

Translate this page into:
Factors determining the enzyme catalytic power caused by noncovalent interactions: Charge alterations in enzyme active sites
⁎Corresponding author. deliang2211@hotmail.com (Deliang Chen)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Enzymes reduce the free energy barrier of a reaction by decreasing positive charges and/or increasing negative charges in the electron-donating center and by decreasing negative charges and/or increasing positive charges in the electron-accepting center of the reaction. Electrostatic interactions and desolvation are the most observed noncovalent interactions through which the charge alterations cause ΔG‡ reduction.
Abstract
Understanding the origin of the enormous catalytic power of enzymes is very important. Electrostatic interactions and desolvation are the phenomena that are most proposed to explain the catalysis of enzymes; however, they also decelerate enzymatic reactions. How enzymes catalyze reactions through noncovalent interactions is still not well-understood. In this study, we explored how enzyme-substrate noncovalent interactions affect the free energy barriers (ΔG‡s) of reactions by using a theoretical derivation approach. We found that enzymes reduce ΔG‡s of reactions by decreasing positive charges and/or increasing negative charges in the electron-donating centers and by decreasing negative charges and/or increasing positive charges in the electron-accepting centers of reactions. Enzyme-substrate noncovalent interactions are essential approaches through which the charge alterations lead to ΔG‡ reductions. Validations with reported experimental data demonstrated that this charge alteration mechanism can explain the catalyses caused by diverse types of noncovalent interactions. Electrostatic interactions and desolvation are the most observed noncovalent interactions essential for ΔG‡ reductions. This mechanism does not contradict any specific enzymatic catalysis and overcomes the shortages of the electrostatic interaction and desolvation mechanisms. This study can provide useful guidance in exploring enzymatic catalysis and designing catalyst.
Keywords
Enzyme catalysis
Biophysics
Free energy barrier reduction
Theoretical derivation
Noncovalent interaction
Charge alteration

1 Introduction
Understanding the fundamental origin of the catalytic power of enzymes is critical to understanding biological systems (Knowles, 1991; Ringe and Petsko, 2008; Singh et al., 2011; Wang et al., 2021), assisting the development of enzyme inhibitors (Hanoian et al., 2015; Montgomery et al., 2017), and exploiting this information for catalyst design (Drienovska et al., 2017; Hilvert, 2013). Enzymes can accelerate reactions significantly without engaging in covalent interactions with their substrates (Lohman et al., 2013; Warshel et al., 2006). In exploring the catalytic puzzle represented by enzymes, the exploration of the question of how enzymes are able to lower the free energy barriers (ΔG‡s) of reactions substantially via noncovalent interactions is a key research objective (Acosta-Silva et al., 2020; Lohman et al., 2013; Warshel et al., 2006; Zhao et al., 2020). Several mechanisms have been put forward to explain why enzymatic reactions have much lower ΔG‡s than their corresponding reference reactions (Fried et al., 2014; Kumar et al., 2019; Lohman et al., 2013; Wu et al., 2000). Electrostatic interactions (Fried et al., 2014; Warshel et al., 2006; Wu et al., 2020; Yi et al., 2021) and desolvation (Healy, 2011; Lohman et al., 2013; Ruben et al., 2013; Sugrue et al., 2016), are the phenomena that are most proposed to explain the catalysis of enzymes. The isomerization of 5-androstene-3,17-dione (5-AND) catalyzed by ketosteroid isomerase (KSI) (Fig. 1A) has been studied extensively to validate the electrostatic interaction and desolvation mechanisms (Fried et al., 2014; Kraut et al., 2006; Layfield and Hammes-Schiffer, 2013; Natarajan et al., 2014; Schwans et al., 2009). The electrostatic interactions between the substrate oxygen atom and the polar hydrogen atoms from Tyr16 and Asp103 of KSI (dashed red lines, Fig. 1A) contribute largely to the catalytic power of KSI, supporting the catalytic role of electrostatic interactions (Fried et al., 2014). The desolvation of the carboxyl group of Asp40 also lowers the reaction's ΔG‡ remarkably, which supports an important catalytic role of desolvation (Ruben et al., 2013). Although experimental data do confirm the validity of the two mentioned proposals, many other experimental data indicate that electrostatic interactions and desolvation decelerate reactions. For example, in the reaction between S-adenosylmethionine and Cl− in aqueous solution (Fig. 1B), the electrostatic interactions between S+/Cl− and water become weaker from the ground state (GS) to transition state (TS) and increase the ΔG‡ of the reaction. In the KSI-catalyzed isomerization of 5-AND, replacing the polar hydrogen atoms from Tyr16 and Asp103 (the atoms colored blue, Fig. 1A) with non-polar atoms, which results in the desolvation of the substrate oxygen atom, increase the ΔG‡s of the reaction from 11.3 kcal/mol to 18.8 kcal/mol (Fried et al., 2014). Thus, in addition to reducing ΔG‡s, electrostatic interactions and desolvation can increase the ΔG‡s of enzymatic reactions as well. Moreover, some enzymatic reactions are catalyzed neither by the electrostatic interactions nor by desolvation. For example, it was reported that orotidine 5′-monophosphate decarboxylase catalyzes the decarboxylation of orotidine 5′-monophosphate through electrostatic stress (Wu et al., 2000). Thus, the effects of electrostatic interactions, desolvation and other noncovalent interactions on ΔG‡s may be quite different or even opposite for different enzymatic reactions. Because of the complexity of enzymatic reactions, it is not easy to use the above-mentioned mechanisms to explain the catalysis of new reactions and the applications of the mechanisms are limited. To solve this problem, it is essential to elucidate what factors determine the catalytic power of enzymes and what roles the noncovalent interactions play in ΔG‡ reduction.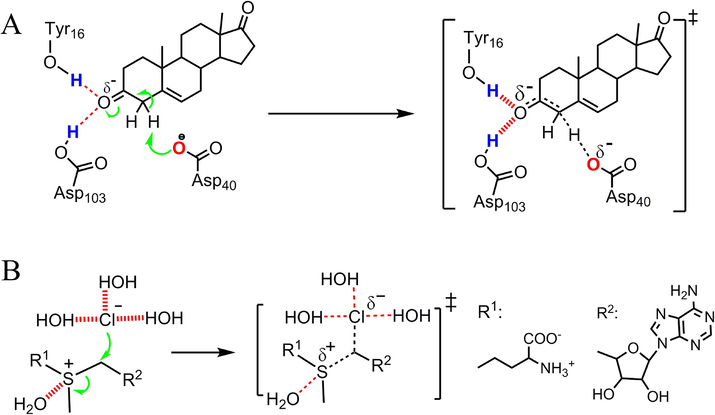
