

Translate this page into:
Green synthesis of 4,6-bisarylpyrimidin-2(1H)-ones and azo-linked 4-arylpyrimidin-2(1H)-ones using NiFe2O4@SiO2nPr@glucose amine as a mild nano catalyst
⁎Corresponding author. nikpassand@iaurasht.ac.ir (Mohammad Nikpassand)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
The clean, environmentally benign and effective synthesis of novel azo-linked 4-arylpyrimidin-2(1H)-one derivatives and 4,6-bisarylpyrimidin-2(1H)-ones via three-component reaction of various aldehydes or synthetized azo-linked aldehydes, urea, and acetophenone promoted by NiFe2O4@SiO2nPr@glucose amine at room temperature (25 °C) was reported. NiFe2O4@SiO2nPr@glucose amine were synthesized and characterized by transmission electron microscope (TEM), fourier transform infrared spectroscopy (FT-IR), vibrating sample magnetometry (VSM), X-ray powder diffraction (XRD) and X-ray spectroscopy (EDX). These compounds were obtained in high yields and short reaction times. The catalyst could be easily recovered and reused for six cycles with almost consistent activity. The structures of the synthesized 4,6-bisarylpyrimidin-2(1H)-one compounds were confirmed by 1H NMR, 13C NMR and FTIR spectral data and elemental analyses.
Keywords
Pyrimidin-2(1H)-one
NiFe2O4@SiO2nPr@glucose amine
Urea
Nanoparticle
Green synthesis

1 Introduction
Heterocyclic compounds play an important role in regulating biological activities and many of them are used as medicines. Nitrogen-containing heterocyclic compounds are of great importance due to their wide range of drug activities, and among the heterocyclic ring systems, pyrimidines have a particular position (Kumar et al., 2011). Pyrimidines exist as constituents of many valuable chemotherapeutic agents (bleomycine), vitamins (vitamin B1), anti-bacterial and anti-malarial drugs, and nucleic acids (cytosine and uracil) (Pothiraj et al., 2008). Dihydropyrimidinones are an important class of heterocyclic compounds due to therapeutic activities such as anti-bacterial, anti-hypertensive, anti-cancer, anti-virus, anti-inflammatory, AIDS inhibitor, calcium channel blocker and anti-calcium channel blocker (Kappe and Fabian, 1997; Overman et al., 1995; Chaudhari and Shah, 2011; Deres et al., 2003; Couto et al., 2011; Duguay et al., 2008).
Different methods have been reported for the synthesis of diphenyl pyrimidines. Three component reaction among benzaldehyde, ethyl acetoacetate and urea in ethanol solvent using a recoverable catalyst SBA-15@Fe3O4 is one of these (Mondal et al., 2012). Synthesis of 3,4-dihydro-pyrimidine-2-(1H)-ions (thions) in solvent-free conditions and in the presence of Fe3O4 (Nasr-Esfahani et al., 2011), pyrido (Pothiraj et al., 2008; Kappe and Fabian, 1997; Overman et al., 1995; Chaudhari and Shah, 2011; Deres et al., 2003; Couto et al., 2011; Duguay et al., 2008; Mondal et al., 2012; Nasr-Esfahani et al., 2011; Rad and Mokhtary, 2015; Ebraheem, 2012; Heravi, 2009; Zhu and Bienayme, 2005; Rakhi et al., 2016; Wang, 2004; Abaszadeh and Seifi, 2017; Bigi et al., 1999; Rani et al., 2001; Yadav et al., 2000; Yadav et al., 2004; Salehi et al., 2003; Martinez et al., 2003; Li et al., 2010; Aghazadeh and Nikpassand, 2019; Zare Fekri et al., 2019; Nikpassand, 2020; Nikpassand and Farokhian, 2016; Nikpassand and Pirdelzendeh, 2016; Nikpassand et al., 2015; Nikpassand et al., 2017; Nikpassand et al., 2018; Heravi et al., 2008; Pourghobadi and Derikvand, 2010) pyrimidines nanoparticles, by direct mononuclear reaction 6-Euroacyl and its derivatives, with aryl aldehydes and malononitrile together with MgO nanoparticles (Rad and Mokhtary, 2015), Preparation of pyrimidine-2-one and pyrimidine-2-thionyl compounds from the combination of chalcone with benzaldehydes with acetophenone in sodium hydroxide and 50% ethanol (Ebraheem, 2012) and direct, one-pot, three component, oxidation reaction among 1,3-diketone, benzaldehyde and ammonium acetate in the presence of some heteropolytic acid as a catalyst (Heravi, 2009) is another example of published scientific reporting methods for the synthesis of various diphenyl pyrimidines.
Various acid catalysts have been proposed for the synthesis of these organic compounds. In the past, traditionally, strong Bronsted acids such as hydrochloric acid and sulfuric acid were utilized. But today, the use of Lewis acids such as BF3OEt2, CuCl, LaCl3, FeCl3, NiCl2, Yb(OTf)3, La(OTf)3, LnCl3, LnBr3, Ln(OTf)3, LiBr, CoCl2, BiCl3, LiClO4, Mn(OAc)3, ZrCl4, Cu(OTf)2, CuCl2, Bi(OTf)3, CeCl3, VCl3, Zn(OTf)2, Sm(NO3)3 and SmCl3 (Zhu and Bienayme, 2005); β-cyclodextrin (Rakhi et al., 2016) KF-Al2O3 (Wang, 2004; Abaszadeh and Seifi, 2017) is common. Solid acid catalysts such as clay, zeolite, ion exchange agents such as amberlite or a heteropolytic acid such as Ag3PW12O40 have also been used today as catalysts (Bigi et al., 1999; Rani et al., 2001; Yadav et al., 2000; Yadav et al., 2004). In addition, materials such as silica sulfuric acid or nanoparticles or silica-iron oxide nanocomposites have also been reported as catalysts (Salehi et al., 2003; Martinez et al., 2003; Li et al., 2010).
Thus, it is essential to design an efficient, green and novel method for the preparation of 4,6-bisarylpyrimidin-2(1H)-ones without those disadvantages such as toxic solvents and catalysts, long reaction times, low yields and use of costly catalysts, we report herein synthesis of 4,6-bisarylpyrimidin-2(1H)-ones with various synthetized azo-linked aldehydes or benzaldehydes, urea and acetophenone in the presence of NiFe2O4@SiO2nPr@glucose amine nanoparticles as an effective and reusable catalyst.
