻⁴ a.u, the HLG for 8a and 8b is 0.26 eV and 0.22 eV, respectively, highlighting that while both structures become nearly gapless, 8b consistently shows a smaller HLG throughout the range of electric fields applied. This indicates that the structure of 8b is more reactive and undergoes greater electronic changes in response to the electric field compared to the 8a structure.
In field-effect molecular switches, the potential barrier (φ) is a crucial factor influencing the electron transfer mechanism within molecular components (φ = HLG/2). This barrier controls the ease with which electrons can move across the molecule when an external electric field is applied, directly affecting the switch’s conductivity and response. A well-tuned potential barrier ensures efficient electron flow, making it essential for optimizing the performance of molecular switches in nano-electronic devices. Accordingly, when φ is less than EF (φ<EF), the molecule is in the ON state, facilitating electron transfer, whereas when φ is greater than EF (φ>EF), the molecule remains in the OFF state, preventing electron transfer.
Table 6 presents the potential barrier (φ) values and corresponding ON/OFF states for two molecular structures (8a-b) under varying applied electric fields. In the absence of an electric field, both structures exhibit relatively high potential barriers, with φ values of 2.25 eV for compound 8a and 2.29 eV for compound 8b, placing them in the OFF state. As the electric field increases, the potential barrier decreases gradually for both structures. At EF>120×10-4 a.u, the compound 8b changes to the on state, while the compound 8a is still in the off state. As the intensity of the electric field increases and at EF>140×10-4 a.u, compound 8a changes its state to the ON state.
| Electrical field(10-4 a.u) | Electrical field (eV) | Compound 8a | Compound 8b | ||
|---|---|---|---|---|---|
| φ (eV) | State | φ (eV) | State | ||
| 0 | 0 | 2.25 | OFF | 2.29 | OFF |
| 20 | 0.05 | 2.19 | OFF | 2.13 | OFF |
| 40 | 0.10 | 2.11 | OFF | 1.98 | OFF |
| 60 | 0.16 | 1.73 | OFF | 1.60 | OFF |
| 80 | 0.21 | 1.37 | OFF | 1.22 | OFF |
| 100 | 0.27 | 0.99 | OFF | 0.83 | OFF |
| 120 | 0.32 | 0.71 | OFF | 0.45 | OFF |
| 140 | 0.38 | 0.61 | OFF | 0.29 | ON |
| 160 | 0.43 | 0.16 | ON | 0.16 | ON |
| 180 | 0.48 | 0.15 | ON | 0.13 | ON |
| 200 | 0.54 | 0.13 | ON | 0.11 | ON |
To better understand this issue, the DOS diagram for compounds 8a-b in some more important electric fields is shown in Figure 6. According to Figure 6, the energy gap changes in electric fields EF>120×10-4 a.u (for compound 8b) and EF>140×10-4 a.u (for compound 8a) clearly show the on and off states in compounds 8a-b. These results indicate that compound 8b is more sensitive to the electric field, potentially making it more efficient for applications in high-sensitivity field-effect molecular switches.
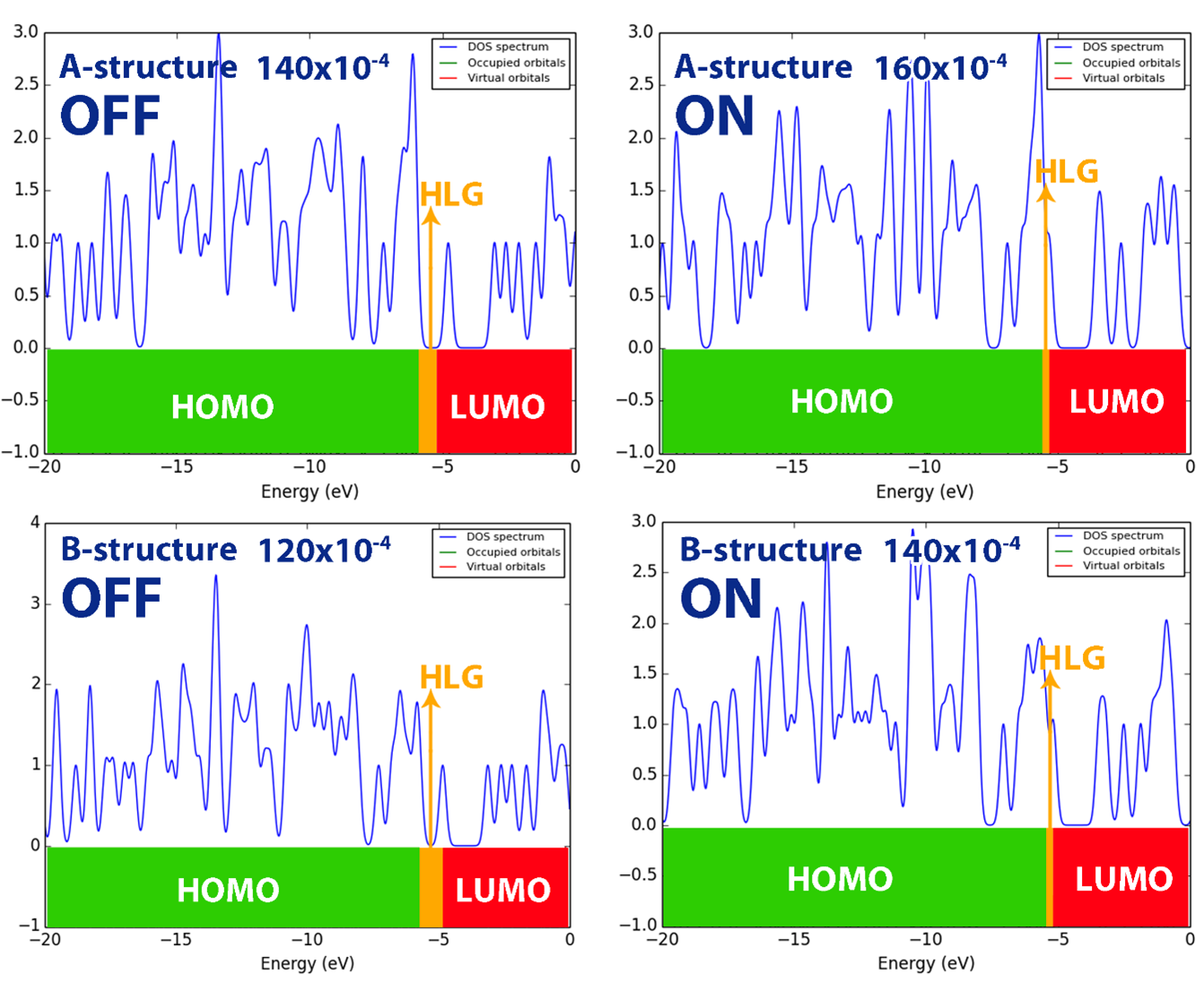
- DOS diagram of studied structures in some electric fields (120X10-4, 140X10-4 and 160X10-4) in ON and OFF states: A for compound 8a and B for compound 8b.
