

Translate this page into:
Hybrid hetarylhydrazones and enamines of Furan-2(3H)-ones as a framework for the synthesis of poly-N-heterocycles
⁎Corresponding author. grinev@ibppm.ru (Vyacheslav S. Grinev)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Substituted furan-2(3H)-ones can act as platform compounds to obtain easily various functionalized derivatives as well as heterocycles with different heteroatoms patterns. In this study, we suppose a simple and effective way to reach poly-N-heterocycles using a set of hybrid hetarylhydrazones and enamines based on furan-2(3H)-ones as a starting material. The presence of a few reaction centers in these the hybrid furan-2(3H)-one derivatives allows them to undergo some intramolecular rearrengements with opening furan ring as well as with keeping it unaffected followed by the increase of complexity of the resulting heterocycles, depending on reaction conditions. It was found that the reaction conditions and the nature of the substituents in the hydrazone fragment affect the direction of the interaction and the nature of the resulting products. Different approaches form a framework which allowed us to create a library of substituted annelated poly-N-heterocycles with highly prominent biological effects. Analysis of the possible biological effects was performed in silico which allows us to reveal leading structures among all synthesized compounds.
Keywords
Hetarylhydrazones
Enamines
Furan-2(3H)-one
Intramolecular cyclocondensation
Poly-N-heterocycles

1 Introduction
Recently, organic chemistry tends to operate with different hybrid structures consisted of two various parts connected by a ‘spacer’ or ‘linker’ which can significantly vary physicochemical properties of studied compounds as well as their biological activity (Pawełczyk et al., 2020). The design of such hybrid systems along with the structure–activity relationships investigations are crucial challenges of the modern synthetic chemistry. Much attention is paid to the study of the biological activity of compounds containing both furan-2(3H)-ones and heterocyclic rings connected via various ‘spacers’ and capable of further modifications through different reactions (Behr et al., 1967; Karci, 2005; Ledenyova et al., 2014; Maiorova et al., 2015a; Maiorova et al., 2015b; Mayorova and Egorova, 2015; Maksimov et al., 2015; Mokhonova et al., 2018). There are two simple ways to obtain these compounds using 5-aryl substituted furan-2(3H)-ones as initial substrates. The first one is an azo coupling reaction of aryldiazonium salts, and the second one is a three-component interaction using an orthoester and an amine under mild conditions both with 5-aryl substituted furan-2(3H)-ones giving rise corresponding hydrazones having anti-inflammatory and analgesic activity (Ledenyova et al., 2015; Butler, 1975; Elnagdi et al., 1983; Makino et al., 1999; Elmaati and El-Taweel, 2004; Deeb and Kotb, 2004) and push–pull enamines (Clark-Lewis and Thompson, 1959; Dorofeenko et al., 1974), respectively, which may be of interest for the investigation of their complexation with transition metal ions (Boyd et al., 1986; Burlov et al., 2015; Ollinger et al., 1975; Bünz et al., 1993). Both hydrazones and enamines based on furan-2(3H)-ones are of interest for studying of their Z/E-tautomeric equilibrium as well as for the creation of molecular switches (Ospiov et al., 2017).
The most preparatively available and synthetically convenient representatives of the class of hydrazones are the hydrazo compounds based on acyclic and aromatic compounds. Acyclic hydrazones are best known, they are convenient synthons in the synthesis of various cyclic systems. They can react with nucleophilic, electrophilic reagents, cycloadditions, and they are capable of various intramolecular cyclization processes and rearrangements (Karci, 2005; Ledenyova et al., 2014; Maiorova et al., 2015a; Maiorova et al., 2015b; Mayorova and Egorova, 2015; Maksimov et al., 2015). Compounds containing a heterocyclic substituent in combination with a hydrazone moiety are significantly less studied and often difficult to obtain.
The interaction of 3-arylhydrazonofuran-2(3H)-ones with nucleophiles was studied. It was shown that the direction of heterocyclization is determined by the polarity of the solvent (Maksimov et al., 2015). The behavior of 3-arylhydrazone-substituted furan-2(3H)-ones under the conditions of acidic and basic catalysis was studied. The influence of the process conditions on the direction of heterocyclization, the nature of the products formed was noticed. It was found that under Fischer conditions a rearrangement for these systems is not implemented, while an intramolecular recyclization via the ANRORC mechanism is observed, the furan ring is opened with subsequent intramolecular C,N-heterocyclization and the formation of the pyrazole ring (Maksimov et al., 2015) (Scheme 1).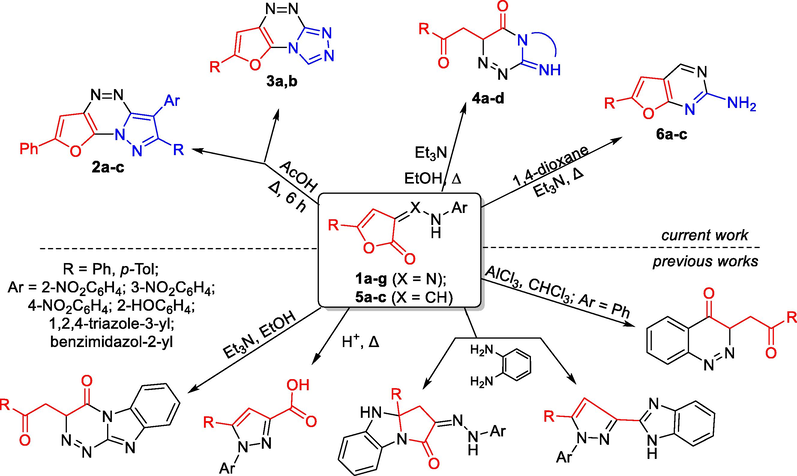
General scheme of syntheses based on (het)arylhydrazones and enaminones.