2 Material and methods
2.1 Apparatus and analysis
Chemicals were purchased from Merck and Fluka and used as purchased. Melting points were measured on an Electro-thermal 9100 apparatus and are uncorrected. 1H NMR spectra were obtained on a Bruker DRX 500Avance spectrometer in DMSO‑d6 as solvent and with TMS as internal standard. FT-IR spectra were recorded on a Shimadzu FT-IR-8400S spectrometer. Elemental analyses were recorded on a Carlo-Erba EA1110CNNO-S analyzer. For the ultrasound reactions, ultrasound apparatus astra 3D (9.5 dm3, 45 KHz frequency, input power with heating, 305 W, number of transducers, 2) from TECNO-GAZ was used.
2.2 Procedure for the synthesis of NiFe2O4@SiO2nPr@glucose amine nanoparticles
2.2.1 Synthesis of NiFe2O4 & NiFe2O4@SiO2nPrCl MNPs
The synthetized NiFe2O4 and NiFe2O4@SiO2nPrCl MNPs were synthesized by Zare Fekri, et al. (Aghazadeh and Nikpassand, 2019; Zare Fekri et al., 2019).
2.2.2 Synthesis of NiFe2O4@SiO2nPr@glucose amine using of ultrasound irradiation
0.5 g NiFe2O4@SiO2nPrCl were dispersed into 50 mL xylene and sonicated for 30 min, followed by the drop wise addition of 0.5 mL glucose amine. Then, the mixture was irradiated in a water bath by ultrasound at room temperature (25 °C) for 20 min under a nitrogen flow. The resulting functionalized NiFe2O4@SiO2nPr@glucose amine was collected by magnetic separation followed by washing with toluene and ethanol several times and drying at 60 °C for 10 h.
2.3 General procedure for preparation of 4,6-Bisarylpyrimidin-2(1H)-ones 4a-n and novel azo-linked 4,6-Bisarylpyrimidin-2(1H)-ones 4o-s using NiFe2O4@SiO2nPr@glucose amine nanoparticles
A mixture of synthetized azo-linked aldehydes or benzaldehydes (1 mmol), acetophenone (1 mmol), urea (1 mmol), 0.05 g NiFe2O4@SiO2nPr@glucose amine and H2O (10 mL) were placed into Pyrex-glass open vessel and stirred at room temperature (25 °C) for the required reaction times. After completion of the reaction, as indicated by TLC (TLC Silica gel 60F₂₅₄, ethyl acetate: n-hexane 1: 2), the resulting mixture was dissolved in hot ethanol (20 mL) and the catalyst separated by a 1.4 Tesla external magnet and washed with hot distilled water (5 mL) and ethanol (5 mL) two times. The resulting 4,6-bisarylpyrimidin-2(1H)-ones was isolated and purified using a thin layer chromatography. The structures of most of the synthesized 4,6-bisarylpyrimidin-2(1H)-one compounds were characterized (1H, 13C NMR and FTIR) and elemental analyses.
4-(2-bromophenyl)-6-phenylpyrimidin-2(1H)-one (4a): White solid, Yield: 94%, mp 215–217 °C; IR (KBr, cm−1) νmax: 3430 (N-H stretching), 2971 (C-H stretching), 1668 (C = O stretching), 1611 and 1522 (C = C stretching), 1371 (C-N stretching), 1023 (C-Br stretching) cm−1. 1H NMR (250 MHz, DMSO‑d6): δH; 6.25 (s, 1H), 6.69 (d, J = 6.3 Hz, 1H), 7.20–7.95 (m, 8H) ppm. 13C NMR (62 MHz, DMSO‑d6): δC; 123.8, 128.9, 129.3, 129.5, 130.1, 130.7, 134.1, 134.6, 137.4, 142.5, 156.5, 158.8, 161.1, 193.2 (C = O) ppm. Anal Calc. for C16H11BrN2O: C, 58.74; H, 3.39; N, 8.56. Found: C, 58.73; H, 3.40; N, 8.58.
4-(3-bromophenyl)-6-phenylpyrimidin-2(1H)-one (4b): White solid, Yield: 94%, mp 246–248 °C; IR (KBr, cm−1) νmax 3408 (N-H stretching), 2983 (C-H stretching), 1672 (C = O stretching), 1632 and 1553 (C = C stretching), 1390 (C-N stretching), 1009 (C-Br stretching) cm−1. 1H NMR (250 MHz, DMSO‑d6): δH; 5.79 (s, 1H), 6.88 (d, J = 7.5 Hz, 1H), 7.29–7.59 (m, 4H), 7.76–7.91 (m, 4H), 8.00 (s, 1H) ppm. 13C NMR (62 MHz, DMSO‑d6): δC; 122.1, 125.6, 128.5, 128.6, 129.0, 129.5, 131.2, 132.1, 133.6, 135.9, 158.2, 160.5, 166.7, 195.7 (C = O) ppm. Anal Calc. for C16H11BrN2O: C, 58.74; H, 3.39; N, 8.56. Found: C, 58.75; H, 3.37; N, 8.55.
4-(4-bromophenyl)-6-phenylpyrimidin-2(1H)-one (4c): White solid, Yield: 93%, mp 238–240 °C; IR (KBr, cm−1) νmax: 3450 (N-H stretching), 2923 (C-H stretching), 1671 (C = O stretching), 1597, 1542 and 1468 (C = C stretching), 1367 (C-N stretching), 1048 (C-Br stretching) cm−1. 1H NMR (250 MHz, DMSO‑d6): δH; 5.76 (s, 1H), 6.05 (s, 1H), 6.81–7.90 (m, 8H), 13.0 (s, br, 1H, NH) ppm. 13C NMR (62 MHz, DMSO‑d6): δC; 127.2, 128.6, 130.4, 131.3, 131.6, 132.0, 133.5, 142.5, 158.2, 160.4, 167.2, 197.6 (C = O) ppm. Anal Calc. for C16H11BrN2O: C, 58.74; H, 3.39; N, 8.56. Found: C, 58.76; H, 3.40; N, 8.58.