3.9. I-V curve
Examining the I-V (current-voltage) diagram of a molecule using DFT (Density Functional Theory) is crucial for understanding its electronic transport properties, which are essential in molecular electronics. The I-V diagram reveals how the molecule responds to varying voltages, indicating its conductive behavior and potential as a component in devices like molecular switches or transistors. DFT allows accurate calculation of electron flow through the molecule under applied bias, providing insights into key factors such as conductance, resistance, and the effect of molecular structure on electron transfer. This analysis helps in optimizing the molecule for specific electronic applications, ensuring efficiency and functionality in nano-electronic systems. Therefore, the I-V curve of each of the molecular structures 8a-b has been shown in Figure 7.
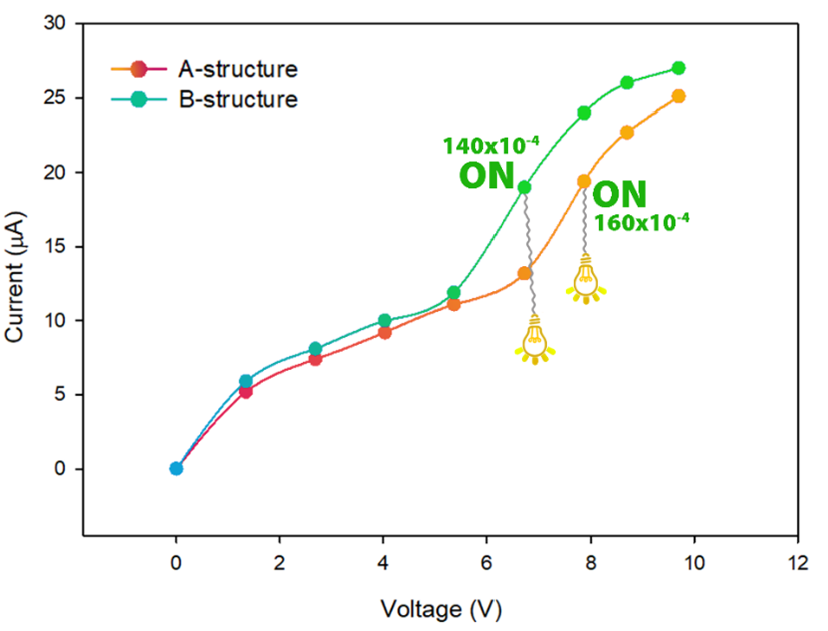
- Current-voltage curve (I-V curve) of 8a-structure, and 8b-structure molecular structures at different electric field intensity.
The analysis of the results shows that in fields higher than 120×10-4 a.u and 160×10-4 a.u (for compound 8b and 8a structures, respectively), the phenomenon of quantum tunneling between the electrode and the molecule is facilitated. This feature makes electron transfer along the length of the molecule easy and ultimately increases the conductivity of the molecule.
3.10. Joule/Peltier like coefficient
Calculating Joule and Peltier heating in a nano-electronic system using QTAIM (Quantum Theory of Atoms in Molecules) is essential for assessing energy dissipation and thermal management at the atomic level. Joule heating, caused by resistance to electron flow, leads to energy loss as heat, affecting the efficiency and reliability of nano-electronic devices. Peltier heating, on the other hand, results from electron transfer across junctions and influences temperature gradients. By applying QTAIM, which precisely analyzes electron density distribution, these heating effects can be accurately evaluated, helping optimize material design to minimize energy loss and improve thermal control in applications such as thermoelectric devices, molecular electronics, and nanoscale circuits.
When a molecular model is exposed to an external field (ε), it can be divided into electron donor/acceptor components, resembling n-p-type behavior. This division allows the study of charge and energy exchange after applying an electric field. To understand pseudo-thermoelectric effects in a molecular nano-electronic system, the molecule is divided into left (SL) and right (SR) sections, similar to an n/p semiconductor (Figure 8). By inducing a temperature gradient or potential difference, charge and energy exchange between the electrode and molecule can lead to thermoelectric-like behavior within the system.
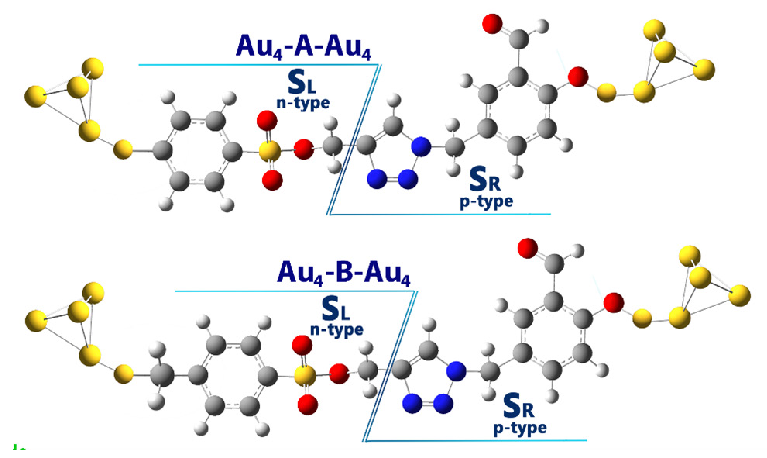
- The investigated molecular structure was separated into two distinct intramolecular regions: the right (SR) and left (SL) sections.
Given that Joule heat is an even function and Peltier heat is an odd function with respect to the reversal of the applied field direction (f/r), the intra-molecular thermoelectric systems, similar to n/p-type thermoelectric systems, can be described by the following expressions (Eq. 12) for Joule-like heat (QJL) and Peltier-like heat (QPL):
In a similar manner, the intramolecular coefficients for Peltier-like (LPL) and Joule-like (LJL) effects can be expressed as follows Eqs. (13-16):
Here
Where
Additionally, it can be demonstrated that the electron kinetic energy of any atomic basin or substrate, according to AIM quantum theory, is given by the following Eq. (17):
In this context, N represents the number of electrons, and dτ denotes the integral over the spin coordinates of the electrons. By applying Eqs. (8-13), the intramolecular thermoelectric coefficients corresponding to the Joule and Peltier effects were computed for both structures under investigation (Figure 9).
