

Translate this page into:
Exploring the interaction of quercetin-3-O-sophoroside with SARS-CoV-2 main proteins by theoretical studies: A probable prelude to control some variants of coronavirus including Delta
⁎Corresponding authors. jbliuzz@163.com (Jianbo Liu), fcchubw@zzu.edu.cn (Bowen Hu), K1akhtari@yahoo.com (Keivan Akhtari), mojtaba.falahati@alumni.ut.ac.ir (Mojtaba Falahati)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
The aim of this study was to investigate the mechanism of interaction between quercetin-3-O-sophoroside and different SARS-CoV-2’s proteins which can bring some useful details about the control of different variants of coronavirus including the recent case, Delta. The chemical structure of the quercetin-3-O-sophoroside was first optimized. Docking studies were performed by CoV disease-2019 (COVID-19) Docking Server. Afterwards, the molecular dynamic study was done using High Throughput Molecular Dynamics (HTMD) tool. The results showed a remarkable stability of the quercetin-3-O-sophoroside based on the calculated parameters. Docking outcomes revealed that the highest affinity of quercetin-3-O-sophoroside was related to the RdRp with RNA. Molecular dynamic studies showed that the target E protein tends to be destabilized in the presence of quercetin-3-O-sophoroside. Based on these results, quercetin-3-O-sophoroside can show promising inhibitory effects on the binding site of the different receptors and may be considered as effective inhibitor of the entry and proliferation of the SARS-CoV-2 and its different variants. Finally, it should be noted, although this paper does not directly deal with the exploring the interaction of main proteins of SARS-CoV-2 Delta variant with quercetin-3-O-sophoroside, at the time of writing, no direct theoretical investigation was reported on the interaction of ligands with the main proteins of Delta variant. Therefore, the present data may provide useful information for designing some theoretical studies in the future for studying the control of SARS-CoV-2 variants due to possible structural similarity between proteins of different variants.
Keywords
SARS-CoV-2
COVID-19
Quercetin-3-O-sophoroside
Molecular docking
Molecular dynamic
Variants

1 Introduction
Given the unique characteristics of the genetic material of coronaviruses (CoV), as well as the mechanism of their proliferation, the emergence of the severe acute respiratory syndrome CoV 2 (SARS-CoV-2) has not been unexpected for virologists (Evangelin et al., 2020). Among these special features, the high genetic recombination rate of this virus has been led to the emergence of new and unknown strains (Deng et al., 2020). The reason for this high recombinant frequency can be attributed to the very long genetic material of the virus compared to other viruses, the complex proliferation, and ultimately to the wide host range of these viruses in humans and animals (Evangelin et al., 2020; Deng et al., 2020; Wu et al., 2020). In order to treat this virus, in addition to sufficient knowledge of this invasive and contagious virus (Zuo et al., 2020; Ashour et al., 2020), it is necessary for different countries to make correct and timely control decisions to take personal protective and hygienic measures (Ong et al., 2020; Ferretti et al., 2020).
Due to severe tissue damage caused by SARS-CoV and middle east respiratory syndrome (MERS-CoV) infection, the mortality rate in infected patients requiring mechanical ventilation (artificial respiration) has been very serious (Yang et al., 2020; Zhou et al., 2020). To date, there is no specific antiviral drug for the treatment of CoV (Zhou et al., 2020; Wu et al., 2020), and the main solutions are based on supportive care, such as maintaining vital signs, regulating oxygen and blood pressure, and reducing side effects such as secondary infections or organ failure (Gheblawi et al., 2020; Abassi et al., 2020; Yan et al., 2020). Based on the previous studies, a combination of protease inhibitors, lopinavir and ritonavir drugs has been shown to significantly improve the condition of COVID-19 patients (Muralidharan et al., 2021; Stower, 2020; Moro et al., 2020). The results of in vitro and in vivo studies show that a combination of lopinavir, ritonavir, and interferon β may be effective against MERS infection (Sheahan et al., 2020).
Recent studies have shown that viral proteins may be potential models for the design of new antiviral drugs to prevent the proliferation of SARS-CoV-2 and possible different variants (Zhang et al., 2020; Hu et al., 2003; Nabavi et al., 2020).
The computational methods are widely used in the discovery, design, and optimization of potential compounds based on their structure, in which small molecules are docked into the structure of the target macromolecules and rated for their binding at the target site (Choudhary et al., 2020; Cavasotto and Filippo, 2020). The study of SARS-CoV-2 inhibitors to develop an effective drug to reduce mortality in COVID-19 patients has been a new topic in recent years (Choudhary et al., 2020; Cavasotto and Filippo, 2020; Zhang et al., 2020; Chowdhury et al., 2020). At present, the lack of appropriate methods to identify inhibitors' mechanism of action on SARS-CoV-2 function and maybe different variants, including Delta and the relationship between their chemical structure and antiviral effect encourages us to further examine their inhibitory effects against COVID-19 (Farinholt et al., 2021; Enmozhi et al., 2021). Software and computational methods of drug design have been a new approach that their emergence and generality have been accompanied by advances in computing power over decades (Farinholt et al., 2021; Enmozhi et al., 2021). These methods are used in conjunction with biological experiments to establish a relationship between the structure and activity of pharmaceutical compounds, the discovery of new compounds, the prediction of the biological activities of designed compounds, and the understanding of biochemical reactions and relative processes (Sonawane et al., 2020; Bobrowskia et al., 2020). In addition, these methods can be used to predict how the designed compounds bind to the target protein, evaluate the energy difference between different compound conformers, and explain the mechanisms of reaction, effect of different groups, decompositions in the chemical structure of compounds, and their performance (Farinholt et al., 2021; Enmozhi et al., 2021; Sonawane et al., 2020; Bobrowskia et al., 2020).
Quercetin-3-O-sophoroside as a major member of quercetin glucoside group is a well-acknowledged anti- inflammatory, anti-oxidant and anti-cancer edible compound (30]. Broccoli, and Poacynum hendersonii leaves have a high content of quercetin glucosides (Jeffery and Araya, 2009; An et al., 2013).