4-(4-chlorophenyl)-6-phenylpyrimidin-2(1H)-one (4d): White solid, Yield: 92%, mp 268–270 °C; IR (KBr, cm−1) νmax: 3446 (N-H stretching), 2962 (C-H stretching), 1680 (C = O stretching), 1594 and 1466 (C = C stretching), 1372 (C-N stretching), 1090 (C-Cl stretching) cm−1. 1H NMR (250 MHz, DMSO‑d6): δH; 5.70 (s, 1H), 7.32–7.66 (m, 6H), 7.90–7.93 (d, J = 8.7 Hz, 3H) ppm. 13C NMR (62 MHz, DMSO‑d6): δC; 128.3, 129.1, 130.1, 131.5, 132.0, 138.2, 142.0, 156.3, 158.1, 160.1, 167.0, 197.3 (C = O) ppm. Anal Calc. for C16H11ClN2O: C, 67.97; H, 3.92; N, 9.91. Found: C, 67.95; H, 3.93; N, 9.89.
4-(2,4-dichlorophenyl) −6-phenylpyrimidin-2(1H)-one (4e): White solid, Yield: 94%, mp 234–236 °C; IR (KBr, cm−1) νmax: 3451 (N-H stretching), 2976, 2925 (C-H stretching), 1671 (C = O stretching), 1592, 1545, 1470 (C = C stretching), 1369 (C-N stretching), 1049 (C-Cl stretching) cm−1. 1H NMR (250 MHz, DMSO‑d6): 6.27 (s, 1H), 6.78–6.87 (m, 3H), 7.43–7.52 (m, 4H), 7.92 (d, J = 6.5 Hz, 1H), 10.4 (s, br, 1H, NH) ppm. 13C NMR (62 MHz, DMSO‑d6): δC; 127.4, 128.5, 128.7, 129.0, 129.1, 129.6, 133.2, 133.6, 138.9, 139.2, 155.6, 157.7, 181.3, 198.9 (C = O) ppm. Anal Calc. for C16H10Cl2N2O: C, 60.59; H, 3.18; N, 8.83. Found: C, 60.60; H, 3.16; N, 8.81.
4-(2,6-dichlorophenyl)-6-phenylpyrimidin-2(1H)-one (4f): White solid, Yield: 93%, mp 250–252 °C; IR (KBr, cm−1) νmax: 3366 (N-H stretching), 1664 (C = O stretching), 1624, 1600, 1482 (C = C stretching), 1377 (C-N stretching), 1134 (C-Cl stretching) cm−1. 1H NMR (250 MHz, DMSO‑d6): δH; 6.67–6.70 (m, 1H), 6.99–7.60 (m, 7H), 7.93 (d, J = 7.0 Hz, 1H), 10.4 (s, br, 1H, NH) ppm. 13C NMR (62 MHz, DMSO‑d6): δc; 128.4, 128.5, 128.8, 129.1, 129.6, 130.2, 130.4, 131.1, 133.6, 133.9, 166.1, 190.0 (C = O) ppm. Anal Calc. for C16H10Cl2N2O: C, 60.59; H, 3.18; N, 8.83. Found: C, 60.57; H, 3.20; N, 8.84.
6-phenyl-4-(m-tolyl)pyrimidin-2(1H)-one (4 g): White solid, Yield: 87%, mp > 300 °C; IR (KBr, cm−1) νmax: 3445 (N-H stretching), 2922 (C-H stretching), 1651 (C = O stretching), 1605, 1552 (C = C stretching) cm−1. 1H NMR (250 MHz, DMSO‑d6): δH; 2.27 (s, 3H), 6.07 (t, J = 7.8 Hz, 1H), 6.65 (d, J = 8.0 Hz, 2H), 7.03–7.95 (m, 7H) ppm. 13C NMR (62 MHz, DMSO‑d6): δc; 20.4, 124.4, 127.9, 129.1, 129.4, 129.8, 130.0, 131.1, 134.8, 138.5, 143.8, 159.2, 161.3, 169.1, 194.6 (C = O) ppm. Anal Calc. for C17H14N2O: C, 77.84; H, 5.38; N, 10.68. Found: C, 77.85; H, 5.36; N, 10.67.
6-phenyl-4-(p-tolyl)pyrimidin-2(1H)-one (4 h): White solid, Yield: 91%, mp 284–286 °C; IR (KBr, cm−1) νmax: 3442 (N-H stretching), 2928 (C-H stretching), 1673 (C = O stretching), 1598, 1462 (C = C stretching) cm−1. 1H NMR (250 MHz, DMSO‑d6): δH; 2.32 (s, 3H), 5.68 (s, 1H), 6.07 (s, 1H), 6.69 (s, 1H), 7.11–7.35 (m, 6H), 7.78 (s, 1H) ppm. 13C NMR (62 MHz, DMSO‑d6): δC; 22.5, 127.3, 129.6, 130.0, 130.5, 130.7, 131.0, 137.6, 140.9, 159.2, 161.3, 169.0, 194.0 (C = O) ppm. Anal Calc. for C17H14N2O: C, 77.84; H, 5.38; N, 10.68. Found: C, 77.85; H, 5.37; N, 10.67.
4-(4-nitrophenyl)-6-phenylpyrimidin-2(1H)-one (4i): Yellow solid, Yield: 96%, mp 343–345 °C; IR (KBr, cm−1) νmax: 3437 (N-H stretching), 2982 (C-H stretching), 1675 (C = O stretching), 1597 and 1542 (C = C stretching), 1538 (NO2 stretching), 1367 (C-N stretching), 1349 (NO2 stretching) cm−1. 1H NMR (250 MHz, DMSO‑d6): δH; 5.73 (s, 1H), 6.11 (s, 1H), 6.68–7.97 (m, 8H), 11.63 (s, br, 1H, NH) ppm. 13C NMR (62 MHz, DMSO‑d6): δC; 124.6, 127.5, 129.4, 131.4, 131.5, 133.3, 133.9, 142.8, 153.5, 158.5, 163.5, 196.1 (C = O) ppm. Anal Calc. for C16H11N3O3: C, 65.53; H, 3.78; N, 14.33. Found: C, 65.55; H, 3.76; N, 14.35.
4-(4-isopropylphenyl)-6-phenylpyrimidin-2(1H)-one (4j): White solid, Yield: 90%, mp 289–291 °C; IR (KBr, cm−1) νmax: 3453 (N-H stretching), 3091 and 2938 (C-H stretching), 1634 (C = O stretching), 1541 and 1563 (C = C stretching) cm−1. 1H NMR (250 MHz, DMSO‑d6): δH; 1.25 (d, J = 6.8 Hz, 6H), 7.51–7.56 (m, 4H), 8.11 (d, J = 7.8 Hz, 2H), 8.17 (d, J = 6.8 Hz, 2H) ppm. 13C NMR (62 MHz, DMSO‑d6): δc; 19.3, 28.5, 122.4, 124.5, 130.4, 131.5, 131.7, 133.5, 139.4, 142.4, 155.7, 158.6, 165.8, 194.3 (C = O) ppm. Anal Calc. for C19H18N2O: C, 78.59; H, 6.25; N, 9.65. Found: C, 78.58; H, 6.27; N, 9.66.