Taking into account the presence in structures of hydrazones and enaminones of highly reactive nucleophilic moieties and groups and the presence of an electrophilic center at carbon atom of the carbonyl group in the furanone ring, several directions of the reaction can be expected depending on the nature of medium and catalyst. Of particular interest are furanones containing a hydrazone or an enamine fragment in their structure, and various poly-N-heterocyclic systems can be obtained by the transformation of these molecules. On the other hand, hydrazones and enamines with typical for such systems tautomerism can bring some stereochemical features to the obtained compounds.
Considering all mentioned features of studied hetarylhydrazones and enaminones we can propose this approach as a framework to obtain poly-N-heterocycles of various structures. In this paper, we provide full NMR spectral characteristics of synthesized compounds as well as their chemoinformatically predicted properties and possible biological activity.
2 Materials and methods
2.1 General analytic techniques
All chemicals purchased from Sigma Aldrich (USA) were of reagent grade and were used without further purification. Analytical TLC was performed using Alugram Sil G UV254 plates (Macherey–Nagel GmbH & Co. KG, Germany) plates (hexane–ethyl acetate–chloroform, 2:2:1; development with iodine vapor). Melting point was determined on a Stuart™ SMP10 melting point apparatus (Cole-Parmer, UK) in open capillaries. The elemental analyses were obtained on a Vario Micro cube Elementar CHNS analyzer (Elementar Analysensysteme GmbH, Germany). FTIR spectra were recorded as KBr pellets on a Nicolet 6700 FTIR spectrophotometer (Thermo Scientific, USA) in the 4000–400 cm−1 range with a spectral resolution of 4 cm−1. The 1H and 13C NMR spectra were recorded at rt on a Varian-400 (Agilent Technologies, USA) spectrometer (400 and 100 MHz, respectively) using CDCl3 as solvent and tetramethylsilane (TMS) as internal standard.
2.2 Synthesis of 6-(4-halophenyl)-2-phenylfuro[2,3-e]pyrazolo[5,1-c][1,2,4]triazines (2a-c)
The samples of corresponding hydrazones 1a-c (10 mmoles) were dissolved in 40–50 mL of glacial acetic acid in a 100 mL round bottom flask equipped with a Liebig reflux condenser and boiled for 6 h. The solution was slowly cooled; the precipitate formed was filtered off, washed with 50 mL of cold 2-propanol, and dried to constant weight. If necessary, it was recrystallized from ice-cold acetic acid.
2.2.1 6-(4-Chlorophenyl)-7-methyl-2-phenylfuro[2,3-e]pyrazolo[5,1-c][1,2,4]triazine (2a)
Yield: 73%. Brown crystals; m.p.: 101–103 °C; FTIR (KBr), ν, cm−1: 1516 (C⚌Ncycl.), 1608 (N⚌N), 1640 (C⚌C); 1H NMR: δ 2.15 (s, 3H), 7.35–7.46 (m, 5H), 7.80 (d, 2H, J = 7.9 Hz), 8.06 (d, 2H, J = 7.9 Hz), 8.45 (s, 1H); 13C NMR: δ 15.0, 103.7, 103.9, 110.8, 125.8, 125.9, 127.0, 127.1, 128.9, 129.0, 129.5, 130.4, 130.5, 131.0, 136.2, 136.3, 140.5, 146.6, 151.7, 157.5; Anal. calcd. for C20H13ClN4O: C 66.67; H 3.61; N 15.56; found, %: C 66.79; H 3.64; N 15.62.
2.2.2 6-(4-Bromophenyl)-7-ethyl-2-phenylfuro[2,3-e]pyrazolo[5,1-c][1,2,4]triazine (2b)
Yield: 75%. Brown crystals; m.p.: 95–97 °C; FTIR (KBr), ν, cm−1: 1521 (C⚌Ncycl.), 1600 (N⚌N), 1638 (C⚌C); 1H NMR: δ 1.15 (t, 3H, J = 4.9 Hz), 2.75 (q, 2H, J = 5.0 Hz), 7.11–7.53 (m, 5H), 7.85 (d, 2H, J = 8.1 Hz), 8.19 (d, 2H, J = 8.1 Hz), 8.67 (s, 1H); 13C NMR: δ 12.8, 22.5, 103.5, 103.6, 111.6, 122.1, 125.7, 125.9, 127.3, 129.0, 129.8, 130.4, 131.0, 132.0, 139.2, 143.2, 156.1, 165.7; Anal. calcd. for C21H15BrN4O: C 60.14; H 3.58; N 13.37; found, %: C 60.29; H 3.53; N 13.12.