Effects of electrostatic interactions on the free energy barriers (ΔG‡s) of reactions. (A) Ground state (GS) and transition state (TS) of the ketosteroid isomerase (KSI)-catalyzed isomerization of 5-androstene-3,17-dione (5-AND). The electrostatic interactions of the substrate oxygen atom reduce the ΔG‡ of the reaction. (B) The GS and TS of the reaction between S-adenosylmethionine and Cl− in aqueous solution. The electrostatic interactions of S+/Cl− with water increase the ΔG‡ of the reaction.
Because of the diversity and complexity of enzymatic reactions, it is challenging to solve the problems by investigating specific enzymatic reactions via experimental and/or computational approaches. Thus, we use a theoretical derivation approach to solve the problems. In the theoretical derivation, the models for quantifying the effects of noncovalent interactions on the ΔG‡s of both enzymatic and solution reactions, which apply to all enzyme-substrate noncovalent interactions (Chen et al., 2019), are used to reveal the factors determining enzyme catalytic power and the roles of noncovalent interactions in enzymatic catalysis. A mechanism is developed in this study for explaining the enormous catalytic power of enzymes. This mechanism can explain the catalyses caused by diverse types of noncovalent interactions, including electrostatic interactions, desolvation, and electrostatic stress.
2 Results and discussion
2.1 Theoretical derivation for exploring the origin of enzyme catalytic power
2.1.1 Model for quantifying the effect of a general enzyme-substrate interaction on enzyme catalytic power
As illustrated by the results of a previously published study (Chen et al., 2019), the noncovalent interactions of the substrate atoms for which no changes in charge density exist between the GSs and TSs have little effects on ΔG‡s. Thus, to investigate the contribution of an enzyme–substrate interaction to the ΔG‡ of an enzymatic reaction, we focus our attention on a substrate atom characterized by a change in charge density between the GS and TS (hereafter called the “reacting atom”. Reacting atoms in a reaction are the substrate atoms that accept or donate electrons and can be determined based on the electron-movement of the reaction). In this study, the reacting atom is represented by “R” in the GS and “R‡” in the TS. We assume that the reacting atom interacts with (i) atom W in the reference reaction (Fig. 2A) or (ii) atom E in the corresponding enzymatic reaction (Fig. 2B, top). Atoms E and W are environmental atoms interacting with reacting atoms and are called “interacting atom” in this study. On the basis of the models for quantifying the reduction in ΔG‡ caused by noncovalent interactions (Chen et al., 2019), we express the reduction in ΔG‡ caused the interaction between R and W (represented with RFEBRW) as
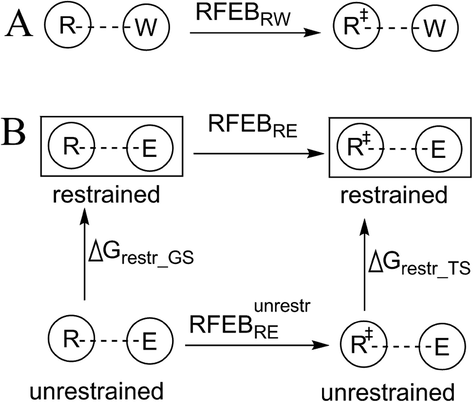
Comparison of an enzyme–substrate interaction with the corresponding reference interaction. The terms R and R‡ represent the GS and TS of a substrate atom characterized by a change in charge density between those two states. E and W represent interacting atoms from the enzyme and from the solution environment. (A) Conversion of R to R‡ in solution. (B) Conversion of R to R‡ in the enzymatic reaction in conditions whereby the interaction between R and E is restrained (top) and the same conversion when the corresponding interaction is unrestrained (bottom).
where HR and HR‡ represent the H-bonding capabilities of R and R‡; HW represents the H-bonding capability of W, the constant 7.02 (unit: kJ/mol) is the hydrogen bonding capability of the hydrogen atom or a lone pair of electrons of water (note: the units for HR‡, HR, and HW are kJ/mol; see Figure S1 for the definition and calculation of H-bonding capability).(Chen et al., 2016) As illustrated in previous studies (Chen et al., 2019; Chen et al., 2016), the H-bonding capability of a polar atom is the water to alkane phase transfer free energy contributed by the polarity of the atom (Figure S1). An atom with higher charge density will have higher H-bonding capability (Figure S2). In a previous study and in Figure S1, we illustrated that H-bonding capability is an ideal parameter for accurately quantifying the effects of the environment-substrate noncovalent interactions on the ΔG‡s of the reactions, no matter the noncovalent interactions are electrostatic interactions, desolvation and/or electrostatic stress. Moreover, the calculation of ΔG‡s of enzymatic and solution reactions is not required for exploring the effects of noncovalent interactions on enzyme catalytic power. Thus, the proper thermal averaging and multiscale simulation, which is required for calculating ΔG‡s in computational enzymology (Warshel and Levitt, 1976), are also not performed in this study.