4-(2-hydroxyphenyl)-6-phenylpyrimidin-2(1H)-one (4 k): White solid, Yield: 89%, mp 295–297 °C; IR (KBr, cm−1) νmax: 3324 and 3216 (N-H stretching), 3075 and 2920 (C-H stretching), 1689 (C = O stretching), 1498 and 1450 (C = C stretching) cm−1. 1H NMR (250 MHz, DMSO‑d6): δH; 2.25 (d, J = 8.7 Hz, 1H), 2.31 (dd, J = 8.7, 2.8 Hz, 1H), 4.05 (d, J = 3.8 Hz, 1H), 6.94–6.99 (m, 2H), 7.26–7.29 (m, 2H), 7.35–7.60 (m, 4H), 7.65–7.72 (m, 2H), 7.57 (s, 1H), 10.58 (s, 1H, OH) ppm.13C NMR (62 MHz, DMSO‑d6): δc; 123.2, 124.4, 129.2, 129.9, 131.7, 131.9, 133.5, 133.8, 141.5, 153.0, 156.6, 157.4, 163.6, 190.2 (C = O) ppm. Anal Calc. for C16H12N2O2: C, 72.72; H, 4.58; N, 10.60. Found: C, 72.74; H, 4.56; N, 10.62.
4-(4-hydroxyphenyl)-6-phenylpyrimidin-2(1H)-one (4 l): White solid, Yield: 90%, mp 259–261 °C; IR (KBr, cm−1) νmax: 3383 (N-H stretching), 3253 (O-H stretching), 2922 (C-H stretching), 1631 (C = O stretching), 1515 and 1450 (C = C stretching) cm−1. 1H NMR (250 MHz, DMSO‑d6): δH; 6.92–7.00 (m, 2H), 7.53–7.68 (m, 3H), 7.69–7.78 (m, 3H), 8.07–8.18 (m, 2H), 10.18 (s, 1H, OH) ppm. 13C NMR (62 MHz, DMSO‑d6): δc; 122.7, 124.7, 129.6, 131.4, 133.0, 133.1, 142.4, 154.6, 158.4, 159.2, 165.0, 193.6 (C = O) ppm. Anal Calc. for C16H12N2O2: C, 72.72; H, 4.58; N, 10.60. Found: C, 72.70; H, 4.59; N, 10.58.
4-(2-hydroxy-5-((3-nitrophenyl)diazenyl)phenyl)-6-phenylpyrimidin-2(1H)-one (4o): Yellow solid, Yield: 93%, mp > 300 °C; IR (KBr, cm−1) νmax: 3393 (N-H stretching), 3251 (O-H stretching), 2959 (C-H stretching), 1668 (C = O stretching), 1542 (NO2 stretching), 1551, 1468 (C = C stretching), 1540 (NO2 stretching) cm−1. 1H NMR (250 MHz, DMSO‑d6): δH; 6.58–6.64 (m, 3H), 7.49–7.72 (m, 5H), 7.78–8.19 (m, 5H), 10.09 (s, 1H, OH), 12.40 (s, 1H, NH) ppm. 13C NMR (62 MHz, DMSO‑d6): δC; 112.8, 118.9, 119.6, 122.7, 124.8, 126.0, 126.6, 127.0, 128.4, 129.5, 130.8, 131.6, 133.4, 133.5, 146.7, 147.9, 153.3, 157.2, 160.6, 190.6 (C = O) ppm. Anal Calc. for C22H15N5O4: C, 63.92; H, 3.66; N, 16.94. Found: C, 63.94; H, 3.64; N, 16.93.
4-(2-hydroxy-5-((4-nitrophenyl)diazenyl)phenyl)-6-phenylpyrimidin-2(1H)-one (4p): Yellow solid, Yield: 94%, mp > 300 °C; IR (KBr, cm−1) νmax: 3452 (N-H stretching), 3237 (O-H stretching), 2928 (C-H stretching), 1670 (C = O stretching), 1540 (NO2 stretching), 1517 and 1461 (C = C stretching), 1351 (NO2 stretching) cm−1. 1H NMR (250 MHz, DMSO‑d6): δH; 6.56–6.68 (m, 2H), 7.66–8.26 (m, 11H), 10.31 (s, 1H, OH), 11.82 (s, 1H, NH) ppm. 13C NMR (62 MHz, DMSO‑d6): δC; 113.8, 118.2, 123.8, 124.9, 126.4, 126.8, 127.0, 127.8, 129.4, 130.1, 133.0, 133.6, 145.7, 147.4, 151.8, 161.0, 161.4, 192.2 (C = O) ppm. Anal Calc. for C22H15N5O4: C, 63.92; H, 3.66; N, 16.94. Found: C, 63.94; H, 3.64; N, 16.93.
4-(5-((4-bromophenyl)diazenyl)-2-hydroxyphenyl)-6-phenylpyrimidin-2(1H)-one (4q): White solid, Yield: 95%, mp > 300 °C; IR (KBr, cm−1) νmax: 3365 (N-H stretching), 3262 (O-H stretching), 2983 (C-H stretching), 1681 (C = O stretching), 1548 and 1473 (C = C stretching) cm−1. 1H NMR (250 MHz, DMSO‑d6): δH; 6.59–6.72 (m, 4H), 7.52–8.09 (m, 7H), 10.09 (s, 1H, OH), 11.92 (s, 1H, NH) ppm. 13C NMR (62 MHz, DMSO‑d6): δC; 115.5, 119.5, 123.6, 124.7, 126.6, 127.5, 127.7, 129.7, 129.9, 130.7, 133.4, 134.8, 143.67, 146.0, 152.5, 161.6, 162.5, 189.3 (C = O) ppm. Anal Calc. for C22H15BrN4O2: C, 59.08; H, 3.38; N, 12.53. Found: C, 59.06; H, 3.40; N, 12.55.
