

Translate this page into:
First report on the QSAR modelling and multistep virtual screening of the inhibitors of nonstructural protein Nsp14 of SARS-CoV-2: Reducing unnecessary chemical synthesis and experimental tests
⁎Corresponding author. sunguohui@bjut.edu.cn (Guohui Sun)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
The QSAR model for the inhibitors of SARS-CoV-2 Nsp14 was built for the first time. Mechanistic analysis identified the main influencing factors for Nsp14 inhibition. The best model was used for virtual screening of 262 untested compounds. Docking and ADMET predictions identified two hit candidates as Nsp14 inhibitors. The developed QSAR model can avoid unnecessary chemical synthesis and test.
Abstract
Corona Virus Disease 2019 (COVID-19), caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), poses a serious threat to human health and life safety. How to effectively prevent and treat COVID-19 is crucial. In this study, we used the inhibitors of nonstructural protein Nsp14 of SARS-CoV-2 to perform the quantitative structure activity relationship (QSAR) modelling for the first time. Based on different dataset division strategies, we selected partial least square (PLS) and multiple linear regression (MLR) methods to develop easily interpretable and reproducible QSAR models with 2D molecular descriptors. All models complied with the strict QSAR validation principles of OECD and internationally recognized validation metrics. The best model contained two molecular descriptors with the following statistical parameters: R2 = 0.7796, = 0.7373, = 0.8539 and CCCtest = 0.9073. Obviously, the model exhibited good prediction performance and can be used for quickly predicting the inhibitory activity of unknown compounds against Nsp14. Mechanistic interpretation identified the detailed relationship between molecular structure information and inhibitory activity. The best QSAR model was used to predict the inhibitory activity of 263 true external compounds without experimental values against Nsp14, and the prediction reliability was analyzed and discussed. Molecular docking and ADMET analyses were conducted for compounds with higher similarity to the modelling compounds. Finally, two compounds were identified as potential candidate drugs of targeting Nsp14. The current work lays a solid theoretical foundation for the discovery of inhibitors targeting Nsp14, and has an important reference significance for the development of anti-COVID-19 drugs.
Keywords
COVID-19
SARS-CoV-2
Nsp14 inhibitor
QSAR
Docking
ADMET

- COVID-19
-
Corona Virus Disease 2019
- SARS-CoV-2
-
severe acute respiratory syndrome coronavirus 2
- QSAR
-
quantitative structure activity relationship
- PLS
-
partial least square
- MLR
-
multiple linear regression
- ADMET
-
absorption, distribution, metabolism, excretion, toxicity
- WHO
-
World Health Organization
- CoVs
-
Coronaviruses
- ORFs
-
open reading frames
- Nsps
-
nonstructural proteins
- S
-
Spike protein
- E
-
Envelope protein
- M
-
Membrane protein
- N
-
Nucleocapsid protein
- 3CLpro
-
3C-likeproteinase
- PLpro
-
papain-like protease
- RTC
-
replication-transcription complex
- Exon
-
N-terminal nucleic acid exonuclease
- N7-MTase
-
N7-methyltransferase
- 3R
-
Reduction, Replacement, Refinement
- OECD
-
Organization for Economic Co-operation and Development
- ORes
-
ordered by response
- OStr
-
ordered by structure similarity
- Rnd
-
randomly
- KS
-
Kennard Stone
- ED
-
Euclidean Distance
- A/P
-
Activity/Property
- GA-VSS
-
Genetic Algorithm-Variable Subset Selection
-
leave-one-out cross-validation correlation coefficient
- QUIK
-
Q under influence of K
- R2
-
coefficient of determination
- CCCtest
-
concordance correlation coefficient for the test set
- RMSE
-
the root mean square error
- MCDM
-
multi-criteria decision making
- AD
-
applicability domain
- PDB
-
Protein Data Bank
- SAH
-
S-adenosyl homocysteine
- VIP
-
variable importance plot
Abbreviations
1 Introduction
In December 2019, Corona Virus Disease 2019 (COVID-19) first emerged in Wuhan, China, and the disease then swept through almost all countries and regions of the world at an alarming rate, rapidly forming a global pandemic (Lu et al., 2020). By the end of December 2022, the cumulative number of confirmed cases and cumulative deaths by the World Health Organization (WHO) had exceeded 650 million and 6.6 million worldwide, respectively (WHO, 2022). Although the WHO no longer keeps statistics, it is still evident from the existing data that COVID-19 has caused great damage to human life and health. The pathogenic virus of COVID-19 is severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which has mutated continuously to produce several strains, such as latest Omicron subvariants BF.7, BA.5.2, BQ.1, BQ.1.1, XBB, XBB.1.5, XBD and XBF, etc. (Chatterjee et al., 2023; Sabbatucci et al., 2023; Wang et al., 2023; Mohammed, 2022). As the virus continues to mutate, self-protection of people and early detection of viral infections seem to be important. It has been demonstrated that various metal and metal oxide nanoparticles showed good antiviral activity and their applications in antiviral mask structures is promising, which can protect the wearer from COVID-19 and other deadly pathogens (Hadinejad et al., 2023). Currently, the common diagnostic methods for SARS-CoV-2 include nucleic acid detection, and immunological detection, however, these methods have their own limitations, and there may be a lack of insufficient cellular materials or errors in the sampling process of nasopharyngeal/oropharyngeal swab collection, thus it is necessary to develop more advanced techniques and materials to develop accurate, ultrafast, and visualized viral detection (Wang et al., 2022). Of course, the later treatment is very important, existing vaccines and therapeutic drugs are not effective in suppressing the activity of the virus, especially in the elderly and children with poor resistance or those with underlying diseases, and the COVID-19 can still be very damaging and even life-threatening to these populations (Pollard et al., 2020; Shahid et al., 2020). Therefore, the development of potent anti-SARS-CoV-2 drugs is still of great importance.
Coronaviruses (CoVs) are a large family of common viruses, most with highly similar structures, that infect humans and cause severe acute and chronic respiratory disease (Perlman and Netland, 2009). About 15 different coronavirus strains have been identified, and seven of them can infect humans, namely, HCoV-229E, HCoV-OC43, HCoV-NL63, HCoV-HKU1, SARS-CoV, MERS-CoV, and SARS-CoV-2 that causes COVID-19 (Ye et al., 2020). SARS-CoV-2 has an approximately 30-kb long RNA genome with 14 open reading frames (ORFs) encoding 29 proteins, including 16 nonstructural proteins (Nsps), 4 structural proteins, and 9 accessory proteins (Kim et al., 2020). The four structural proteins in SARS-CoV-2 include Spike protein (S), Envelope protein (E), Membrane protein (M) and Nucleocapsid protein (N). These proteins are responsible for host invasion, viral envelope formation, stability and interaction with the RNA genome (Chan et al., 2020). Meanwhile, 16 Nsps (Nsp1 to Nsp16) in SARS-CoV-2 play an important role in maintaining normal viral vital signs (Arya et al., 2021). A significant proportion of existing marketed drugs target Nsps, such as remdesivir that targets RNA polymerase (Nsp12), which has been approved for the treatment of critically ill patients, but its safety and efficacy for COVID-19 needs further validation (Beigel et al., 2020; Grein et al., 2020). Similarly, 3CLpro (3C-likeproteinase/Nsp5) and PLpro (papain-like protease/Nsp3) are also very popular drug targets (Zhang et al., 2020; Yang and Rao, 2021; Calleja et al., 2022; Shen et al., 2022). Unfortunately, no perfect performing small molecule drugs with efficient inhibition of SARS-CoV-2 have been developed from them. With the increasing research on SARS-CoV-2, novel drug targets have been developed, especially the Nsps that play an important role in the life cycle of SARS-CoV-2 have great prospects for development as drug targets (Nunes et al., 2022; Yadav et al., 2021; Samdani et al., 2022).