Lee et al. [a] studied the inhibitory activity of quercetin-3-O-sophoroside against H1N1, H3N2, and H5N1 viruses. They suggested that the size of sugar moiety on quercetin-3-sophoroside and other flavonoids can affect the antiviral effect of these compounds.
Hollman et al. (1999) studied the catabolism of quercetin-3-O-sophoroside in the small intestine. Also, it is reported that during this process the sugar moiety is removed (Wang et al., 2020). Since in this study the molecule has been studied in its intact form, non-oral methods should be prescribed in its possible medical use for the treatment of viral disease. Accessibility of the target protein and lack of closely related host cell counterparts which improve the inhibitory potency and fewer off-target effects against host proteins, are two important aspects in selecting favorable receptors among a wide variety of target proteins.
Therefore, in this study, in order to identify the exact mechanism of quercetin-3-O-sophoroside performance and identify the amino acid residues involved in this process and the optimal energy of quercetin-3-O-sophoroside upon interaction with different SARS-CoV-2’s proteins including, E protein (ion channel), helicase ADP site, helicase NCB site, main protease, N protein NCB site, Nsp14 (ExoN), Nsp14 (N7-MTase), Nsp15 (endoribonuclease), Nsp16 (2′-O-MTase), papain-like protease, RdRp with RNA, and RdRp without RNA, molecular, docking and molecular dynamic studies were done. This information may provide some potential information about the control of SARS-CoV-2 and reported variants.
2 Material and methods
2.1 Global reactivity descriptors
The quercetin-3-O-sophoroside molecule geometry optimization was carried out using the PM3 semi-empirical method and a single point energy calculation was performed using Becke3-Parameter hybrid function which combined with the Lee–Yang– Parr correlation functional, abbreviated as B3LYP (Stephens et al., 1994; Lee et al., 1988) with the standard 6-31G(d,p) basis both implemented in the GAUSSIAN98W suite of programs (Frisch et al., 1998).
2.2 Molecular docking
In order to investigate the affinity of quercetin-3-O-sophoroside toward covid-19 virus's receptors molecular docking calculations were carried out using COVID-19 Docking Server (Kong et al., 2020). We used all proteins provided by this server, which uses the JSMol (http://jmol.sourceforge.net/) for molecular visualization, where the plots of complex configurations were extracted from this page.
2.3 Molecular dynamics study
In order to study the adsorption effects of quercetin-3-O-sophoroside on target proteins, E protein was selected as the receptor model. The molecular dynamics calculation was performed using HTMD software (Doerr et al., 2016). The GAFF2 method (Mayo et al., 1990) was employed for parameterization of the quercetin-3-O-sophoroside. Conditions were set at 1 atm pressure and 310 K. The software generated parameters in the AMBER-compatible format. The flipping of side chains and placement of missing hydrogen atoms at biological pH was done using Protein Prepare application (Doerr et al., 2016). All mentioned applications are available on PlayMolecule site (www.playmolecule.org). Also, visualization of the structures was done by using CHIMERA tool (www.cgl.ucsf.edu/chimera).
3 Results
3.1 Optimized geometry of quercetin-3-O-sophoroside
Fig. 1a depicts the optimized geometry of the quercetin-3-O-sophoroside molecule. As can be seen, this molecule consists of a quercetin part and a sugar moiety. Fig. 1b illustrates the molecular electrostatic potential as a descriptor for charge-controlled reactions. The negative electronic potential is shown in red, while the positive electrostatic potential is depicted in blue.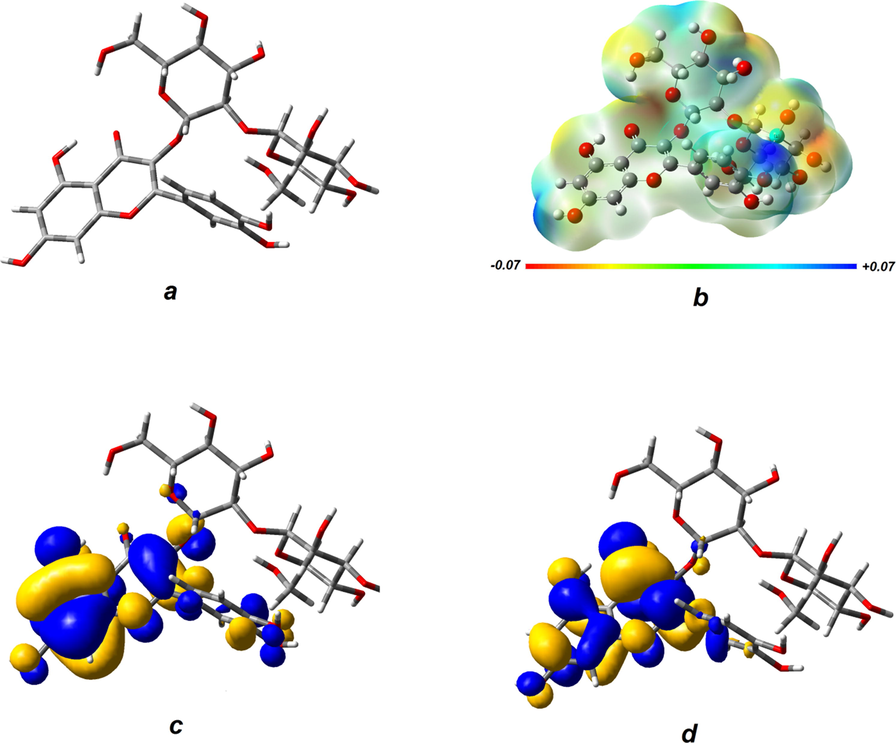
Optimized geometry of the quercetin-3-O-sophoroside (a) Electrostatic potential surface of the molecule (b) The plot of HOMO (c) and LUMO (d).
As shown in Fig. 1b, the most intense electrostatic potential regions are located on sugar moiety which can play an important role in interaction of the molecule with electrostatic active sites on the biomolecules. The calculated molecular orbitals (Fig. 1c and d) show the more favorable sites for redox reactions. The Highest Occupied Molecular Orbital (HOMO) plot shows electron donor sites and the Lowest Unoccupied Molecular Orbital (LUMO) plot shows electron acceptor sites. Although both orbitals mainly located on quercetin part, it has been proven that sugar moiety in this kind of compounds plays a vital role in biological activities.