4-(5-((3-chlorophenyl)diazenyl)-2-hydroxyphenyl)-6-phenylpyrimidin-2(1H)-one (4r): White solid, Yield: 96%, mp > 300 °C; IR (KBr, cm−1) νmax: 3338 (N-H stretching), 3268 (O-H stretching), 2993 (C-H stretching), 1675 (C = O stretching), 1562 and 1474 (C = C stretching) cm−1. 1H NMR (250 MHz, DMSO‑d6): δH; 6.70–6.79 (m, 3H), 7.37–7.77 (m, 6H), 7.84–8.21 (m, 4H), 10.24 (s, 1H, OH), 12.09 (s, 1H, NH) ppm. 13C NMR (62 MHz, DMSO‑d6): δC; 113.5, 118.8, 119.4, 123.1, 124.4, 124.7, 126.4, 128.4, 128.5, 129.4, 130.7, 131.4, 131.8, 133.8, 147.4, 149.3, 151.9, 155.7, 158.6, 185.5 (C = O) ppm. Anal Calc. for C22H15ClN4O2: C, 65.59; H, 3.75; N, 13.91. Found: C, 65.61; H, 3.76; N, 13.89.
4-(5-((4-chlorophenyl)diazenyl)-2-hydroxyphenyl)-6-phenylpyrimidin-2(1H)-one (4 s): White solid, Yield: 95%, mp > 300 °C; IR (KBr, cm−1) νmax: 3379 (N-H stretching), 3205 (O-H stretching), 2984 (C-H stretching), 1674 (C = O stretching), 1583 and 1448 (C = C stretching) cm−1. 1H NMR (250 MHz, DMSO‑d6): δH; 6.51–6.70 (m, 4H), 7.73–8.05 (m, 9H), 9.97 (s, 1H, OH), 12.47 (s, 1H, NH) ppm. 13C NMR (62 MHz, DMSO‑d6): δC; 113.5, 119.0, 121.7, 123.5, 125.6, 126.0, 127.4, 129.4, 129.7, 130.3, 131.4, 133.5, 142.5, 145.7, 151.5, 160.4, 161.4, 190.5 (C = O) ppm. Anal Calc. for C22H15ClN4O2: C, 65.59; H, 3.75; N, 13.91. Found: C, 65.58; H, 3.76; N, 13.89.
3 Results and discussion
The morphology and size of the NiFe2O4@SiO2nPr@glucose amine MNPs was investigated by TEM spectrum as shown in Figs. 1 and 2. In order to investigate the reusing performance of NiFe2O4@SiO2nPr@glucose amine, the recyclability of catalyst was tested in 4a.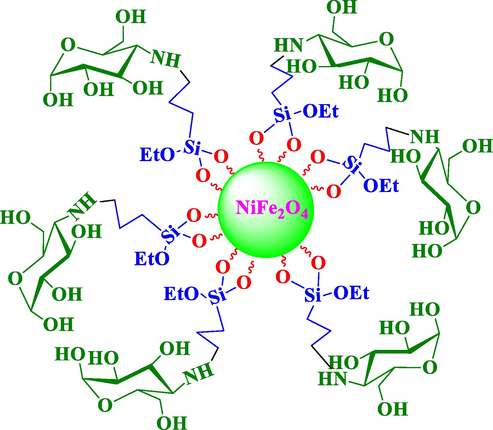
The structure of amino glucose‐functionalized silica‐coated NiFe2O4 nanoparticles.

The TEM image of the synthesized NiFe2O4@SiO2nPr@amino glucose.
The FT-IR spectra of NiFe2O4, NiFe2O4@SiO2 and NiFe2O4@SiO2nPr@amino glucose MNPs are shown in Fig. 3. The absorption band in 594 cm−1 is related to the stretching vibration of the Fe–O bond of bare NiFe2O4 that was appeared in 590 cm−1 in the FT-IR spectrum of NiFe2O4@SiO2nPr@amino glucose MNPs. The absorption bands of Si-O-Si vibrations in SiO2 shell were appeared in 1045 and 1130 cm−1 in NiFe2O4@SiO2 spectra that seemed at 1033, 1137 and 767 cm−1 in the spectrum of the NiFe2O4@SiO2nPr@amino glucose MNPs. Also, the peaks at 1645 and 3429 cm−1 (in the spectrum of the NiFe2O4) and at 1645 and 3423 cm−1 (in the spectrum of the NiFe2O4@SiO2nPr@amino glucose MNPs) are attributed to the stretching vibrations of the hydroxyl (–OH) groups on the surface of MNPs. The peaks in region 2950 and 3120 cm−1 refer to the stretching band of C-H aliphatic and NH stretching in NiFe2O4@SiO2nPr@amino glucose MNPs (Fig. 3).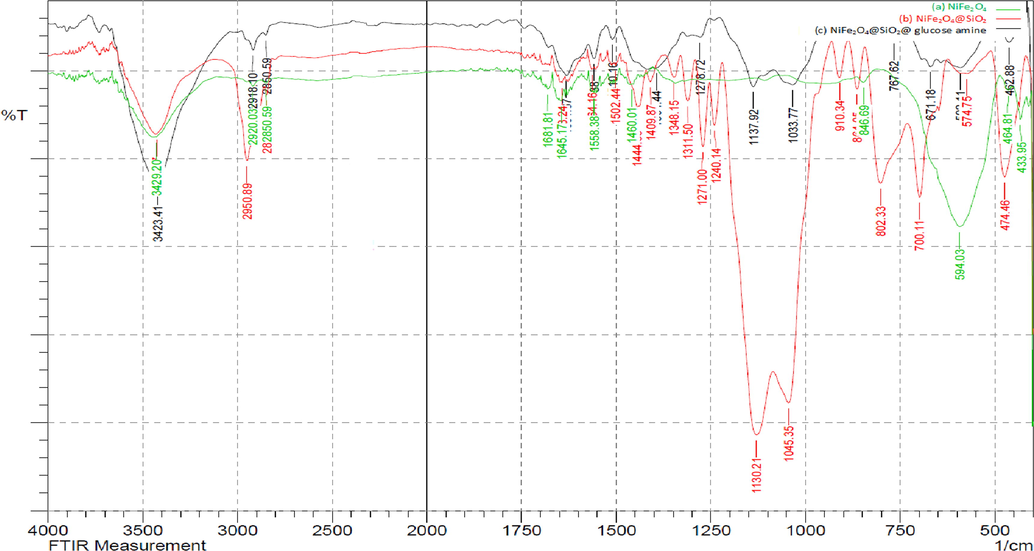
FT‐IR spectra of (a) NiFe2O4, (b) NiFe2O4@SiO2, (c) NiFe2O4@SiO2nPr@amino glucose MNPs.
