

Translate this page into:
Greener pastures in evaluating antidiabetic drug for a quinoxaline Derivative: Synthesis, Characterization, Molecular Docking, in vitro and HSA/DFT/XRD studies
⁎Corresponding author. y.ramli@um5s.net.ma (Youssef Ramli)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The physicochemical, pharmacokinetic and toxicological properties of MOQTA have been evaluated.
Abstract
A new quinoxaline containing triazole MOQTA is prepared via azide − alkyne cycloaddition “click” procedure. The MOQTA is characterized by XRD and DFT. Frontier Molecular Orbitals (FMOs) and Molecular Electrostatic Potential (MEP) surfaces of MOQTA were generated. Hirshfeld surface analysis (HSA) and 2D fingerprint plots were presented. Compound MOQTA has moderate anti-diabetic activity and excellent anti-oxidant activity. Molecular docking were carried to examine the binding mode between MOQTA and α-glucosidase / α-amylase.
Abstract
In an effort to develop a potent antidiabetic drug, new quinoxaline derivative, 2-(4-((3-methyl-2-oxoquinoxalin-1(2H)-yl)methyl)-4,5-dihydro-1H-1,2,3-triazol-1-yl)-N-(p-tolyl)acetamide (MOQTA) was synthesized and characterized by XRD and various spectroscopic tools (IR, 1H &13C NMR, ESI-MS). The geometric optimization of the molecule was calculated with Density Functional Theory (DFT) method by B3LYP with a 6–311++G(d,p) basis set. Frontier Molecular Orbitals (FMOs) and Molecular Electrostatic Potential (MEP) surfaces of the title compound were generated. Furthermore, Hirshfeld surface analysis (HSA) and 2D fingerprint plots were presented. The calculated MEP and HSA surface interactions were compared in terms of hydrogen bonds and π-π stacking interactions obtained by X-ray packing analyses. X-ray crystallographic structure analysis revealed that the N—HN, C—HO and C—HN intermolecular hydrogen bonds were in agreement with those obtained by HSA. Moreover, MOQTA was assessed for its in vitro anti-diabetic activity. Likewise, molecular docking analyses were conducted to examine the binding mode between MOQTA and the enzymes α-glucosidase and α-amylase. Finally, the physicochemical, pharmacokinetic and toxicological properties of MOQTA have been evaluated by using in silico absorption, distribution, metabolism, excretion and toxicity analysis prediction.
Keywords
Quinoxaline
1,2,3-triazole
XRD
Hirshfeld Surface Analysis (HSA), DFT
Anti-diabetic activity
Molecular docking

1 Introduction
Small heterocycles ring's therapeutic and industrial importance, having nitrogen atoms, has attracted much attention. Examples of heterocycles studied as candidates in drug discovery are: triazoles, quinoxalines and acetamides. They have an important role in medicinal chemistry. Quinoxaline and its derivatives have attracted significant considerations due to their pharmacological activities (Ramli and Essassi, 2015; Ramli et al., 2014; Abad et al., 2020) and industrial properties (Allali et al., 2019; Lgaz, 2015; Laabaissi et al., 2020). This heterocycle already preset in variety of biological active substance and present anti-microbial (Badran et al., 2003; Singh et al., 2010), anti-fungal (Wagle et al., 2008), anti-malarial (Hui et al., 2006), anti-bacterial (Griffith et al., 1992; El-Sabbagh et al., 2009), anti-HIV (Loriga et al., 1997; Balzarini et al., 2000), anti-tuberculosis (Carta et al., 2006), anti-protozoal (Ancizu et al., 2010), anti-inflammatory (Carta et al., 2001), anthelmintic (Guillon et al., 2004) and anti-cancer (Gupta et al., 2005) properties. In addition, quinoxaline derivatives possess anticorrosion characteristics (Lgaz et al., 2016; Zarrok et al., 2012; El Ouali et al., 2010; Abboud et al., 2007; Zarrok et al., 2012; Tazouti et al., 2016; el Aoufir et al., 2016). Similarly, the triazole ring system is linked to biological and pharmacological activities as anti-inflammatory (Abdel-Megeed et al., 2009), antifungal (Nowaczyk and Modzelewska-Banachiewicz, 2008), antibacterial (Foroumadi et al., 2003), antihypertensive (Sato et al., 2009), anti-Alzheimer’s disease (Missioui et al., 2022), anti-COVID-19 (Missioui et al., 2022; Zhang et al., 2020) and anticancer (Shivarama Holla et al., 2003) activity. Also, this heterocyclic scaffold can tolerate reductive/oxidative and basic/acid hydrolysis environments, displaying high-level aromatic endurance to metabolic degradation (Tron et al., 2008; Ferreira et al., 2010; Lauria et al., 2014). Diabetes affects around a quarter of the World's population, and this figure is expected to increase to 700 million by 2045 (Nazir et al., 2018). The identified α‐amylase and α-glucosidase inhibitors, as miglitol, voglibose, and acarbose, have been tried to treat type 2 diabetes(T2DM). Nevertheless, it was interesting to discover the novel α‐amylase and α-glucosidase inhibitors because of the undesirable effects (for example, diarrhea and stomachache) of the existing α‐amylase and α-glucosidase inhibitors. Therefore, the therapeutic use of inhibitors of α‐amylase and α‐glucosidase in the medication of T2DM side-effect has been considered (Gu et al., 2020; Alomari et al., 2021; Hussain et al., 2021; Tavaf et al., 2020). A variety of molecules, including quinoxaline (Ramli and Essassi, 2015; Missioui et al., 2021) N-arylacetamides (Missioui et al., 2021; Wang et al., 2017; Moghimi et al., 2020; Asath et al., 2016) triazole (Ye et al., 2019) have been reported as potential antidiabetic agents. Because of this application and as part of our ongoing work (Zaoui et al., 2021; Abad et al., 2021; Abad et al., 2021; Guerrab et al., 2021; Guerrab et al., 2019; , xxxx), more recent work has reported synthesizing a novel N-arylacetamides quinoxaline-based compound as an anti-diabetic drug (Missioui et al., 2021). Continuing our ongoing research to discover novel quinoxaline-based compounds and improve this antidiabetic activity, the title quinoxaline compound including triazole was synthesized, charecterized by crystallographic techniques., and assessed as an antidiabetic agent. Molecular docking, geometric optimization, MEP surfaces, FMOs, and HSA were calculated for the title compound. These studies highlighted the interaction of the title compound as a promising α‐amylase and α‐glucosidase inhibitor.
2 Experimental section
2.1 Synthesis and crystallization
All chemicals, including solvents, were obtained from commercial sources of high purity and used as received. Follow-up of the reactions were made by TLC. In a Büchi Melting Point SMP-20 apparatus, the compound's melting point was determined and uncorrected. 1H NMR and 13C NMR spectra were collected using a Bruker Avance 300 NMR Spectrometer in DMSO‑d6 and a « JNM-ECZ500R/S1 FT NMR System de JEOL » spectrometer, using TMS as the internal standard and CHLOROFORM-D as solvent. FT-IR spectra were collected using the Bruker-VERTEX 70 device. Mass spectra were recorded on an API 3200 LC/MS/MS mass spectrometer using electrospray ionization (ESI).
2.1.1 3-Methyl-1H-quinoxalin-2-one (MQO)
Compound MQO was synthesized following a procedure similar to that of Hinsberg (Hinsberg, 1887); using O-phenylenediamine (15 mmol) and ethyl pyruvate (22.5 mmol) in HCl 4.4 N at room temperature for 30 min (Scheme 1).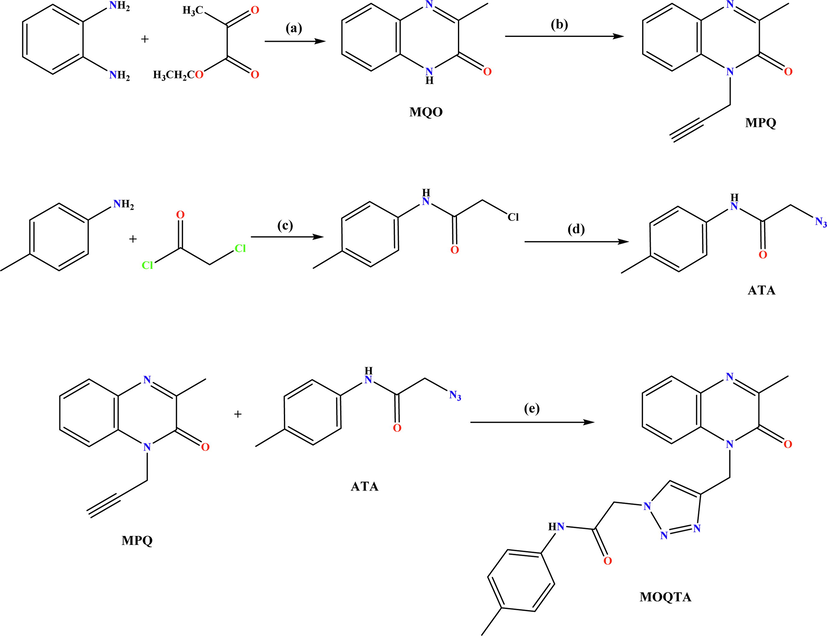
Synthesis pathway of 2-(4-((3-methyl-2-oxoquinoxalin-1(2H)-yl)methyl)-1H-1,2,3-triazol-1-yl)-N-(p-tolyl) acetamide (MOQTA). Reagents and conditions: (a) HCl 4 N,rt; (b) K2CO3, propargyl bromide, TBAB, DMF, rt, 24 h; (c) THF, 0 °C; (d); NaN3, DMF, 40 °C; (e) CuSO4, Sodium ascorbate, EtOH / H2O.
2.1.2 3-methyl-1-(prop-2-yn-1-yl)quinoxalin-2(1H)-one (MPQ)
To 15 mL of DMF and MQO (0.5 g, 3.11 mmol) was added potassium carbonate (0.5 g, 3.73 mmol), propargyl bromide (0.41 mL, 3.11 mmol), and tetra-nbutylammonium bromide. The solution was stirred at 25 0C for 24 h. After filtration, all volatiles were removed under low pressure. The recrystallization is carried out in ethanol to afford MPQ as colorless crystals in a good (68%) yield. (Benzeid et al., 2009) (Scheme 1).
2.1.3 2-azido-N-(p-tolyl)acetamide (ATA)
A previous procedure (Missioui et al., 2022) was used to afford 2-chloro-N-(p-tolyl)acetamide, precursor. 2-chloro-N-(p-tolyl)acetamide (0.011 mol) and sodium azide (0.015 mol) were dissolved in a mixture of ethanol/water (2:1 ratio) then refluxed for 24 h at 80 °C. After completing the reaction, the compound ATA: 2-azido-N-(p-tolyl)acetamide precipitated, filtered, washed with cold water, and recrystallized from ethanol.