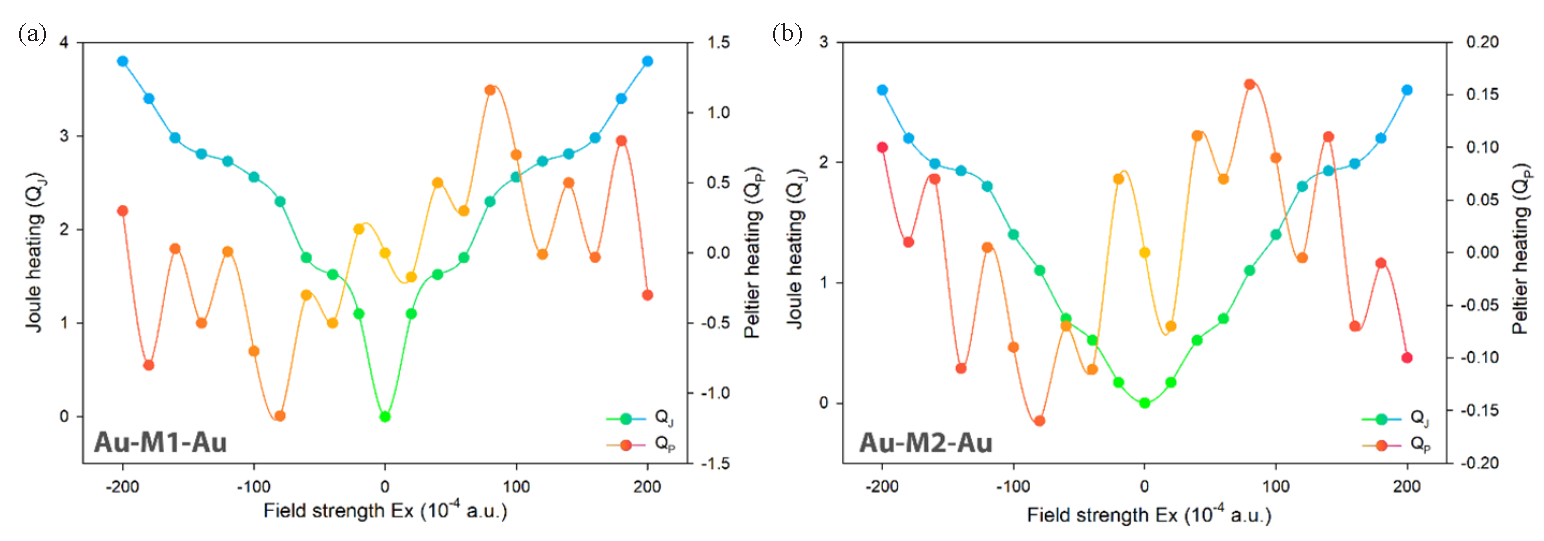
- Effect of external electric field on Peltier/Joule coefficients in molecular structures (a) for 8a and (b) for 8b derivatives.
The results indicate that Joule and Peltier heating are lower in the 8b-structure compared to the A-structure (Figure 9). The methyl substitution in Structure 8b reduces heat generation, enhancing energy efficiency in electronic devices by minimizing energy loss as heat. This also allows for better temperature control, which can improve the performance and reliability of thermoelectric and cooling systems.
3.11. In-silico study
3.11.1. Network pharmacology
The potential target genes for the synthesized Triazole Benzene Sulfonate derivatives 8a-b (a total of 144 genes) were first retrieved from the Swiss Target database. In addition, a dataset of 721 genes linked to Breast Cancer was sourced from the Disgenet database. To identify common targets between the cancer-related genes and those corresponding to the synthesized compounds, Venn diagram analysis was performed. From this, 37 genes were selected as potential key anti-cancer targets for Breast Cancer, warranting further investigation.
3.11.2. Development of the compound-target interaction network
Furthermore, four triazole benzene sulfonate (TBS) derivatives (8a-b) were chosen, along with 37 primary targets and the pathways most strongly linked to Breast Cancer genes, to create a network diagram that illustrates the connections between active compounds, target genes, and their related pathways. The involvement of multiple targets for each compound suggests a potential synergistic effect, highlighting the capability of TBS derivatives to function as effective anti-BC agents. This implies that the compounds’ action on several targets may work together to improve their therapeutic impact in Breast Cancer treatment. The common targets have been shown in Figure 10.
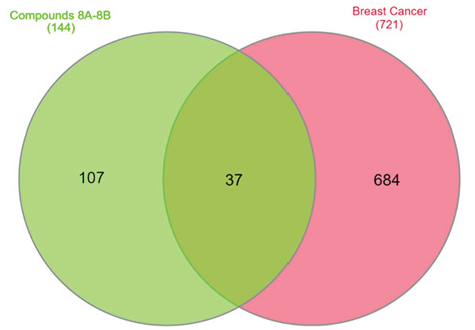
- Venn plot of Disease and designed compound overlapping targets.
3.11.3. Construction of the PPI network and identification of critical genes
The top 10 overlapping genes were submitted to the STRING database to construct a Protein-Protein Interaction (PPI) network, where nodes and their connections represent interactions among targets involved in disease progression. Analysis of the PPI network identified key genes ranked by their interaction scores, with the top 10 genes listed in Table 7 and Figure 11. Among these, AKT1 ranked highest with a score of 33, indicating its critical role. EGFR followed with a score of 31, while SRC and HSP90AA1 shared the third position with scores of 28. MTOR (25), PIK3CA (24), and IGF1R, GSK3B, KDR, and PARP1 (21 each) also emerged as significant targets. This ranking underscores the functional relevance of these genes, with a high degree of interaction suggesting their central roles in breast cancer pathogenesis. The analysis highlights AKT1 as a particularly important target due to its pivotal role in cellular signaling.
| Rank | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Gene name | AKT1 | EGFR | SRC | HSP90AA1 | MTOR | PIK3CA | IGF1R | GSK3B | KDR | PARP1 |
| Degree score | 33 | 31 | 28 | 28 | 25 | 24 | 21 | 21 | 21 | 21 |
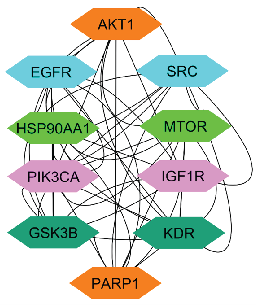
- Diagram illustrating the network of the top 10 ranked genes. AKT1: Alpha serine/threonine-protein kinase, SRC: Proto-oncogene tyrosine-protein kinase, EGFR: Epidermal growth factor receptor, MTOR: Mammalian target of rapamycin, HSP90AA1: Heat shock protein 90α, IGF1R: Insulinlike growth factor1 receptor, PIK3CA: phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha, KDR: Kinase insert domain receptor, GSK3B: Glycogen synthase kinase-3 beta, PARP1: Poly (ADP-ribose) polymerase 1.