The structure of NiFe2O4@SiO2nPr@amino glucose was also confirmed by XRD analysis. In Fig. 4, the XRD patterns of NiFe2O4@SiO2nPr@amino glucose MNPs and pure NiFe2O4 (from JCPDS No. 54–0964) are illustrated. The comparison of the XRD patterns indicated that both patterns exhibits peaks with 2θ at 30°,36°, 45°, 50°, 54°, 58° and 62° which are representative of the structure and broad peak in 10-30° is related to NiFe2O4 covered by SiO2 (Fig. 4).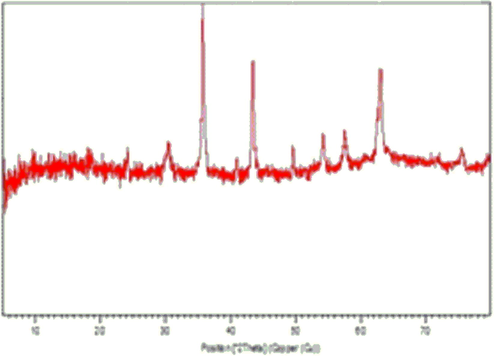
The XRD patterns of a) NiFe2O4@SiO2nPr@amino glucose and b) NiFe2O4.
The results of energy dispersive X-ray spectroscopy (EDX) analysis of the synthesized NiFe2O4@SiO2nPr@amino glucose MNPs (Fig. 5) proved existence of Fe (30.17 w/w%), O (16.21 w/w%), Si (2.2 w/w%), N (0.45 w/w%), C (5.30 w/w%) and Ni (31.12 w/w %) atoms in the structure that confirms the presence of NiFe2O4 core in the structure of MNP.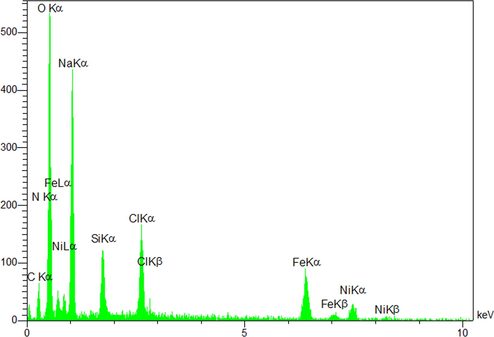
EDX results of NiFe2O4@SiO2nPr@amino glucose nanocatalyst.
Fig. 6 shows the hysteresis loop of the synthesized NiFe2O4@SiO2nPr@amino glucose MNPs. VSM measurements were carried out at room temperature (25 °C) by taking the solid sample on the tips of the vibrating rod and analyzing in an applied magnetic field sweeping from − 20 to 20 kOe (Fig. 6).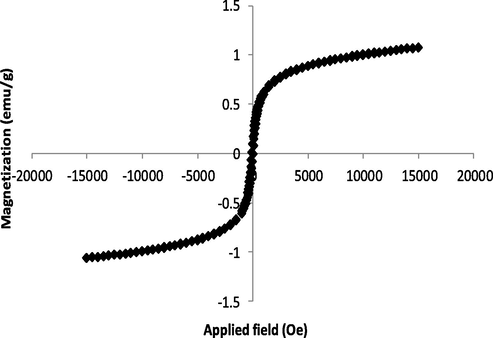
Vibrating scanning magnetometery (VSM) curve of NiFe2O4@SiO2nPr@amino glucose nanocatalyst.
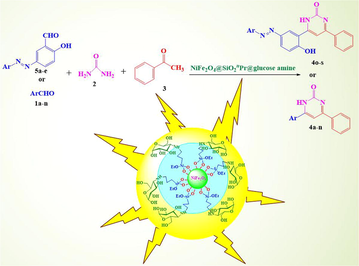
In order to achieve a more efficient synthetic process, improving the yields and reaction times minimize by-products, decrease the number of separate reaction steps and also in extending our research on the synthesis and study of heterocyclic and pharmaceutical compounds (Nikpassand, 2020; Nikpassand and Farokhian, 2016; Nikpassand and Pirdelzendeh, 2016; Nikpassand et al., 2015; Nikpassand et al., 2017; Nikpassand et al., 2018; Heravi et al., 2008; Pourghobadi and Derikvand, 2010), in this work, the first synthesis of some derivatives of 4,6-bisarylpyrimidin-2(1H)-ones from various aldehydes, urea, acetophenone and NiFe2O4@SiO2nPr@glucose amine as a catalyst is reported (Scheme 1).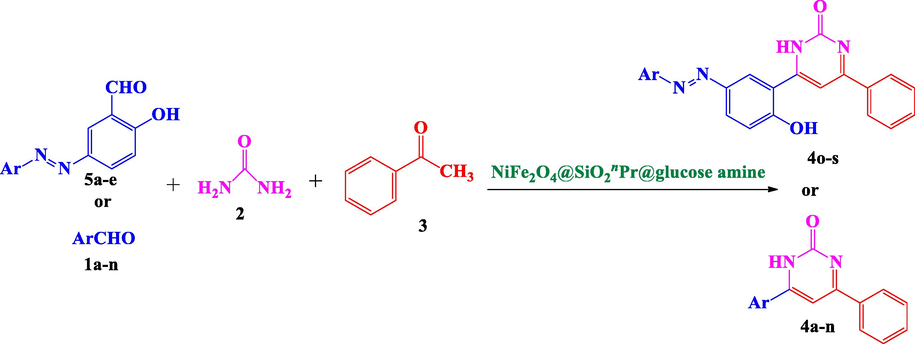
Preparation of 4,6-bisarylpyrimidin-2(1H)-ones using NiFe2O4@SiO2nPr@glucose amine.
The structure of NiFe2O4@SiO2nPr@glucose amine magnetic nanoparticle that was synthesized in three steps from commercially available materials. The Fe3O4@SiO2 core‐shell structures were then sequentially treated with 3‐chloropropyltrimethoxysilane. Next, it was treated with Tannic acid to obtain the Glucose amine‐functionalized silica‐coated NiFe2O4nanoparticles (NiFe2O4@SiO2nPr@glucose amine).