Nsp14 is one of the Nsps. After the virus enters the host cell, 16 Nsps form the replication-transcription complex (RTC) necessary for viral genome replication and transcription. Nsp14 is one of the RTC components (Yan et al., 2021), it is a bifunctional protein with a proofreading function for the N-terminal nucleic acid exonuclease (ExoN) and a capping modification function for the C-terminal guanine N7-methyltransferase (N7-MTase) mRNA (Chen et al., 2009; Ogando et al., 2020). It has been suggested that Nsp14 is responsible for the evolution and maintenance of the large genome of coronaviruses, and that its expression is important for the stability of viral mRNA, which may help the virus to evade degradation by host immunity to a certain extent (Zaffagni et al., 2022). N-terminal ribose exonuclease activity of Nsp14 enables coronavirus to resist the inhibition by many nucleoside analogs, including Ribavirin, and partially reduces its susceptibility to Remdesivir (Xu et al., 2020). N7-methylation of the viral RNA cap plays a key role in the translation of viral RNA into proteins. Inhibition of SARS-CoV-2 Nsp14 N7-MTase also blocks the enzymatic coupling of viral RNA methylation, as 2′-O-MTase (Nsp16) recognizes only N7-methylated cap substrates (Decroly et al., 2008; Bouvet et al., 2010; Chen et al., 2009; Yan et al., 2021). Expression of Nsp14 triggers a dramatic remodeling of the transcriptome, altering the splicing of more than 1000 genes, leading to a dramatic increase in the number of circular RNAs associated with innate immunity, while allowing activation of the NF-kB pathway (Zaffagni et al., 2022). These effects are closely related to the guanine N7-MTase structural domain (Zaffagni et al., 2022; Yan and Chen, 2020). Therefore, it is clear that Nsp14 is a very promising drug target, and some compounds have been tested with significant inhibitory activity to Nsp14, which may bring new ideas for the development of potent anti-SARS-CoV-2 drugs.
For a large database of compounds, it is impractical to use experimental methods to determine the inhibitory effect of compounds on Nsp14 one by one, which is undoubtedly costly in terms of human, material and financial resources, as well as time cost. With the worldwide commitment to reduce animal experiments and support the 3R (Reduction, Replacement, Refinement) principles, traditional experimental assays are no longer suitable for the identification of a large number of compounds (Tosca et al., 2023). Chemoinformatics and computational biology have proven extremely useful in developing leads for severe diseases. These in silico approaches become an essential part of drug development process. They are commonly used to find new medications or improve the therapeutic efficacy (or pharmacokinetic features) of chemical series (Roy, 2022; Kar and Roy, 2013). Among them, Quantitative Structure Activity Relationship (QSAR) is one of the hotspots of in silico approaches in the field of medicinal chemistry, and it plays an important role as an efficient computerized screening and prediction tool for predicting drug activity, toxicity and permeability, as well as the mechanism of action of target molecules (Gramatica, 2020; Sharma and Bhatia, 2021; Kumar et al., 2022; Perkins et al., 2003). Compared to traditional drug development, QSAR can not only significantly shorten the development cycle, but also save significant economic costs and avoid a large number of unnecessary in vitro and in vivo experiments (Worth et al., 2007; Ford, 2016). To increase the acceptance of QSAR models, the Organization for Economic Co-operation and Development (OECD) has specifically developed five QSAR validation principles for regulatory use, including (1) a defined endpoint; (2) an unambiguous algorithm; (3) a defined domain of applicability; (4) appropriate measures of goodness-of-fit, robustness and predictivity; and (5) a mechanistic interpretation, if possible (OECD, 2007).
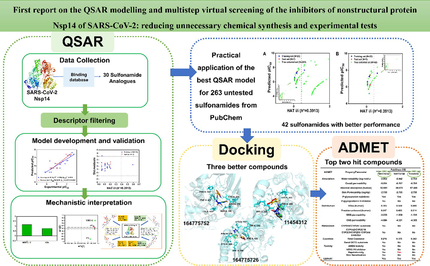
To ensure that one molecule can be successfully developed into a clinical antiviral drug, after QSAR prediction, it needs to be further validated for drug-like properties. Molecular docking and ADMET (absorption, distribution, metabolism, excretion, and toxicity) prediction are essential. Molecular docking allows exploring the interaction pattern of protein–ligand complexes, providing binding free energy (docking score) and key amino acid residues responsible for inhibitory activity (Ferreira et al., 2015). The predictions of pharmacokinetics and bioavailability are also very important for drug screening. For example, the internationally accepted standard “Lipinski’s principle of five” of an orally active drug includes (Lipinski et al., 2001; Lipinski, 2004): (1) the number of hydrogen donors (the number of hydrogen atoms connected to N and O) is less than 5; (2) the number of hydrogen receptors (the number of N and O) is less than 10; (3) the relative molecular weight is less than 500; (4) the octanol–water partition coefficient (logP) is less than 5; (5) 10 or fewer rotatable bonds. This rule of thumb describes the molecular properties vital for the pharmacokinetics of a molecule in human body, including its ADME properties (O'Brien and Fallah, 2013), has been widely used in the initial screening of compound libraries since it was published (Alam et al., 2021). The ADMET properties can determine the efficacy and toxicity of a drug, is a remarkable indicator to evaluate whether a compound exhibits drug-likeness. In this study, we developed an easily interpretable and reproducible QSAR model based on Nsp14 inhibitors. Both multiple linear regression (MLR) and partial least squares (PLS) methods were used to build different types of QSAR models to identify the key structural features of compounds responsible for SARS-CoV-2 Nsp14 inhibition, as well as to discover new Nsp14 inhibitors. Molecular docking and ADMET prediction were performed on the priority compounds screened by QSAR model to further explore their binding modes with target protein Nsp14 and the druggability, thus provide valuable reference for the development of potent anti-SARS-CoV-2 drugs. The complete workflow in this study is shown in Fig. 1.
Schematic illustration of the workflow of the present study.
2 Materials and methods
2.1 Data collection and curation
In the current work, a set of 30 sulfonamide-based bisubstrate analogs were collected from the Binding database (https://www.bindingdb.org/) as SARS-CoV-2 Nsp14 inhibitors, and these molecules target the N7-MTase of SARS-CoV-2 Nsp14 with significant inhibitory activity (Table S1). According to OECD principle 1, “a QSAR should be associated with a defined endpoint” (OECD, 2007), the experimental activity of each compound was expressed as IC50 (μM). The IC50 values are determined based on the same bioassay protocol (based on the radioactive N7-MTase assay) according to previous study (Ahmed-Belkacem et al., 2022). Firstly, the compounds were dissolved in 5 % DMSO, and the SARS-CoV-2 Nsp14 proteins were dissolved in 3 % DMSO. At 30 °C, the compounds with different concentrations were incubated with 50 nM SARS-CoV-2 Nsp14 in the reaction mixture [40 mM Tris-HCl (pH 8.0), 1 mM DTT, 1 mM MgCl2, 2 μM SAM and 0.1 μM 3H-SAM (Perkin Elmer)] for 30 min. The final enzymatic activity (IC50) was determined by filter-binding assay (Ahmed-Belkacem et al., 2022). For model development, we converted the IC50 (μM) values into molar units (M) and then further converted them into pIC50 (pIC50 = -logIC50) values, which is a customary practice in QSAR modelling (Gramatica et al., 2016; Gramatica, 2020; Li et al., 2022, 2023; Chen et al., 2023). The pIC50 value was positively correlated with the inhibitory activity compared to the IC50 value. We carefully examined all structures in the dataset before calculating the descriptors in order to develop statistically significant QSAR models.
2.2 Descriptor calculation and dataset division
After downloading the structures of the modelling compounds from the Binding database, the structures were optimized by the 3D module of ChemBioDraw Ultra 14.0 software in preparation for the next step of descriptor calculation. Dragon (https://chm.kode-solutions.net/) (Dragon, 2021) and PaDEL descriptors (Yap, 2011) were combined to characterize the structural features of the compounds, including functional group counts, topological indices, ring descriptors, connectivity indices, atomic center fragments, 2D atomic pairs, atomic type E states, molecular properties, etc., to more fully extract the hidden important structural information. To facilitate mechanistic interpretation, only 2D descriptors with clear physicochemical significance were calculated for model building, and constant or near-constant and highly correlated descriptors (correlation coefficient > 0.95) were removed to reduce redundant variable information (Gramatica et al., 2013; Gramatica et al., 2014).