In the enzymatic reaction, atom E cannot move as freely as the atoms in solution and is restrained so that its interaction with R is close to the optimal interaction geometry (e.g. H-bond geometry) between the GS and TS. In previous studies, this phenomenon was referred to as “electrostatic preorganization”(Fuxreiter and Mones, 2014; Kamerlin et al., 2010). On the basis of the thermodynamic cycle drawn to compare the reduction in ΔG‡ caused by the restrained R…E (represented by RFEBRE, Fig. 2B, top) with that caused by the unrestrained R…E interaction (represented by RFEBREunrest, Fig. 2B, bottom), we express RFEBRE as
As demonstrated in a previous study (Chen et al., 2019), the effect of electrostatic preorganization on ΔG‡ is small. Moreover, if the reacting atom has a lower charge density in the TS than in the GS, the term ΔGreorg_GS − ΔGreorg_TS is negative and the corresponding electrostatic preorganization does not contribute to the enzyme’s catalytic power. Thus, the catalytic power contributed by R…E roughly equals to (HR‡ − HR) (HE − HW) /7.02, which is the model used for exploring the origin of enzyme-catalytic power contributed by enzyme-substrate noncovalent interactions.
2.1.2 Exploring the origin of enzyme catalytic power on the basis of equation (4)
Equation (4) indicates that the catalytic power contributed by the R…E interaction can be enhanced by adjusting the charge density (or H-bonding capability) of E. Fig. 3A shows the generic enzyme-substrate interactions occurring in a reaction in which electrons move from one reacting atom (R1) to another (R2). In the electron-donating center, R‡1 (TS of R1) is either negative or positive. If R‡1 is negative, it has a lower negative charge density than R1 (HR1‡ < HR1, see Fig. 3B). To make the interaction favorable to enzyme catalytic power (RFEBRE - RFEBRW > 0), E1 should have a lower positive charge density than the corresponding atom in the reference reaction in aqueous solution (HE1 < HW, HW is 7.02 in most cases) or have a negative charge density (HE1 < 0. HE1 is less than zero because E1 is negative and has negative-negative electron repulsion with R‡1). If R‡1 is positive, it has a larger positive charge density than R1 (HR1‡ > HR1, see Fig. 3C). Then, E1 should have a larger negative charge density than the corresponding atom in the reference reaction in aqueous solution (HE1 > HW). Thus, the catalytic power contributed by R1…E1 originates from the charge difference between E1 and the corresponding atom in the reference reaction. The charge differences shown in Fig. 3B&C are summarized in Fig. 3F (left), which indicates that the catalytic power of a reaction involving electron-movement can be enhanced by decreasing positive charges and/or increasing negative charges in the electron-donating center. Similarly, the relationships between HE2 and the catalytic power contributed by R2…E2 (Fig. 3D&E) indicate that the catalytic power can also be enhanced by decreasing negative charges and/or increasing positive charges of the electron-accepting center (right, Fig. 3F).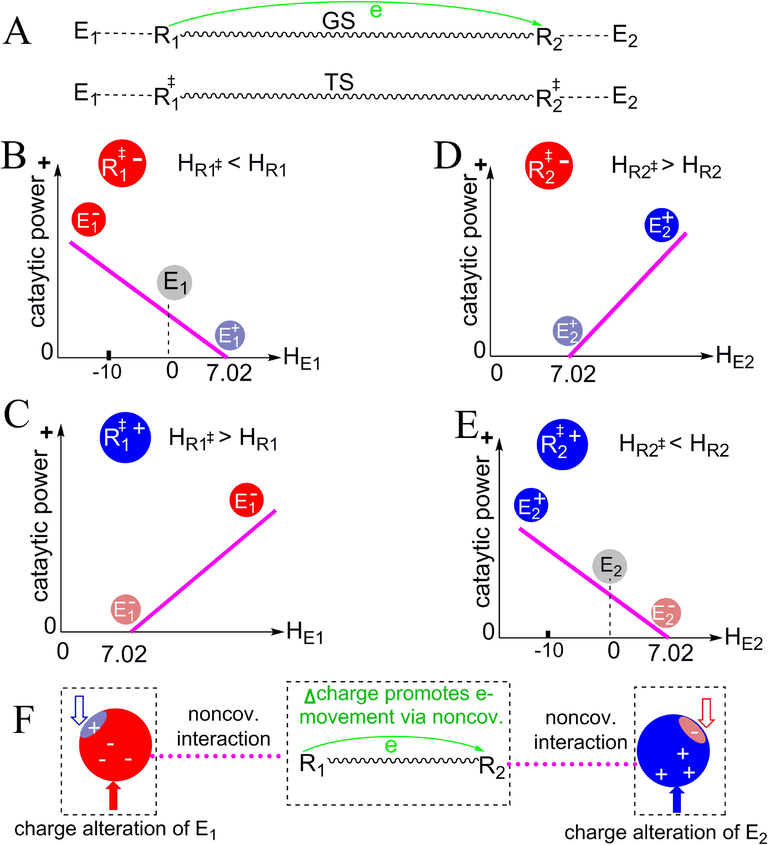
Effects of the charge alterations of the enzyme-based interacting atoms on enzyme catalytic power. R1 and R2 represent substrate atoms; R1‡ and R2‡ are the TSs of R1 and R2, respectively. E1 and E2 indicate the interacting atoms from the enzyme. (A) GS and TS of the reaction in which electrons move from R1 to R2. (B, C) The relationships between HE1 (H-bonding capability of E1) and the catalytic power contributed by R1…E1 when R1‡ is negative as shown in (B) or positive as shown in (C). (D, E) The relationships between HE2 (H-bonding capability of E2) and the catalytic power contributed by R2…E2 when R2‡ is negative as shown in (D) or positive as shown in (E). (F) Summary of the approaches for enhancing enzyme catalytic powers: in the electron-donating center, decreasing positive charges and increasing negative charges; in the electron-accepting center, decreasing negative charges and increasing positive charges.