3.1 Catalytic studies
In order to evaluate the catalytic capability of the synthesized heterogeneous catalyst (NiFe2O4@SiO2nPr@glucose amine) in organic reactions, we chose to examine its activity in an one‐pot reaction for the reaction between various aldehydes, acetophenone and urea (Scheme 1).
3.2 Effect of solvent type
In order to optimize the model process and the efficiency of solvents in this reaction, for the sake of comparisons some commonly available solvents such as Et2O, H2O, EtOH, hexane, CHCl3 and DMF were studied. In these experiments, 4-bromobenzaldehyde 1a (1.0 mmol), acetophenone (1 mmol) and urea (1.0 mmol) were mixed with 10 mL solvent (Table 1). Among the tested solvents, we found that H2O was the most appropriate solvent for these reactions (77% yields in 8 h).
Entry
Solvent
Time (h)
Yield (%)
1
Et2O
24
53
2
H2O
8
77
3
EtOH
8
68
4
Hexane
24
54
5
CHCl3
24
59
6
DMF
36
72
3.3 Effect of catalyst type and temperature on reaction time
To find the appropriate catalyst, in order to synthesize the derivatives of 4,6-bisarylpyrimidin-2(1H)-one 4a from the reaction of 2-bromobenzaldehyde 1a (1 mmol), acetophenone 2 (1 mmol), urea 3 (1 mmol) in H2O (10 mL) using of 0.1 g of available catalysts at room temperature (25 °C) were used and the efficiency and reaction rate were compared. In comparison to other catalysts studied, NiFe2O4@SiO2nPr@glucose amine catalyst at room temperature (25 °C) showed the best results and efficiency and time for product 4a preparation (entry 10, Table 2). *Reaction conditions: 2-bromobenzaldehyde 1a (1 mmol), acetophenone 2 (1 mmol), urea 3 (1 mmol) were used in H2O (10 mL) as solvent.# 2 mL of ionic liquid were used for ionic liquids in entries 13 and 14.
Entry
catalyst
Time (min)
Yield (%)a,b
1
–
480
77
2
K10
360
81
3
KSF
360
78
4
Fe+3-montmorillonite K10
360
82
5
L-Proline
720
59
6
nano-Fe3O4
240
72
7
nano-SiO2
360
70
8
NiFe2O4@SiO2
240
74
9
Glucose amine
180
80
10
NiFe2O4@SiO2nPr@glucose amine
120
94
11
NiFe2O4@SiO2nPr@glucose amine (60 °C)
120
94
12
NiFe2O4@SiO2nPr@glucose amine (100 °C)
120
94
13
[BBIM]Br
1200
76
14
[BBIM]HSO4
900
83
3.4 Effect of NiFe2O4@SiO2nPr@glucose amine catalyst value
Synthesis of product 4a with different amounts of NiFe2O4@SiO2nPr@glucose amine at room temperature (25 °C) was investigated and it was found that using of 0.05 g of the desired catalyst per 1 mmol of primary reaction aldehyde with better yield and at shorter time (Table 3).
Entry
Product
Structure
Time (min)
Yield (%)a,b
Observed Melting Point (°C)
Reported Melting Point (°C)
1
4a
120
94
215–217
–
2
4b

90
94
246–248
–
3
4c
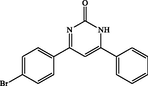
60
93
238–240
251–254 (Heravi et al., 2008)
4
4d
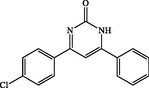
60
92
268–270
258–260 (Pourghobadi and Derikvand, 2010)
5
4e
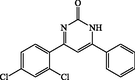
60
94
234–236
–
6
4f
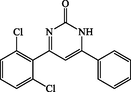
90
93
250–252
–
7
4g
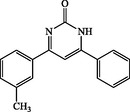
120
87
>300
–
8
4h
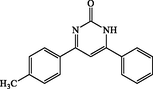
120
91
284–286
287–290 (Heravi et al., 2008)
9
4i
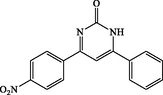
90
96
243–245
–
10
4j
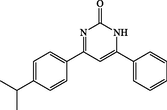
180
90
289–291
287–288 (Heravi et al., 2008)
11
4k
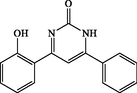
180
89
295–297
–
12
4l
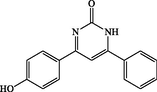
180
90
259–261
–
13
4m
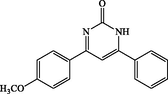
120
92
253–255
258–260 (Heravi et al., 2008)
14
4n
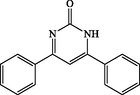
120
90
233–245
238–242 (Pourghobadi and Derikvand, 2010)
15
4o
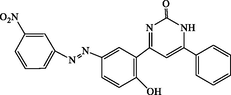
120
93
>300
–
16
4p
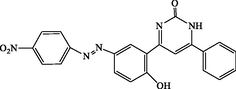
90
94
>300
–
17
4q
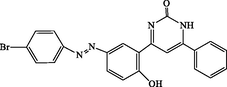
90
95
>300
–
18
4r
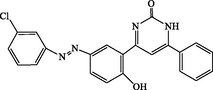
90
96
>300
–
19
4s
120
95
>300
–
According to the results of the steps to evaluate the optimal conditions for the synthesis of 4,6-bisarylpyrimidin-2(1H)-ones from various benzaldehydes, acetophenone and urea, It was found that the best reaction conditions to use NiFe2O4@SiO2nPr@glucose amine in aqueous medium at room temperature (25 °C) is the best conditions, so the same method was used to synthesize other derivatives of bisarylpyrimidin-2(1H)-one compounds (Scheme 1 and Table 3).