To obtain QSAR models with higher prediction quality, a total of six division techniques in QSARINS 2.2.4 software and Dataset Division GUI software (http://dtclab.webs.com/softwaretools) were used to divide the dataset into training and test sets in a ratio of approximately 3:1. Three division techniques in QSARINS, including (1) ordered by response (ORes), (2) ordered by structure similarity (OStr), and (3) randomly (Rnd) (Gramatica et al., 2013). Dataset Division GUI software includes three division techniques, namely, (1) Kennard Stone based method (KS), (2) Euclidean Distance based method (ED), (3) Activity/Property based method (A/P) (Ambure et al., 2019). The training set was used to build the QSAR model and the test set was used to validate the developed model in terms of its predictive power and to avoid possible biases.
2.3 Descriptor selection and model building
According to OECD principle 2, “a QSAR should be associated with an unambiguous algorithm” (OECD, 2007). Identifying important descriptors from a large number of initial descriptors using different variable selection methods is an important step in QSAR modelling. Due to the small dataset in this study, two different software were used to construct the QSAR models in order to filter a more reliable model. First, the Genetic Algorithm-Variable Subset Selection (GA-VSS) module included in the QSARINS software was used for variable screening, and MLR was used to build the QSAR model. In the descriptor selection process, the leave-one-out cross-validation correlation coefficient ( ) was used as the fitness function to evaluate the model robustness. The QUIK (Q under influence of K) rule was applied to avoid multicollinearity with a threshold of 0.05 (Todeschini et al., 1999). Secondly, the three dataset division results generated by Dataset Division GUI software were further used by the genetic algorithm (GA) in the Small Dataset Modeler software (https://sites.google.com/view/smalldatasetmodelling) as a variable selection method to build the MLR model and its corresponding PLS model (GA-PLS model using the same descriptors selected in the GA-MLR analysis) (Ambure et al., 2019). The final MLR/PLS models were selected based on the MAELOO(95%) after running GA-MLR for at least 10 times and the selected model was evaluated using the test set predictions (Roy et al., 2016).
2.4 Model performance evaluation
OECD principle 4 states that “a QSAR should be associated with appropriate measures of goodness-of-fit, robustness and predictivity” (OECD, 2007), and statistical validation is a key step in testing the eligibility of the model. In this study, internationally accepted internal and external validation metrics were used to check the robustness and predictability of the generated models.
In internal validation, we evaluated the robustness and goodness-of-fit of the model mainly through and the coefficient of determination R2 (including adjusted ), with larger values indicating better model quality (Tropsha, 2010). As for the external validation, we calculated various statistical parameters to verify the model prediction performance, such as , , , , , Δ , concordance correlation coefficient for the test set (CCCtest), and the root mean square error (RMSE) (Tropsha, 2010; Gramatica and Sangion, 2016). The parameter scales to be satisfied by a better model are as follows (Chirico and Gramatica, 2011, 2012): R2 > 0.7, > 0.6, > 0.6; CCCtest > 0.85; > 0.65; Δ < 0.2; MAEtest and RMSEtest should be as small as possible. For more detailed information on these statistical parameters, please refer to Table S2.
The models generated by QSARINS software were ranked using the multi-criteria decision making (MCDM) module (Gramatica et al., 2013). MCDMfit was obtained based on the maximum values of R2, Radj2, CCCtrain and the minimum value of R2-Radj2. MCDMtest was selected based on the maximum values of QF12, QF22, QF32, and CCCtest. Finally, we ranked the generated QSAR models based on MCDMfit and MCDMtest values. For the models generated by Small Dataset Modeler software (Ambure et al., 2019), the MAE95% value of the test set of each model was compared, the model with the smallest MAE95% value was selected.
2.5 Applicability domain (AD)
According to OECD principle 3, “a QSAR should be associated with a defined domain of applicability” (OECD, 2007). For this purpose, the applicability domain (AD) analysis was performed to define the scope of application for a QSAR model. The prediction of a compound can be considered reliable only if it falls within the AD. In this study, we used the leverage method combined with the standardized residual method to define the AD. It can be simply understood that a compound should be identified as a structural outlier if its leverage value h is larger than the threshold value h* (h*=3(p + 1)/n; p: the number of descriptors in the model; n: the number of compounds in the training set); while a compound with predicted residual higher than ± 3 standardized residuals, it should be considered a response outlier (Gramatica et al., 2013). The results of the AD analysis can be visualized by Williams plot, which directly shows the structural and response outliers, but it should be noted that some compounds may not be considered as outliers even if h > h*, because their standardized residual values may be small.
2.6 Screening of untested compounds using developed QSAR models
One of the main purposes of QSAR modelling in this study was to screen compounds having significant inhibitory effects on SARS-CoV-2 Nsp14 from the large database of compounds. We downloaded 263 compounds from the PubChem website (https://pubchem.ncbi.nlm.nih.gov/) using the sulfonamide structural fragment (Fig. 2A) as template molecule 1 to form a large true external set. Notably, the structures of these 263 unknown true external compounds have the same substructures with the 30 compounds used for QSAR modelling. Meanwhile, in order to select compounds with more similar structures to the 30 modelling compounds, a benzene ring was added to template 1 as template 2 (Fig. 2B). Therefore, a total of 42 compounds were extracted from the 263 compounds to form a small true external set. The detailed information of true external set was listed in Table S1. In order to verify the reliability of our chosen QSAR model for the prediction of unknown compounds, certain measures will be chosen to further evaluate the prediction results, which will help us to prioritize potential compounds in expensive in vitro or in vivo experiments. We used “Prediction Reliability Indicator (PRI)” tool (https://dtclab.webs.com/software-tools) to check the prediction confidence of each true external compound (Roy et al., 2018). On the basis of the composite score of each compound, the prediction quality was classified into three groups: “good” (composite score 3), “moderate” (composite score 2) and “bad/unreliable” (composite score 1) (Khan and Roy, 2019; Roy et al., 2018).
(A) Template 1 for large true external set (263 compounds); (B) Template 2 for small true external set (42 compounds).
2.7 Molecular docking study
In this paper, we used molecular docking technique to determine the interaction pattern of small molecule ligands with the target protein Nsp14. The crystal structure of Nsp14 protein was retrieved from the Protein Data Bank (PDB) with the code 7R2V (available at: https://www.rcsb.org/structure/7R2V) (Czarna et al., 2022). 7R2V is a protein–ligand complex with S-adenosyl homocysteine (SAH) bound to it. Molecular docking studies were performed using Discovery Studio 4.0 (Accelrys Inc.). Before docking, the ligand SAH bound to the protein was removed, and then the protein preparation was completed by the removal of deletion residues, hydrogenation and docking site generation from the protein. The sites were generated using the “from current selection” program in the receptor-ligand interaction module, referring to the study by Ahmed-Belkacem et al. using the four residues TYR420, PHE506, PHE426, ARG310 to generate active sites (Ahmed-Belkacem et al., 2022). The selected compounds were subjected to ligand preparation through the small molecule module of the Discovery Studio platform to generate a series of ligand conformations. Each of these generated conformations will next be used for molecular docking in the CDOCKER module. The CDOCKER binding energy parameters (kcal/mol) of all the receptor-ligand complexes were examined and the highest scores (most negative) favored the binding of the receptor to the ligand.
2.8 ADMET profiling
After molecular docking study, we tested the screened compounds again to determine their drug-likeness and ADMET properties. We used pkCSM (Pires et al., 2015) (http://biosig.unimelb.edu.au/pkcsm/), SwissADME (http://www.swissadme.ch/) (Daina et al., 2017), and the ADMET and TOPKAT® module embedded in Biovia Discovery Studio software (Kumar et al., 2022). Three different methods were used to predict the ADMET properties of the compounds. The aim of using multiple software is to maximize the assessment of all possible toxicities of the compounds as well as key drug-likeness information, which can increase the chance of reaching anti-SARS-CoV-2 drug candidates.