2.2.3 6-(4-Fluorophenyl)-2-phenylfuro[2,3-e]pyrazolo[5,1-c][1,2,4]triazine (2c)
Yield: 77%. Brown crystals; m.p.: 105–107 °C; FTIR (KBr), ν, cm−1: 1524 (C⚌Ncycl.), 1610 (N⚌N), 1637 (C⚌C); 1H NMR: δ 6.52 (s, 1H), 7.10–7.50 (m, 5H), 8.12 (d, 2H, J = 8.2 Hz), 8.26 (d, 2H, J = 8.2 Hz), 8.60 (s, 1H); 13C NMR: δ 103.6, 115.9, 116.9, 125.2, 127.2, 128.9, 129.0, 130.3, 130.5, 130.6, 130.9, 131.0, 139.9, 143.2, 147.0, 147.1, 157.3, 163.1; Anal. calcd. for C19H11FN4O: C 69.09; H 3.36; N 16.96; found, %: C 69.19; H 3.44; N 16.63.
2.3 Synthesis of 2-arylfuro[2,3-e][1,2,4]triazolo[3,4-c][1,2,4]triazines (3a,b)
The method described above for compounds 2a-c was used with hydrazones 1d,e as a starting material.
2.3.1 2-Phenylfuro[2,3-e][1,2,4]triazolo[3,4-c][1,2,4]triazine (3a)
Yield: 45%. Brown crystals; m.p.: 137–139 °C; FTIR (KBr), ν, cm−1: 1524 (C⚌Ncycl.), 1537 (C⚌Ncycl.), 1602 (N⚌N), 1635 (C⚌C), 1642 (C⚌C); 1H NMR (400 MHz, CDCl3) δ 7.20 (s, 1H), 7.98 (s, 1H), 7.51–7.69 (m, 5H); 13C NMR: δ 101.9, 125.3, 127.2, 129.0, 130.3, 130.9, 139.9, 144.2, 149.3, 154.5; Anal. calcd. for C12H7N5O: C 60.76, H 2.97, N 29.52; found: C 61.00, H 3.18, N 29.88.
2.3.2 2-(p-Tolyl)furo[2,3-e][1,2,4]triazolo[3,4-c][1,2,4]triazine (3b)
Yield: 42%. Brown crystals; m.p.: 152–154 °C; FTIR (KBr), ν, cm−1: 1520 (C⚌Ncycl.), 1531 (C⚌Ncycl.), 1612 (N⚌N), 1630 (C⚌C), 1646 (C⚌C); 1H NMR (400 MHz, CDCl3) δ 2.48 (s, 3H), 7.26 (s, 1H), 7.94 (s, 1H), 7.49–7.58 (m, 4H); 13C NMR: δ 21.4, 102.2, 125.6, 127.2, 129.2, 130.1, 130.9, 139.5, 144.1, 149.2, 154.7; Anal. calcd. for C13H9N5O: C 62.15, H 3.61, N 27.87; found: C 62.51, H 3.82, N 27.75.
2.4 Synthesis of 6-(2-oxo-2-arylethyl)-[1,2,4]hetaryl[1,2,4]triazinones (4a-d)
The samples of corresponding hydrazones 1d-g (3.92 mmol) was placed in a 100 mL round bottom flask equipped with a Liebig reflux condenser, and 20 mL of ethanol and catalytic amounts of triethylamine were added. The reaction mixture was heated for 5 h. The content of the flask was then acidified with hydrochloric acid to pH 7 followed by subsequent filtering off and recrystallization from ethanol.
2.4.1 6-(2-Oxo-2-phenylethyl)-[1,2,4]triazolo[3,4-c][1,2,4]triazin-5(6H)-one (4a)
Yield: 87%. Brown crystals; m.p.: 102–103 °C; FTIR (KBr), ν, cm−1: 1538 (C⚌N), 1614 (N⚌N), 1716 (C⚌O), 1784 (C⚌O); 1H NMR (400 MHz, CDCl3) δ 4.01 (d, 1H, J = 8.4 Hz), 4.51 (d, 1H, J = 8.4 Hz), 6.15 (t, 1H, J = 11.8 Hz), 7.49–8.14 (m, 5H), 9.77 (s, 1H); 13C NMR: δ 38.4, 56.8, 128.9, 129.0, 154.3, 157.1, 167.5, 198.8; Anal. calcd. for C12H9N5O2: C 56.47, H 3.55, N 27.44; found: C 56.83, H 3.39, N 27.12.
2.4.2 6-[2-Oxo-2-(p-tolyl)ethyl]-[1,2,4]triazolo[3,4-c][1,2,4]triazin-5(6H)-one (4b)
Yield: 86%. Brown crystals; m.p.: 115–116 °C; FTIR (KBr), ν, cm−1: 1536 (C⚌N), 1612 (N⚌N), 1716 (C⚌O), 1782 (C⚌O); 1H NMR (400 MHz, CDCl3) δ 2.34 (s, 3H), 3.98 (d, 1H, J = 8.2 Hz), 4.52 (d, 1H, J = 8.2 Hz), 6.15 (t, 1H, J = 11.8 Hz), 7.30 (d, 2H, J = 8.0 Hz), 8.11 (d, 2H, J = 8.0 Hz), 9.78 (s, 1H); 13C NMR: δ 21.4, 38.3, 57.1, 128.7, 129.2, 141.8, 154.5, 157.1, 167.2, 198.9; Anal. calcd. for C13H11N5O2: C 57.99, H 4.12, N 26.01; found: C 58.21, H 4.40, N 25.84.