2.1.3 Mechanism developed in this study
Based on Fig. 3F, we conclude that enzymes reduce the ΔG‡s of reactions by decreasing positive charges and/or increasing negative charges in the electron-donating centers and by decreasing negative charges and/or increasing positive charges of the electron-accepting centers of the reactions. This charge alteration mechanism is the mechanism developed in this study for explaining the catalytic power contributed by enzyme-substrate noncovalent interactions. It can be simplified as: Enzymes reduce ΔG‡s of reactions by increasing the net negative charges in the electron-donating centers and by increasing the net positive charges in the electron-accepting centers of reactions.
2.2 Validation of the charge alteration mechanism using diverse enzymatic reactions
Using diverse and well-characterized examples of enzymatic reactions, we validate that the enzyme catalytic power originates from the charge alterations as shown in Fig. 3F. All the catalyses of enzymatic reactions used for validation were proved via experimental studies and/or via joint experimental and theoretical investigations in literature. The first example is the isomerization of 5-AND, whereby electrons move from the substrate's α-H to the oxygen atom (Fig. 4A). The charge alterations in the enzymatic reaction can be identified based on the charge difference between enzymatic and reference reactions in the GS. In the electron-donating center, the COO− interacts with the hydrogen atoms from bulk water in the reference reaction and with nonpolar atoms in the KSI-catalyzed reaction, indicating that the charge alteration is reducing positive charges. It was reported that the desolvation of the COO− of Asp40 contributes to the catalytic power of KSI (Ruben et al., 2013), supporting that reducing positive charges in the electron-donating center enhances the catalytic power of KSI. In the electron-accepting center, the hydrogen atoms for Tyr16 and Asp103 have larger positive charge density than the hydrogen atoms from water (Figure S3), indicating that the charge alteration is increasing positive charges. It was reported that the electrostatic interactions in the electron-accepting center of KSI contribute to the catalytic power of KSI (Fried et al., 2014; Wu et al., 2020), supporting that increasing positive charges in the electron-accepting center enhances the catalytic power of KSI. The catalytic power contributed by the electrostatic interactions in the electron-accepting center of KSI includes the small contribution contributed by electrostatic preorganization. This catalytic power can also be explained by the charge alteration mechanism because the preorganization of the positively charged atoms increase the positive charge density in the electron-accepting center (Figure S4). Changing the polar hydrogen atoms for Tyr16 and Asp103 to nonpolar atoms increases the ΔG‡ of the reaction from 11.5 kcal/mol to 18.8 kcal/mol (Fried et al., 2014), indicating that decreasing positive charges in the electron-accepting center, which is opposite to the charge alterations shown in Fig. 3F, increase ΔG‡s of reactions.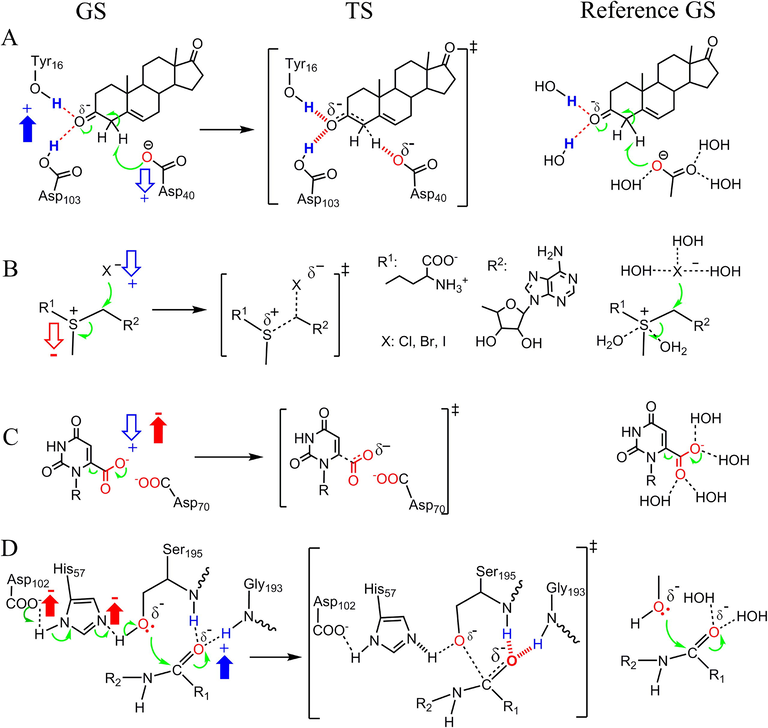
Validating that charge alterations in the electron-donating and –accepting centers of enzymatic reactions determines enzyme catalytic power. Charge alterations are determined by comparing the GSs of enzymatic reactions (left) with the GSs of the reference reactions (right), and are represented with blue (for positive charge) and red (for negative charge) arrows. The up and down arrows represent an increase and decrease in charges, respectively. (A) The isomerization of 5-androstene-3,17-dione. (B) The reaction of S-adenosylmethionine with X− (X−= Br−, Cl−, or I−). (C) The electrostatic stress in the decarboxylation of orotidine 5′-monophosphate catalyzed by orotidine 5′-monophosphate decarboxylase. (D) The catalytic triad in the peptide bond cleavage catalyzed by serine proteases.