The process of performing this reaction for the synthesis of 4,6-bisarylpyrimidin-2(1H)-ones using a NiFe2O4@SiO2nPr@glucose amine nanocatalyst is presented in Scheme 2. Initially, NiFe2O4@SiO2nPr@glucose amine nanocatalyst seems to activate the enolization of acetophenone by forming a hydrogen bond with the carbonyl functional group. Next, the aldol condensation reaction of activated enolate 5 with aldehyde 1 leads to production chalcone 7 during dehydration. Subsequently, the urea is reacted with intermediate 7 and the corresponding imine 8 is produced. Then, a heterocyclic ring 9 is formed during the Michael addition within the molecular ring, and finally by oxidizing the compound 9, the final product 4 is formed (Scheme 2).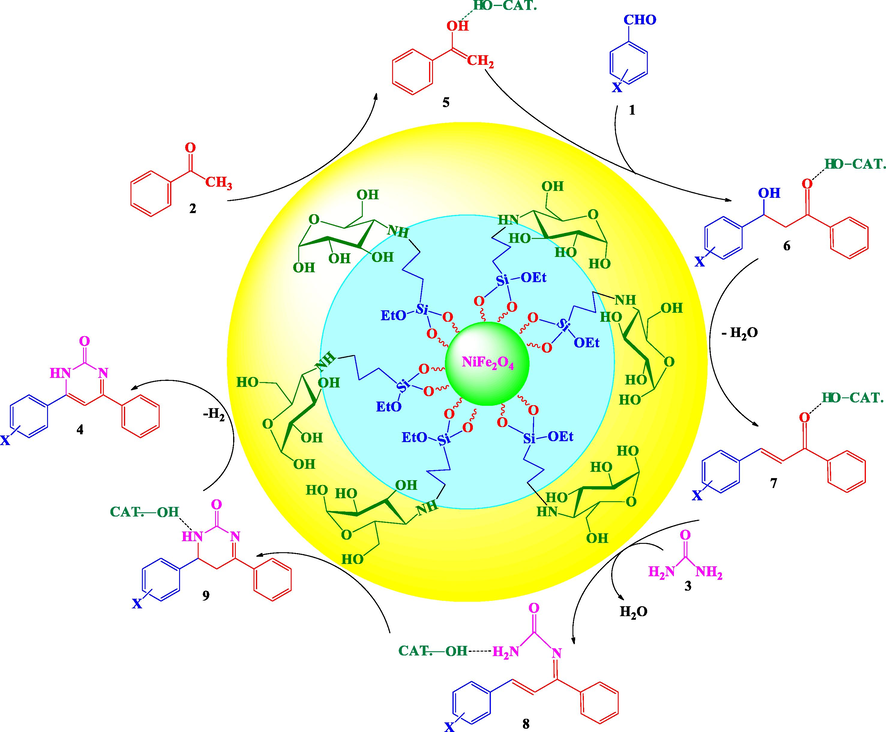
Proposed mechanism for synthesis of 4,6-bisarylpyrimidin-2(1H)-ones.
The recyclability and reusability of catalyst were studied in the model one‐pot reaction among various benzaldehyde, acetophenone and urea. At the end of the reaction, the separated catalyst can be reused after being washed with warm EtOH and drying at 80 °C. NiFe2O4@SiO2nPr@glucose amine was used again for subsequent experiments under similar reaction conditions. The catalyst could be reused for the next cycle without any notable loss of its activity. Yields of the product decreased only slightly after reusing the catalyst six times (Table 4).
Run
1
2
3
4
5
6
7
Yield (%)
94
94
92
93
93
92
83
Time (min)
120
120
120
120
120
120
300
Mp (°C)
215–217
215–217
213–215
214–216
212–214
215–217
211–213
4 Conclusions
In conclusion, we have investigated NiFe2O4@SiO2nPr@glucose amine as a new, eco-friendly, inexpensive, mild and reusable catalyst for the synthesis of 4,6-bisarylpyrimidin-2(1H)-ones and novel azo-linked 4-arylpyrimidin-2(1H)-ones at room temperature (25 °C). High yield, a simple work‐up procedure, ease of separation and recyclability of the magnetic catalyst, adherence to the basics of green chemistry, eco-friendly and based on natural ingredients and waste reduction are some advantages of this method.
Acknowledgements
Financial support from the Research Council of Islamic Azad University, Rasht Branch is sincerely acknowledged.
References
- Chem. Pharm. Res.. 2011;3:234-252.
- Soc. Mycology. 2008;36:66-69.
- Tetrahedron. 1997;53:2803-2816.
- J. Am. Chem. Soc.. 1995;117:2657-2658.
- Adv. Dev. Res.. 2011;2:54-63.
- K. Deres, C.H. Schroeder, A. Paessens, S. Goldmann, H.J. Hacker, O. Weber, T. Kramer,. U. Niewoehner, U. Pleiss, J. Stoltefuss, E. Graef, D. Koletzki, R.N.A. Masantschek, A. Reimann, R. Jaeger, R. Grob, B. Beckermann, K.H. Schlemmer, D. Haebich, H.R. Waigmann, Science 299 (2003) 893-896.
- Arkivoc. 2011:115-126.
- Cent. Eur. J. Chem.. 2008;6:562-568.
- Dalton Trans.. 2012;41:6173-6181.
- Chin. J. Catal.. 2011;32:1484-1489.
- Int Nano Lett. 2015;5:109-123.
- Rafidain j S. 2012;24:120-127.
- M.M. Heravi, S. sadjadi, H.A. Oskooie, R. Hekmat shoar, F.F. Bamoharram, Tetrahedron Lett, 50 (2009) 662-666.
- WILEY-VCH Verlag GmbH & Co KGaA, Weinheim.. 2005;110:1581-1587.
- Tetrahedron Lett.. 2016;57:1656-1660.
- X.S. Wang, Zh.s. Zeng, D.Q. Shi, X.y. Wei, Zh.m. ZonS, Synth. commun, 34 (2004) 4331-4338.
- Iran. J. Chem. Chem. Eng.. 2017;36:35-43.
- Tetrahedron Lett.. 1999;40:3465.
- Green Chem... 2001;3:305-306.
- J. Chem. Res. 2000:354-355.
- Eur. J. Org. Chem.. 2004;3:552-557.
- Tetrahedron Lett.. 2003;44:2889-2891.
- Tetrahedron. 2003;59:1553-1556.
- Ultrason. Sonochem.. 2010;17:363-1266.
- Carbohy. Res.. 2019;483:107755.
- J. Organomet. Chem.. 2019;894:18-27.
- Dyes Pigm.. 2020;173:107936.
- Ultrason. Sonochem.. 2016;28:341-345.
- Dyes Pigm.. 2016;130:314-318.
- M. Nikpassand, L. Zare Fekri, L. Karimian, M. Rassa, Curr. Org. Synth. 12 (2015) 358-362.
- Dyes Pigm.. 2017;136:140-144.
- J. CO2 Util.. 2018;27:320-325.
- Mol. Div.. 2008;12:191-196.
- Chin. Chem. Lett.. 2010;21:269-272.
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2020.10.022.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