2.4.3 3-(2-Oxo-2-phenylethyl)benzo[4,5]imidazo[2,1-c][1,2,4]triazin-4(3H)-one (4c)
Yield: 83%. Brown crystals; m.p.: 117–118 °C; FTIR (KBr), ν, cm−1: 1525 (C⚌N), 1610 (N⚌N), 1720 (C⚌O), 1780 (C⚌O); 1H NMR (400 MHz, CDCl3) δ 4.00 (d, 1H, J = 8.1 Hz), 4.52 (d, 1H, J = 8.1 Hz), 6.15 (t, 1H, J = 11.9 Hz), 7.51–7.69 (m, 9H); 13C NMR: δ 38.6, 57.5, 115.9, 116.9, 125.3, 128.8, 129.0, 133.3, 136.7, 142.1, 167.3, 198.8; Anal. calcd. for C17H12N4O2: C 67.10, H 3.98, N 18.41; found: C 66.97, H 4.01, N 18.00.
2.4.4 3-(2-Oxo-2-(p-tolyl)ethyl)benzo[4,5]imidazo[2,1-c][1,2,4]triazin-4(3H)-one (4d)
Yield: 82%. Brown crystals; m.p.: 120–121 °C; FTIR (KBr), ν, cm−1: 1528 (C⚌N), 1614 (N⚌N), 1718 (C⚌O), 1782 (C⚌O); 1H NMR (400 MHz, CDCl3) δ 2.35 (s, 3H), 3.99 (d, 1H, J = 8.0 Hz), 4.51 (d, 1H, J = 8.0 Hz), 6.16 (t, 1H, J = 12.0 Hz), 7.50–7.71 (m, 8H); 13C NMR: δ 21.3, 38.4, 57.6, 115.2, 115.9, 126.3, 128.8, 129.1, 133.7, 136.7, 142.2, 167.5, 199.1; Anal. calcd. for C18H14N4O2: C 67.91, H 4.43, N 17.60; found: C 68.03, H 4.26, N 17.38.
2.5 Synthesis of 6-arylfuro[2,3-d]pyrimidin-2-amine (6a-c)
The samples of corresponding enamines 5a-c (3.46 mmol) and 20 mL of 1,4-dioxane were placed in a round bottom flask equipped with a Liebig reflux condenser, and the reaction mixture was boiled for 2 h. The precipitated crystals were filtered off, washed with 1,4-dioxane and dried.
2.5.1 6-Phenylfuro[2,3-d]pyrimidin-2-amine (6a)
Yield: 76%. White crystals; m.p.: 187–189 °C; FTIR (KBr), ν, cm−1: 1242 (C—O—C), 1515 (C⚌N), 3319 (NH2); 1H NMR (400 MHz, CDCl3) δ 6.94 (s, 1H), 7.65–8.10 (m, 5H), 8.65 (s, 1H), 9.26 (s, 2H); 13C NMR: δ 104.7, 111.5, 126.4, 129.1, 131.1, 132.2, 149.8, 153.9, 154.3, 166.7; Anal. calcd. for C13H11N3O: C 68.24; H 4.92; N 19.89; found: C 68.52H 4.83; N 19.59.
2.5.2 6-(p-Tolyl)furo[2,3-d]pyrimidin-2-amine (6b)
Yield: 73%. White crystals; m.p.: 190–192 °C; FTIR (KBr), ν, cm−1: 1242 (C—O—C), 1517 (C⚌N), 3320 (NH2); 1H NMR (400 MHz, CDCl3) δ 2.35 (s, 3H), 6.93 (s, 1H), 7.28 (d, 2H, J = 8.6 Hz), 7.88 (d, 2H, J = 8.6 Hz), 8.66 (s, 1H), 9.24 (s, 2H); 13C NMR: δ 21.4, 104.4, 110.5, 126.4, 129.1, 131.1, 132.1, 149.8, 154.4, 161.2, 168.6; Anal. calcd. for C13H11N3O: C 69.32; H 4.92; N 18.66; found: C 68.92H 4.82; N 18.99.
2.5.3 6-(4-Bromophenyl)furo[2,3-d]pyrimidin-2-amine (6c)
Yield: 78%. White crystals; m.p.: 185–188 °C; FTIR (KBr), ν, cm−1: 1242 (C—O—C), 1515 (C⚌N), 3000–2950 (CH3), 3318 (NH2); 1H NMR (400 MHz, CDCl3) δ 6.95 (s, 1H), 7.55 (d, 2H, J = 8.7 Hz), 7.78 (d, 2H, J = 8.6 Hz), 8.67 (s, 1H), 9.23 (s, 2H); 13C NMR: δ 104.8, 112.4, 126.4, 129.1, 131.1, 132.1, 154.2, 154.4, 167.3; Anal. calcd. for C12H8BrN3O: C 49.68; H 2.78; N 14.48; found: C 49.72; H, 2.77; N, 14.50.