2.1.4 2-(4-((3-methyl-2-oxoquinoxalin-1(2H)-yl)methyl)-1H-1,2,3-triazol-1-yl)-N-(p-tolyl) acetamide (MOQTA)
To a solution of MPQ (0.5 mmol), in a water–ethanol mixture (5/15 mL), 2-azido-N-phenyl)acetamide (0.65 mmol) was added with sodium ascorbate and a catalytic amount of copper sulphate. After stirring, under reflux for 24 h, the solution was concentrated. The residue was washed with water and purified by column chromatography on silica gel using a hexane/ethyl acetate (9/1) mixture as eluent. The solid product isolated was recrystallized from ethanol to afford a light yellow block-like specimen of compound MOQTA. (Scheme 1).
2.2 X-Ray crystal data collection
A light-yellow block-like sample of MOQTA of dimensions 0.197 mm × 0.250 mm × 0.268 mm, was mounted on a polymer loop in a cold nitrogen stream on a Bruker Smart APEX CCD diffractometer equipped with a fine-focus sealed tube (Mo-Kα, λ = 0.71073 Å) and a graphite monochromator. The complete sphere of data was processed using SAINT (Bruker APEX3, SAINT, SADABS SHELXTL, Bruker AXS, Inc., Madison, WI., 2016). The structure was solved by direct methods and refined by the full-matrix least-squares method on F2 using SHELXT and SHELXL programs (Sheldrick, 2015; Sheldrick, 2015). The molecular and packing diagrams were generated using DIAMOND (Brandenburg and Putz, 2012). A group of 1565 reflections having I/σ(I) ≥ 13 was analyzed with CELL_NOW (Sheldrick, 2008) which showed the crystal to be twinned by a 180° rotation about the a-axis. The data were integrated with SAINT (Bruker-AXS and Madison, 2019) using the two-component orientation matrix generated by CELL_NOW. The structure is in (Fig. 1), and crystal and refinement (refined as a 2-component twin) details are presented in Table 1.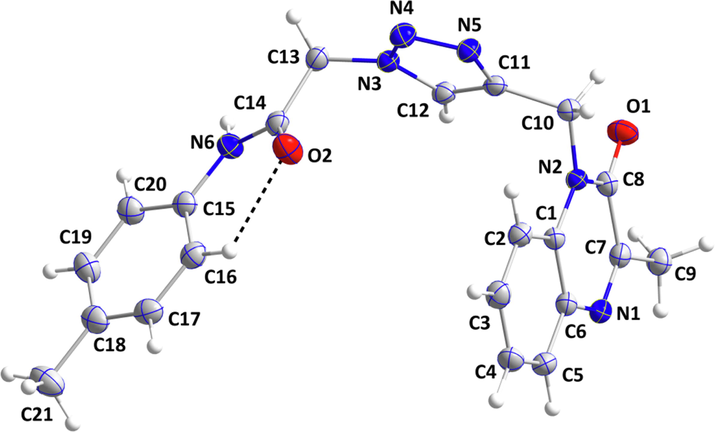
The MOQTA molecule with labeling scheme and 50% probability ellipsoids.
Value
Parameter
Chemical formula
C21H20N6O2
Crystal system, space group
Monoclinic, P21/n
Temperature (K)
150
a, b, c (Å)
7.1831 (7), 18.8662 (17), 13.6756 (12)
β (°)
96.113 (1)
V (Å3)
1842.7 (3)
Z
4
Radiation type
Mo Ka
µ (mm−1)
0.10
Crystal size (mm)
0.27 × 0.25 × 0.20
Absorption correction
Multi-scan
TWINABS (Sheldrick, 2009) (Sheldrick, 2009)
Tmin, Tmax
0.97, 0.998
No. of measured, independent and
observed [I > 2σ(I)] reflections53140, 53140, 39,130
Rint
0.042
(sin θ/λ)max (Å−1)
0.668
R[F2 > 2σ(F2)], wR(F2), S
0.053, 0.145, 1.07
No. of reflections
8723
No. of parameters
265
No. of restraints
2
H-atom treatment
H-atom parameters constrained
Δρmax, Δρmin (e Å−3)
0.29, −0.22
The structure was solved and refined using the Bruker SHELXTL Software Package, using the space group P 1 21/n 1, with Z = 4 for the formula unit, C21H20N6O2. The final anisotropic full-matrix least-squares refinement on F2 with 265 variables converged at R1 = 5.27%, for the observed data and wR2 = 14.53% for all data. The goodness-of-fit was 1.067. The largest peak in the final difference electron density synthesis was 0.344 e-/Å3, and the largest hole was −0.230 e-/Å3 with an RMS deviation of 0.053 e-/Å3. Based on the final model, the calculated density was 1.400 g/cm3 and F(0 0 0), 816 e-.
The supplementary crystallographic data for this paper are available: CCDC 2073886.
2.3 Biological activity
All chemicals and solvents were purchased from Sigma–Aldrich and used as received.
2.3.1 Enzyme inhibitory activities
-
α-Glucosidase inhibition assay.
The α-glucosidase inhibitory activity was performed using ρ-nitrophenyl-α-D-glucopyranoside as a substrate (Kee et al., 2013; Mortada et al., 2022) with minor modifications. Also, the detailed protocol was described in our previous work (Hashim et al., 2013).
2.3.2 Antioxidant activity
For the protocol used to evaluate the antioxidant activity, see the reported method by Mortada et al. (Hashim et al., 2013), Tuberoso et al. (Tuberoso et al., 2013).
2.4 Computational procedure
Geometrical optimization of the title compound was calculated using Gaussian 03 program package (Frisch et al., 2004). This operation used the density functional theory by Becke's three-parameter hybrid functional (Becke, 1993) with Lee–Yang–Parr correlation functional (Lee et al., 1988; Miehlich et al., 1989) (B3LYP) approach in conjunction with the 6–311++G(d,p) basis set (Pople et al., 1986). The data obtained by XRD was used as initial values. FMO, MEP map and harmonic vibrational bands were generated using GaussView molecular visualization program (Frisch et al., 2007).
HSA (Spackman and Byrom, 1997; McKinnon et al., 1998) of the mole AutoDock Vina program in the crystal structure and 2D fingerprint (Spackman and McKinnon, 2002) plots were formed using Crystal Explorer 3.0 computer program (Wolff et al., 2012) using the CIF file.
2.5 Docking methodology
The docking study was carried out against α -glucosidase and α -amylase enzymes. The crystal structure of α-amylase (PDB Id: 4GQR; resolution 1.2 Å) (Williams, 2012) and α-glucosidase (PDB Id: 5NN5; resolution 2.0 Å) (xxxx) were downloaded from the PDB database. Auto Dock tool (ADT) was employed to prepare proteins (Trott and Olson, 2010). The 3D structure of the studied compound (MOQTA) was obtained using a CIF file. And acarbose was used as a reference drug for comparison in the in vitro study. Docking grids box was established to cover the catalytic nucleophile and acid/base sites assigned to D518 and D616, respectively (Hermans, 1991; Roig-Zamboni, 2017) And were prepared using Auto Grid with a spacing of 0.375 Å and dimensions 70; 42; 70 Å (x = -11.303; y = -30.54; z = 93.287) and 64; 42; 46 Å (x = 5.024; y = 13.128; z = 60.575) respectively for α-amylase and α-glucosidase. The grids were created around the enzyme active site. Docking was performed using AutoDock Vina and employed the Lamarck Genetic Algorithm (LGA) and the Solis and Wets search methods. Other docking parameters were set at their default values. The best docking poses for the studied compound were visually analyzed using Discovery Studio 4.5. The docking procedure was first validated by re-docking the 1-DEOXYNOJIRIMYCIN and 3,5,7-TRIHYDROXY-2-(3,4,5-TRIHYDROXYPHENYL)-4H-CHROMEN-4-ONE successively into its binding site of the crystal structure of the α-glucosidase and α-amylase enzyme.
3 Result and discussion
The preparation of MOQTA was described in Scheme1. The starting material, MQO was made by treating o-phenylenediamine with ethyl pyruvate in chloridric acid 4.4 N. MOQTA shows a good synthon for various pharmacologically active compounds (Ramli et al., 2010). The lactam function of MQO is very reactive, so it is condensed by alkylation with propargyl bromide to afford the dipolarophile MPO. An example, it is used to synthesize 1,2,3-triazole. With an intense dipole moment, this scaffold could actively participate in hydrogen bonding and π -stacking interactions (Ferreira et al., 2010); π-stacking, hydrogen bonding and hydrophobic effect play critical roles in promoting both enzymes' inhibitory activity (Liu et al., 2007; Li et al., 2011). Due to the importance of the 1;2;3 -triazole and N-arylacetamide moieties in the therapeutic area and have suitable inhibitory activities towards α‐amylase and α‐glucosidase (Ye et al., 2019; Wang et al., 2016; Wang et al., 2017), it seems interesting to associate these heterocycles with the 3-methylquinoxalin-2-one derivative MPQ. Thus, we have studied the copper-catalyzed 1,3-dipolar cycloaddition reaction of 2-azido-N-phenylacetamide and the compound MPO. The structures of MPQ and MOQTA were elucidated based on spectral data.
3.1 3-methyl-1-(prop-2-yn-1-yl)quinoxalin-2(1H)-one (MPQ)
Yield 73%, FT‐IR (ATR, υ, cm‐1): 1264.90 υ (N-C amide), 1650.10 υ (C = O amide), (1571.54–1598.51) υ(C = Carom), (1051.91–1265.29) υ(C-Harom), 2114 υ (C≡C propargyl). 1H NMR (chloroform-d) δ ppm: 2.6 (3H, s,CH3); 2.3 (1H, s, CH); 5.1 (2H,s, CH2); [7.2–7.8] (4H, m, CHarom); 13C NMR (chloroform-d) δ ppm: 21.6 (CH3); 31.6 (CH2); 75.6 (CH); 78.5 (C≡C propargyl); 157.58 (C = O quinoxaline).
3.2 2-(4-((3-methyl-2-oxoquinoxalin-1(2H)-yl)methyl)-1H-1,2,3-triazol-1-yl)-N- (p-tolyl)acetamide (MOQTA)
Yield 66%, mp = 222.7–224.5 °C, FT‐IR (ATR, υ, cm‐1): (636.86–675.05) υ(C-H CH3qinoxaline), 951.46 υ(CH2), (1057.91–1369.13) υ(C-Harom), 1307 υ(N = Ntriazole), 1362.13 υ(N-Hacetaamide), (1423.31–1467.21) υ(CH3 Tolylacetaamide), (1509.78–1534) υ(C = Carom), 1661.85 υ(C = O amide), 1701.38 υ(C = O acetaamide); 1H NMR (DMSO‑d6) δ ppm: 5.55 (2H, s, CH2qui); 5.26 (2H, s, CH2triazol); 8.1 (1H, s, CHTriazol); 11.37 (1H, s, NH); 2.5 (3H, s, CH3 quinoxaline); 2.26 (3H, s, CH3 tolyl); 6.48–7.79 (m, J = 7.5 Hz, 13HAr); 13C NMR (DMSO‑d6) δppm: 37.30 (CH2-NQuin); 52.13 (CH2-NTriazol); 21.17 (CH3 Quin); 20.40 (CH3); 129.24 (CHTriazol); 165 (C = O amid); 157.58 (C = O quinoxaline); HRMS (ESI-MS) (m/z) calculated for C21H20N6O2 388,16 found 389.16.