3 Results and discussion
The main objective of the current study is to develop statistically significant QSAR models that use easily interpretable descriptors, then use the validated models to identify potent Nsp14 inhibitors of SARS-CoV-2 from compound databases. The predictions of the QSAR models will be further screened by molecular docking and ADMET prediction. In general, we performed the following four main tasks: (1) develop a validated QSAR model with excellent predictive performance; (2) the QSAR model as a preliminary screening stage, was utilized to predict the inhibitory potency of true external compounds against Nsp14; (3) the better performing 42 compounds in step 2 were docked to the protein active site, and analyze the docking pattern; (4) perform ADMET predictions on compounds with higher docking scores and predicted inhibitory activity. After the above four steps of screening, the most promising potential Nsp14 inhibitors were finally identified.
3.1 QSAR analysis
According to the OECD's QSAR validation guidelines (OECD, 2007), QSAR models that meet various parameter criteria should be selected. In this study, a variety of dataset division methods combined with two computational methods (PLS and MLR) were used to construct nine different classes of models. As shown in Table 1, the nine models were able to explain training set variance (R2) between 0.7150 and 0.7796 (0.6130 ∼ 0.7373 in terms of
). In addition,
ranged between 0.5573 and 0.8070,
ranged between 0.5547 and 0.8023, CCCtest ranged between 0.7700 and 0.9073, and MAEtest ranged between 0.2323 and 0.4155. These parameters indicated that all nine models exhibited accepted but with different degrees of predictive ability.
Ntr
Ntest
R2
CCCtest
RMSEtest
MAEtest
Δ
Model 1 (Eq.1 for OStr-MLR): pIC50 = 4.1823 + 0.5138nCb- + 0.5865MDEC-12
23
7
0.7796
0.7373
0.8539
0.8036
0.8023
0.8932
0.9073
0.2773
0.2323
0.7890
0.0464
Model 2 (Eq.2 for ORes-MLR): pIC50 = 2.8404 + 13.4451MATS2v + 0.8287MLFER_E
23
7
0.7750
0.6988
0.7390
0.7346
0.7214
0.7465
0.8345
0.4065
0.3089
0.5779
0.2407
Model 3 (Eq.3 for Rnd-MLR): pIC50 = 6.3284 + 0.3654VE3sign_B(v) + 0.8287MDEC-12
24
6
0.7722
0.7287
0.6784
0.5573
0.5547
0.6226
0.8109
0.5002
0.4155
0.5513
0.2162
Model 4 (Eq.4 for ED-MLR): pIC50 = 2.207 + 9.705SpMAD_EA(ri) −12.243GATS2v + 3.474SpMin8_Bh(s)
24
6
0.7440
0.6480
–
0.7830
0.7600
–
0.8560
–
0.2780
0.6520
0.1790
Model 5 (Eq.5 for KS-MLR): pIC50 = 4.167 + 0.117VE3sign_X + 1.219Eig11_AEA(dm) + 11.37MATS2v
22
8
0.7150
0.6130
–
0.7280
0.7270
–
0.8580
–
0.3720
0.6580
0.0200
Model 6 (Eq.6 for A/P-MLR): pIC50 = -3.259 + 7.062SpMin2_Bh(s) + 12.288MATS2v + 0.25nCb-
24
6
0.7470
0.6820
–
0.8070
0.7930
–
0.9000
–
0.3140
0.7440
0.0990
Model 7 (Eq.7 for ED-PLS): pIC50 = 1102.311 + 3.102SpMin6_Bh(m) − 1107.098ChiA_X + 0.602SpMax3_Bh(m)
24
6
0.7480
0.6860
–
0.8000
0.7790
–
0.8760
–
0.2720
0.6980
0.1570
Model 8 (Eq.8 for KS-PLS): pIC50 = 3.598 + 0.407SsCH3 + 0.12X3sol + 0.001P_VSA_i_3
22
8
0.7360
0.6620
–
0.7020
0.7010
–
0.7700
–
0.3430
0.3620
0.3660
Model 9 (Eq.9 for A/P-PLS): pIC50 = -12.835 + 11.794MATS2v − 0.926LLS_02 + 5.43SpMax4_Bh(v)
24
6
0.7580
0.7040
–
0.7630
0.7450
–
0.8640
–
0.3620
0.7700
0.1250
In order to find new potential Nsp14 inhibitors using the best QSAR model and to identify the structural features in the compounds that have important influence on Nsp14 inhibition, we comprehensively compared the validation parameters of the nine models in Table 1. It could be found that the internal and external parameters of Model 1 (Eq.1) were better than those of other models, thus being the best QSAR model. Model 1 (Eq.1) was constructed based on the OStr division method combined with the MLR method. The model contained only two 2D descriptors, namely nCb- and MDEC-12, the correlation between these two descriptors was very weak (Fig. 3A). The typical statistical parameters of Model 1 were R2 = 0.7796,
= 0.7373,
= 0.8539,
= 0.8023 ∼ 0.8932 and CCCtest = 0.9073. It is evident that the best model meets the OECD guidelines in terms of robustness, predictivity and adaptability (OECD, 2007). The detailed validation parameters of Model 1 were given in Table 1. The scatter plot (Fig. 3B) and William plot (Fig. 3C) provided a visual representation of the excellent performance of the best model. As shown in Fig. 3B, the predicted and experimental pIC50 values of modelling compounds exhibited a good linear relationship, indicating good goodness of fit and predictive ability. From the William plot (Fig. 3C), it was found that only compound 30 in the training set had an h value higher than the warning h*, thus being considered a structural outlier. However, its prediction residual was very small and acceptable (<0.12 log units) (see Table S3), also indicating the good predictive reliability of the model. We also found one response outlier (compound 5) with a standardized residual greater than three standard deviation units, thus affecting the model's goodness-of-fit to some extent.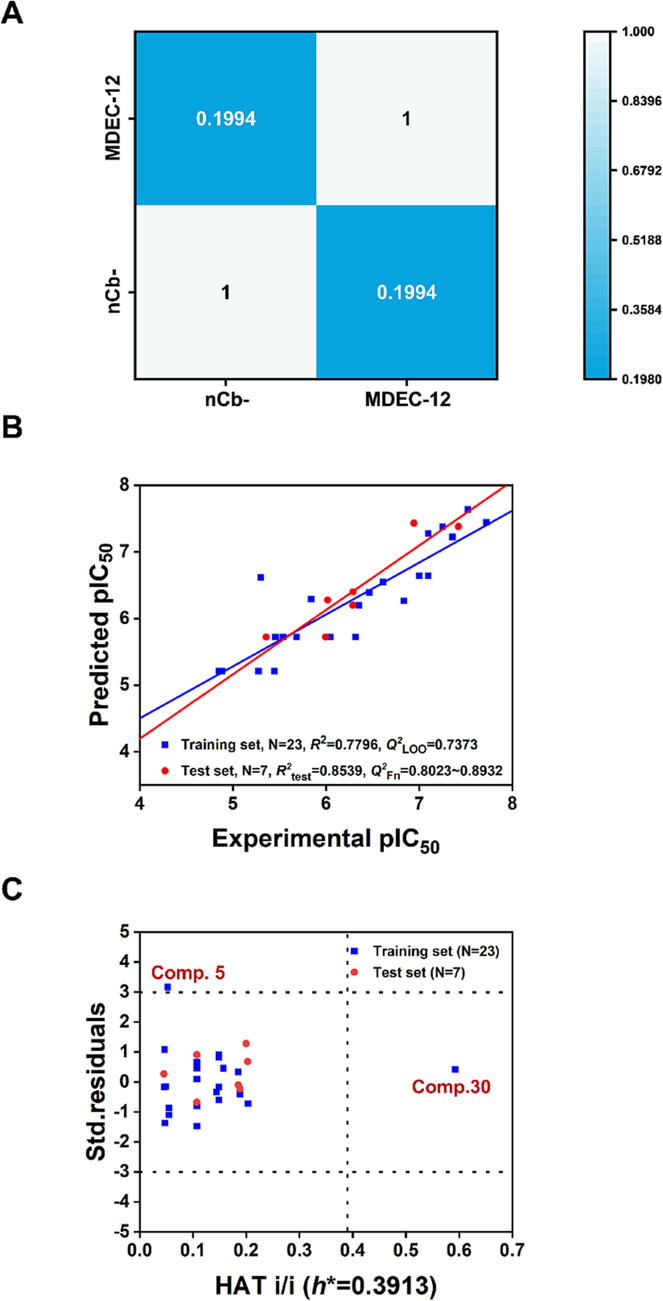
(A) The weak inter-correlation of nCb- and MDEC-12; (B) Scatter plot of predicted and experimental values of modelling compounds in the best QSAR model; (C) Williams plot of the best QSAR model.