3.11.4. Top 10 genes: a network pharmacology approach
The network pharmacology analysis highlighted the top 10 genes based on their interaction degree within the PPI network. The degree score reflects the number of direct connections each gene has with others in the network. These genes and their respective scores have been listed in Table 7.
3.11.5. Impact of the top 10 genes on cancer
Through network pharmacology analysis, several important genes have been recognized for their significant involvement in cancer, impacting various stages of tumor progression and treatment resistance.
A central player in the PI3K/AKT signaling pathway, AKT1 is essential for regulating cell growth, survival, and metabolism. It promotes cell proliferation and inhibits apoptosis, making it a vital target in cancer treatment. The EGFR is involved in the regulation of cell growth and division. Aberrant activation of EGFR signaling is linked to several cancers, particularly non-small cell lung cancer, and targeting EGFR can enhance therapeutic efficacy. Proto-oncogene tyrosine-protein kinase (SRC) is a non-receptor tyrosine kinase regulates various signaling pathways that control cell growth, differentiation, and migration. Overexpression or aberrant activity of SRC is often associated with tumor progression and metastasis. Heat shock protein 90 alpha (HSP90AA1) is crucial for protein folding and stabilization of client proteins, many of which are involved in cancer progression. Inhibiting HSP90 can disrupt multiple oncogenic pathways simultaneously. The mechanistic target of rapamycin (mTOR) is a key regulator of cell growth, proliferation, and survival in response to nutrients and growth factors. Its dysregulation is implicated in various cancers, making it a significant therapeutic target. The PIK3CA gene encodes a subunit of the phosphoinositide 3-kinase (PI3K) and is frequently mutated in cancers, leading to enhanced signaling that promotes cell survival and growth. Targeting PIK3CA has therapeutic potential in cancer treatment. The IGF1R is involved in cell proliferation and survival. It plays a critical role in tumor growth and resistance to therapies, highlighting its importance in cancer biology. Glycogen synthase kinase 3 beta (GSK3B) is involved in various signaling pathways, including those regulating the cell cycle and apoptosis. Its role in tumor suppression makes it a potential target for cancer therapy. Kinase insert domain receptor (KDR), also known as VEGFR2, is crucial for angiogenesis and lymphangiogenesis, processes essential for tumor growth and metastasis. Inhibition of KDR can disrupt the blood supply to tumors, providing a therapeutic avenue. Poly (ADP-ribose) polymerase 1 (PARP1) is involved in DNA repair mechanisms. Its inhibition has gained traction in cancer therapy, particularly in tumors with homologous recombination deficiencies, as it can lead to synthetic lethality. These results lay the groundwork for further investigation of these genes as potential therapeutic targets by exploring their roles and interactions within the cancer network. KEGG pathway enrichment analysis and overview of biological process (BP) in Blue, cellular component (CC) in Red, and molecular function (MF) in Green as shown in Figures 12 and 13 respectively.
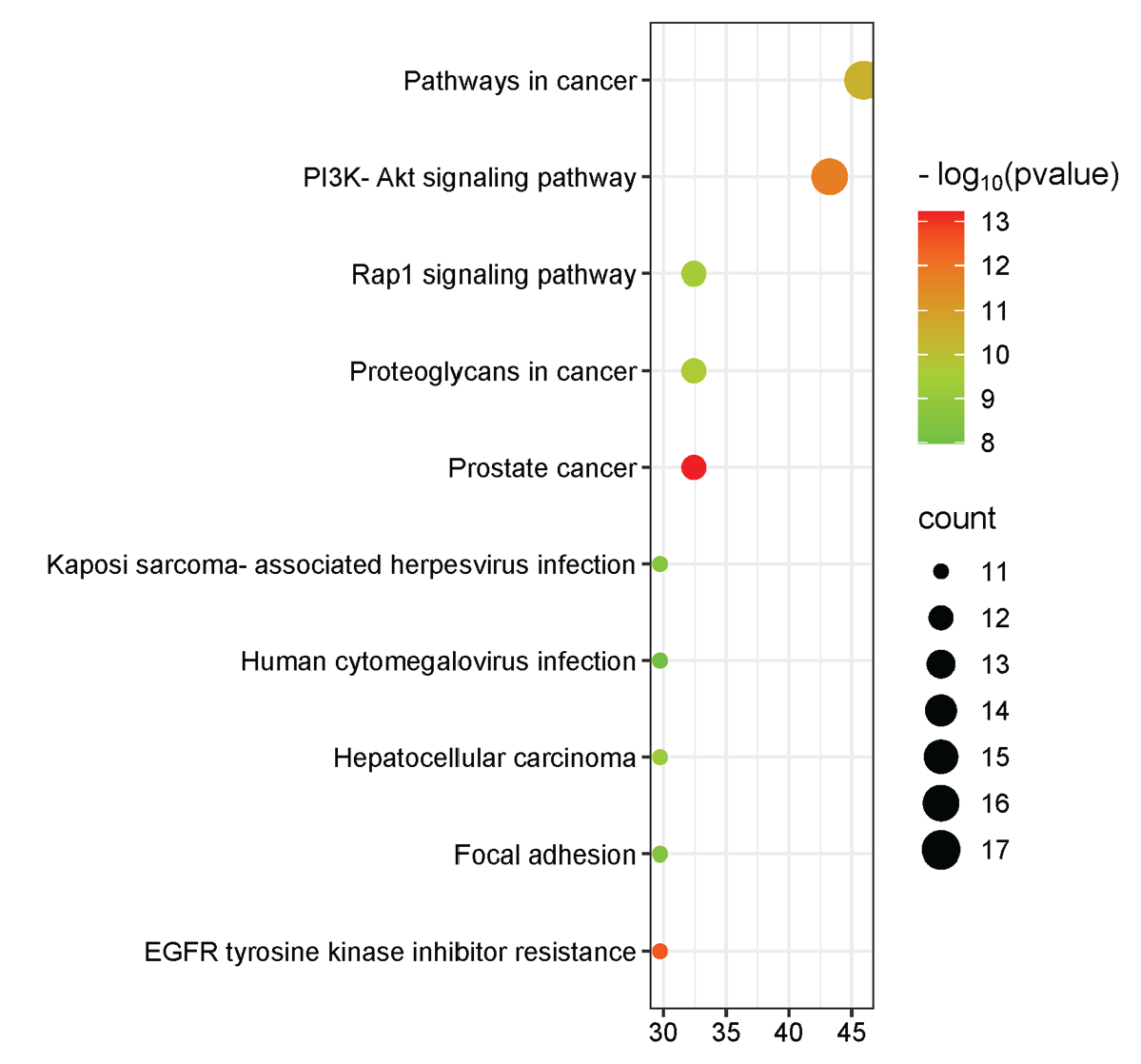
- KEGG pathway enrichment analysis.