The 1H NMR spectrum of MPQ revealed three signals at δ 2.59, 2.27, and 5.04 ppm due to the methyl of quinoxaline, CH of propargyl group, and CH2 attached to the quinoxaline nitrogen, respectively. Confirming the alkylation reaction of 3-methylquinoxaline MQO with propargyl bromide. Also, 1H NMR shows the signals relating to aromatic protons between 7.24 and 7.81 ppm. (Abad et al., 2020).
1H NMR spectrum of the title compound MOQTA, exhibited five signals at δ 2.26, 5.26, 5.55, 8.1, and 11.37, referring to CH3 tolyl group, CH2 triazole adjacent, CH2 quinoxaline adjacent, CH of triazole group and NH of acetamide group, respectively. It also revealed the absence of signals at δ 3.3 et 5.1 ppm corresponding to the propargyl hydrogen, which confirms the cycloaddition reaction between MPQ and the azide ATA. The 13C NMR spectrum presented signals at δ at 20.40, 37.30, 52.13, 129,24 and 165 ppm referring to CH3 of tolyl group, CH2 triazol adjacent, CH2 quinoxaline adjacent, CH of triazole group and carbon of acetamide, respectively. Furthermore, in addition to the presence of the CH3 quinoxaline signal at 21.17 ppm. IR spectrum of MOQTA revealed a band at 1308.19 cm−1 for the N = Ntriazole, a band at 1362.13 cm−1 for the NH group and an absorption band at 1769.6 cm−1 characteristics for the carbonyl group. These data are in agreement with the characteristics of the analogous compounds of MOQTA (Abad et al., 2020; Missioui et al., 2022). Its mass spectrum demonstrated a molecular ion peak (MH+, m/z = 389.16) which conforms to C21H20N6O2.
3.3 X-ray crystallography
As indicated by the 1.33(10)° dihedral angle between the constituent planes, the quinoxaline unit is not planar. The dihedral angle between the C1/C6/N1/C7/C8/N2 and N3/N4/N5/C11/C12 planes is 88.77(10)° while that between the latter ring and the C15···C20 ring is 89.97(5)° giving the molecule an approximate “U”-shaped conformation. The orientation of the C15···C20 ring is partially determined by the weak, intramolecular C16 –H16…O2 interaction (Table 2 and Fig. 1). The bond lengths and interbond angles appear as expected for the given formulation.
D—H…A
D—H
H…A
D…A
D—H…A
N6—H6…N5i
0.91
2.25
3.142 (2)
165
C3—H3…Cg4ii
0.95
2.69
3.516 (2)
146
C4—H4…N4iii
0.95
2.53
3.356 (3)
145
C9—H9C…Cg2iv
0.98
2.61
3.364 (2)
134
C10—H10A…O2v
0.99
2.57
3.439 (2)
146
C12—H12…N1iv
0.95
2.54
3.379 (3)
147
C13—H13B…O1vi
0.99
2.37
3.174 (2)
138
C16—H16…O2
0.95
2.28
2.868 (3)
120
C19—H19…O2i
0.95
2.55
3.157 (3)
122
C20—H20…O2i
0.95
2.52
3.133 (3)
123
Symmetry codes: (i) x − 1, y, z; (ii) x + 1, y, z; (iii) − x + 3/2, y + 1/2, −z + 1/2; (iv) − x + 1, −y + 1, −z + 1; (v) x + 1/2, −y + 1/2, z + 1/2; (vi) x − 1/2, −y + 1/2, z − 1/2.
In the crystal, N6—H6···N5 H-bonds with graph sets C1,1(7) form chains of molecules extending along the a-axis direction which are linked into layers parallel to the ac plane by C13—H13B···O1 H-bonds having graph sets C1,1(9). These interactions are reinforced by weaker C10—H10A···O2, C19—H19···O2 and C20—H20···O2 hydrogen bonds having graph sets C1,1(8), C1,1(7) and C1,1(6), respectively plus) C3—H3···Cg4 interactions (Table 2 and Fig. 2). Each layer has an inversion-related counterpart which is linked to it via C12—H12···N1 hydrogen bonds (graph sets C1,1(9)) and slipped π-stacking interactions between quinoxaline units (centroid···centroid = 3.5495(12) Å, dihedral angle = 1.33°) Table 2 and Fig. 3). The layers are connected by C4—H4···N4 hydrogen bonds with graph sets C1,1(10) (Table 2 and Fig. 4).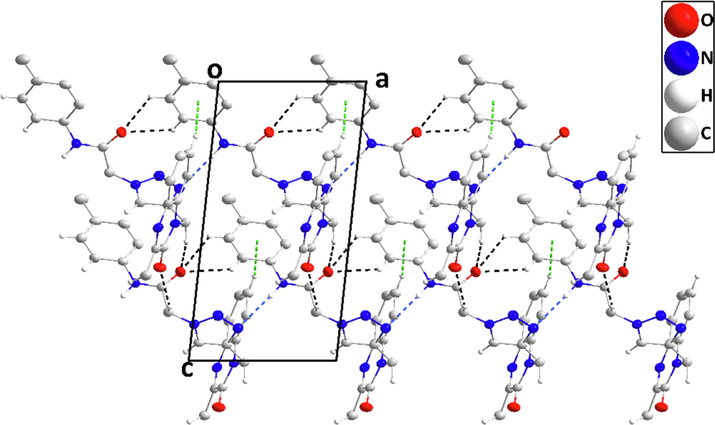
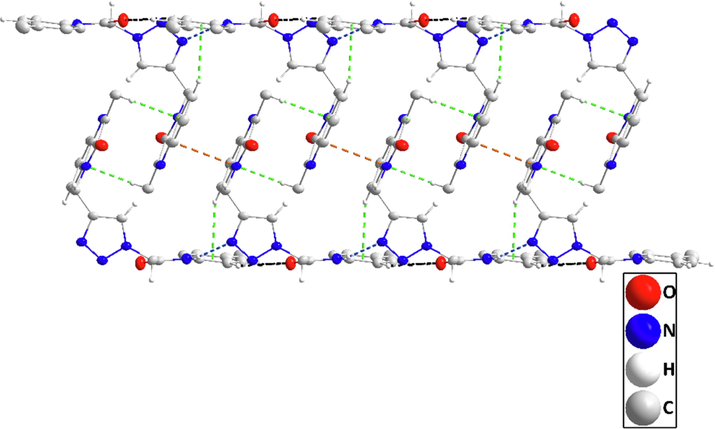
Detail of a portion of two chains seen along the b-axis direction. N—H···N, and C—H···O H-bonds are depicted, respectively, by blue and black dashed lines. C—H···π(ring) interactions are depicted by green dashed lines.
Detail of portions of two inversion-related layers projected on
showing the π-stacking interactions (orange dashed lines). C—H···π(ring) interactions and C—H···O hydrogen bonds are depicted, respectively, by green and black dashed lines.
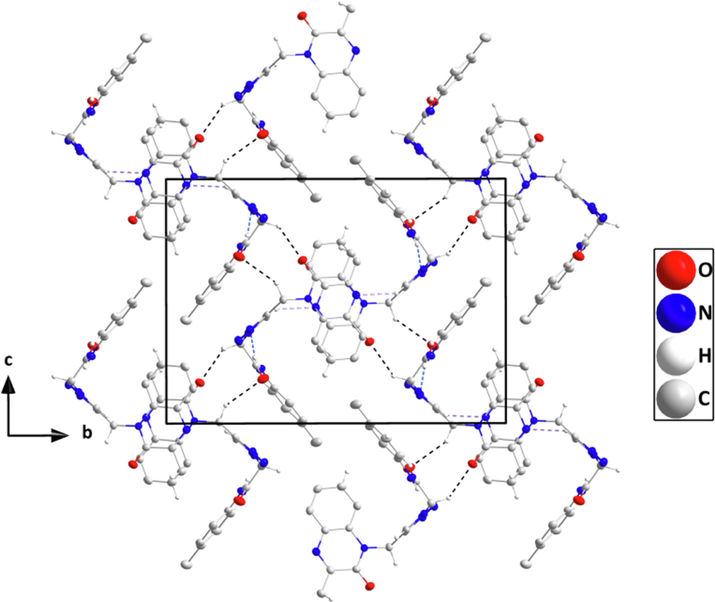
Packing seen along the a-axis direction giving an end view of several chains and their linkage through C—H···O hydrogen bonds (black dashed lines). π-stacking and C—H···π(ring) interactions are omitted for clarity.
3.4 Computational ad theoretical study
3.4.1 Geometric optimization of MOQTA
The optimized molecular parameters of the molecule using DFT/B3LYP/6–311++G(d,p) method and X-ray data for bond angles and bond distances were given in Table 3 and Table 4, respectively. For bond distances, the error values range up to 0.0241 Å. While the significant error was found for N4—N5 bond distance with 0.0241 Å, whereas the smallest error was found for C10—C11 bond distance. It can be considered the theoretical results are consistent with the experimental results.