3.2 Mechanistic interpretation of the modelled descriptors
The selected descriptors in the model equation reflect the role of specific structural and physicochemical properties of the compounds in controlling the response endpoint. These properties will be crucial for the development of potential Nsp14 inhibitors. In line with OECD principle 5 (“a mechanistic interpretation, if possible”) (OECD, 2007), we provided a detailed mechanistic explanation for both descriptors appearing in the best QSAR model, including nCb- and MDEC-12.
Fig. 4A showed the variable importance plot (VIP), from which we found that the most important descriptor in the model (VIP score ≥ 1.0) was MDEC-12 and the second important descriptor (VIP score < 1.0) was nCb-. The loading plot was also used to assess the influence of the two descriptors on the inhibitory activity of Nsp14. The distances between the descriptors and the Y-response or the origin were taken into account, respectively. If one descriptor is close to the Y-response and far from the origin, it is considered more important (Kumar et al., 2020). Thus, it was obvious that from the Fig. 4B, MDEC-12 showed a greater effect on the inhibitory activity against Nsp14, which was consistent with the VIP results.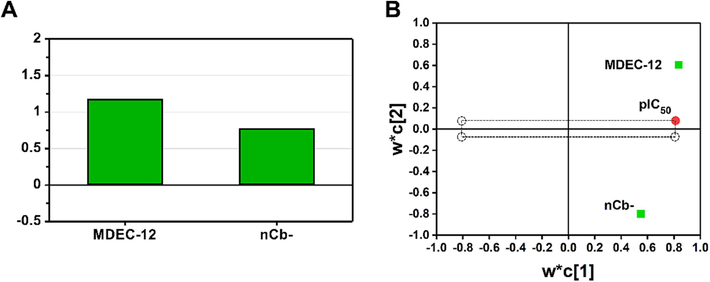
(A) Variable importance plot (VIP); (B) loading plot for the two descriptor variables in the best QSAR model.
Both two descriptors included in the best model showed a positive correlation with Nsp14 inhibitory activity, implying that the higher the nCb- and MDEC-12 values of a compound, the more significant the inhibitory potency on Nsp14. The nCb- descriptor refers to the number of substituted benzene moieties present in the molecule. As shown in Fig. 5, compound 30 had the highest nCb- value (5) in the modelling dataset, indicating that compound 30 has five substituent groups on the benzene ring. Therefore, it exhibited a high pIC50 value (7.523) and was the most active compound in inhibiting Nsp14 in the modelling dataset. On the other hand, the nCb-value of compound 5 (3) was smaller than that of compound 30, implying only three substituent groups on its benzene ring, leading to a relatively decreased pIC50 value (5.301) (Fig. 5). The second descriptor MDEC-12 represents the molecular distance edge between all primary and secondary carbons. As shown in Fig. 5, compound 28 had the largest MDEC-12 value (2.915), thus it showed high inhibitory activity with pIC50 value of 6.943. It is interesting to note that the nCb-value of compound 28 is exactly equal to that of compound 5, both being 3. However, in terms of the MDEC-12 value, compound 28 (2.915) exceeded than compound 5 (1.524), thus, it was observed that compound 28 (pIC50 = 6.943) had a higher pIC50 value than that of compound 5 (pIC50 = 5.301). In the modelling dataset, the pIC50 value (4.85) of compound 1 was the smallest, which was due to both the two descriptors had very small values (nCb-=2; MDEC-12 = 0). The mechanistic interpretation provided a more transparent and understandable relationship between the structural features of a compound and its inhibitory effect on Nsp14. For more information on the descriptors of the compounds, please see Table S3.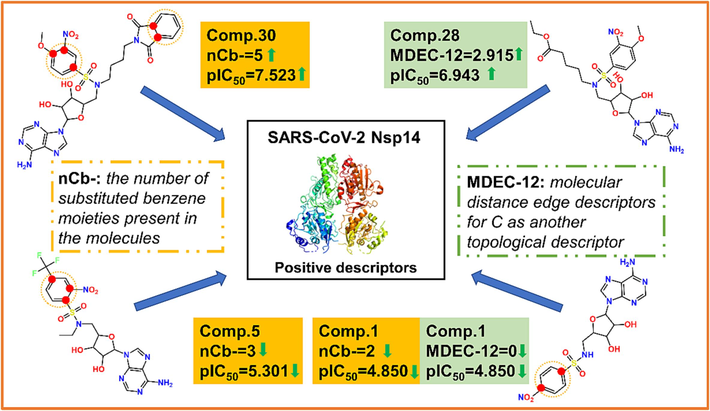
Schematic diagram of the selected descriptors in controlling the inhibitory activity against Nsp14 in the best QSAR model.
3.3 Initial screening using the best QSAR model
We used the best QSAR model to predict the true external compounds without experimental values, with the aim to look for compounds that might have a significant inhibitory effect on Nsp14 activity. For the 263 untested compounds collected from the PubChem website (https://pubchem.ncbi.nlm.nih.gov/), the best model was used to predict their inhibitory activity against Nsp14. Insubria plot, constructed by the leverage values versus the predicted toxicity, can show whether the true external compounds are within the AD range of the model (Gramatica et al., 2013; Gramatica et al., 2014). As shown in Fig. 6A, the coverage of the model for 263 compounds did not seem to be acceptable, reaching only 47 %, with a significant proportion of compounds being outside the AD range. Therefore, we further screened the true external set and selected 42 structurally most similar compounds from the 263 compounds to as a small true external set (see section 2.6). Obviously, the model showed a significant coverage (81 %) for the 42 compounds (Fig. 6B). At the same time, to further validate the prediction results given by the best model, the prediction quality of each compound was evaluated using the “prediction reliability indicator” tool developed by Roy's team (Roy et al., 2018). For the 263 untested compounds, most were “good” or “moderate” (>90 %), while 26 compounds were “bad” (Table S4). Among the small true external set consisting of 42 untested compounds, 39 compounds were “good” or “moderate” (>92 %) and only 3 compounds were “bad” (Table S5). Therefore, the best model we developed can predict the inhibitory activity of compounds without experimental values to a certain extent. If the structural similarity between the query compounds and the source compounds is better, the predicted results should be more reliable.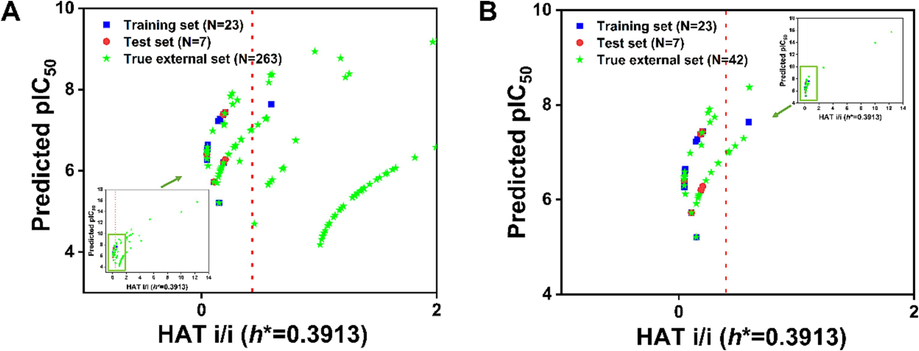
(A) Insubria plot of 263 untested true external compounds; (B) Insubria plot of 42 untested, structurally most similar compounds.