2.6 Predicted biological activity analysis
Calculation of molecular parameters such as lipophilicity (Log P), solubility in water (Log S) and topological polar surface area (TPSA) has been determined by two cheminformatic tools available online for free: Molinspiration Chemoinformatics ( https://www.molinspiration.com/services/) and OSIRIS Property Explorer ( https://www.organic-chemistry.org/prog/peo/). In particular, Molinspiration Chemoinformatics software was used to calculate Log P and TPSA values. OSIRIS Property Explorer (Organic Chemistry Portal) was used to calculate also Log P and TPSA and to determine pharmacokinetic parameters such as toxicity risks, solubility and overall drug-likeness of synthesized poly-N-heterocycles. Molecular properties were calculated to evaluate the drug-likeness (DL) and drug score (DS) which combines DL, Log P, Log S, molecular weight and toxicity in one handy value than may be used to estimate the compound's overall potential to qualify for a drug of the synthesized compounds.
3 Results and discussion
3.1 Synthesis of phenylfuro[2,3-e]pyrazolo[5,1-c][1,2,4]triazines (2a–c)
The hydrazones (1a–c) obtained by azo coupling with 5-arylfuran-2(3H)-ones, as intermediate reaction products, can be isolated and characterized instead of the reaction with six-membered azo components (Mokhonova et al., 2018). They can be considered as hybrid compounds containing two heterocyclic portions linked by a ‘spacer’, hydrazo group. Under acid catalysis, compounds 1a–c were shown to undergo intramolecular cyclocondensation reaction to form new polyheterocyclic derivatives of halophenyl substituted phenylfuro[2,3-e]pyrazolo[5,1-c][1,2,4]triazine series 2a–c. The reaction was carried out by boiling in glacial acetic acid solution for 6 h in 73–77% yields (Scheme 2).![Synthesis and representatives of the furo[2,3-e]pyrazolo[5,1-c][1,2,4]triazine series 2a–c.](/content/184/2021/14/2/img/10.1016_j.arabjc.2020.102950-fig2.png)
Synthesis and representatives of the furo[2,3-e]pyrazolo[5,1-c][1,2,4]triazine series 2a–c.
The 1H NMR spectra of the obtained compounds 2a,b show the lack of signals of hydrazone protons as well as the signals of protons at pyrazole ring. Due to the aromatic nature of the whole tricyclic system, the singlet of the furan ring proton at C-3 is significantly shifted from its typical position (at approximately 6.50 ppm) towards lower field (at 8.45–8.67 ppm). In the 13C NMR spectra the most low-field signals of the carbon atom bounded with halogen atoms are noticed at 157.5–165.1 ppm in contrast to initial hydrazones having the carbonyl carbon. The NMR spectral data allowed us to make a choice in favor of the formation of tricyclic systems 2a-c.
As a proposed mechanism of the formation of compounds 2a-c under acidic conditions, we suggested that hydrazones 1a-c undergo a protonation of the furan ring followed by the intramolecular rearrangement with subsequent nucleophilic attack of the electron pair of the nitrogen atom of the pyrazole ring at the carbon atom bearing positive charge of the carbonyl group to form bicyclic 3-(2-hydroxy-2-phenylvinyl)-7,8-disubstituted pyrazolo[5,1-c][1,2,4]triazin-4(1H)-ones, which can transform via tautomeric intramolecular proton transfer into corresponding 3-(2-hydroxy-2-phenylvinyl)-7,8-disubstituted pyrazolo[5,1-c][1,2,4]triazin-4-ols. Further, they undergo a condensation with the furan ring closure to form tricyclic systems identified by us as 6-(4-halogenophenyl)-7-alkyl-2-phenylfuro[2,3-e]pyrazolo[5,1-c][1,2,4]triazines 2a–c (Scheme 3).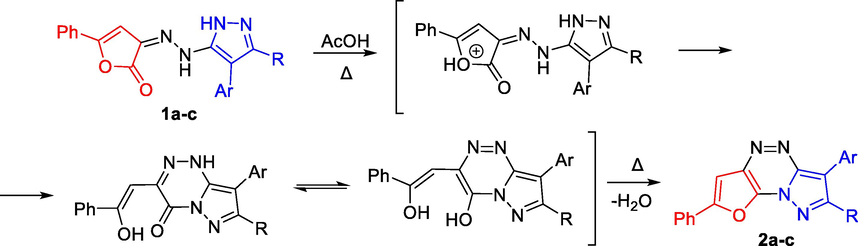
Proposed mechanism of the formation of 2a-c.
3.2 Synthesis of phenylfuro[2,3-e][1,2,4]triazolo[3,4-c][1,2,4]triazines (3a,b)
For hydrazones 1d,e bearing a 1,2,4-triazole moiety in the hydrazone part of the molecule, the formation of the tricyclic systems is also observed under acidic conditions (Maksimov et al., 2015), namely, boiling in glacial acetic acid for 6 h with the formation of phenylfuro[2,3-e][1,2,4]triazolo[3,4-c][1,2,4]triazines (3a,b) in 42–45% yields (Scheme 4).![Synthesis and representatives of the phenylfuro[2,3-e][1,2,4]triazolo[3,4-c][1,2,4]triazines 3a,b.](/content/184/2021/14/2/img/10.1016_j.arabjc.2020.102950-fig4.png)
Synthesis and representatives of the phenylfuro[2,3-e][1,2,4]triazolo[3,4-c][1,2,4]triazines 3a,b.