Bond Distances
Atoms
X-ray
DFT
Error
Atoms
X-ray
DFT
Error
O1-C8
1.226(2)
1.2276
−0.0016
N1-C6
1.390(2)
1.3834
0.0066
N1-C7
1.298(2)
1.2915
0.0065
N2-C1
1.401(2)
1.3949
0.0061
N2-C8
1.375(2)
1.3886
−0.0136
N3-N4
1.341(2)
1.3546
−0.0136
N2-C10
1.476(2)
1.4757
0.0003
N3-C13
1.455(2)
1.4498
0.0052
N3-C12
1.348(2)
1.3538
−0.0058
N5-C11
1.364(2)
1.3641
−0.0001
N4-N5
1.323(2)
1.2989
0.0241
N6-C15
1.426(2)
1.4179
0.0081
N6-C14
1.347(2)
1.3661
−0.0191
C1-C2
1.391(3)
1.4045
−0.0135
C1-C6
1.409(3)
1.4149
−0.0059
C2-C3
1.379(3)
1.3874
−0.0084
C7-C9
1.488(3)
1.4977
−0.0097
C3-C4
1.387(3)
1.4006
−0.0136
C10-C11
1.497(3)
1.497
0
C4-C5
1.379(3)
1.3833
−0.0043
C15-C20
1.386(3)
1.3986
−0.0126
C5-C6
1.396(3)
1.4042
−0.0082
C16-C17
1.387(3)
1.3919
−0.0049
C7-C8
1.487(3)
1.4856
0.0014
C17-C18
1.386(3)
1.3986
−0.0126
C11-C12
1.364(3)
1.3783
−0.0143
C18-C19
1.390(3)
1.3977
−0.0077
C13-C14
1.526(3)
1.5401
−0.0141
C19-C20
1.385(3)
1.3903
−0.0053
C15-C16
1.393(3)
1.3995
−0.0065
O2-C14
1.216(2)
1.2163
−0.0003
C18-C21
1.507(3)
1.5093
−0.0023
Bond Angles
Atoms
X-ray
DFT
Atoms
X-ray
DFT
C7-N1-C6
118.86(16)
119.4641
C19-C20-C15
120.3(2)
120.4785
C8-N2-C10
117.96(16)
116.4543
C8-N2-C1
121.83(16)
121.8952
N4-N3-C12
111.22(16)
110.8914
C1-N2-C10
120.20(16)
121.6489
C12-N3-C13
128.38(17)
129.8309
N4-N3-C13
120.09(16)
119.2652
N4-N5-C11
108.52(16)
109.4753
N5-N4-N3
106.98(15)
107.2333
C2-C1-N2
122.54(17)
122.8732
C14-N6-C15
126.89(17)
128.9826
N2-C1-C6
117.97(17)
117.6003
C2-C1-C6
119.48(18)
119.5256
C2-C3-C4
121.14(19)
121.1275
C3-C2-C1
119.91(19)
119.6938
C4-C5-C6
120.65(19)
120.7363
C5-C4-C3
119.45(19)
119.4402
N1-C6-C1
122.02(17)
122.0814
N1-C6-C5
118.61(17)
118.4638
N1-C7-C8
123.44(18)
123.2729
C5-C6-C1
119.36(18)
119.4542
C8-C7-C9
116.44(17)
116.3535
N1-C7-C9
120.12(17)
120.3735
O1-C8-C7
121.79(18)
122.6526
O1-C8-N2
122.56(18)
121.8852
N2-C10-C11
112.40(15)
114.019
N2-C8-C7
115.65(17)
115.4554
N5-C11-C12
108.55(17)
108.2603
N5-C11-C10
121.18(17)
121.7624
C12-C11-C10
130.27(18)
129.976
N3-C12-C11
104.73(17)
104.1396
N3-C13-C14
110.25(15)
111.9846
O2-C14-C13
120.37(18)
120.6494
O2-C14-N6
125.25(19)
125.5932
C20-C15-C16
119.29(19)
119.2427
N6-C14-C13
114.37(17)
113.7077
C16-C15-N6
123.26(18)
123.4533
C20-C15-N6
117.41(18)
117.3039
C18-C17-C16
122.3(2)
122.2655
C17-C16-C15
119.3(2)
119.3124
C17-C18-C21
121.6(2)
121.0374
C17-C18-C19
117.3(2)
117.4913
C20-C19-C18
121.5(2)
121.2089
C19-C18-C21
121.1(2)
121.4692
For bond angles, the biggest difference between X-ray and optimized data was calculated for C14-N6-C15 bond angle with −2.0926° error value. The lowest error value belongs to C5-C4-C3 bond angle with 0.0098°. Like bond distances, bond angles calculated by the DFT method are close to experimental values.
3.4.2 Molecular Electrostatic map
Molecular Electrostatic Potential (MEP) map of the MOQTA was generated by DFT/B3LYP/6–311++G(d,p) basis set and given in Fig. 5. MEP reveals electrophilic and nucleophilic centers and helps us understand non-bonding interactions (Hathwar et al., 2015; Okulik and Jubert, 2005; Luque et al., 2000). In the MEP, while the red areas show electrophilic attack centers, the blue areas show nucleophilic attack centers (Scrocco and Tomasi, 1978). It can be seen that the electrophilic centers in the MEP are in the vicinity of the oxygen and nitrogen atoms. These values are −0.0510 a.u. for O1 atom, −0.0417 a.u., for O2 atom, −0.0383 a.u. for N1 atom and −0.0461 a.u. for the region between N4 and N5 atom. The most nucleophilic region was detected at the vicinity of H6 atom with + 0.0709 a.u. The crystal structure contains intermolecular N6—H6N5, C4—H4N4, C10—H10AO2, C12—H12N1, C13—H13O1, C19—H19O2, C20—H20O2 hydrogen bonds and intramolecular C16—H16O2 hydrogen bonds. This is a sign that all electrophilic and nucleophilic attack centers participate in hydrogen bonds.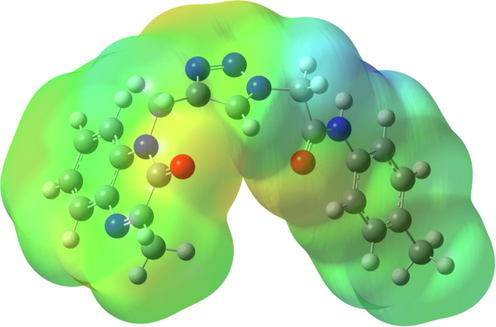
The Molecular Electrostatic Potential (MEP) map of MOQTA.
3.4.3 Hirshfeld surface analysis (HSA)
HSA and 2D fingerprint plots were created using Crystal Explorer 3.0 computer program and Hirshfeld surfaces of the molecule with the normalized contact distance (dnorm) was given in Fig. 6. The dnorm can take negative and positive values according to intermolecular contacts are closer or longer than VDW radii, respectively. In the Hirshfeld surface negative values are given by red areas, and positive values are presented by blue areas. dnorm range from 0.366 Å to 1.258 Å.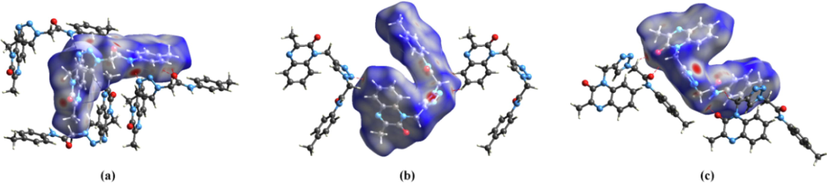
Hirshfeld surface with intermolecular hydrogen bonds.
In Fig. 5, the Hirshfeld surface of the molecule with adjacent molecules is given. Fig. 6 (a) represents intermolecular N6—H6···N5, C12—H12···N1, C19—H19···O2 and C20—H20···O2 interactions between the central molecule and adjacent molecules, Fig. 6 (b) represents intermolecular C4—H4···N4 hydrogen bond between the central molecule and adjacent molecules, Fig. 6 (c) represents intermolecular C10—H10A···O2 and C13—H13B···O1 interactions between the central molecule and adjacent molecules. In the Hirshfeld surface, the environment of C4—H4, C10—H10A, C12—H12, C13—H13B, C19—H19, C20—H20, N6—H6 groups and N1, N4, N5, O1, O2, atoms are seen like red areas that these regions are active regions for intermolecular interactions. X-ray diffraction results show that intermolecular N6—H6N5, C4—H4N4, C10—H10AO2, C12—H12N1, C13—H13BO1, C19—H19···O2, C20—H20O2 hydrogen bonds are present in these regions.
In Fig. 7, OH, HO, NH and HN interactions and the 2D fingerprint plot with hydrogen bonds were given and it is seen that OH / HO and NH / HN interactions in the 2D fingerprint plot are seen like two distinct spikes. While the spikes in the lower right and upper left of the 2D fingerprint plot correspond to NH / HN interactions resulting from intermolecular C12—H12···N1, C4—H4···N4 and N6—H6···N5 interactions. The spikes in the middle right and middle left of the 2D fingerprint plot correspond to OH/ HO interactions resulting from intermolecular C10—H10A···O2, C13—H13BO1, C19—H19···O2, C20—H20O2 hydrogen bonds. The contributions of these interactions are 12.1% and 15.8% to the Hirshfeld surface, respectively.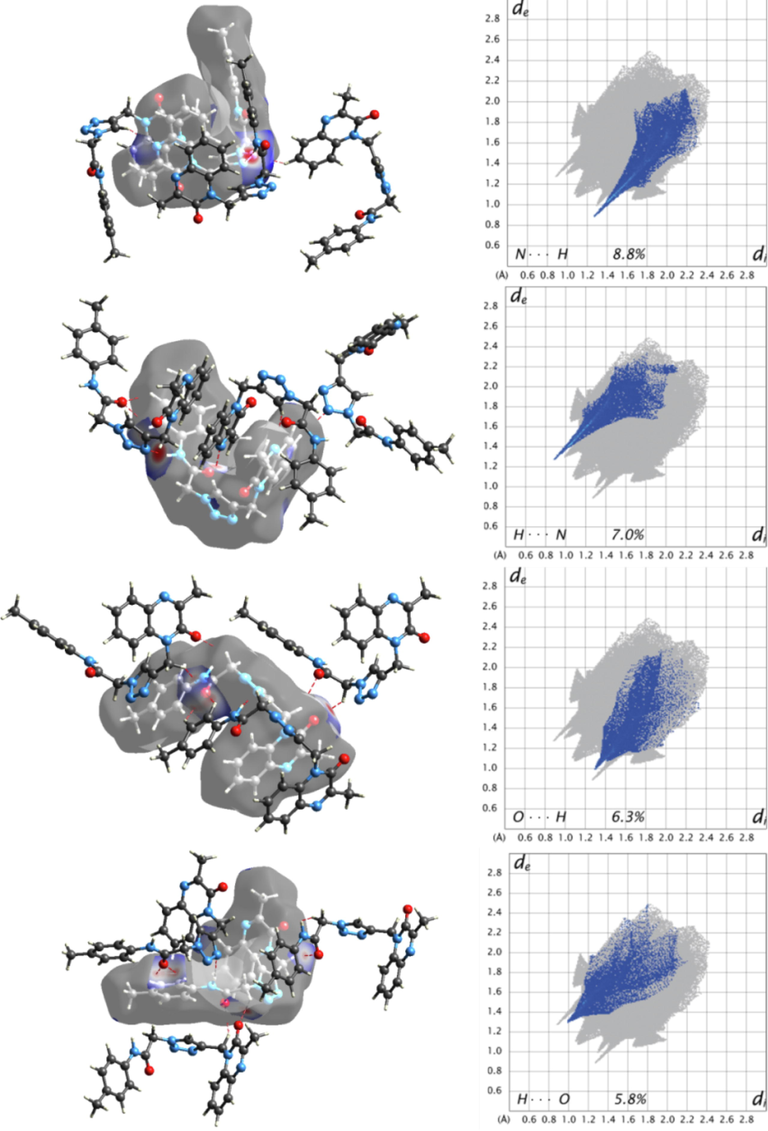
2D fingerprint plot and 3D Hirshfeld surface mapped with dnorm with the intermolecular interactions between the molecule and neighbor molecules.