3.4 Molecular docking analysis
The 42 structurally most similar compounds were docked to the N7-MTase active pocket of Nsp14, of which 17 molecules were successfully docked, and detailed molecular docking analysis was reported in Table S6. Based on the binding energy and predicted inhibitory activity, the top three molecules were evaluated together, including 164775752, 164775726, and 11454312. The predicted pIC50 of compound 164775752 was 6.120, and the binding energy was −29.761 kcal/mol. The predicted pIC50 of 164775726 was 6.432, and the binding energy was –23.199 kcal/mol. Obviously, the comprehensive performance of these two compounds was good. In contrast, for compound 11454312, although its binding energy (-18.219 kcal/mol) was not very satisfactory, its predicted inhibitory activity (pIC50 = 6.767) was the second highest among these 17 molecules, thus it was used as the third target molecule for further study. Although compound 125506109 exhibited the highest predicted activity (pIC50 = 6.9923), it possessed an undesirable binding energy (-8.626 kcal/mol), thus was not selected for further analysis. As shown in Table 2, a detailed summary of the binding energy (CDocker energy), interacting residues, forces involved, and predicted activities of the three compounds was available. At the same time, we also discussed the detailed docking interactions of the selected three compounds with the active site residues of Nsp14.
No.
PubChem CID
SMILES
Binding energy (kcal/mol)
Interacting residues
Interacting forces
Predicted pIC50
1
164,775,752
COC1 = NC = NC2 = C1N = CN2[C@H]3[C@@H]([C@@H]([C@H](O3)CNS(=O)(=O)C4 = CC = CC(=C4O)C(=O)N)O)O
−29.7610
ASN306, PRO335, CYS309, ARG310, LYS423, PHE426
Carbon hydrogen bond, π-Sulfur bond, Conventional hydrogen bond, Attractive charge, Alkyl bond, π-Alkyl bond, π-π Stacked
6.1200
2
164,775,726
CN(C[C@@H]1[C@H]([C@H]([C@@H](O1)N2C = NC3 = C(N = CN = C32)N)O)O)S(=O)(=O)C4 = CC = CC(=C4O)C(=O)N
–23.1989
PHE426, ARG310, LYS423
π-π Stacked, π-π T-shaped, Attractive charge, Conventional hydrogen bond
6.4315
3
11,454,312
CC1 = CC = C(C = C1)S(=O)(=O)NC(=O)[C@@H]2[C@@H]3[C@H]([C@@H](O2)N4C = NC5 = C(N = CN = C54)N)OC(O3)(C)C
−18.2187
PHE 426, TRP292, ARG310, LYS423, CYS309, PHE506
π-π Stacked, Alkyl bond, π-π T-shaped, π-Sulfur bond, Attractive charge, π-Cation, Conventional hydrogen bond, π-Alkyl bond, π-Donor hydrogen bond
6.7674
The best docking conformations of the three compounds 164775752, 164775726, 11454312 with Nsp14 were shown in Fig. 7. The 3D figures (left) highlighted the classical hydrogen bonds (pink) formed between the compounds and amino acid residues. The three compounds also formed non-classical hydrogen bonds and other bonds with amino acid residues, these interactions were shown in the 2D figures (right). The compound with the best binding energy in the screening list was compound 164775752 (Fig. 7A), which interacted with residues ASN306, PRO335, ARG310 through hydrogen bonding, including one conventional hydrogen bond with ARG310 (pink), two carbon hydrogen bonds with ASN306 and PRO335. The compound also formed π-Sulfur bond with CYS309, attractive charge with ARG310, π-π Stacked with PHE426, and π-alkyl hydrophobic bond with LYS423.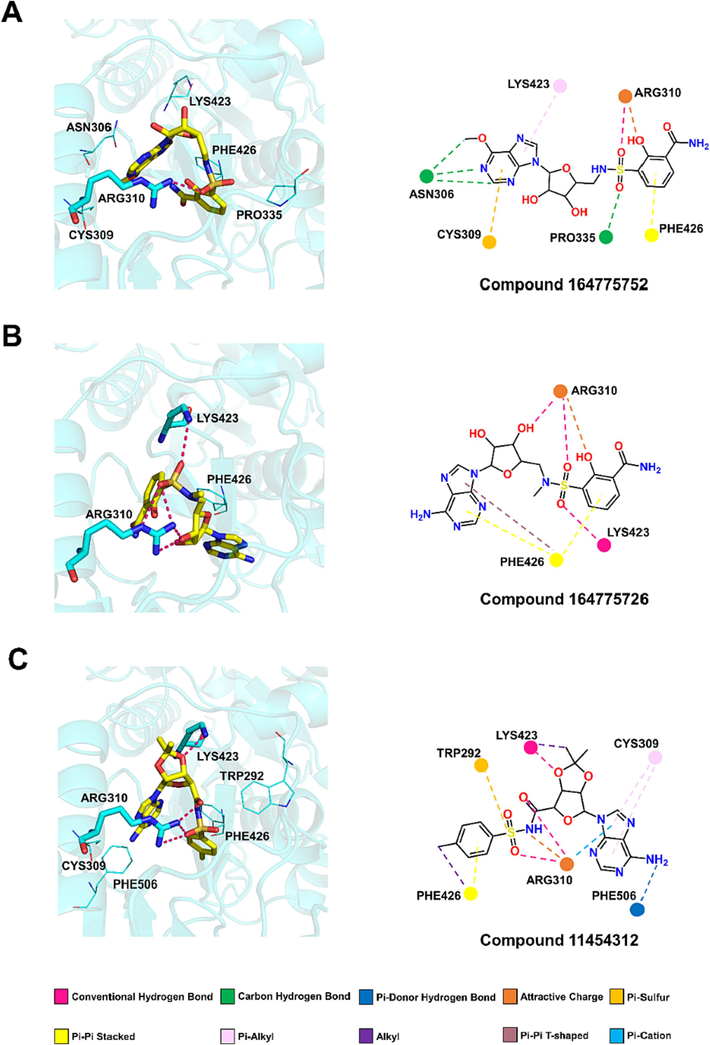
Molecular docking interactions of compounds (A) 164,775,752 (B) 164,775,726 and (C) 11,454,312 with the active site of Nsp14 N7-MTase domain.
The second-ranked binding energy was compound 164775726 (Fig. 7B). It interacted with residues ARG310 and LYS423 through hydrogen bonding, which included three conventional hydrogen bonds with ARG310 and other one with LYS423 (pink). Compound 164775726 also formed π-π Stacked bond and π-π T-shaped bond with PHE 426, and attractive charge with ARG310.
Compound 11454312 had the highest predicted inhibitory activity (pIC50 = 6.7674) although it exhibited the worse binding energy (Fig. 7C). It interacted with residues ARG310, LYS423 and PHE506 through hydrogen bonding, which included three conventional hydrogen bonds with ARG310 and other one with LYS423 (pink). Furthermore, it formed π-π Stacked bond and alkyl hydrophobic bond with PHE426, π-Sulfur bond with TRP292, two π-alkyl hydrophobic bonds with CYS309. ARG310 was also involved in the formation of attractive charge and π-Cation. As well, LYS423 participated the formation of alkyl hydrophobic bond.
The docking analysis of the three top compounds with N7-MTase active site of Nsp14 showed that they could interact with important amino acid residues such as PHE426 and ARG310. Especially the key amino acid residue ARG310, all the three candidates could interact with it by hydrogen bonding interactions. Therefore, the three compounds may be potential Nsp14 inhibitor candidates.