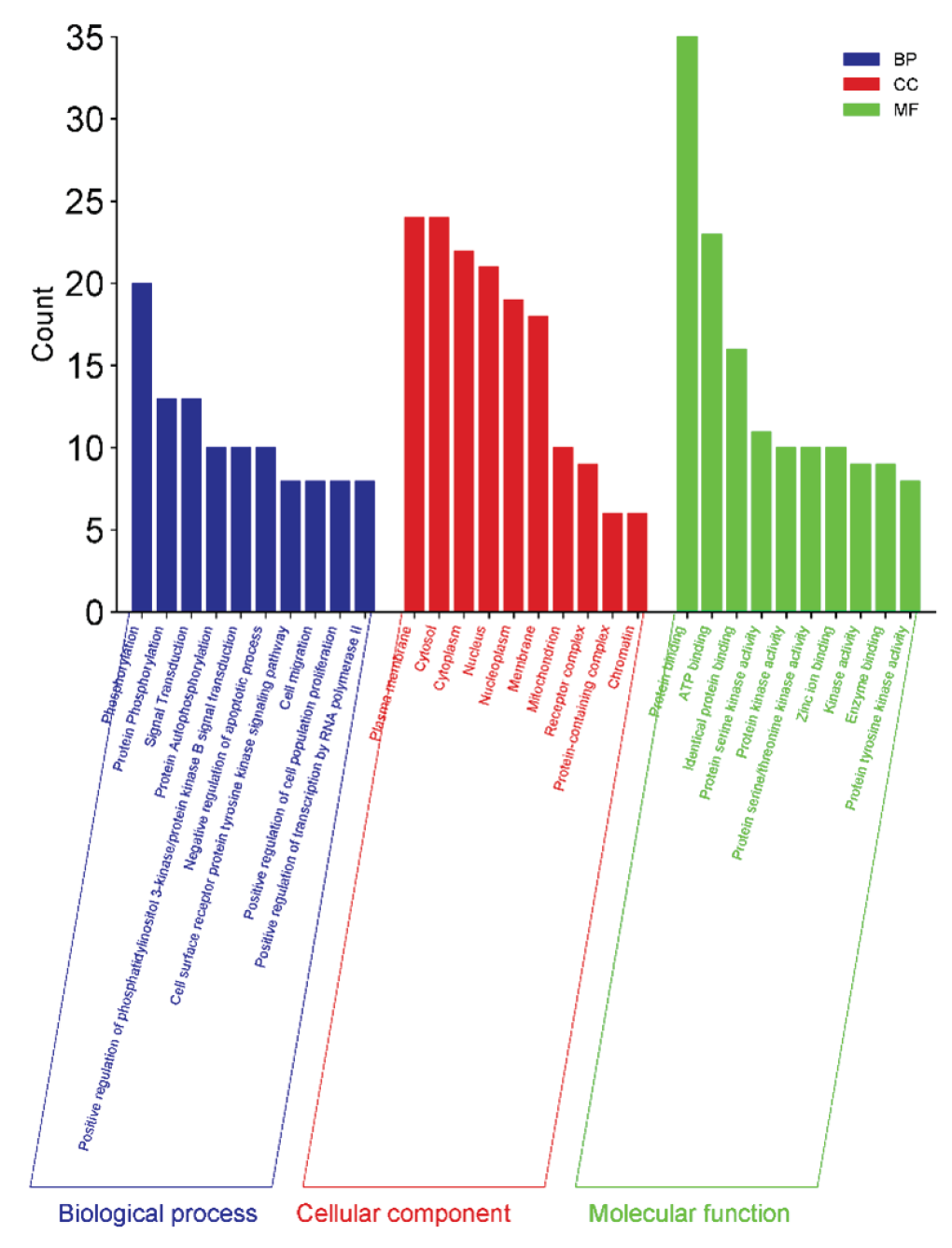
- A summary of biological processes (BP) in blue, cellular components (CC) in red, and molecular functions (MF) in green.
3.11.6. Network pharmacology analysis of designed triazole benzene sulfonate derivatives (8a-b) targeting breast cancer
The potential of Triazole-benzenesulfonate derivatives as anticancer agents has gained significant attention due to their diverse pharmacological activities. In this research, derivatives 8a-b were developed with a focus on targeting breast cancer. Network pharmacology was applied to investigate the interactions between these compounds and breast cancer-related genes. Key genes were identified, underscoring their involvement in important breast cancer pathways. The top 10 genes, ranked according to their degree in the network, are pivotal in regulating cellular signaling and have profound relevance in the context of breast cancer. A comparison with enrichment and KEGG pathway analyses revealed AKT1 as the central target. Further functional annotation and enrichment analyses provided insights into the roles of these targets, which were subsequently selected for ADMET and molecular docking investigations.
3.11.7. Physicochemical properties
The physico-chemical parameters of the designed ligands 8a and 8b, as shown in Table 1S, indicate favorable drug-like properties. The Log P values for 8a and 8b are 2.26 and 2.70, respectively, suggesting good lipophilicity. Both compounds have identical TPSA values of 111.39 Å2, which is within the acceptable range for oral bioavailability. The Molecular Weights (MW) of 8a (373.39 g/mol) and 8b (387.42 g/mol) are below 500 g/mol, complying with Lipinski’s rule. Each compound has 7 HBA and 1 HBD, along with 7 rotatable bonds, indicating moderate flexibility. Notably, neither compound violates any of Lipinski’s rules.