In order to study the structural-biological activity, at first the global reactivity descriptors of the quercetin-3-O-sophoroside molecule were calculated and the affinity of the molecule toward the SARS-CoV-2’s receptors was evaluated using molecular docking study. The global parameters of the quercetin-3-O-sophoroside molecule were calculated as following:
The Ionization potential (I) and electron affinity (A) are given by:
Which EHOMO and ELUMO represent the energies of the respective molecular orbitals. The chemical potential μ and the hardness η are expressed in terms of the ionization potential I and the electron affinity A (Koulgi et al., 2021):
The calculated global reactivity parameters are gathered in Table 1.
Parameter
Calculated result
EHOMO (eV)
−5.943
ELUMO (eV)
−1.393
ΔELUMO-HOMO (eV)
4.550
I=- EHOMO (eV)
5.943
A=- ELUMO (eV)
1.393
(eV)
2.275
(eV)
3.668
1.0693
The relatively larger value of the quercetin-3-O-sophoroside molecular hardness with respect to similar value for other antioxidants which were calculated by similar method reveals a remarkable stability of the molecule.
3.2 Molecular docking results
In this study, molecular docking simulations between quercetin-3-O-sophoroside and different proteins such as E protein (ion channel) (Fig. 2a), helicase ADP site (Fig. 2b), helicase NCB site (Fig. 2c), main protease (Fig. 2d), N protein NCB site (e), Nsp14 (ExoN) (Fig. 2f), Nsp14 (N7-MTase) (Fig. 2g), Nsp15 (endoribonuclease) (Fig. 2h), Nsp16 (2′-O-MTase) (Fig. 2h), papain-like protease (Fig. 2j), RdRp with RNA (Fig. 2k), and RdRp without RNA (Fig. 2l) were performed to obtain the respective binding energies, involved amino acid residues, and corresponding linkages between the ligand and the receptors. In this method, the binding energy or docking energy calculated is a set of intracellular energy and ligand internal energy.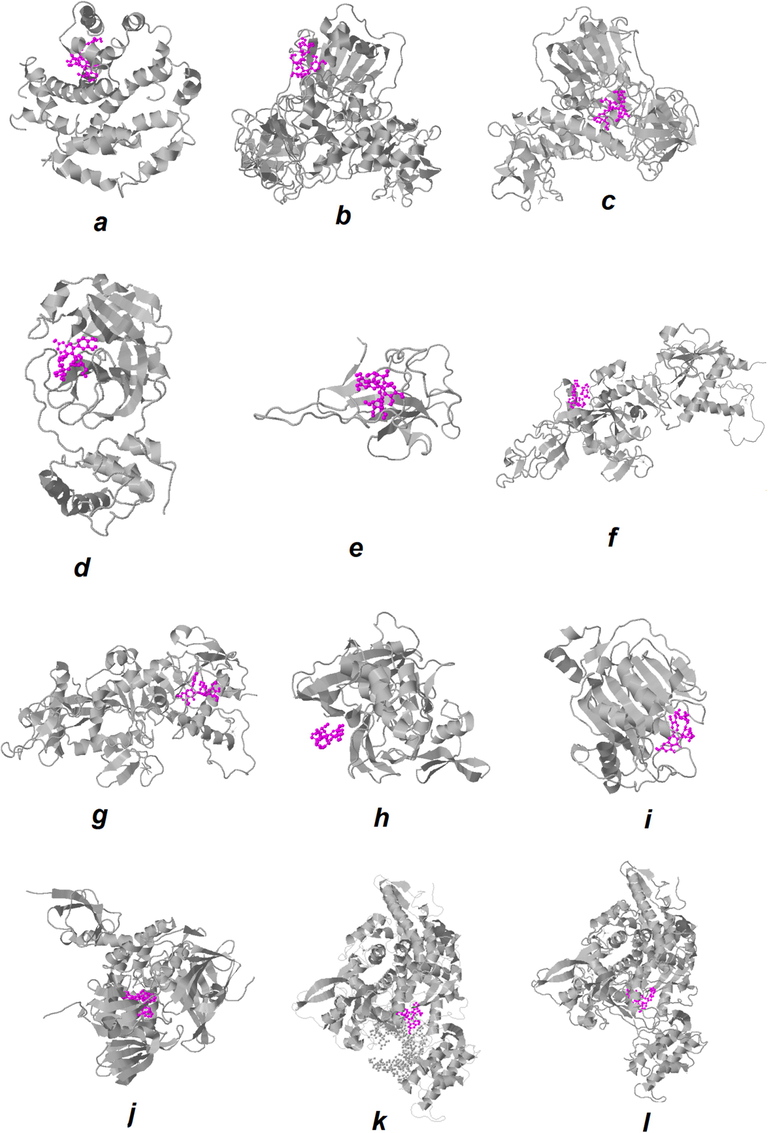
The best docked models’ visualization of the quercetin-3-O-sophoroside with different target proteins of SARS-CoV-2; the ligand is shown in purple. E protein (ion channel) (a), helicase ADP site (b), helicase NCB site (c), MmainpProtease (d), N protein NCB site (e), Nsp14 (ExoN) (f), Nsp14 (N7-MTase) (g), Nsp15 (endoribonuclease) (h), Nsp16 (2′-O-MTase) (i), papain-like protease (j),RdRp with RNA (k), and RdRp without RNA (l).
The negative binding energy, the proper interactions, and the strong bond with the main amino acid residues in the active site are observed for most proteins upon interaction with quercetin-3-O-sophoroside (Table 2).