The second example is the halide alkylation catalyzed by halogenases (Fig. 4B), in which electrons move from X− (X−= Br−, Cl−, or I−) to S+. Halogenases significantly accelerate this reaction via the desolvation of both X − (in the electron-donating center) and S+ (in the electron-accepting center) (Lohman et al., 2013). By comparing the substrate-environment noncovalent interactions in the GS of the enzymatic reaction with those of the reference reaction, we can find that halogenase catalyses the reaction by decreasing positive charges in the electron-donating center and by decreasing negative charges in the electron-accepting center of the reaction. The third example is the catalysis by electrostatic stress, which exists in the decarboxylation of orotidine 5′-monophosphate catalyzed by orotidine 5′-monophosphate decarboxylase (Fig. 4C) (Desai et al., 2014; Wu et al., 2000). In the electron-donating center of the reference reaction, the substrate COO− group interacts with hydrogen atoms of water. By contrast, the substrate COO− group in the enzymatic reaction does not have electrostatic interactions with positively charged atoms, but have negative-negative repulsive interactions with the COO– group of Asp70. This difference indicates that the catalytic power of electrostatic stress originates from both decreasing positive charges and increasing negative charges in the electron-donating center. The forth example is the catalysis by catalytic triad, which exists in the electron-donating center of the peptide bond cleavage catalyzed by serine proteases (Fig. 4D). (Hedstrom, 2002; Sprang et al., 1987). By comparing the electron-donating centers in the enzymatic and reference reactions, we find that the charge alteration is the increase in negative charges (COO− of Asp102 and the N with a lone pair of electrons in His57). Thus, the catalytic power of the catalytic triad originates from the charge alteration of increasing negative charges in electron-donating centers.
Reported experimental data also support that the charge alterations as shown in Fig. 3F enhance the catalytic power of metalloenzymes and the opposite charge alterations largely decrease the catalytic power of metalloenzymes. Fig. 5 shows that the GSs and TSs of CO2 hydration catalyzed by wild type carbonic anhydrase II (CAII) and by E117Q CAII at pH 8.9 (Huang et al., 1996; McCall et al., 2000). The electron-movements of the reactions show the zinc complexes are the electron-accepting centers of the reactions. As demonstrated in Figure S5 and in previous studies (Chen et al., 2018a; Huang et al., 1996), the His119 in wild-type CAII is neutral while the His119 in E117Q CAII is negatively charged. Thus, the negative charge density in the electron-accepting center of the reaction catalyzed by wild-type CAII is lower than that catalyzed by E117Q CAII. The second-order rate constant for CO2 hydration catalyzed by wild-type CAII at pH 8.9 is > 105 folds larger than that catalyzed by E117Q CAII, supporting that decreasing the negative charge in the electron-accepting center enhances the catalytic power of metalloenzymes or increasing the negative charge in the electron-accepting center reduces the catalytic power of metalloenzymes.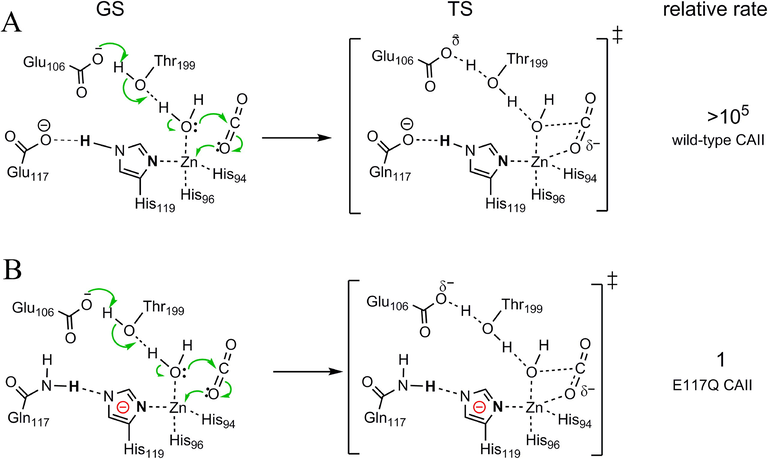
Effect of charge alterations on the catalytic power of metalloenzymes. (A) The GS, TS and the relative second-order rate constant for CO2 hydration catalyzed by wild type carbonic anhydrase II (CAII) at pH 8.9.(Huang et al., 1996; McCall et al., 2000) (B) the GS, TS and the relative second-order rate constant for CO2 hydration catalyzed by E117Q CAII at pH 8.9.