3.5 ADMET profiling of the top selected screened compounds
The three compounds were further tested for their ADMET properties. As can be seen in Table 3, the ADME properties of all three compounds fell within the acceptable range (O'Brien and Fallah, 2013). Although all three compounds violated one principle (the number of hydrogen receptors were greater than 10) about “Lipinski’s principle of five”, they were acceptable. Regarding toxicity, compound 164775726 was predicted to be potentially hepatotoxic, thus it was excluded. The other two compounds were predicted without significant toxicity, suggesting that they are amenable to being pursued as therapeutic candidates. More details of the ADMET results were provided in the Table S7. To date, no specific drug targeting SARS-CoV-2 Nsp14 on the market, we were unable to find available data to use as references. As a result, compounds 164775752, 11454312 as candidate Nsp14 inhibitors that passed the ADMET predictions need to undergo a series of rigorous clinical trials. a: ADMET is an important criterion for screening drug candidates for drug-like properties, including absorption, distribution, metabolism, excretion, and toxicity. “Lipinski's principle of five” is also an important criterion, where a potential drug can only violate one rule. b: Properties/Parameters are used to characterize the drug-like properties of compounds and have strict value ranges. Absorption consists of six items, especially intestinal absorption (human), in which the high value represents that a molecule can easily traverse the digestive system barrier. Distribution consists of four items, which evaluates the effect of a molecule on the blood–brain-barrier (BBB), plasma, etc. Metabolism evaluates whether a compound can act as a substrate or inhibitor of related proteases. Excretion means that a molecule can be eliminated from the body in an efficient manner without negatively affecting the accumulation of the drug in the organism. Toxicity consists of four items, qualifying drugs should all be free of toxicity. c: The compounds collected from the PubChem database were screened by QSAR modeling and molecular docking. As a result, we identified three candidate compounds, and the drug properties of all three compounds were evaluated. *: ≫ means a higher value is better for a candidate drug.
ADMETa
Property/Parameterb
Value Rangeb
Three Candidatesc (PubChem CID)
164775752
164775726
11454312
Absorption
Water solubility (log mol/L)
≫*
−2.862
−2.942
−2.764
Caco2 permeability
≫*
−0.554
−0.107
−0.704
intestinal absorption (human)
>30 %
52.691
49.875
67.499
Skin Permeability (log Kp)
<-2.5
−2.735
−2.735
−2.735
P-glycoprotein substrate
Yes/No
Yes
Yes
Yes
P-glycoprotein I/ II inhibitor
Yes/No
No
No
No
Distribution
VDss (human)
>0.7
0.763
0.559
0.906
Fraction unbound (human)
≫*
0.547
0.483
0.514
BBB permeability
<-1
−2.238
−1.856
−1.134
CNS permeability
<-3
−4.684
−4.321
−4.365
Metabolism
CYP2D6/CYP3A4 substrate
Yes/No
No
No
No
CYP1A2/CYP2C19 CYP2C9/CYP2D6/ CYP3A4 inhibitior
Yes/No
No
No
No
Excretion
Total Clearance
≫*
0.278
0.175
0.392
Renal OCT2 substrate
Yes/No
No
No
No
Toxicity
AMES toxicity
Yes/No
No
No
No
hERG I/II inhibitor
Yes/No
No
No
No
Hepatotoxicity
Yes/No
No
Yes
No
Skin Sensitisation
Yes/No
No
No
No
Lipinski
Yes/No
Yes
Yes
Yes
4 Conclusions
Non-structural proteins of SARS-CoV-2 have been very popular targets during drug development, and Nsp14 shows great potential as the drug target because it plays an important role in viral replication and transcription. In this study, QSAR models were successfully developed based the OECD principles using 30 sulfonamide-based bisubstrate analogs as the Nsp14 inhibitors for the first time. All the models were evaluated by the strict internal and external validation criteria, the mechanistic interpretation revealed the relationship between structural information and the inhibitory activity of compounds against Nsp14. The best model (Model 1) was applied to the large compound database (263 untested compounds) to find new potential inhibitors of Nsp14. Molecular docking studies were used to explore the interaction pattern of the QSAR-based pre-screened compounds with the active site of the target Nsp14 protein. By comprehensively considering the predicted inhibitory activity and the docking results, three molecules were evaluated as the top HIT candidates. Meanwhile, to ensure that the three compounds can be further developed as oral drugs in humans, we performed ADMET predictions. Through stepwise screening, compounds 164775752, 11454312 can be finally identified as potential candidate Nsp14 inhibitors that being promising for further clinical studies.
CRediT authorship contribution statement
Qianqian Wang: Data curation, Investigation, Writing – original draft. Tengjiao Fan: Software. Runqing Jia: Formal analysis, Resources. Na Zhang: Validation, Software. Lijiao Zhao: Formal analysis, Software, Resources. Rugang Zhong: Writing – review & editing. Guohui Sun: Conceptualization, Methodology, Formal analysis, Funding acquisition, Writing – review & editing.
Acknowledgements
This work was supported by the Beijing Natural Science Foundation (No. 7242193, 7222016), the National Natural Science Foundation of China (No. 82003599), the Project of Cultivation for young top-motch Talents of Beijing Municipal Institutions (No. BPHR202203016), and Science and Technology General Project of Beijing Municipal Education Commission (No. KM202110005005). The authors thank Prof. P. Gramatica (University of Insubria, Varese, Italy) for authorizing the use of QSARINS 2.2.4 software, and thank Prof. K. Roy (Jadavpur University, Kolkata, India) for authorizing the use of “Prediction Reliability Indicator” tools (available at http://dtclab.webs.com/software-tools).
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Potent inhibition of SARS-CoV-2 nsp14 N7-Methyltransferase by sulfonamide-based bisubstrate analogues. J. Med. Chem.. 2022;65:6231-6249.
- [Google Scholar]
- Detection of natural inhibitors against human liver cancer cell lines through QSAR, molecular docking and ADMET studies. Curr. Top. Med. Chem.. 2021;21:686-695.
- [Google Scholar]
- New workflow for QSAR model development from small data sets: Small dataset curator and small dataset modeler. Integration of data curation, exhaustive double cross-validation, and a set of optimal model selection techniques. J. Chem. Inf. Model.. 2019;59:4070-4076.
- [Google Scholar]
- ACTT-1 study group members. Remdesivir for the treatment of Covid-19 - final report. N. Engl. J. Med.. 2020;383:1813-1826.
- [Google Scholar]
- In vitro reconstitution of SARS-coronavirus mRNA cap methylation. PLoS Pathog.. 2010;6:e1000863.
- [Google Scholar]
- Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect.. 2020;9:221-236.
- [Google Scholar]
- A detailed overview of SARS-CoV-2 omicron: Its sub-variants, mutations and pathophysiology, clinical characteristics, immunological landscape, immune escape, and therapies. Viruses. 2023;15:167.
- [Google Scholar]
- Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc. Natl. Acad. Sci. U. S. A.. 2009;106:3484-3489.
- [Google Scholar]
- Ecotoxicological QSAR study of fused/non-fused polycyclic aromatic hydrocarbons (FNFPAHs): Assessment and priority ranking of the acute toxicity to Pimephales promelas by QSAR and consensus modeling methods. Sci. Total Environ.. 2023;876:162736
- [Google Scholar]
- Real external predictivity of QSAR models: how to evaluate it? Comparison of different validation criteria and proposal of using the concordance correlation coefficient. J. Chem. Inf. Model.. 2011;51:2320-2335.
- [Google Scholar]
- Real external predictivity of QSAR models. Part 2. New intercomparable thresholds for different validation criteria and the need for scatter plot inspection. J. Chem. Inf. Model.. 2012;52:2044-2058.
- [Google Scholar]
- Refolding of lid subdomain of SARS-CoV-2 nsp14 upon nsp10 interaction releases exonuclease activity. Structure. 2022;30:1050-1054.
- [Google Scholar]
- SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep.. 2017;7:42717.
- [Google Scholar]
- Coronavirus nonstructural protein 16 is a cap-0 binding enzyme possessing (nucleoside-2'O)-methyltransferase activity. J. Virol.. 2008;82:8071-8084.
- [Google Scholar]
- Dragon. Dragon for Windows (Software for Molecular Descriptor Calculation) Version 7.0.6, Talete srl, Milan, Italy. https://chm.kode-solutions.net/ (Accessed 3 September 2021).
- Molecular docking and structure-based drug design strategies. Molecules. 2015;20:13384-13421.