3.11.8. Bioactivity
The bioactivity scores of ligands 8a and 8b across a variety of biological targets, including GPCRs, ion channels, kinases, nuclear receptors, proteases, and enzymes, have been summarized in Table 2S. Ligand 8a shows notable bioactivity as an enzyme inhibitor with a score of 0.22, and marginal activity as a protease inhibitor (PI) (0.01). Its scores for GPCR ligand (-0.01), ion channel modulator (ICM) (-0.23), kinase inhibitor (KI) (-0.15), and nuclear receptor ligand (NRL) (-0.23) indicate less potential in these categories. Ligand 8b also exhibits some bioactivity as an enzyme inhibitor with a score of 0.16 but shows weaker potential in other categories, including GPCR ligand (-0.05), ICM (-0.29), KI (-0.18), NRL (-0.25), and PI (-0.05).
3.11.9. Toxicity
Table 3S presents the toxicity scores of the designed compounds 8a and 8b across various toxicity categories, including hepatotoxicity, carcinogenicity, immunotoxicity, mutagenicity, and cytotoxicity. For both compounds, all toxicity types were predicted to be inactive. Specifically, compound 8a shows probabilities of 0.54 for hepatotoxicity, 0.56 for carcinogenicity, 0.86 for immunotoxicity, 0.60 for mutagenicity, and 0.78 for cytotoxicity. Similarly, compound 8b demonstrates probabilities of 0.55 for hepatotoxicity, 0.55 for carcinogenicity, 0.84 for immunotoxicity, 0.59 for mutagenicity, and 0.79 for cytotoxicity.
3.11.10. Molecular docking
Compound 8a demonstrated a docking score of -7.50 kcal/mol, forming interactions with 9 amino acid residues. Key hydrogen bonds were observed with Asn 54, Leu 295, Cys 296, and Glu 298. Pi-Alkyl interactions involved Val 270, Arg 273, Ile 84, and Cys 310, while Gln 79 exhibited an Amide Pi-stacked interaction, contributing to the compound’s overall stability within the active site. Compound 8b achieved a docking score of -7.61 kcal/mol, interacting with 8 residues. Hydrogen bonds were formed with Asn 54 and Thr 211, while Pi-Alkyl interactions occurred with Ile 84, Leu A264, Val 270, and Cys 310. Pi-cation interactions were observed with Asp 292 and Arg 273, indicating multiple stabilizing interactions. Capivasertib (standard) exhibited the highest docking score of -7.96 kcal/mol, with interactions involving 8 amino acids. Hydrogen bonds were established with Ser 205, Phe 293, and Asp 274, while Pi-Alkyl interactions occurred with Arg 273, Ile 84, Leu 210, and Lys 268. Additionally, a Pi-Pi stacking interaction with Trp 80 was noted, further strengthening its binding affinity (Figure 14 and Table 8).
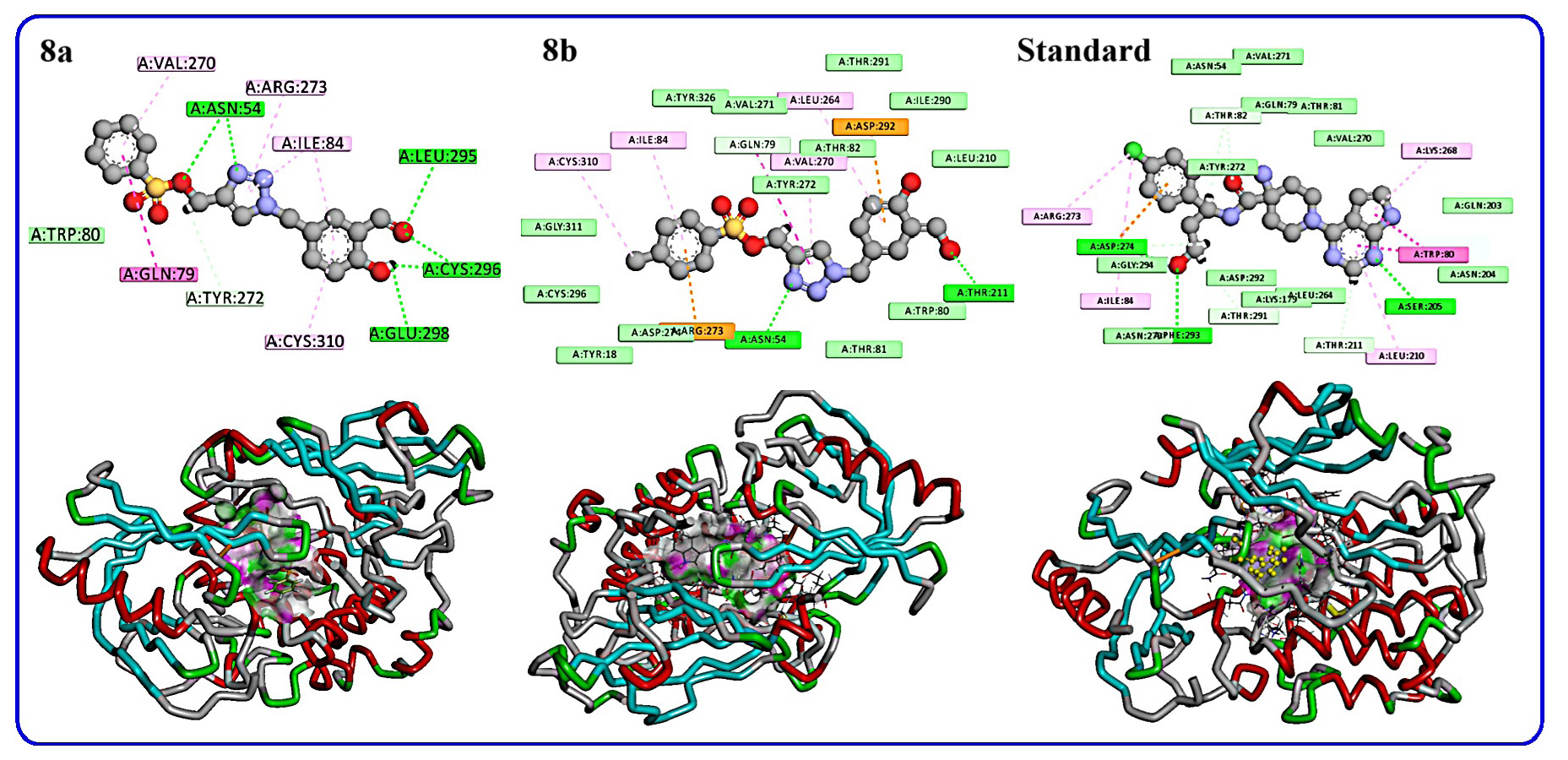
- 2/3D representations of products 8a-b and standard.