Fingerprint plots of crystal and dominant interactions can be seen in Fig. 8. In the 2D fingerprint plot, characteristic wings at the top of left and bottom of right are defined like C-Hπ interactions (Seth et al., 2011; Luo and Sun, 2013). As shown in Fig. 8, while the upper left wing corresponds to the spots in the vicinity of C-H donor, the lower right wing corresponds to the spots in the vicinity of π acceptor. The crystal structure has C3—H3π, C9—H9Cπ interactions and the contribution of C···H / H···C interactions is 18.5% to Hirshfeld surface. H-H contacts with 44.7% give the most dominant contribution to the Hirshfeld surface. The contributions of intermolecular interactions with rations to the total Hirshfeld surface were given in Table 5.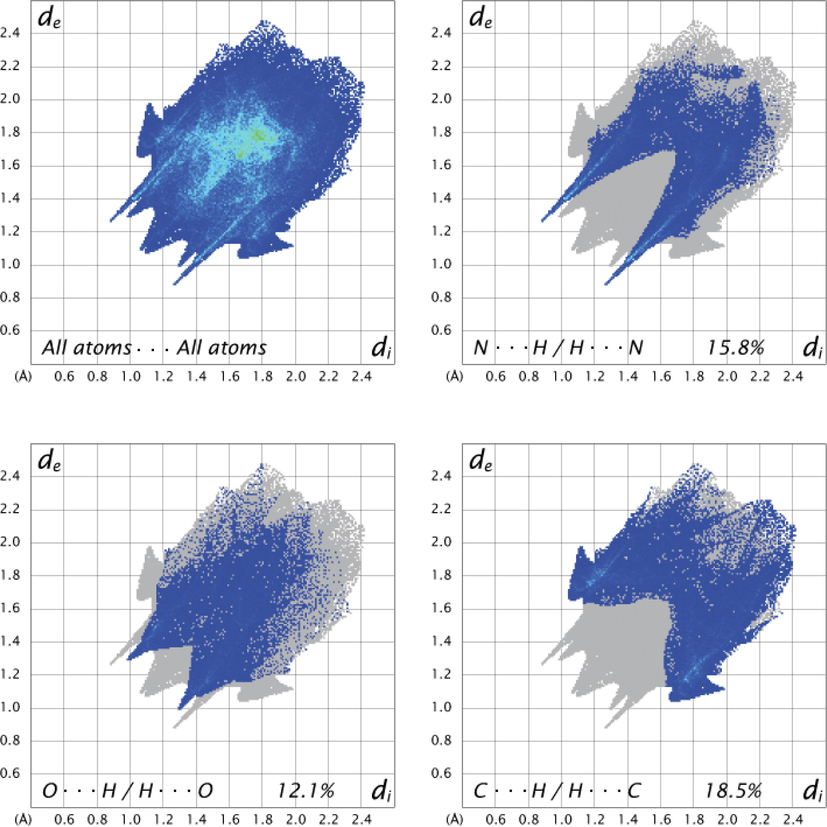
2D fingerprint plot of All atoms···All atoms, N···H / H···N, O···H / H···O, C···H / H···C interactions.
H···H
C···H
N···H
O···H
C···C
C···N
C···O
N···O
N···N
44.7
18.5
15.8
12.1
3.9
3.3
1.2
0.3
0.2
3.4.4 Frontier Molecular orbitals
Frontier molecular orbitals (FMOs) play an essential role in molecular orbitals in chemical, electrical, and optical properties (Fleming and Wiley, 1979). Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) from these orbitals are very important for the biological activities of molecules. Because of charge transfer, while a small energy gap between HOMO and LUMO increases the biological activity of molecules, low energy gap decreases the biological activity of molecules (Yousef et al., 2016; Agrawal et al., 2016; Wu et al., 2020; Pearson, 2005). According to Koopmans' theorem (Koopmans, 1934), ionization potential,
, and electron affinity,
, are given by.
Chemical hardness,
, is defined as the resistance of chemical potential against to change electron number (Pearson, 2005; Parr and Pearson, 1983) and is defined in terms of ionization potential and electron affinity.
While soft molecules have a low energy gap, hard molecules have high energy gap (Fleming and Wiley, 1979).
Therefore, FMOs were created using DFT with B3LYP/6–311++G(d,p) basis set and are given in Fig. 9. In molecule, HOMO and LUMO orbitals are mainly concentrated on 3-methylquinoxalin-2(1H)-one part of a molecule. While HOMO-1 orbitals are concentrated on N-p-tolylformamide group, LUMO + 1 orbitals are concentrated on 2-(4,5-dihydro-1H-1,2,3-triazol-1-yl)-N-(p-tolyl)acetamide group of the molecule. The energy gap between LUMO and HOMO orbitals is 4.3824 eV, too.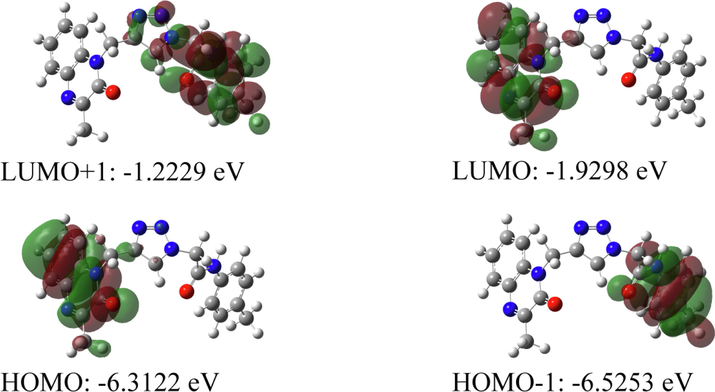
Frontier molecular orbitals of the compound.
3.5 Biological activity
3.5.1 Antidiabetic activity
The in vitro antidiabetic activity of hybrid quinoxaline derivative MOQTA was screened for two enzymes inhibitory properties. Effects were compared with acarbose. The obtained results proved that the MOQTA has an inhibitory effect on the digestive enzymes, as shown in Table 6. The results showed that MOQTA had an average inhibitory effect on α-glucosidase activity (IC50 288.6 ± 0.965 µM), which exceeds the acarbose taken as a reference with IC50 value was activity (IC50 72.58 ± 0.682 µM). MOQTA is a statistically highly significant as P < 0.001, a similar result was found for α-amylase. Table 6 demonstrated an excellent activity (IC50246.6 ± 1.16 µM), which exceeds the acarbose taken as a reference (IC50 115.6 ± 0.574 µM). Therefore, it is considered an active compound.
(IC50 µmol/ml)
α-glucosidase
α-amylase
MOQTA
288.6 ± 0.965****
246.6 ± 1.1****
NPOQA
83.78 ± 0.888
199.7 ± 0.952 (Missioui et al., 2021)
Standard
72.58 ± 0.682
115.6 ± 0.574
Values represent mean ± standard deviation (n = 3).
3.5.2 Antioxidant activity
The antioxidant activity of MOQTA complex was evaluated in vitro using radical methods, ABTS, DPPH, FRAP and H2O2. The antioxidant property of MOQTA was assessed in comparison with ascorbic acid as standard. The obtained result of DPPH assay showed good antioxidant activity, with IC50 value (101 ± 1.01 μM), similar to the action of NPOQA, a novel N-arylacetamides quinoxaline-based compound recently reported (Missioui et al., 2021). While IC50 value of ascorbic acid (78.11 ± 0.68 μM). In the ABTS test, MOQTA has developed an important antioxidant activity compared to the activity of NPOQA (Fig. 10) (Missioui et al., 2021). The antioxidant activity presented in the DPPH test, Table 7, displayed the average antioxidant ability (59.61 ± 0.65 µM). Furthermore, the FRAP method observed the highest reducing power for MOQTA (133,9 ± 0,52 µM). The synthesized compound MOQTA demonstrated activity in H2O2, with IC50 value (44.83 ± 1.32 ± μM), while IC50 value of ascorbic acid (7.45 ± 1.11 μM).
The structure of NPOQA.
IC50 µM)
DPPH
ABTS
FRAP
H2O2
MOQTA
NPOQA
101 ± 1.01
104 ± 4.65
59.61 ± 0.65
330.30 ± 3.44
133.9 ± 0.52
298.54 ± 6.59
44.83 ± 1.32
5.06 ± 0.48 (Missioui et al., 2021)
Ascorbic Acid
78.11 ± 0.68
22.49 ± 0.59
88.12 ± 0.23
7.45 ± 1.11
3.6 Molecular docking results
Molecular docking aims to predict the binding types of the studied compound against α-amylase enzymes and α-glucosidase. Docking studies have been reported in previous studies where many compounds were docked with the α-glucosidase/amylase enzymes (Missioui et al., 2021). The docking study of MOQTA and acarbose (standard) is represented in Fig. 11, 12, 13. The binding energy of the compounds and implicated residues of α-glucosidase and α-amylase are shown in Table 8. The results showed that both MOQTA and standard were able to bind to the active site of α-glucosidase, the calculated binding energy was found to be −7.5 and −6.5 kcal/mol successively for MOQTA and standard (Table 8), significantly improved compared to the binding affinity of NPOQA (Missioui et al., 2021). MOQTA showed three hydrogen bonds interactions with the main chains of residues SER 566, ARG 189, TYR191 as appeared in Fig. 11 and stabilized in the active site pocket with hydrophobic interactions with residues LEU565, ASP243, ARG190, and TYR191. Many H-bonded interactions were formed between different hydroxyl groups of acarbose and amino acids Arg252, Pro312, Ser3, Arg421, Gly403, Arg398, Thr11, Arg5, Asn5, and Gly334 of the α-glucosidase enzyme (Fig. 13). The studied compound MOQTA and standard shear the same hydrophobic interactions with α-glucosidase residues arg190, which may explain the crucial binding affinity compared to the standard drug acarbose.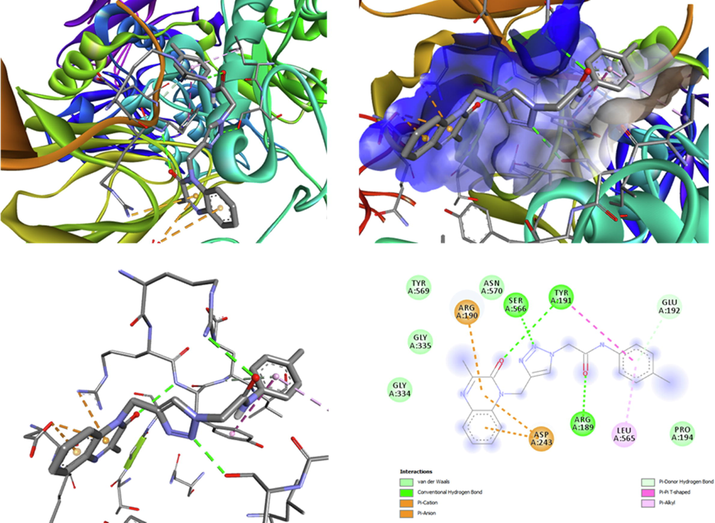
The ligand MOQTA is docked into the binding site of glucosidase. (A) Binding of MOQTA with glucosidase, (B) (3D) amino acid residues and interactions in MOQTA-glucosidase hydrophobic pocket. (C) (3D) amino acid residues and interactions in MOQTA-glucosidase complex. (D) (2D) amino acid residues and many interactions formed in MOQTA-glucosidase compound.