- [Google Scholar]
- Refinement, reduction, and replacement of animal toxicity tests by computational methods. ILAR J.. 2016;57:226-233.
- [Google Scholar]
- On the development and validation of QSAR models. Methods Mol. Biol.. 2013;930:499-526.
- [Google Scholar]
- Principles of QSAR modeling: comments and suggestions from personal experience. Int. J. Quant. Struct.-Prop Relat.. 2020;5:61-97.
- [Google Scholar]
- QSARINS: A new software for the development, analysis, and validation of QSAR MLR models. J. Comput.. 2013;34:2121-2132.
- [Google Scholar]
- QSARINS-chem: Insubria datasets and new QSAR/QSPR models for environmental pollutants in QSARINS. J. Comput. Chem.. 2014;35:1036-1044.
- [Google Scholar]
- A historical excursus on the statistical validation parameters for QSAR models: A clarification concerning metrics and terminology. J. Chem. Inf. Model.. 2016;56:1127-1131.
- [Google Scholar]
- Aquatic ecotoxicity of personal care products: QSAR models and ranking for prioritization and safer alternatives' design. Green Chem.. 2016;18(16):4393-4406.
- [Google Scholar]
- Compassionate use of remdesivir for patients with severe Covid-19. N. Engl. J. Med.. 2020;382:2327-2336.
- [Google Scholar]
- A novel vision of reinforcing nanofibrous masks with metal nanoparticles: antiviral mechanisms investigation. Adv. Fiber Mater.. 2023;5:1273-1317.
- [Google Scholar]
- How far can virtual screening take us in drug discovery? Expert Opin. Drug Discov.. 2013;8:245-261.
- [Google Scholar]
- Ecotoxicological QSAR modelling of organic chemicals against Pseudokirchneriella subcapitata using consensus predictions approach. SAR QSAR Environ. Res.. 2019;30:665-681.
- [Google Scholar]
- Exploring 2D-QSAR for prediction of beta-secretase 1 (BACE1) inhibitory activity against Alzheimer's disease. SAR QSAR Environ. Res.. 2020;31:87-133.
- [Google Scholar]
- Identification of potential antivirals against 3CLpro enzyme for the treatment of SARS-CoV-2: A multi-step virtual screening study. SAR QSAR Environ. Res.. 2022;33:357-386.
- [Google Scholar]
- Systematic QSAR and iQCCR modelling of fused/non-fused aromatic hydrocarbons (FNFAHs) carcinogenicity to rodents: reducing unnecessary chemical synthesis and animal testing. Green Chem.. 2022;24(13):5304-5319.
- [Google Scholar]
- Ecotoxicological QSAR modelling of the acute toxicity of fused and non-fused polycyclic aromatic hydrocarbons (FNFPAHs) against two aquatic organisms: Consensus modelling and comparison with ECOSAR. Aquat. Toxicol.. 2023;255:106393
- [Google Scholar]
- Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov. Today Technol.. 2004;1:337-341.
- [Google Scholar]
- Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev.. 2001;46:3-26.
- [Google Scholar]
- Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565-574.
- [Google Scholar]
- Manal Mohammed. 2022. COVID-19: What we know about new omicron variant BF.7. https://medicalxpress.com/news/2022-12-covid-omicron-variant-bf7.html.
- Antivirals virtual screening to SARS-CoV-2 non-structural proteins. J. Biomol. Struct. Dyn.. 2022;40:8989-9003.
- [Google Scholar]
- Small molecule kinase inhibitors approved by the FDA from 2000 to 2011: a systematic review of preclinical ADME data. Expert Opin. Drug Metab. Toxicol.. 2013;9:1597-1612.
- [Google Scholar]
- OECD (Organization for Economic Co-Operation and Development). 2007. Guidance Document on the Validation of (Quantitative) Structure-Activity Relationship [(Q)SAR] Models. OECD Series on Testing and Assessment, No. 69. OECD Publishing, Paris https://www.oecd.org/env/guidance-document-on-the-validation-of-quantitative-structure-activity-relationship-q-sar-models-9789264085442-en.htm.
- The enzymatic activity of the nsp14 exoribonuclease is critical for replication of MERS-CoV and SARS-CoV-2. J. Virol.. 2020;94:e01246-e10320.
- [Google Scholar]
- Quantitative structure-activity relationship methods: perspectives on drug discovery and toxicology. Environ. Toxicol. Chem.. 2003;22:1666-1679.
- [Google Scholar]
- Coronaviruses post-SARS: update on replication and pathogenesis. Nat. Rev. Microbiol.. 2009;7:439-450.
- [Google Scholar]
- pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem.. 2015;58:4066-4072.
- [Google Scholar]
- Applications of chem-bioinformatic, chemometric and machine learning approaches for COVID-19 related research. Struct. Chem.. 2022;33:1389-1390.
- [Google Scholar]
- Be aware of error measures. Further studies on validation of predictive QSAR models. Chemom. Intell. Lab. Syst.. 2016;152:18-33.
- [Google Scholar]
- How precise are our quantitative structure-activity relationship derived predictions for new query chemicals? ACS Omega. 2018;3:11392-11406.
- [Google Scholar]
- Omicron variant evolution on vaccines and monoclonal antibodies. Inflammopharmacology 2023:1-10.
- [Google Scholar]
- Targeting SARS-CoV-2 non-structural protein 13 via helicase-inhibitor-repurposing and non-structural protein 16 through pharmacophore-based screening. Mol. Divers. 2022:1-19.
- [Google Scholar]
- Recent trends in QSAR in modelling of drug-protein and protein-protein interactions. Comb. Chem. High Throughput Screen.. 2021;24:1031-1041.
- [Google Scholar]
- Design of SARS-CoV-2 PLpro inhibitors for COVID-19 antiviral therapy leveraging binding cooperativity. J. Med. Chem.. 2022;65:2940-2955.
- [Google Scholar]
- The K correlation index: theory development and its application in chemometrics. Chemom. Intell. Lab. Syst.. 1999;46:13-29.
- [Google Scholar]
- Replacement, reduction, and refinement of animal experiments in anticancer drug development: The contribution of 3D in vitro cancer models in the drug efficacy assessment. Biomedicines. 2023;11:1058.
- [Google Scholar]
- Best practices for QSAR model development, validation, and exploitation. Mol. Inform.. 2010;29:476-488.
- [Google Scholar]
- Alarming antibody evasion properties of rising SARS-CoV-2 BQ and XBB subvariants. Cell. 2023;186:279-286.
- [Google Scholar]
- Visual detection of COVID-19 from materials aspect. Adv. Fiber Mater.. 2022;4:1304-1333.
- [Google Scholar]
- WHO. 2022. WHO Coronavirus (COVID-19) Dashboard. https://covid19.who.int/data (Accessed on 30 December 2022).
- The role of the European Chemicals Bureau in promoting the regulatory use of (Q)SAR methods. SAR QSAR Environ. Res.. 2007;18:111-125.
- [Google Scholar]
- Role of structural and non-structural proteins and therapeutic targets of SARS-CoV-2 for COVID-19. Cells. 2021;10:821.
- [Google Scholar]
- Circular RNAs in immune response and viral infection. Trends Biochem. Sci.. 2020;45:1022-1034.
- [Google Scholar]
- Cryo-EM structure of an extended SARS-CoV2 replication and transcription complex reveals an intermediate state in cap synthesis. Cell. 2021;184:184-193.
- [Google Scholar]
- Coupling of N7-methyltransferase and 3'-5' exoribonuclease with SARS-CoV-2 polymerase reveals mechanisms for capping and proofreading. Cell. 2021;184:3474-3485.
- [Google Scholar]
- Structural biology of SARS-CoV-2 and implications for therapeutic development. Nat. Rev. Microbiol.. 2021;19:685-700.
- [Google Scholar]
- PaDEL-descriptor: an open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem.. 2011;32:1466-1474.
- [Google Scholar]
- SARS-CoV-2 Nsp14 mediates the effects of viral infection on the host cell transcriptome. Elife. 2022;11:e71945.
- [Google Scholar]
- Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science. 2020;368:409-412.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2024.105614.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