Receptors
Binding energy (kcal/mol)
quercetin-3-O-sophoroside
1
E protein (ion channel)
−6.70 ± 0.58
2
Helicase ADP site
−7.10 ± 0.91
3
Helicase NCB site
−8.80 ± 0.45
4
Main protease
−8.70 ± 1.08
5
N protein NCB site
−8.70 ± 0.94
6
Nsp14 (ExoN)
−7.10 ± 0.67
7
Nsp14 (N7-MTase)
−9.10 ± 0.89
8
Nsp15 (endoribonuclease)
−7.10 ± 0.37
9
Nsp16 (2′-O-MTase)
−8.70 ± 0.92
10
Papain-like protease
−9.5 ± 0.67
11
RdRp with RNA
−9.70 ± 1.58
12
RdRp without RNA
−8.40 ± 1.12
Also, as can be seen in Table 2, the binding energies for quercetin-3-O-sophoroside are potentialy high in all cases which reveals the potency of the quercetin-3-O-sophoroside in inhibiting SARS-CoV-2’s enzymes and disruption in function of target proteins.
From the binding energies presented in Table.2, we observed that the highest affinity of quercetin-3-O-sophoroside is related to the RdRp with RNA, the binding energy for this case is a considerable value of −9.70 ± 1.58 kcal/mol. On the other hand, E protein (ion channel), Helicase ADP site, Nsp14 (ExoN), and Nsp15 (endoribonuclease) with binding energies of −6.70 ± 0.58 kcal/mol, −7.10 ± 0.91 kcal/mol, −7.10 ± 0.67 kcal/mol, and −7.10 ± 0.37 kcal/mol, respectively showed the lowest binding energy among other proteins. Visualization of the docked pose was done using PyMOL (http://pymol.sourceforge.net/) tool. The ligand with surrounding active site residues within 4 Å is shown in Fig. 3 for E protein (ion channel) (Fig. 3a), helicase ADP site (Fig. 3b), helicase NCB site (Fig. 3c), main protease (Fig. 3d), N protein NCB site (Fig. 3e), Nsp14 (ExoN) (Fig. 3f), Nsp14 (N7-MTase) (Fig. 3g), Nsp15 (endoribonuclease) (Fig. 3h), Nsp16 (2′-O-MTase) (Fig. 3i), papain-like protease (Fig. 3j), RdRp with RNA (Fig. 3k), and RdRp without RNA (Fig. 3l). The nearest interacting residues are summarized in Table 3.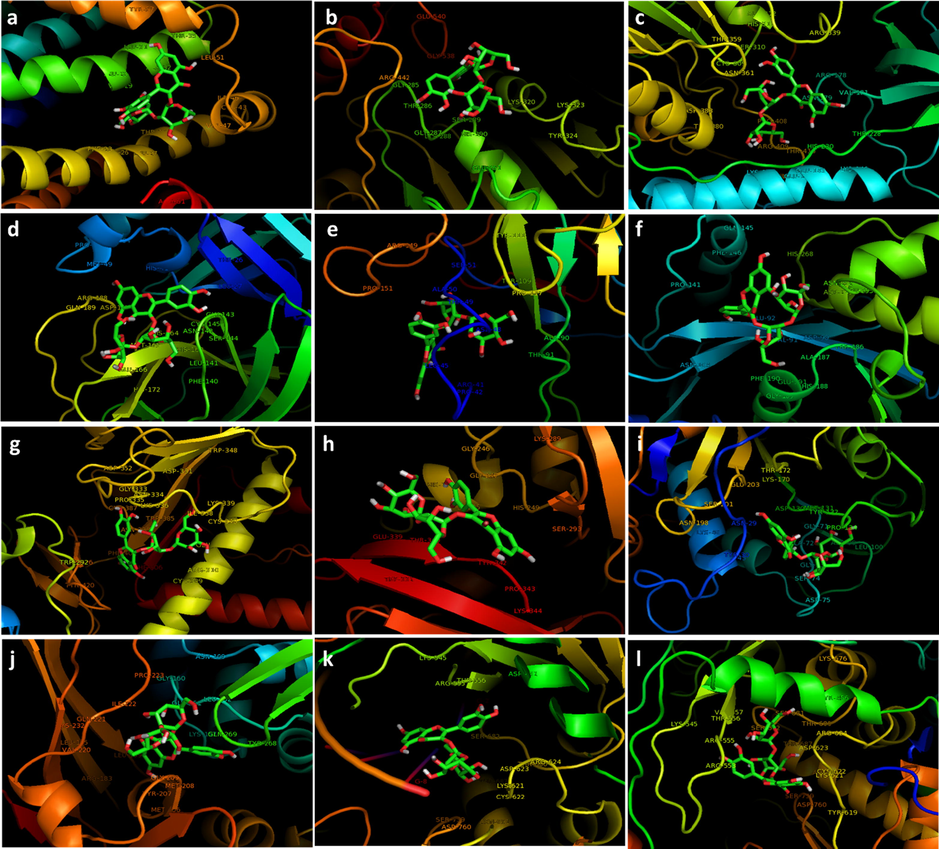
The interacting residues of the receptors with quercetin-3-O-sophoroside; E protein (ion channel) (a), helicase ADP site (b), helicase NCB site (c), main protease (d), N protein NCB site (e), Nsp14 (ExoN) (f), Nsp14 (N7-MTase) (g), Nsp15 (endoribonuclease) (h), Nsp16 (2′-O-MTase) (i), apain-like protease (j), RdRp with RNA (k), and RdRp without RNA (l).