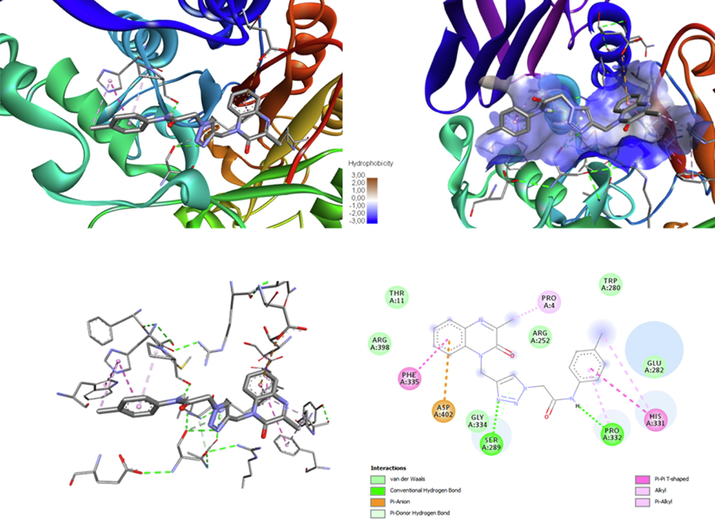
The ligand MOQTA is docked well into the binding site of amylase. (A) The interaction of MOQTA with amylase, (B) (3D) amino acid residues interactions in acarbose-amylase hydrophobic pocket. (C) (3D) amino acid residues and interactions in MOQTA- amylase complex. (D) (2D) amino acid residues and interactions formed in MOQTA-amylase.
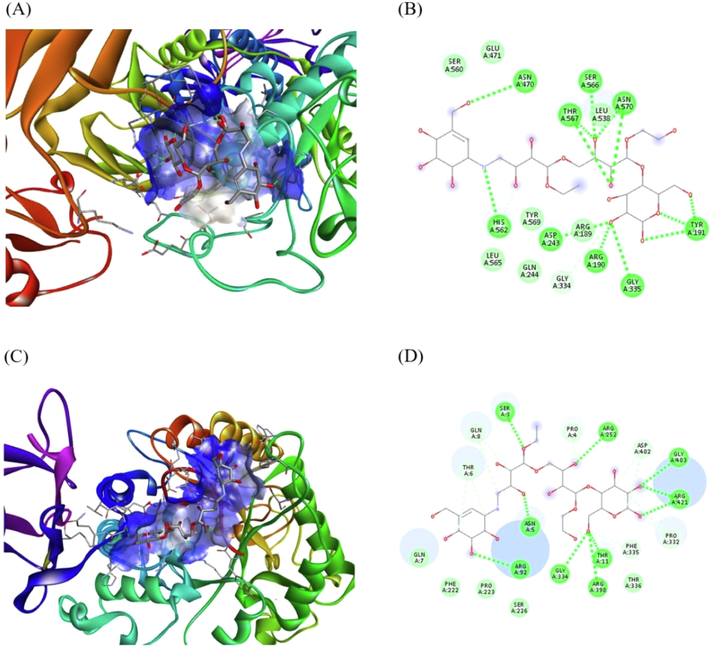
Acarbose was docked with –amylase and glucosidase. (A) A display of binding of acarbose with glucosidase (B) amino acid residues and interactions in Acarbose-glucosidase complex, (C) A display of binding acarbose with -amylase (D) A display of amino acid residues and interactions formed in acarbose-amylase complex.
α-Glucosidase
α-Amylase
Affinity kcal/mol
Residues
Affinity kcal/mol
Residues
Standard
−6.5
THR567, ASN570, SER566, HIS562,, LEU538, ASN470, GLY335, ASP243, ARG190, TYR191, ARG189
−6.6
ARG421, GLY403, ASP402, ARG252, GLN8, THR6, PRO4, SER3
PRP332, PHE335, THR336, THR11, ARG398, GLY334, ASN5, ARG92
NPOQA
−6.9
LEU538, ASP243, ASN570, TYR191, SER566, THR56
−7.6
PRP332, PHE335, THR336, THR11, ARG398, GLY334, ASN5, ARG92ASP402,ARG398, SER289, TYR333, ARG421, PRO332
MOQTA
−7.5
LEU565, ASP243, ARG190, TYR191,
TYR191, SER 566, ARG 189−7.7
PRO332 SER289
HIS331 PRO4 PHE335 ASP 402
We have also performed a docking study of MOQTA and the standard with α-amylase. The calculated binding affinity was predicted to be-7.7 and −6.6 Kcal/mol successively for MOQTA and acarbose, as shown in Table 8, which is better than the binding affinity of NPOQA (Missioui et al., 2021). Compound MOQTA occupied the hydrophobic pocket of the enzyme and showed two hydrogen bonds interactions with residues PRO332 SER289 and several other types of interactions with residues HIS331 PRO4 PHE335 ASP 402. Interactions analysis showed that the formed ring 1,2,3-triazole allowed the studied compound to establish strong N…H hydrogen interactions, which is consistent with the literature (Ferreira et al., 2010); Consequently, the formation of a stable complex ligand-receptor with a low free energy ΔG (Fig. 11). The comparison of the obtained results with the binding of NPOQA (Missioui et al., 2021) showed an increase of affinity between ligand and studied enzyme that can also be allowed to the heterocyclic structure of triazole.
The binding energy of MOQTA with α-amylase and α-glucosidase is very encouraging. This suggests that MOQTA merits further assessment in the context of in vitro/ in vivo of α-glucosidase and α-amylase studies.
3.7 Lipinski's rule and ADMET prediction
The Lipinski rule and ADMET parameters(Kerflani et al., 2022; Lipinski et al., 2001) of the investigated molecule were performed using the web servers Swissadme (‘SwissADME’. http://www.swissadme.ch/ (accessed Dec. 15, 2021) and ADMETlab2.0 (‘ADMETlab 2.0′. https://admetmesh.scbdd.com/ (accessed Sep. 24, 2021), respectively. The Lipinski rule including molecular weight, number of rotational bonds, number of acceptor hydrogen bonds, number of donor hydrogen bonds and log(P) (Adedotun, 2022) were determined. Molecules that violate more than one of these parameters may have bioavailability problems and a high probability of not being similar to a drug (Ferreira and Andricopulo, 2019).
The server (ADMETlab) was utilized to predict the absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of MOQTA. Pioglitazone was used as a standard drug.
As observed in Table 9 MOQTA respects all the conditions mentioned in Lipinski's rule with Lipophilicity (MLogP) < 5, H-bond Donors < 5, and H-bond Acceptors < 10. As reported in Table 10, logD value is in the range of 2.423, suggesting a stabilized distribution. Thus, the obtained value of logP 1.938 indicates that the compound may be slightly permeable to the biological barriers of the physiological system but still has lipophilicity estimated as ideal in the intestinal absorption phase. The PSA evaluated at 94.7 presents a good permeability.
Lipinski’s Rule
Compound
Molecular Weight (g/mol)
Lipophilicity (MLogP)
H-bond Donors
H-bond Acceptors
Rule Violations
Drug-Likeness
<500
<5
<5
<10
<2
MOQTA
388.42
1.85
1
8
0
yes
Pioglitazone
356.4
2.16
2
4
0
yes
Physicochemical descriptor
Value
Lipophilicity(LogP)
1.938
Distribution at pH 7.4 (logD)
2.423
Polar Surface Area (PSA)
94.70
Plasma protein binding (PPB)
94.32%
The selected molecule has a low BBB (blood–brain barrier penetration), with a MDCK permeability of 2.6e-05 and a Caco-2 permeability value of −4.747 for the metabolism. The MOQTA could inhibit the cytochromes CYP2C19, CYP2C9, and CYP3A4. (Table11). Non-Rat Acute Oral Toxicity effect has been observed.
ADMET prediction
Value
Absorption
HIA
0.005
MDCK Permeability
2.6e-05
Caco-2 Permeability
−4.747
P-glycoprotein inhibitior
0.977
P-glycoprotein substrate
0.011
F20%
0.007
F30%
0.005
Distribution
PPB
94.32%
VD
0.232
BBB
Penetration
0.29
Fu
4.345%
Metabolism
CYP2C19 substrate
CYP2C19 inhibitor
0.419
0.786
CYP2C9 inhibitor
0.917
CYP2C9 substrate
0.796
CYP2D6 inhibitor
0.578
CYP2D6 substrate
0.461
CYP3A4 inhibitor
0.67
Excretion
CL
8.93
T1/2
0.537
Rat Acute Oral Toxicity
0.034
The molecule satisfied the conditions mentioned in Lipinski's rule and has good ADMET properties; thus, MOQTA could be orally active in humans.
4 Conclusions
Herein, a straightforward synthesis of a new quinoxaline derivative MOQTA is reported. The XRD analysis illustrates that the quinoxaline unit is not entirely planar, as revealed by the dihedral angle between the constituent planes, 1.33 (10)°. The optimized geometry of the molecule was obtained by the DFT method. Both bond distances and bond angles of an optimized molecule are consistent with the experimental values. Based on the theoretical calculation resuls, the oxygen atom is the main reaction site for electrophilic attack and H6 atom is the most important reaction site of the nucleophilic attack. Hirshfeld surface analysis study also confirms the regions which are active for intermolecular interactions. In the 2D fingerprint plot, NH / HN and OH / HO interactions are present like two distinct spikes and CH / HC interactions are seen like the lower right-wing and the upper left wing. The contributions of these interactions to Hirshfeld surface are 15.8% for NH / HN, 12.1% for OH / HO and 18.5% for CH / HC. The data obtained X-ray diffraction show that N6—H6N5, C4—H4N4, C10—H10AO2, C12—H12N1, C13—H13BO1, C19—H19O2, C20—H20O2 interactions are present in these active regions. The energy gap between LOMO and HUMO orbitals is 4.3824 eV. The compound MOQTA demonstrated moderate anti-diabetic activity and excellent anti-oxidant activity. Besides, molecular docking analyses were carried out to examine the binding mode between MOQTA and the two enzymes, α-glucosidase and α-amylase. Thus, this study explores, theoretical prediction, antidiabetic, antioxidant activities and docking studies of triazole-quinoxaline- N-arylacetamide hybrid system for the development of potential multifunctional medicinal candidates. Finally, physicochemical properties, as well as drug-likeness, demonstrated that MOQTA had interesting ADMET properties.