We have examined a large number of enzymatic reactions catalysed by diverse enzyme-substrate noncovalent interactions and found that their catalytic power originates from the charge alterations shown in Fig. 3F. Evidence indicates, therefore, that enzymes reduce the ΔG‡s of reactions via the charge alteration mechanism as shown in Fig. 3F.
2.3 Comparison between this mechanism and some widely accepted mechanisms
Several mechanisms have been developed to explain the enzyme catalytic power contributed by enzyme-substrate noncovalent interactions. The electrostatic interaction and desolvation mechanisms are the most widely accepted mechanisms. We take the interaction between a substrate atom R and an enzyme atom E (R•••E) to illustrate the difference between this charge alteration mechanism and the two widely accepted mechanisms. The electrostatic interaction mechanism indicates that the enzyme catalytic power contributed by R•••E originates from the electrostatic interaction between R and E. The desolvation mechanism indicates that the enzyme catalytic power originates from the desolvation of R or E. This charge alteration mechanism indicates that the enzyme catalytic power originates from the charge difference between E and the corresponding atom in the reference solution reaction. The noncovalent interaction between E and R, e.g. electrostatic interaction, electrostatic stress, is essential for the charge difference to cause the enzyme catalysis. For example, the polar hydrogen atoms from Tyr16 and Asp103 in KSI promote the electron movement from the substrate α-H to the substrate oxygen atom through their electrostatic interactions with the substrate oxygen atom (Fig. 4A). Without the electrostatic interactions, the polar hydrogen atoms cannot catalyze the reaction. Thus, noncovalent interactions (including electrostatic interaction) are the essential approaches through which the charge alterations lead to enzyme catalysis. However, noncovalent interactions are also the approaches through which the opposite charge alterations of Fig. 3F decelerate reactions. Desolvation is an approach of charge alterations. Whether a charge alteration contributes to enzyme catalytic power depends on the charge alteration of R. Thus, desolvation can enhance, decrease or have no effect on enzymatic power.
This charge alteration mechanism has a few advantages as compared to the electrostatic interaction and desolvation mechanisms. First, the charge alteration mechanism can explain the catalyses caused by diverse enzyme-substrate noncovalent interactions, including electrostatic interactions, desolvation, and electrostatic stress. However, neither the electrostatic interaction mechanism nor the desolvation mechanism can explain all the catalyses shown in 4–5. The electrostatic interaction mechanism applies only to the catalysis caused by electrostatic interactions and desolvation mechaism applies only to the catalysis caused by desolvation. Second, the charge alteration mechanism does not contradict any specific enzymatic catalysis because no reaction can be catalysed via the opposite charge alterations shown in Fig. 3F. However, the electrostatic interaction mechanism contradicts the catalysis caused by desolvation or electrostatic stress; the desolvation and electrostatic stress mechanisms contradict the catalysis caused by electrostatic interactions. Thus, the charge alteration mechanism can be easily used to explain the catalysis of new reactions involving electron-movements and can provide useful guidance for reducing the ΔG‡s of the reactions involving electron-movements.
It is also reported that enzymes can catalyze reactions by polarizing substrate bonds caused by enzyme-substrate electrostatic interactions.(Deng et al., 2017; Peng et al., 2014) The charge alteration mechanism in this manuscript is quite different from the polarization mechanism in literature. First, the targets for the “polarization” in literature are the substrate bonds that are polarized by enzyme-substrate H-bonds; while the “charge alteration” in this manuscript is targeted for the atoms in the electron–donating and –accepting centers of reactions. Second, many enzymatic catalyses, e.g. the catalysis by desovation and the catalysis by electrostatic stress, cannot be explained by the “polarization” in literature, but can be explained by the “charge alteration” mechanism in this manuscript.
The merits of the charge alteration mechanism originate from the model for quantifying the contribution of an enzyme-substrate interaction to enzyme catalytic power [equation (3)]. This model is a general model that applies to all noncovalent interactions. However, the electrostatic interaction, desolvation and electrostatic stress mechanisms are developed by investigating specific enzymatic reactions through experimental and/or computational approaches. Because enzymatic catalysis is very complex, it is impossible to consider all possible enzyme-substrate interactions by investigating specific enzyme-substrate reactions. Thus, the theoretical derivation used in this study plays an irreplaceable role in exploring the origin of enzyme catalytic power.
2.4 Effects of noncovalent interactions on enzyme-substrate bindings
The effects of enzyme-substrate noncovalent interactions on ΔG‡s are different from their effects on reaction rates because the interactions also affect enzyme-substrate bindings. Electrostatic interactions and hydrophobic interactions are favourable to enzyme-substrate bindings. When the enzyme-substrate bindings are weak, enzyme active sites are not saturated. Increasing the enzyme-substrate binding strengths by electrostatic interactions and/or hydrophobic interactions will enhance reaction rates considerably. By contrast, when the enzyme active sites are almost saturated, further enhancing the enzyme–substrate interactions by electrostatic interactions and/or hydrophobic interactions will have little effect on reaction rates, which is in agreement with the conclusions of Singh and coworkers (Singh et al., 2011). Desolvation and electrostatic stress are unfavourable to enzyme-substrate bindings and can reduce reaction rates largely. The negative effects of desolvation and electrostatic stress on enzyme-substrate bindings are usually cancelled by the favorable hydrophobic interactions and/or electrostatic interactions in enzymatic reactions. Other factors such as steric strain and dynamic effects also have contributions to some enzymatic reactions (Glowacki et al., 2012; Kovermann et al., 2015; Swiderek et al., 2015). Dynamic effects are important for the entering of substrates to the active sites of enzymes. Because enzyme catalytic power is based on the comparison between enzymatic and reference solution reactions, the dynamical effects do not have obvious contribution to enzyme catalytic power, which were demonstrated in many studies (Olsson et al., 2006a; Olsson et al., 2006b; Warshel and Bora, 2016; Warshel et al., 2006). To fully understand the enormous catalytic power of enzymes, it is also important to study the effects of other factors on enzyme catalytic power.