Ligand
Receptors
Residue interacted
The minimum distance between ligand and receptors atoms (dm)
dm < 4Ȧ
Quercetin-3-O-sophoroside
E protein (ion channel)
Phe-23, Phe-26, Leu-27, Thr-30, Val-47, Cys-43, Ile-46, Leu-51, Thr-35, Ala-32, Leu-31, Leu-28, Val-29
Helicase ADP site
Arg-442, Thr-440, Thr-286, Gly-285, Gly-287, Ser-289, His-290, Lys-320, Lys-322, Tyr-324, Glu-261
Helicase NCB site
Arg-409, Thr-410, Pro-408, His-230, Glu-143, Thr-228, Asn-179, Arg-178, Val-181, Arg-339, Ser-310, Asn-361, Cys-309, His-311
Maiprotease
Glu-166, Gln-189, Met-165, His-164, Cys-145, Gly-143, Asn-142, Asn-119, Asp-187
N protein NCB site
Arg-41, Pro-42, Leu-45, Asn-48, Thr-49, Ala-50, Ser-51, Pro-151, Thr-91, Ala-90, Pro-117, Tyr-109
Nsp14 (ExoN)
Phe-190, Glu-191, Ala-178, Val-91, Asp-90, Glu-92, Asn-252, Asp-273, Leu-235, His-268, Gln-145, Pro-141, Asn-104
Nsp14 (N7-MTase)
Cys-309, Arg-310, Cys-310, Ile-338, Lys-339, Gln-313, Phe-401, Trp-385, Lys-336, Cys-387, Asn-334, Pro-335, Gly-333
Nsp15 (endoribonuclease)
Trp-332, Lys-344, Pro-343, Tyr-342, Thr-340, Glu-339, Ser-293, His-249, Gly-247, Gly-246, His-234
Nsp16 (2′-O-MTase)
Asp-75, Ser-74, Gly-73, Leu-100, Asp-99, Gly-71, Ala-72, Pro-134, Asp-130, Tyr-132, Met-131, Tyr-30, Asn-29, Glu-203, Lys-170, Lys-46
Papain-like protease
Met-206, Tyr-207, Met-208, Gly-209, Tyr-268, Gln-269, Lys-157, Leu-162, Glu-161, Gly-160, Ile-222, Gln-221, Leu-199
RdRp with RNA
Cys-622, Lys-621, Asp-623, Ser-682, Arg-555, Thr-556, Asp-452, G-8
RdRp without RNA
Tyr-619, Asp-760, Ser-759, Arg-553, Arg-555, Thr-556, Val-557, Ser-682, Ser-681, Thr-680, Arg-624, Thr-687, Cys-622, Lys-621, Asp-623
Molecular docking studies have shown that the quercetin-3-O-sophoroside binds to RdRp with RNA protein by hydrogen bonding and electrostatic interactions. Indeed, the structure of quercetin-3-O-sophoroside forms hydrophilic interactions with the amino acid residues of Cys-622, Lys-621, Asp-623, Ser-682, Arg-555, Thr-556, Asp-452 as well as RNA G-8 nucleotide including. Indeed, due to the presence of sophoroside group in the ring structure of quercetin, the lipophilicity of this compound increases. The high lipophilicity of sophoroside group can affect the interaction of hydrophobic quercetin with the receptor as well as the possibility of virus penetration (host cell membrane permeability).
In addition to the mentioned interactions, this drug is able to create π-π interactions, which is the connection between the phenyl ring and the phenyl amino acid ring Tyr-619 in the case of RdRp without RNA. The number of reversible bonds affects the physicochemical properties of the quercetin-3-O-sophoroside in oral administration as well as the spatial orientation of the compound at the site of protein binding.
In other hand, it was revealed that in the case of RdRp without RNA, the binding energy was reduced to −8.40 kcal/mol, relative to RdRp with RNA. Therefore, it may be indicated that the dominance of hydrophilic residues and presence of G nucleotides provide a favorable environment for the interaction of RdRp with quercetin-3-O-sophoroside. RdRp is a nonstructural viral protein. This enzyme, which plays an important role in the virus cycle, is important in two ways: i), for its accessibility, ii) as an attractive target for antiviral compounds (Zhu et al., 2020; Aftab et al., 2020).
The structure of the RdRp of SARS-CoV-2 is consisting of three subdomains and the functionally important catalytic sites of the RdRp are TRP 617, CYS 622, SER 759, ASP 760, ASP 761, CYS 813 (Koulgi et al., 2021). As can be seen in Table 1, some of ligand's neighboring sites are located in the catalytic region, which can terminate base pair chain elongation (Yin et al., 2020).
A number of drugs approved by FDA which contain nucleotide analogues and non-nucleotide compounds have been tested in order to inhibit RdRp (Chien et al., 2020; Elfiky, 2020; Zhang and Zhou, 2020; Zhao and Bourne, 2020). In addition to these compounds, other ones containing phytochemicals and their metabolites have been also suggested to inhibit this enzyme (da Silva et al., 2020; Saakre et al., 2021).
E protein (ion channel) has shown the weakest binding energy among the compounds studied, possibly due to the dominance of hydrophobic residues involved in the binding pocket with quercetin-3-O-sophoroside, namely Phe-23, Phe-26, Leu-27, Thr-30, Val-47, Cys-43, Ile-46, Leu-51, Thr-35, Ala-32, Leu-31, Leu-28, Val-29. These hydrophobic residues cause a reduction in hydrophilic interactions, which is not a potential target for the system. E protein as the smallest corona virus's structural protein, has an essential role in envelope formation, budding, assembly, and modulating viral pathogenesis (Ashour et al., 2020; Schoeman and Fielding, 2019). Ye and Hogue (2007), which its disruption can significantly cripple the virus. Therefore, it is important to explore some compounds that can interact with this protein. Several compounds have been proposed to inhibit corona virus's E protein (Singh Tomar and Arkin, 2020; Gupta et al., 2021). This protein has a large hydrophobic transmembrane domain, However, modeling this protein without considering the membrane may provide useful information on how it interacts with compounds that are introduced as drug candidates (Gupta et al., 2021).
3.3 Molecular dynamic study
The prediction of the conformational changes of the protein is the analysis of the structural status of each residue in the protein sequence in three possible forms: helix, sheet, and coil. Structural prediction is based on the fact that the protein structure has a regular arrangement of amino acids that are stabilized by different bonding patterns. The regularity of the structure is the main basis of predictive algorithms.
The plot of root mean square deviation (RMSD) was shown in Fig. 4. The RMSD values increased gradually in the first 9 ns, followed by a sharp increase for 4 ns, and after that, the system reached a point of equilibrium. The difference between the maximum and the minimum RMSD values over the rest of the time is about 1 nm. It might be attributed to a substantial conformational change of the protein.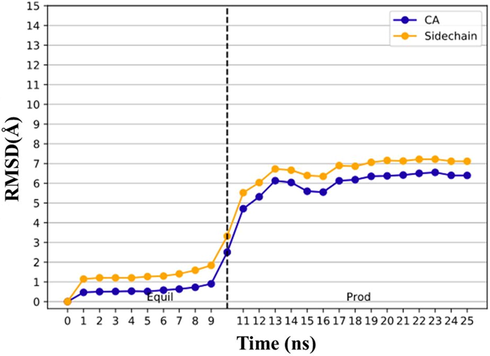
RMSD analysis for quercetin-3-O-sophoroside/E protein backbone (CA: blue) and side chain (yellow) during the molecular dynamic simulation.