CRediT authorship contribution statement
Mohcine Missioui: . Salma Mortada: . Walid Guerrab: . Güneş Demirtaş: Writing – original draft. Joel T. Mague: . M'hammed Ansar: Conceptualization, Methodology. My El Abbes Faouzi: Supervision. E.M. Essassi: Methodology. Yassin T.H. Mehdar: Conceptualization, Writing – review & editing. Faizah S. Aljohani: Conceptualization, Writing – review & editing. Musa A. Said: Conceptualization, Writing – review & editing. Youssef Ramli: Conceptualization, Methodology, Writing – review & editing, Supervision.
Acknowledgements
Authors would like to thank Mohammed V University and the Ondokuz Mayıs University Research Fund for financial support for this study. The support of NSF-MRI Grant #1228232 for the purchase of the diffractometer and Tulane University for support of the Tulane Crystallography Laboratory is gratefully acknowledged. MAS is also thankful to AvH for its continuous support.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Y. Ramli, E. M. Essassi, Advances in synthetic approaches, functionnalization and biological properties of quinoxaline derivatives. In: Advances in Chemistry Research Ed. James C. Taylor Nova Science Publishers, New York. 27 (2015) 109–160.
- Y. Ramli, A. Moussaif, K. Karrouchi, E. M. Essassi, Pharmacological profile of quinoxalinone. J. Chem. 2014 Article ID 563406 (2014) 1–21. 10.1155/2014/563406.
- N. Abad, Y.Ramli, W. Ettahiri, S. Ferfra, E. M. Essassi, Quinoxaline derivatives: syntheses, reactivities and biological properties les derives de la quinoxaline: syntheses, reactivites et proprietes biologiques, J. Mar. Chim. Heterocycl., 19 (2020) 1-12. 10.48369/IMIST.PRSM/jmch-v19i2.22352.
- M. Allali and A. Zarrouk T. Laabaissi, F. Benhiba, Z. Rouifi, M. Missioui, K. Ourrak, H. Oudda, Y. Ramli, I. Warad, New quinoxaline derivative as a green corrosion inhibitor for mild steel in mild acidic medium: Electrochemical and theoretical studies, Int. J. Corros. Scale Inhib.8 (2019) 241–256. 10.17675/2305-6894-2019-8-2-6.
- H. Lgaz, Y. ELaoufir, Y. Ramli, M. Larouj, H. Zarrok, R. Salghi, A. Zarrouk, A. Elmidaoui, A. Guenbour, E.M. Essassi, H. Oudda, Synergistic effect of potassium iodide with (E)-3-(4 methoxystyryl) quinoxalin-2 (1H)-one on the corrosio n inhibition of carbon steel in 1.0 M HCl, Der Pharma Chemica 7 (2015) 36-45, http://derpharmachemica.com/archive.html.
- Coupling of chemical, electrochemical and theoretical approach to study the corrosion inhibition of mild steel by new quinoxaline compounds in 1 M HCl. Heliyon. 2020;6:e03939
- [CrossRef] [Google Scholar]
- Arch Pharm Res. 2003;26(2):107-113.
- [CrossRef]
- Pharmaceuticals. 2010;3(8):2416-2425.
- [CrossRef]
- Ind J Chem. 2008;47(3):439-448.
- Bioorg. Med. Chem. Lett.. 2006;16(4):815-820.
- [CrossRef]
- Med. Chem. Res.. 1992;2:467-473.
- Med. Chem. Res.. 2009;18(9):782-797.
- [CrossRef]
- M. Loriga, S. Piras, P. Sanna, Paglietti (1997) Farmaco 52(3):157–166.
- AIDS Res. Hum. Retroviruses. 2000;16(6):517-528.
- Med. Chem.. 2006;2:113-122.
- [CrossRef]
- New 3-methylquinoxaline-2-carboxamide 1,4-di-n-oxide derivatives as anti- mycobacterium tuberculosis agents. Bioorg Med Chem. 2010;18:2713-2719.
- [CrossRef] [Google Scholar]
- Farmaco. 2001;56(12):933-938.
- [CrossRef]
- J Med Chem. 2004;47:1997-2009.
- [CrossRef]
- Synthesis and pharmacological evaluation of substituted 5-[4-[2-(6,7-dimethyl-1,2,3,4-tetrahydro-2-oxo-4-quinoxalinyl)ethoxy]phenyl]methylene]thiazolidine-2,4-dione derivatives as potent euglycemic and hypolipidemic agents. Bioorg. Med. Chem. Lett.. 2005;15:1019-1022.
- [CrossRef] [Google Scholar]
- Understanding the Adsorption of Quinoxaline Derivatives as Corrosion Inhibitors for Mild Steel in Acidic Medium: Experimental, Theoretical and Molecular Dynamic Simulation Studies. J. Steel. Struct. Constr.. 2016;2:1000111J.
- [CrossRef] [Google Scholar]
- Int. J. Electrochem. Sci.. 2012;7:8958-8973.
- J. Mater. Envir. Sci.. 2010;1:1-8.
- 2,3-Quinoxalinedione as a novel corrosion inhibitor for mild steel in 1 M HCl. Mater. Chem. Phys.. 2007;105:1-5.
- [CrossRef] [Google Scholar]
- Electrochim. Acta. 2012;30:405-417.
- J. Mol. Liq.. 2016;221:815-832.
- J. Mater. Environ. Sci.. 2016;7:4330-4347.
- Eur. J. Med. Chem.. 2009;44:117.
- [CrossRef]
- Triazole derivatives with antifungal activity: a pharmacophore model study. Acta Poloniae Pharmaceutica n Drug Research. 2008;65:795.
- [Google Scholar]
- Synthesis and in vitro antibacterial evaluation of N-[5-(5-nitro-2-thienyl)-1,3,4-thiadiazole-2-yl] piperazinyl quinolones. Eur. J. Med. Chem.. 2003;38:851.
- [CrossRef] [Google Scholar]
- J. Med. Chem.. 2009;23(8):927.
- Docking of Disordered Independent Molecules of Novel Crystal Structure of (N-(4-methoxyphenyl)-2-(3-methyl-2-oxo-3,4-dihydroquinoxalin-1(2H)-yl)acetamide as anti-Covid-19 and anti-Alzheimer's disease. Crystal structure, HSA/DFT/XRD, Journal of Molecular Structure. 2022;1247:131420
- [CrossRef] [Google Scholar]
- Design, synthesis, and biological evaluation of novel thiazolidinone– containing quinoxaline-1,4-di-N-oxides as antimycobacterial and antifungal agents. Front. Chem.. 2020;8:598.
- [CrossRef] [Google Scholar]
- Boja Poojary, Synthesis characterization and anticancer activity studies on some Mannich bases derived from 1,2,4-triazoles. Eur. J. Med. Chem.. 2003;38:759.
- [CrossRef] [Google Scholar]
- Click chemistry reactions in medicinal chemistry: applications of the 1,3-dipolar cycloaddition between azides and alkynes. Med. Res. Rev.. 2008;28:278e308.
- [CrossRef] [Google Scholar]
- Synthesis, biological activity, and molecular modeling studies of 1H–1,2,3-triazole derivatives of carbohydrates as a-glucosidases inhibitors. J. Med. Chem.. 2010;53:2364e2375.
- [CrossRef] [Google Scholar]
- 1,2,3-Triazole in heterocyclic compounds, endowed with biological activity, through 1,3-dipolar cycloadditions. Eur. J. Org. Chem.. 2014;16:3289e3306.
- [CrossRef] [Google Scholar]
- M. Nazir, M. A. Abbasi, Aziz-ur-Rehman , S. Z. Siddiqui , K. M. Khan, U. Salar , M. Shahid , M. Ashraf , M. A. Lodhi , F. A. Khan, Bioorganic Chemistry 81(2018) 253-263. 10.1016/j.bioorg.2018.08.010.
- S. Afr. J. Bot.. 2020;133:151-160.
- [CrossRef]
- M. Alomari, M. Taha, F.Rahim, M. Selvaraj, N. Iqbal, S. Chigurupati , S. Hussain, N. Uddin, N. B. Almandil, M. Nawaz, R. K. Farooq, K. M. Khan, Synthesis of indole-based-thiadiazole derivatives as a potent inhibitor of α-glucosidase enzyme along with in silico study, Bioorgan. Chem. 108(2021)104638. 10.1016/j.bioorg.2021.104638.
- J. Mol. Struct. 2021
- [CrossRef]
- Carbohydr. Res.. 2020;494:108069
- [CrossRef]
- Novel antioxidant quinoxaline derivative: Synthesis, crystal structure, theoretical studies, antidiabetic activity and molecular docking study. J. Mol. Struct.. 2021;1239:130484
- [CrossRef] [Google Scholar]
- Synthesis, molecular docking and a-glucosidase inhibition of 2-((5,6-diphenyl-1,2,4-triazin-3-yl) thio)-N-arylacetamides. Bioorg. Med. Chem. Lett.. 2017;27:1115-1118.
- [CrossRef] [Google Scholar]
- Bioorg. Chem.. 2020;102:104071
- [CrossRef]
- R. M. Asath, T.N. Rekha, S. Premkumar, T. Mathavan, A. Milton Franklin Beni, Journal of Molecular Structure 1125(2016) 633–642. 10.1016/j.molstruc.2016.07.064.
- G-J. Ye, T. Lan, Z-X. Huang, X-N Cheng, C-Y. Cai, S-M. Ding, M-L. Xie, B. Wang, Eur. J. Med. Chem. 177 (2019) 362e373, 10.1016/j.ejmech.2019.05.045.
- Synthesis, structural characterisation and theoretical studies of a novel pyridazine derivative: Investigations of anti-inflammatory activity and inhibition of α-glucosidase. J. Mol. Struct.. 2021;1234:130177
- [CrossRef] [Google Scholar]
- Ramli Synthesis, crystal structure, DFT calculations, Hirshfeld surface analysis, energy frameworks, molecular dynamics and docking studies of novel isoxazolequinoxaline derivative (IZQ) as anti-cancer drug. J. Mol. Struct.. 2021;1232:130004
- [CrossRef] [Google Scholar]
- Synthesis and crystal structure of 1-octyl-3-phenylquinoxalin-2 (1H)-one, C22H26N2O. Zeitschrift für Kristallographie-New Crystal Structures. 2021;236:173-175.
- [CrossRef] [Google Scholar]
- Synthesis and crystal structure of 2-azido-N-phenylacetamide, C8H8N4O. Zeitschrift für Kristallographie-New Crystal Structures. 2021;236:133-134.