The spectral data of the obtained compounds are similar to those obtained on pyrazolotriazine systems, with the exception of the singlet in the region of 7.26 ppm related to the proton of the fused triazole ring, which also allows you to make a choice in favor of the formation of polyfused systems. In the 13C NMR spectra the signal of the carbon atom of the C-3 furan ring noticed at 101.9–102.2 ppm.
The proposed mechanism of the formation is completely the same as for 2a-c presented on Scheme 3.
3.3 Synthesis of [1,2,4]triazolo[3,4-c][1,2,4]triazin-5(6H)-ones (4a,b) and benzo[4,5]imidazo[2,1-c][1,2,4]triazin-4(3H)-ones (4c,d)
For the purpose to investigate the behavior of the hydrazones under basic conditions, another synthetic approach was used. The reaction under milder conditions such as in boiling ethanol with triethylamine as a catalysis leads to the formation of 2-oxo-2-arylethyl substituted [1,2,4]triazolo[3,4-c][1,2,4]triazin-5(6H)-ones (4a,b) and benzo[4,5]imidazo[2,1-c][1,2,4]triazin-4(3H)-ones (4c,d) depends on the structure of heterocyclic substituent in 82–87% yields (Scheme 5).![Synthesis of 2-oxo-2-arethyl substituted [1,2,4]triazolo[3,4-c][1,2,4]triazin-5(6H)-ones (4a,b) and benzo[4,5]imidazo[2,1-c][1,2,4]triazin-4(3H)-ones (4c,d).](/content/184/2021/14/2/img/10.1016_j.arabjc.2020.102950-fig5.png)
Synthesis of 2-oxo-2-arethyl substituted [1,2,4]triazolo[3,4-c][1,2,4]triazin-5(6H)-ones (4a,b) and benzo[4,5]imidazo[2,1-c][1,2,4]triazin-4(3H)-ones (4c,d).
The appearance in the 1H NMR spectra of characteristic signals as two doublets of magnetically non-equivalent protons of the methylene group (in ranges of 3.98–4.01 ppm and 4.51–4.52 ppm, respectively) and a triplet of the proton at the tertiary carbon atom at 6.15–6.16 ppm confirms the intramolecular heterocyclization of compounds 1d–g under conditions of basic catalysis and the formation of 2-oxo-2-phenylethyl substituted hetaryl[1,2,4]triazinones series 4a–d. In the 13C NMR spectra characteristic signals are the peaks of the carbon atoms of two carbonyl groups, cyclic ones at 167.3–167.5 ppm and exocyclic at 198.8–199.1 ppm.
Obviously, the presence of triethylamine shifts the tautomeric equilibrium towards azo form of the hydrazones and increases the nucleophilicity of the nitrogen atom in the heterocyclic moiety of hydrazones 1d-g, which allows them to attack the carbon atom of the carbonyl group, and the reaction stops at the stage of bicyclic systems with 2-oxo-2-phenylethyl substituent. Further condensation requires acidic conditions to implement the enolization followed by the second nucleophilic attack and the formation of tricyclic system, which under basic conditions cannot occur (Scheme 6).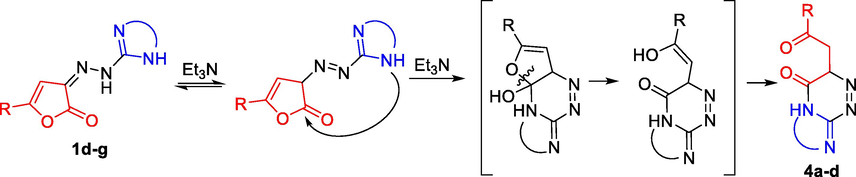
Proposed mechanism of the formation of 4a-d.
We believe that triethylamine can significantly polarize the hydrazone molecule by elongation their N-H bonds making possible the formation of a resonance structure with an electron-rich heteroring. The resulting partially negative charge can be stabilized by delocalization in hydrazoheterocyclic moiety, analogously with 3-amino-1,2,4-triazole, which exhibits similar, kind of acidic properties (Scheme 7).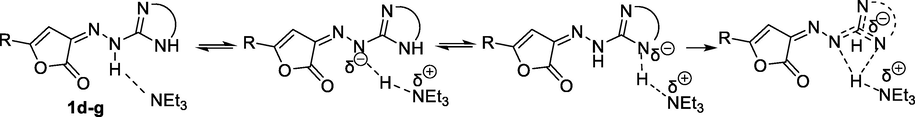
Proposed alternative mechanism of the polarization of 1d-g.