Fig. 5 shows the plot of root mean square fluctuations (RMSF) of E protein backbone and side chains in the presence of quercetin-3-O-sophoroside, which is considered as the analysis of the thermal motions of each amino acid residue around its average position. The larger RMSF values indicate the substantial random motions of the amino acid residues. In fact as shown in Fig. 6, more flexible residues (Evangelin et al., 2020; Deng et al., 2020; Wu et al., 2020; Zuo et al., 2020) are located in C-terminal tail of α -helix 1 (H1).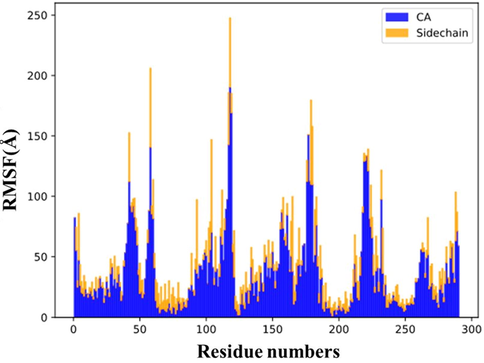
RMSF analysis for quercetin-3-O-sophoroside/E protein backbone (CA: blue) and side chain (yellow) during the molecular dynamic simulation.
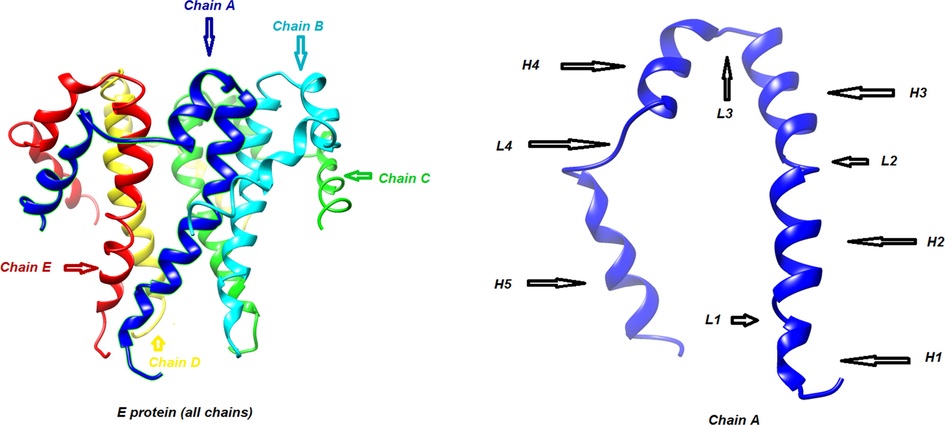
Three-dimensional structure: of α-helix (H) and loop (L) in E protein.
The second and third peaks in the graph are related to residues (40-45) and (55-59) located in loop 4 (L4) and N-terminal of chain A, respectively. In the chain B, residues (102–105) located in loop 3 (L3) have a moderate intensity in the RMSF graph while the most intense peak in the graph is related to residues (110–119) which are the constituent amino acids of α -helix 4 (H4). This instability can be induced by presence of the ligand in the vicinity of these residues. The other peaks in the RMSF graph are related to similar locations in adjacent chains. Fig. 7 also shows the structural changes of E protein (Fig. A) after interaction with quercetin-3-Osophoroside complex during 25 ns evolution (Fig. 7B).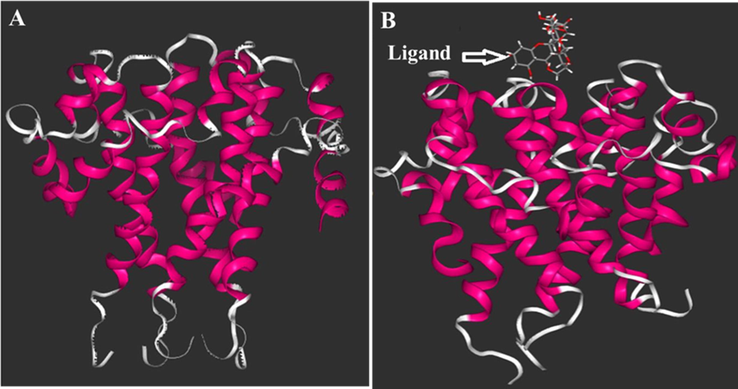
The structure of ligand/E protein complex in the initial (A) and after 25 ns evaluation (B) woth quercetin-3-O- sophoroside as a ligand.
Therefore, this study provides a clear reason for the more unstable or disintegration of the E protein structure in the presence of the quercetin-3-O-sophoroside. The change in protein shape and its denaturation can disrupt the functionality of the protein in the biological and non-biological environments.
Based on this result and already published data (Babadaei et al., 2021; Babadaei et al., 2021; Sk et al., 2020; Basit et al., 2021), it can be indicated that some potential small molecules can be used in clinical settings to inhibit the main SARS-CoV-2’s proteins and enzymes. It should be also noted that cellular and molecular assays (Taheri et al., 2021) along with some classified information for determining protective practices (Barzinjy et al., 2021) should be considered to further obtain potential details about the control of this disease.
4 Conclusion
In this study, the interaction of quercetin-3-O-sophoroside with different SARS-CoV-2′ proteins including E protein (ion channel), helicase ADP site, helicase NCB site, main protease, N protein NCB site, Nsp14 (ExoN) Nsp14 (N7-MTase), Nsp15 (endoribonuclease), Nsp16 (2′-O-MTase), papain-like protease, RdRp with RNA, and RdRp without RNA and its effect on the structure of target proteins, main protease was studied by different molecular docking and molecular dynamic simulations to may provide useful information for control of SARS-CoV-2 and maybe its other variants, including Delta. The results of these studies suggested that the hydrophilic side chains of the proteins show the greatest tendency to interact with quercetin-3-O-sophoroside. In addition, it was shown that aromatic amino acids with a flat π-π structure can be adsorbed on the surface of quercetin-3-O-sophoroside. Regarding the changes in the secondary structure of protein, the helical structures, β-sheets and the loops have been changed in the structure of the protein. Although simulation in this study has yielded interpretable results, there are some limitations to such simulations, which are important barriers to a better understanding of how proteins are adsorbed and its dynamics on different surfaces. The first limitation is that these calculations begin with a specific protein orientation, while other possible orientations can lead to different protein behaviors and affect the final results. The second limitation is related to the simulation time, because longer simulation times can better represent system behavior, and ultimately the results are based on the parameters of the simulation field and other possible force fields can show other behaviors of the system. Therefore, considering the results of this study and based on the mentioned limitations, more studies are needed to prove the accuracy of the reported outcomes.