3 Conclusion
In this study, we used a theoretical derivation approach to explore how noncovalent interactions affect the catalytic power of enzymes. Enzymes reduce the ΔG‡s of reactions by decreasing positive charges and/or increasing negative charges in the electron-donating centers and by decreasing negative charges and/or increasing positive charges of the electron-accepting centers of the reactions. Electrostatic interactions and desolvation are the most observed noncovalent interactions through which the charge alterations lead to ΔG‡ reductions. This charge alteration mechanism applies to the catalyses caused diverse noncovalent interactions. The findings in this study can find useful applications in the fields of enzyme catalysis and can provide guidance for the design of catalysts.
4 Methods
4.1 General procedure for exploring how enzyme-substrate noncovalent interactions affect enzyme catalysis.
Electrostatic interactions and desolvation are the most observed enzyme-substrate noncovalent interactions affecting the free energy barriers (ΔG‡s) of enzymatic reactions. Because enzymatic reactions are diverse and complex, the effects on enzyme-substrate noncovalent interactions on enzyme catalysis are complicated. It is challenging to reveal how enzyme-substrate noncovalent interactions affect enzyme catalysis merely by investigating specific enzymatic reactions via experimental and/or computational approaches. Thus, in this study, we explored how enzyme-substrate noncovalent interactions affect enzyme catalysis via a theoretical derivation. The theoretical derivation is not targeted for any specific enzymatic reaction, but we validated our findings with various specific enzymatic reactions. Below are the processes of the theoretical derivation:
The models reported in a previous study (Chen et al., 2019) for quantifying the effects of noncovalent interactions on the ΔG‡s of enzymatic and solution reactions were used to explore how enzyme-substrate noncovalent interactions reduce the ΔG‡s of enzymatic reactions. Then the origin of enzyme catalytic power and the roles of electrostatic interactions and desolvation in enzyme catalysis were derived.
The proposal for explaining how enzyme-substrate noncovalent interactions catalyze reaction was validated with various enzymatic reactions, including the reactions that were previously explained by electrostatic interactions, desolvation, electrostatic stress and catalytic triads and the reactions catalyzed by metalloenzymes.
4.2 Methods for calculating H-bonding capabilities
The H-bonding capabilities of polar hydrogen atoms used in Figure S1 were calculated from experimental water to alkane partition coefficients and water to n-octanol partition coefficients of the organic compounds. The details for the calculation were described in previous studies (Chen et al., 2018b; Chen et al., 2020).
CRediT authorship contribution statement
Deliang Chen: Methodology, Investigation. Yibao Li: Investigation, Writing – original draft. Xun Li: Writing – original draft. Tor Savidge: Methodology. Yiping Qian: Writing – original draft. Xiaolin Fan: Methodology.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China, China (21763002, 21762003) and the Natural Science Foundation of Jiangxi Province, China (20202ACBL203008).
References
- Catalytic Effect of Electric Fields on the Kemp Elimination Reactions with Neutral Bases. ChemPhysChem. 2020;21:2594-2604.
- [Google Scholar]
- The shielding effect of metal complexes on the binding affinities of ligands to metalloproteins. PCCP. 2018;21:205-216.
- [Google Scholar]
- Effective lead optimization targeting the displacement of bridging receptor-ligand water molecules. PCCP. 2018;20:24399-24407.
- [Google Scholar]
- Quantitative Effects of Substrate-Environment Interactions on the Free Energy Barriers of Reactions. J. Phys. Chem. C. 2019;123:13586-13592.
- [Google Scholar]
- Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci. Adv.. 2016;2:e1501240
- [Google Scholar]
- A general linear free energy relationship for predicting partition coefficients of neutral organic compounds. Chemosphere. 2020;247:125869
- [Google Scholar]
- Difference FTIR Studies of Substrate Distribution in Triosephosphate Isomerase. J. Phys. Chem. B. 2017;121:10036-10045.
- [Google Scholar]
- Investigating the role of a backbone to substrate hydrogen bond in OMP decarboxylase using a site-specific amide to ester substitution. P Natl. Acad. Sci. USA. 2014;111:15066-15071.
- [Google Scholar]
- Design of an enantioselective artificial metallo-hydratase enzyme containing an unnatural metal-binding amino acid. Chem. Sci.. 2017;8:7228-7235.
- [Google Scholar]
- Extreme electric fields power catalysis in the active site of ketosteroid isomerase. Science. 2014;346:1510-1514.
- [Google Scholar]
- The role of reorganization energy in rational enzyme design. Curr. Opin. Chem. Biol.. 2014;21:34-41.