3.4 Synthesis of 6-arylfuro[2,3-d]pyrimidin-2-amines (6a-c)
As alternative substrates based on aryl substituted furan-2(3H)-ones enaminones 5a-c were used. Enaminones 5 and hydrazones 1 may be considered as partly isosterical analogues, especially in furanone ring and in the ‘spacer’ part which may be a hydrazo group as well as enamine unit. Enaminones 5 can be easily obtained by a three-component reaction of 5-arylfuran-2(3H)-one, triethyl orthoformate and corresponding amine and may contain acyclic or cyclic nitrogen containing moiety depending on the initial amine used. The transformation of enamine derivatives of furan-2(3H)-ones (5a-c) under basic conditions by refluxing them in solution of 1,4-dioxane with the presence of catalytic amounts of triethylamine lead to bicyclic furopyrimidine structures 6a-c containing a free primary amino group in 73–78% yields (Scheme 8).![Synthesis of 6-arylfuro[2,3-d]pyrimidin-2-amines 6a-c.](/content/184/2021/14/2/img/10.1016_j.arabjc.2020.102950-fig8.png)
Synthesis of 6-arylfuro[2,3-d]pyrimidin-2-amines 6a-c.
The 1H NMR spectra of the compounds 6a-c contain three singlets of the aromatic proton at 8.65–8.67 ppm, low-field slightly broaded peak of the amino group at 9.23–9.26 ppm. In the 13C NMR spectra the most low-field signal is the singlet of the carbon atom at primary amino group at 166.7–168.6 ppm.
Compounds 5a-c contain in their structure a bifunctional enamine ‘spacer’ –NH-C⚌C–, amino and imino groups. The formation of the furopyrimidine structures 6a-c implements via ANRORC mechanism. The nucleophilic attack of the nitrogen atom of the imino group at the carbon atom of the carbonyl group of the furan-2(3H)-one system with subsequent hydrogen transfer from the cyclic secondary amino group to the oxygen atom of the carbonyl and the furan ring closure accompanied by the loss of water lead to the formation of compounds 6a-c (Scheme 9).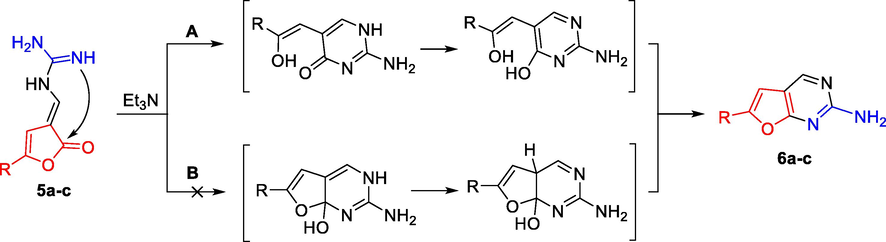
Proposed mechanism of the formation of 6a-c.
It is impossible to exclude both directions in the probable scheme of the formation of compounds 6a-c; however, in our opinion, path B without opening the furan ring cannot be implemented in the presence of a strong nucleophile hydrazine. Thus, direction A seems to be more preferable.
3.5 Predicted biological activity analysis
To assess the difference of some crucial for biological activity parameters such as bioavailability and aqueous solubility of all synthesized compounds we carried out calculations the lipophilicity indices (Log P), which are defined as the decimal logarithm of the 1-octanol/water partition coefficient and solubility indices (Log S), which reflect its absorption and distribution characteristics and defined as the decimal logarithm of the solubility measured in mol L-1. The topological polar surface area, TPSA, is a parameter, which reflects drug transport properties and correlates with the human intestinal absorption, Caco-2 monolayers permeability, and blood–brain barrier penetration very well. TPSA is defined as a sum of surfaces of polar atoms (e.g. oxygens, nitrogens and attached hydrogens) in a molecule. These characteristics for organic compounds can be predicted quite well based on their structure. To evaluate lipophilicity (MI-Log P and OPE-Log P indices) and TPSA (MI-TPSA and OPE-TPSA indices) we used two algorithms. DL and DS scores were calculated using OPE approach. The results of the calculations are presented in Table 1.
Comp-d
MW
MI-Log P
OPE-Log P
MI-TPSA
OPE-TPSA
OPE-Log S
OPE-DL
OPE-DS
2a
360.80
5.42
4.00
56.23
56.22
−7.85
0.61
0.17
2b
419.28
6.12
4.54
56.23
56.22
−8.11
−2.37
0.09
2c
330.32
4.68
3.10
56.23
56.22
−7.06
−0.64
0.16
3a
237.22
2.07
1.25
69.12
69.11
−3.57
−1.09
0.26
3b
251.25
2.52
1.59
69.12
69.11
−3.91
−1.99
0.23
4a
255.24
1.14
0.89
89.59
89.57
−3.41
−0.18
0.31
4b
269.26
1.59
1.24
89.59
89.57
−3.75
1.39
0.37
4c
304.31
3.18
2.72
76.70
76.68
−4.67
−0.87
0.22
4d
318.34
3.63
3.07
76.70
76.68
−5.02
0.75
0.27
6a
211.22
2.38
2.27
64.95
64.94
−4.74
−0.31
0.53
6b
225.25
2.83
2.62
64.95
64.94
−5.08
−1.00
0.44
6c
290.12
3.19
3.00
64.95
64.94
−5.57
−2.29
0.34
First of all, it should be noticed that the OPE algorithm returned smaller values of the Log P indices in contrast to MI, but keeping the tendencies, while the TPSA values were quite equal by these two approaches. The most lipophilic substances were found among halogen-substituted furo[2,3-e]pyrazolo[5,1-c][1,2,4]triazines 2a-c with bromine-contained derivative as a leader. Nevertheless, the higher DL as well as DS values were demonstrated by chlorine-substituted compound 2a. The lack of halophenyl substituent at the position 6 as well as the presence of the additional nitrogen atom in furo[2,3-e][1,2,4]triazolo[3,4-c][1,2,4]triazines 3a,b led to significantly increase in lipophilicity along with the TPSA values which resulted in an increase in their DS indices. A combination of minimal Log P values with maximal TPSA (near 90) among all compounds under study for 2-oxo-2-arylethyl substituted [1,2,4]triazolo[3,4-c][1,2,4]triazin-5(6H)-ones 4a,b reflects relatively high drug scores and, in less magnitude, this is true for 2-oxo-2-arylethyl substituted benzo[4,5]imidazo[2,1-c][1,2,4]triazin-4(3H)-ones 4c,d. Transition to 6-arylfuro[2,3-d]pyrimidin-2-amines 6a-c allowed us to balance all key parameters which resulted in maximal DS values among synthesized compounds 2a-6c.