Funding
The authors gratefully acknowledge the China Postdoctoral Science Foundation research grant No. 2020M672291, Hepatobiliary Foundation of Henan Charity General Federation GDXZ2019006, and Qatar National Research Fund (a part of Qatar Foundation) grant No. NPRP10-0120-170211. The authors would also like to acknowledge GCC-2017-005 (Qatar University) and NPRP12S-0310-190276 (QNRF) grants. The statements made here are the sole responsibility of authors.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Letter to the Editor: Angiotensin-converting enzyme 2: an ally or a Trojan horse? Implications to SARS-CoV-2-related cardiovascular complications. Am. J. Physiol.-Heart Circulatory Physiol.. 2020;318(5):H1080-H1083.
- [Google Scholar]
- Analysis of SARS-CoV-2 RNA-dependent RNA polymerase as a potential therapeutic drug target using a computational approach. J. Transl. Med.. 2020;18(1):275.
- [Google Scholar]
- Simultaneous qualitative and quantitative analysis of phenolic acids and flavonoids for the quality control of Apocynum venetum L. leaves by HPLC–DAD–ESI–IT–TOF–MS and HPLC–DAD. J. Pharm. Biomed. Anal.. 2013;1(85):295-304.
- [Google Scholar]
- Insights into the recent 2019 novel coronavirus (SARS-CoV-2) in light of past human coronavirus outbreaks. Pathogens.. 2020 Mar;9(3):186-190.
- [Google Scholar]
- Insights into the recent 2019 novel coronavirus (SARS-CoV-2) in light of past human coronavirus outbreaks. Pathogens. 2020;9(3):186-195.
- [Google Scholar]
- Development of remdesivir repositioning as a nucleotide analog against COVID-19 RNA dependent RNA polymerase. J. Biomol. Struct. Dyn.. 2021;39(10):3771-3779.
- [Google Scholar]
- The expression level of angiotensin-converting enzyme 2 determines the severity of COVID-19: Lung and heart tissue as targets. J. Biomol. Struct. Dyn.. 2021;39(10):3780-3786.
- [Google Scholar]
- Fear of COVID-19 as a precautionary measure to prevent the epidemic among the population of the Kurdistan Region/Iraq: based on a questionnaire survey. Journal of Public Health.. 2021 May;3:1-8.
- [Google Scholar]
- Truncated human angiotensin converting enzyme 2; a potential inhibitor of SARS-CoV-2 spike glycoprotein and potent COVID-19 therapeutic agent. J. Biomol. Struct. Dyn.. 2021;39(10):3605-3614.
- [Google Scholar]
- Bobrowskia, T., Alvesb, V.M., Melo-Filhoa, C.C., Korna, D., Auerbachd, S., Schmittb, C., Muratova, E.N., Tropshaa, A., 2020. Computational models identify several FDA approved or experimental drugs as putative agents against SARS-CoV-2 2. 1, 1–8.
- Cavasotto, C., Filippo, J.D., 2020. In silico Drug Repurposing for COVID-19: Targeting SARS-CoV-2 Delta protein s through Docking and Quantum Mechanical Scoring 1, 1–5.
- Nucleotide analogues as inhibitors of SARS-CoV-2 polymerase, a key drug target for COVID-19. J. Proteome Res.. 2020 Jul;21(1):1-5.
- [Google Scholar]
- Identification of SARS-CoV-2 cell entry inhibitors by drug repurposing using in silico structure-based virtual screening approach. Front. Immunol.. 2020 Jul;10(11):1664-1669.
- [Google Scholar]
- Chowdhury, T., Roymahapatra, G., Mandal, S.M., 2020. In Silico Identification of a Potent Arsenic Based Approved Drug Darinaparsin against SARS-CoV-2: Inhibitor of RNA dependent RNA polymerase (RdRp) and Necessary Proteases. 1, 1–8.
- Flavonoid glycosides and their putative human metabolites as potential inhibitors of the SARS-CoV-2 main protease (Mpro) and RNA-dependent RNA polymerase (RdRp) Mem. Inst. Oswaldo Cruz.. 2020 Sep;30(115):e200207-e200212.
- [Google Scholar]
- Serological survey of SARS-CoV-2 for experimental, domestic, companion and wild animals excludes intermediate hosts of 35 different species of animals. Transboundary Emerg. Dis.. 2020;67(4):1745-1749.
- [Google Scholar]
- HTMD: high-throughput molecular dynamics for molecular discovery. J. Chem. Theory Comput.. 2016;12(4):1845-1852.
- [Google Scholar]
- SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: an in silico perspective. J. Biomol. Struct. Dyn.. 2020 May;6(1):1-9.
- [Google Scholar]
- Andrographolide as a potential inhibitor of SARS-CoV-2 main protease: An in silico approach. J. Biomol. Struct. Dyn.. 2021 Jun 13;39(9):3092-3098.
- [Google Scholar]
- Farinholt, T., Doddapaneni, H., Qin, X., Menon, V., Meng, Q., Metcalf, G., Chao, H., Gingras, M.C., Farinholt, P., Agrawal, C., Muzny, D.M., 2021. Transmission event of SARS-CoV-2 Delta variant reveals multiple vaccine breakthrough infections. medRxiv. 1, 1–5.
- Quantifying SARS-CoV-2 transmission suggests epidemic control with digital contact tracing. Science. 2020 Mar;31(1):1-5.