- [Google Scholar]
- Protein dynamics and enzyme catalysis: the ghost in the machine? Biochem. Soc. Trans.. 2012;40:515-521.
- [Google Scholar]
- Perspectives on electrostatics and conformational motions in enzyme catalysis. Acc. Chem. Res.. 2015;48:482-489.
- [Google Scholar]
- The effect of desolvation on nucleophilic halogenase activity. Comput. Theor. Chem.. 2011;964:91-93.
- [Google Scholar]
- Reversal of the hydrogen bond to zinc ligand histidine-119 dramatically diminishes catalysis and enhances metal equilibration kinetics in carbonic anhydrase II. Biochemistry. 1996;35:3439-3446.
- [Google Scholar]
- Ketosteroid isomerase provides further support for the idea that enzymes work by electrostatic preorganization. Proc. Natl. Acad. Sci. U S A. 2010;107:4075-4080.
- [Google Scholar]
- Structural basis for catalytically restrictive dynamics of a high-energy enzyme state. Nat. Commun.. 2015;6:7644.
- [Google Scholar]
- Testing electrostatic complementarity in enzyme catalysis: hydrogen bonding in the ketosteroid isomerase oxyanion hole. PLoS Biol.. 2006;4:e99
- [Google Scholar]
- Low-Barrier and Canonical Hydrogen Bonds Modulate Activity and Specificity of a Catalytic Triad. Angew. Chem. Int. Ed.. 2019;58:16260-16266.
- [Google Scholar]
- Calculation of vibrational shifts of nitrile probes in the active site of ketosteroid isomerase upon ligand binding. J. Am. Chem. Soc.. 2013;135:717-725.
- [Google Scholar]
- Catalysis by desolvation: the catalytic prowess of SAM-dependent halide-alkylating enzymes. J. Am. Chem. Soc.. 2013;135:14473-14475.
- [Google Scholar]
- Function and mechanism of zinc metalloenzymes. J. Nutr.. 2000;130(5S Suppl):1437S-1446S.
- [Google Scholar]
- Computational Glycobiology: Mechanistic Studies of Carbohydrate-Active Enzymes and Implication for Inhibitor Design. Adv. Protein. Chem. Struct. Biol.. 2017;109:25-76.
- [Google Scholar]
- Using unnatural amino acids to probe the energetics of oxyanion hole hydrogen bonds in the ketosteroid isomerase active site. J. Am. Chem. Soc.. 2014;136:7643-7654.
- [Google Scholar]
- Transition state theory can be used in studies of enzyme catalysis: lessons from simulations of tunnelling and dynamical effects in lipoxygenase and other systems. Philos. Trans. R. Soc. Lond. B Biol. Sci.. 2006;361:1417-1432.
- [Google Scholar]
- Dynamical contributions to enzyme catalysis: critical tests of a popular hypothesis. Chem. Rev.. 2006;106:1737-1756.
- [Google Scholar]
- Energy Landscape of the Michaelis Complex of Lactate Dehydrogenase: Relationship to Catalytic Mechanism. Biochemistry. 2014;53:1849-1857.
- [Google Scholar]
- Ground state destabilization from a positioned general base in the ketosteroid isomerase active site. Biochemistry. 2013;52:1074-1081.
- [Google Scholar]
- Determining the catalytic role of remote substrate binding interactions in ketosteroid isomerase. Proc. Natl. Acad. Sci. U S A. 2009;106:14271-14275.
- [Google Scholar]
- The evolution of catalytic function in the HIV-1 protease. J. Mol. Biol.. 2011;408:792-805.
- [Google Scholar]
- The three-dimensional structure of Asn102 mutant of trypsin: role of Asp102 in serine protease catalysis. Science. 1987;237:905-909.
- [Google Scholar]
- Active Site Desolvation and Thermostability Trade-Offs in the Evolution of Catalytically Diverse Triazine Hydrolases. Biochemistry. 2016;55:6304-6313.
- [Google Scholar]
- Protein Conformational Landscapes and Catalysis. Influence of Active Site Conformations in the Reaction Catalyzed by L-Lactate Dehydrogenase. ACS Catal.. 2015;5:1172-1185.
- [Google Scholar]
- The potential antioxidant ability of hydroxytyrosol on Caenorhabditis elegans against oxidative damage via the insulin signaling pathway. Arab J. Chem.. 2021;14:103149
- [Google Scholar]
- Perspective: Defining and quantifying the role of dynamics in enzyme catalysis. J. Chem. Phys.. 2016;144:180901
- [Google Scholar]
- Theoretical studies of enzymic reactions: dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol.. 1976;103:227-249.
- [Google Scholar]
- Electrostatic stress in catalysis: structure and mechanism of the enzyme orotidine monophosphate decarboxylase. Proc. Natl. Acad. Sci. U S A. 2000;97:2017-2022.
- [Google Scholar]
- A Preorganized Electric Field Leads to Minimal Geometrical Reorientation in the Catalytic Reaction of Ketosteroid Isomerase. J. Am. Chem. Soc.. 2020;142:9993-9998.
- [Google Scholar]
- Electrostatic Interactions Accelerating Water Oxidation Catalysis via Intercatalyst O-O Coupling. J. Am. Chem. Soc.. 2021;143:2484-2490.
- [Google Scholar]
- Noncovalent Interaction-Assisted Ferrocenyl Phosphine Ligands in Asymmetric Catalysis. Accounts Chem Res. 2020;53:1905-1921.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2021.103611.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