| Compound | Docking score (Kcal mol−1) | Number of interacting residues | Amino acid and type of interaction |
|---|---|---|---|
| 8a | -7.50 | 9 | Asn 54; Leu 295; Cys 296; Glu 298 (H-bond); Val 270; Arg 273; Ile 84; Cys 310(Pi-Alkyl); Gln 79 Amide Pi-stacked |
| 8b | -7.61 | 8 | Asn 54; Thr 211(H-bond); Ile 84, Leu A 264, Val 270, Cys 310(Pi-Alkyl); Asp 292, Arg 273 (Pi-cation) |
| STD capivasertib | -7.96 | 8 | Ser 205, Phe 293, Asp 274(H-bond); Arg 273, Ile 84, Leu 210, Lys 268(Pi-Alkyl); Trp 80 (Pi-Pi stacked) |
3.12. Discussion
The physico-chemical profile of ligands 8a and 8b suggests that they possess drug-like characteristics suitable for further exploration in drug discovery. The Log P values indicate adequate lipophilicity, which could enhance membrane permeability. The TPSA values, being under the critical threshold, point towards good absorption and permeation potential. The molecular weight of both compounds falls within the range recommended by Lipinski’s rule, ensuring favorable pharmacokinetics. Additionally, the balance between HBA and HBD ensures that these ligands could form stable hydrogen bonds with biological targets, enhancing binding affinity. The absence of rule violations reinforces the potential of these ligands for oral bioavailability, making them promising candidates for further biological evaluations.
The bioactivity scores of ligands 8a and 8b suggest that both compounds exhibit the highest potential as enzyme inhibitors, with scores of 0.22 and 0.16, respectively, which are close to or above the general threshold (0.00) for bioactivity. This indicates that the designed ligands may effectively inhibit enzyme targets. Ligand 8a also displays minimal protease inhibitory activity (0.01), suggesting potential in that area, though its activity across other categories, such as GPCR ligands, ion channel modulators, kinase inhibitors, and nuclear receptor ligands, is limited. Similarly, ligand 8b shows comparable trends, with enzyme inhibition as the most promising bioactivity, though its scores in other categories suggest a lower likelihood of efficacy in those biological pathways. These findings highlight the potential of both compounds as enzyme inhibitors, warranting further exploration in enzyme-targeted studies.
The toxicity analysis of compounds 8a and 8b suggests that both ligands exhibit an inactive toxicity profile across all categories, including hepatotoxicity, carcinogenicity, immunotoxicity, mutagenicity, and cytotoxicity. The probability values for each category are generally moderate to high, indicating a strong likelihood that these compounds are non-toxic in these areas. Compound 8a shows a slightly higher probability of immunotoxicity (0.86) and cytotoxicity (0.78), whereas 8b follows a similar trend, with high probabilities for immunotoxicity (0.84) and cytotoxicity (0.79). Despite these elevated probabilities, the “inactive” toxicity status suggests a favorable safety profile for both compounds, making them promising candidates for further development with minimal toxicity concerns.
The docking results revealed that compound 8b had a slightly better docking score than 8a (-7.61 vs. -7.50 kcal/mol), indicating a stronger binding affinity. The presence of Pi-cation interactions in 8b with Asp 292 and Arg 273 likely contributed to this enhanced binding. Meanwhile, compound 8a exhibited multiple stabilizing interactions through hydrogen bonds and Pi-Alkyl interactions, though its binding affinity was slightly lower than 8b. Capivasertib, serving as the reference compound, demonstrated the highest docking score (-7.96 kcal/mol), supported by diverse interactions including hydrogen bonds, Pi-Alkyl, and Pi-Pi stacking. This robust interaction profile highlights its superior inhibitory potential, setting a benchmark for the designed compounds. While both 8a and 8b show promise, they fall short of Capivasertib’s binding efficiency.
4. Conclusions
This research presents two extensively characterized new derivative of 1,2,3-triazole-O-tosyls 8a-b, employing various spectroscopic techniques such as NMR and HRMS. The molecular structure of the 1,2,3-triazole-O-tosyl derivatives 8a-b was optimized in the gas phase using the B3LYP functional combined with the 6-311++G(d,p) basis set. The experimental data from NMR spectroscopy were aligned with computational results, confirming consistency between the two approaches. The results show that while both structures experienced a decrease in the energy gap and electron potential barrier with increasing electric field intensity, compound 8b exhibited a more significant response. Landauer’s theory confirmed its superior conductivity, and analysis revealed that 8b could switch to the ON state at EF > 120 × 10⁻⁴ (a.u.), acting as a molecular switch. QTAIM studies also predicted lower Joule/Pelletier-like coefficients for 8b. In addition, pharmacological network analysis and molecular docking simulations were performed in silico, along with ADMET predictions, to investigate the interactions of these derivatives with AKT1. The results emphasized the robust binding affinity of the synthesized 1,2,3-triazole hybrid 8b to AKT1, exhibiting a binding energy score of -7.61 kJ/mol.
Acknowledgment
This study is supported via funding from Prince Sattam bin Abdulaziz University project number (PSAU/2024/03/31599).
CRediT authorship contribution statement
Yassine Riadi: Conceptualization; Data curation; Methodology; Project administration; Funding acquisition; Resources; Supervision; Validation; Writing - Review & Editing; Mohammed H. Geesi: Conceptualization; Data curation; Investigation; Methodology; Formal analysis; Validation; Writing - Original Draft.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Declaration of Generative AI and AI-assisted technologies in the writing process
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Supplementary data
Supplementary material to this article can be found online at https://doi.org/10.25259/AJC_32_2025.