- [Google Scholar]
- Frisch, M., Trucks, G., Schlegel, K.H., Scuseria, G., Robb, M.A., Cheeseman, J.R., Zakrzewski, V.G., Riul, A., Stratmann, Jr. R.E., Burant, J., Dapprich, S., 1998. Gaussian 98: revision A. 7. 1, 1–2.
- Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. Circ. Res.. 2020;126(10):1456-1474.
- [Google Scholar]
- In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel. J. Biomol. Struct. Dyn.. 2021;39(7):2617-2627.
- [Google Scholar]
- The sugar moiety is a major determinant of the absorption of dietary flavonoid glycosides in man. Free Radical Res.. 1999;31(6):569-573.
- [Google Scholar]
- Hu, F., Jiang, J., Yin, P., 2020. Prediction of potential commercially inhibitors against SARS-CoV-2 by multi-task deep model. arXiv preprint arXiv:2003.00728. 1, 1–5.
- Kong, R., Yang, G., Xue, R., Liu, M., Wang, F., Hu, J., Guo, X., Chang, S., 2020. COVID-19 Docking Server: An interactive server for docking small molecules, peptides and antibodies against potential targets of COVID-19. arXiv preprint arXiv:2003.00163. 1, 1–10.
- Natural plant products as potential inhibitors of RNA dependent RNA polymerase of Severe Acute Respiratory Syndrome Coronavirus-2. PLoS One.. 2021 May 13;16(5):e0251801-9.
- [Google Scholar]
- Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B.. 1988;37(2):785-789.
- [Google Scholar]
- DREIDING: a generic force field for molecular simulations. J. Phys. Chem.. 1990;94(26):8897-8909.
- [Google Scholar]
- Targeting the Coronavirus SARS-CoV-2: computational insights into the mechanism of action of the protease inhibitors Lopinavir. Ritonavir, Nelfinavir.. 2020;1:1-5.
- [Google Scholar]
- Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 protease against COVID-19. J. Biomol. Struct. Dyn.. 2021;39(7):2673-2678.
- [Google Scholar]
- Lessons learned from SARS-CoV and MERS-CoV: FDA-approved Abelson tyrosine-protein kinase 2 inhibitors may help us combat SARS-CoV-2. Arch Med Sci.. 2020;16(3):519-521.
- [Google Scholar]
- Air, surface environmental, and personal protective equipment contamination by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) from a symptomatic patient. JAMA. 2020 Mar;4(1):1-5.
- [Google Scholar]
- Perspectives on plant flavonoid quercetin-based drugs for novel SARS-CoV-2. Beni. Suef. Univ. J. Basic Appl. Sci.. 2021;10(1):21-29.
- [Google Scholar]
- Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nature Commun.. 2020 Jan 10;11(1):1-4.
- [Google Scholar]
- SARS-CoV-2 E protein is a potential ion channel that can be inhibited by Gliclazide and Memantine. Biochem. Biophys. Res. Commun.. 2020;530:10-14.
- [Google Scholar]
- Elucidating biophysical basis of binding of inhibitors to SARS-CoV-2 main protease by using molecular dynamics simulations and free energy calculations. J. Biomol. Struct. Dyn.. 2020 May;27(1):1-3.
- [Google Scholar]
- Homology Modeling and Docking Studies of TMPRSS2 with Experimentally Known Inhibitors Camostat Mesylate. Nafamostat Bromhexine Hydrochloride to Control SARS-Coronavirus-2.. 2020;1:1-8.
- [Google Scholar]
- Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem.. 1994;98(45):11623-11627.
- [Google Scholar]
- Evaluation of expression of VDR-associated lncRNAs in COVID-19 patients. BMC Infect. Dis.. 2021 Dec;21(1):1.
- [Google Scholar]
- A comparison of the absorption and metabolism of the major quercetin in brassica, quercetin-3-O-sophoroside, to that of quercetin aglycone, in rats. Food Chem.. 2020 May;1(311):125880-125888.
- [Google Scholar]
- Prolonged presence of SARS-CoV-2 viral RNA in faecal samples. Lancet Gastroenterol. Hepatol.. 2020;5(5):434-435.
- [Google Scholar]
- Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharmaceutica Sinica B.. 2020;10(5):766-788.
- [Google Scholar]
- The first 75 days of novel coronavirus (SARS-CoV-2) outbreak: recent advances, prevention, and treatment. Int. J. Environ. Res. Public Health. 2020 Jan;17(7):2323.
- [Google Scholar]
- The deadly coronaviruses: The 2003 SARS pandemic and the 2020 novel coronavirus epidemic in China. J. Autoimmun.. 2020;109:102434.
- [CrossRef] [Google Scholar]
- Role of the coronavirus E viroporin protein transmembrane domain in virus assembly. J. Virol.. 2007;81(7):3597-3607.
- [Google Scholar]
- Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science. 2020;368(6498):1499-1504.
- [Google Scholar]
- Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368(6489):409-412.
- [Google Scholar]
- In Silico Screening of Potential Spike Glycoprotein Inhibitors of SARS-CoV-2 with Drug Repurposing. Strategy.. 2020;1:1-5.
- [Google Scholar]
- Structural Basis of the Potential Binding Mechanism of Remdesivir to SARS-CoV-2 RNA-Dependent RNA Polymerase. J. Phys. Chem. B.. 2020;124(32):6955-6962.
- [Google Scholar]
- Structural insights into the binding modes of viral RNA-dependent RNA polymerases using a function-site interaction fingerprint method for RNA virus drug discovery. J. Proteome Res.. 2020;19(11):4698-4705.
- [Google Scholar]
- Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discovery. 2020 Mar 16;6(1):1-8.
- [Google Scholar]
- Coronavirus disease 2019 (COVID-19): a clinical update. Front. Med.. 2020;14(2):126-135.
- [Google Scholar]
- RNA-dependent RNA polymerase as a target for COVID-19 drug discovery. SLAS Discov.. 2020;25(10):1141-1151.
- [Google Scholar]
- Expert recommendations for tracheal intubation in critically ill patients with noval coronavirus disease 2019. Chin. Med. Sci. J.. 2020 Feb;27:10.
- [Google Scholar]