- [CrossRef] [Google Scholar]
- Synthesis of a novel phenytoin derivative: crystal structure, Hirshfeld surface analysis and DFT calculations. J. Mol. Struct.. 2019;1205:127630
- [CrossRef] [Google Scholar]
- W. Guerrab, M.Jemli, J. Akachar, G.Demirtaş , J.T. Mague, A. Ibrahimi, Jamal Taoufik, M. Ansar, K. Alaoui & Y. Ramli, Design, synthesis, structural and molecular characterization, toxicity, psychotropic activity and molecular docking evaluation of a novel phenytoin derivative: 3-decyl-5,5-diphenylimidazolidine-2,4-dione, Journal of Biomolecular Structure and Dynamics, In press, 10.1080/07391102.2021.1922096.
- Liebigs Ann. Chem.. 1887;237:1228.
- Bruker APEX3, SAINT, SADABS & SHELXTL, Bruker AXS, Inc., Madison, WI. (2016).
- Acta Cryst. A. 2015;71:3-8.
- [CrossRef]
- Acta Cryst. C. 2015;71:3-8.
- [CrossRef]
- DIAMOND. Bonn, Germany: Crystal Impact GbR; 2012.
- G. M. Sheldrick, Univ. of Göttingen, Göttingen, Germany (2008).
- Bruker-AXS, LLC, Madison, WI, USA (2019).
- TWINABS. Göttingen, Germany: University of Göttingen; 2009.
- Screening culinary herbs for antioxidant and α-glucosidase inhibitory activities. Int J Food Sci Technol. 2013;48:1884-1891.
- [CrossRef] [Google Scholar]
- New styrylquinoxaline: synthesis, structural, biological evaluation, ADMET prediction and molecular docking investigations. J. Biomol. Struct. Dyn. 2022
- [CrossRef] [Google Scholar]
- Antioxidant and α -Amylase Inhibitory Property of Phyllanthus virgatus L.: An In Vitro and Molecular Interaction Study. Biomed Res. Int.. 2013;2013:1-12.
- [CrossRef] [Google Scholar]
- Antioxidant capacity and vasodilatory properties of Mediterranean food: The case of Cannonau wine, myrtle berries liqueur and strawberry-tree honey. Food Chem.. 2013;140:686-691.
- [CrossRef] [Google Scholar]
- J.R.C. M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, S.S.I. J.A. Montgomery, Jr., T. Vreven, K.N. Kudin, J.C. Burant, J.M. Millam, G.A.P. J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, T. H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, H.P.H. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J.E. Knox, A.J. J.B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, G.A. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, P.Y. Ayala, K. Morokuma, A.D.D. Voth, P. Salvador, J.J. Dannenberg, V.G. Zakrzewski, S. Dapprich, J.B.F. M.C. Strain, O. Farkas, A.D. Rabuck, D.K. Malick, K. Raghavachari, A. J.V. Ortiz, Q. Cui, A.G. Baboul, S. Clifford, J. Cioslowski, B.B. Stefanov, G. Liu, M.A.A.-L. Liashenko, P. Piskorz, I. Komaromi, D.J. Fox, R.L. Martin, T. Keith, W.C. C.Y. Peng, A. Nanayakkara, B. Johnson, M. Challacombe, P.M.W. Gill, J.A.P. M.W. Wong, C. Gonzalez, Gaussian 03, Revision C.02, Gaussian, Inc., Wallingford CT, (2004).
- Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys.. 1993;98:5648-5652.
- [CrossRef] [Google Scholar]
- Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B.. 1988;37:785-789.
- [CrossRef] [Google Scholar]
- Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett.. 1989;157:200-206.
- [CrossRef] [Google Scholar]
- J.A. Pople, P. V. Schleyer, W.J. Hehre, L. Radom, AB INITIO molecular orbital theory, 1986. 10.1016/s0022-328x(00)99651-7.
- Æ. Frisch, R.D. Dennington II, T.A. Keith, J. Millam, A.B. Nielsen, A.J. Holder, J. Hiscocks, GaussView 4, (2007). www.gaussian.com.
- A novel definition of a molecule in a crystal. Chem. Phys. Lett.. 1997;267:215-220.
- [CrossRef] [Google Scholar]
- Visualising intermolecular interactions in crystals: naphthalene vs. terephthalic acid. Chem. Commun. 1998:2071-2072.
- [CrossRef] [Google Scholar]
- Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm. 2002;4:378-392.
- [CrossRef] [Google Scholar]
- D. S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, J. and M.A. Spackman, CrystalExplorer (Version 3.1), Univ. West. Aust. (2012). 10.1039/b704980c.
- Order and disorder: differential structural impacts of myricetin and ethyl caffeate on human amylase, an antidiabetic target. J. Med. Chem. ACS Publications. 2012;55:10177-10186.
- [Google Scholar]
- V. Roig-Zamboni, et al. ‘Structure of human lysosomal acid α-glucosidase–a guide for the treatment of Pompe disease’, Nature communications. Nature Publishing Group, 8(1), pp. 1–10.
- AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem.. 2010;31(2017):455-461.
- [CrossRef] [Google Scholar]
- Human lysosomal alpha-glucosidase Characterization of the catalytic site’. J. Biol. Chem.. 1991;266(21):13507.
- [CrossRef] [Google Scholar]
- Structure of human lysosomal acid α-glucosidase–a guide for the treatment of Pompe disease. Nature communications. Nature Publishing Group. 2017;8(1):1-10.
- [CrossRef] [Google Scholar]
- Y. Ramli, H. Benzeid, R. Bouhfid, Y. Kandri Rodi, S. Ferfra, E.M. Essassi, Synthesis, reactivity and biological activity of quinoxalin-2-one derivatives, Studiişi Cercetări Ştiinţifice Chimieşi Inginerie Chimică, Biotehnologii, Industrie Alimentară 11 (2010) 67-90, CSCC6201011V01S01A0005 [0002970].
- Synthesis of xanthone derivatives with extended p systems as a-glucosidase inhibitors: insight into the probable binding mode. Bioorg. Med. Chem.. 2007;15:2810e2814.
- [CrossRef] [Google Scholar]
- Toward potent aglucosidase inhibitors based on xanthones: a closer look into the structureactivity correlations. Eur. J. Med. Chem.. 2011;46:4050e4055.
- [CrossRef] [Google Scholar]
- Synthesis and biological evaluation of novel 2,4,5-triarylimidazole-1,2,3-triazole derivatives via click chemistry as α-glucosidase inhibitors. Bioorg. Med. Chem. Lett. 2016;26:5719e5723.
- [CrossRef] [Google Scholar]
- Synthesis, in vitro evaluation and molecular docking studies of novel triazine-triazole derivatives as potential α-glucosidase inhibitors. Eur. J. Med. Chem.. 2017;125:423e429.
- [CrossRef] [Google Scholar]
- A newly synthesized nitrogen-rich derivative of bicyclic quinoxaline—Structural and conceptual DFT reactivity study. J. Phys. Org. Chem.. 2020;33:e4055
- [CrossRef] [Google Scholar]
- Synthesis of novel hybrid quinoxaline containing triazole and acetamide moieties by azide-alkyne click chemistry: Experimental and theoretical characterization. J. Mol. Struct.. 2022;1253:132132
- [CrossRef] [Google Scholar]
- Quantitative analysis of intermolecular interactions in orthorhombic rubrene. IUCrJ.. 2015;2:563-574.
- [CrossRef] [Google Scholar]
- Theoretical Analysis of the Reactive Sites of Non–steroidal Anti–inflammatory Drugs. Internet Electron. J. Mol. Des.. 2005;4:17-30.
- [Google Scholar]
- Perspective on “Electrostatic interactions of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects”. Theor. Chem. Accounts Theory, Comput. Model. (Theoretica Chim. Acta).. 2000;103:343-345.
- [CrossRef] [Google Scholar]
- Electronic Molecular Structure, Reactivity and Intermolecular Forces: An Euristic Interpretation by Means of Electrostatic Molecular Potentials. in 1978:115-193.
- [CrossRef] [Google Scholar]
- Structural elucidation, Hirshfeld surface analysis and quantum mechanical study of para-nitro benzylidene methyl arjunolate. J. Mol. Struct. 2011
- [CrossRef] [Google Scholar]
- Pharmaceutical co-crystals of pyrazinecarboxamide (PZA) with various carboxylic acids: Crystallography, hirshfeld surfaces, and dissolution study. Cryst. Growth Des. 2013
- [CrossRef] [Google Scholar]
- Frontier Orbitals and Organic Chemical Reactions. Front. Orbitals Org. Chem. React. 1979
- [CrossRef] [Google Scholar]
- Structural, DFT and biological studies on Cr(III) complexes of semi and thiosemicarbazide ligands derived from diketo hydrazide. J. Mol. Struct.. 2016;1125:788-799.
- [CrossRef] [Google Scholar]
- Evaluation of structure-reactivity descriptors and biological activity spectra of 4-(6-methoxy-2-naphthyl)-2-butanone using spectroscopic techniques. Spectrochim. Acta A. 2016;167:142-156.
- [CrossRef] [Google Scholar]
- 1,2,4-triazole-3-thione Schiff bases compounds: Crystal structure, hirshfeld surface analysis. DFT Stud. Biol. Eval.. 2020;1219:128591
- [CrossRef] [Google Scholar]
- Chemical hardness and density functional theory. J. Chem. Sci.. 2005;117:369-377.
- [CrossRef] [Google Scholar]
- Über die Zuordnung von Wellenfunktionen und Eigenwerten zu den Einzelnen Elektronen Eines Atoms. Physica.. 1934;1:104-113.
- [CrossRef] [Google Scholar]
- Absolute hardness: companion parameter to absolute electronegativity. J. Am. Chem. Soc.. 1983;105:7512-7516.
- [CrossRef] [Google Scholar]
- A. Kerflani, K. Si Larbi, A. Rabahi, A. Bouchoucha, S. Zaater, and S. Terrachet-Bouaziz, ‘Novel palladium (II) complexes with iminocoumarin ligands: Synthesis, characterisation, electrochemical behaviour, DFT calculations and biological activities, ADMET study and molecular docking’, Inorganica Chimica Acta, vol. 529, p. 120659, Jan. 2022, 10.1016/j.ica.2021.120659.
- Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3-26.
- [CrossRef] [Google Scholar]
- ‘SwissADME’. http://www.swissadme.ch/ (accessed Dec. 15, 2021).
- ‘ADMETlab 2.0’. https://admetmesh.scbdd.com/ (accessed Sep. 24, 2021).
- Molecular docking, ADMET analysis, and bioactivity studies of phytochemicals from Phyllanthus niruri as potential inhibitors of hepatitis C virus NSB5 polymerase. J. Indian Chem. Soc.. 2022;99(2):100321
- [CrossRef] [Google Scholar]
- ADMET modeling approaches in drug discovery. Drug Discovery Today. 2019;24(5):1157-1165.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2022.103851.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