References
- Anti-methicillin resistant staphylococcus aureus activity, synergism with oxacillin and molecular docking studies of metronidazole-triazole hybrids. European Journal of Medicinal Chemistry. 2016;115:426-437. https://doi.org/10.1016/j.ejmech.2016.03.041
- [Google Scholar]
- Regulation of inflammation in cancer by eicosanoids. Prostaglandins & Other Lipid Mediators. 2011;96:27-36. https://doi.org/10.1016/j.prostaglandins.2011.08.004
- [Google Scholar]
- Inflammation by remote control. Nature. 2005;435:752-3. https://doi.org/10.1038/435752a
- [Google Scholar]
- Eicosanoids and cancer. Clinics (Sao Paulo, Brazil). 2018;73:e530s. https://doi.org/10.6061/clinics/2018/e530s
- [Google Scholar]
- The multifaceted link between inflammation and human diseases. Journal of Cellular Physiology. 2018;233:6458-6471. https://doi.org/10.1002/jcp.26479
- [Google Scholar]
- Predicting the future burden of cancer. Nature Reviews. Cancer. 2006;6:63-74. https://doi.org/10.1038/nrc1781
- [Google Scholar]
- New insights into chemistry and anti-infective potential of triazole scaffold. Current Medicinal Chemistry. 2011;18:3265-3297. https://doi.org/10.2174/092986711796391615
- [Google Scholar]
- Synthesis, characterization and antimicrobial activity of some substituted 1,2,3-triazoles. European Journal of Medicinal Chemistry. 2005;40:1173-8. https://doi.org/10.1016/j.ejmech.2005.02.013
- [Google Scholar]
- Design and synthesis of novel 1,2,3-triazole-pyrimidine hybrids as potential anticancer agents. European Journal of Medicinal Chemistry. 2014;86:368-380. https://doi.org/10.1016/j.ejmech.2014.08.010
- [Google Scholar]
- Synthesis, characterization and antimicrobial activity of 4-((1-benzyl/phenyl-1H-1,2,3-triazol-4-yl)methoxy)benzaldehyde analogues. Medicinal Chemistry Research. 2016;25:644-652. https://doi.org/10.1007/s00044-016-1515-0
- [Google Scholar]
- The application of click chemistry in the synthesis of agents with anticancer activity. Drug Design, Development and Therapy. 2015;9:1585-1599. https://doi.org/10.2147/DDDT.S56038
- [Google Scholar]
- Hybrid molecules: The privileged scaffolds for various pharmaceuticals. European Journal of Medicinal Chemistry. 2016;124:500-36. https://doi.org/10.1016/j.ejmech.2016.08.039
- [Google Scholar]
- 4-quinolone hybrids and their antibacterial activities. European Journal of Medicinal Chemistry. 2017;141:335-345. https://doi.org/10.1016/j.ejmech.2017.09.050
- [Google Scholar]
- Thiazolidinone-linked1,2,3-triazoles with monoterpenic skeleton as new potential anticancer agents: Design, synthesis and molecular docking studies. Bioorganic Chemistry. 2021;115:105184. https://doi.org/10.1016/j.bioorg.2021.105184
- [Google Scholar]
- New 1,2,3-triazoles from (R)-carvone: Synthesis, DFT mechanistic study and in vitro cytotoxic evaluation. Molecules (Basel, Switzerland). 2022;27:769. https://doi.org/10.3390/molecules27030769
- [Google Scholar]
- Novel isoxazoline-linked 1,3,4-thiadiazole hybrids as anticancer agents: Design, synthesis, biological evaluation, molecular docking, and molecular dynamics simulation. Archiv der Pharmazie. 2022;355:e2200066. https://doi.org/10.1002/ardp.202200066
- [Google Scholar]
- Pterocarpanquinones, aza-pterocarpanquinone and derivatives: Synthesis, antineoplasic activity on human malignant cell lines and antileishmanial activity on leishmania amazonensis. Bioorganic & Medicinal Chemistry. 2011;19:6885-6891. https://doi.org/10.1016/j.bmc.2011.09.025
- [Google Scholar]
- 11a-n-tosyl-5-deoxi-pterocarpan (LQB-223), a promising prototype for targeting MDR leukemia cell lines. European Journal of Medicinal Chemistry. 2014;78:190-197. https://doi.org/10.1016/j.ejmech.2014.03.039
- [Google Scholar]
- 11a-n-tosyl-5-carbapterocarpans: Synthesis, antineoplastic evaluation and in silico prediction of ADMETox properties. Bioorganic Chemistry. 2018;80:585-590. https://doi.org/10.1016/j.bioorg.2018.07.004
- [Google Scholar]
- Design, synthesis and cytotoxic activity of novel salicylaldehyde hydrazones against leukemia and breast cancer. International Journal of Molecular Sciences. 2023;24:7352. https://doi.org/10.3390/ijms24087352
- [Google Scholar]
- Aldehyde functionalized ionic liquid on electrochemically reduced graphene oxide as a versatile platform for covalent immobilization of biomolecules and biosensing. Biosensors & Bioelectronics. 2018;103:104-112. https://doi.org/10.1016/j.bios.2017.12.030
- [Google Scholar]
- SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Research. 2019;47:W357-W364. https://doi.org/10.1093/nar/gkz382
- [Google Scholar]
- Towards a universal SMILES representation-A standard method to generate canonical based on the InChI. Journal of Cheminformatics. 2012;4:22. https://doi.org/10.1186/1758-2946-4-22
- [Google Scholar]
- STITCH: Interaction networks of chemicals and proteins. Nucleic Acids Research. 2008;36:D684-D688. https://doi.org/10.1093/nar/gkm795
- [Google Scholar]
- DAVID: Database for annotation, visualization, and integrated discovery. Genome Biology. 2003;4:P3. https://doi.org/ 10.1186/gb-2003-4-9-r60
- [Google Scholar]
- Exploring the cyclization of thiosemicarbazone to 1,3,4-thiadiazole: Synthesis, characterization and in-silico study. Journal of Molecular Structure. 2025;1322:140385. https://doi.org/10.1016/j.molstruc.2024.140385
- [Google Scholar]
- Ligand based pharmacophore modelling and integrated computational approaches in the quest for small molecule inhibitors against hCA IX. RSC Advances. 2024;14:3346-3358. https://doi.org/10.1039/d3ra08618f
- [Google Scholar]
- Targeting cyclin-dependent kinase 2 CDK2: Insights from molecular docking and dynamics simulation - A systematic computational approach to discover novel cancer therapeutics. Computational Biology and Chemistry. 2024;112:108134. https://doi.org/10.1016/j.compbiolchem.2024.108134
- [Google Scholar]
- Multitargeted molecular docking and dynamics simulation of thymol-based chalcones against cancer protein markers: Synthesis, characterization, and computational study. Journal of Molecular Structure. 2024;1317:139116. https://doi.org/10.1016/j.molstruc.2024.139116
- [Google Scholar]