4 Conclusion
A simple and effective method to construct poly-N-heterocycles using a set of hybrid hetarylhydrazones and enamines based on furan-2(3H)-ones as a platform was proposed. The presence of reaction centers in the hybrid furan-2(3H)-one derivatives makes possible to use some intramolecular rearrengements to obtain various heterocycles of different complexity, depending on reaction conditions. All spectral features as well as possible synthesis schema were discussed. A library of synthesized substituted annelated with the furan ring poly-N-heterocycles was screened for predicted lipophilicity and solubility indices as well as TPSA values using two chemoinformatic approaches. The analysis of possible biological activity reveals some leading structures of various series which allowed us to recommend them for further investigation.
Funding
The study was supported by a grant from the Russian Foundation for Basic Research (Project 19-33-60038).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Behr, L.C., Fusco, R., Jarboe, C.H., 1967. Pyrazoles and reduced and condensed pyrazoles. In: Wiley, R.H. (Ed.), Chem. Het. Comp., Vol. 22. Wiley, New York, p 888.
- Boyd, D.R., Jennings, W.B., Waring, L.C., 1986. Dynamic stereochemistry of imines and derivatives. 19. Mutarotation and E-Z isomerization of chiral imines in [2H4]methanol solution. J. Org. Chem. 51, 992–995. DOI 10.1021/jo00357a007, URL https://pubs.acs.org/doi/abs/10.1021/jo00357a007.
- E-Z isomerism of anilinomethylenemalonic acid derivatives. Magn. Reson. Chem.. 1993;31:371-374. https://onlinelibrary.wiley.com/doi/abs/10.1002/mrc.1260310412
- [CrossRef] [Google Scholar]
- Burlov, A.S., Mashchenko, S.A., Vlasenko, V.G., Lysenko, K.A., Antsyshkina, A.S., Sadikov, G.G., Sergienko, V.S., Koshchienko, Yu.V., Zubavichus, Ya.V., Uraev, A.I., Garnovskii, D.A., Korshunova, E.V., Levchenkov, S.I., 2015. Metal complexes of azomethine compounds bearing an azo group in the amine fragment: Syntheses, structures, and properties. Russ. J. Coord. Chem. 41, 376–386. doi: 10.1134/S1070328415060019, URL https://link.springer.com/article/10.1134/S1070328415060019.
- Diazotization of heterocyclic primary amines. Chem. Rev.. 1975;75:241-257. https://pubs.acs.org/doi/abs/10.1021/cr60294a004
- [CrossRef] [Google Scholar]
- Clark-Lewis, J.W., Thompson, M.J., 1959. 481. 5-Aminomethylene-1:3-dimethylbarbituric acids. J. Chem. Soc. 2401–2408. doi: 10.1039/JR9590002401, URL https://pubs.rsc.org/en/content/articlelanding/1959/jr/jr9590002401.
- Reactions of 3-diazopyrazolo[3,4-c]pyridazine with reactive methylene compounds and other groups. Heterocycles. 2004;63:1143-1151. https://www.heterocycles.jp/newlibrary/downloads/PDF/01198/63/5
- [CrossRef] [Google Scholar]
- Reaction of ethoxyvinyl derivatives of pyrylium and pyridinium salts with primary and secondary amines. Chem. Heterocycl. Compd.. 1974;10:1166-1171. https://link.springer.com/article/10.1007/BF00470156
- [CrossRef] [Google Scholar]
- New trends in the chemistry of 5-aminopyrazoles. J. Heterocycl. Chem.. 2004;41:109-134. https://onlinelibrary.wiley.com/doi/abs/10.1002/jhet.5570410201
- [CrossRef] [Google Scholar]
- Recent developments in chemistry of 3(5)-aminopyrazoles. Heterocycles. 1983;20:2437-11470. https://www.heterocycles.jp/newlibrary/downloads/PDF/10478/20/12
- [CrossRef] [Google Scholar]
- Synthesis of disazo dyes derived from heterocyclic components. Color Technol.. 2005;121:275-280. URL: https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1478-4408.2005.tb00286.x
- [CrossRef] [Google Scholar]
- The chemistry of pyrazole-3(5)-diazonium salts. Chem. Heterocycl. Compd.. 2014;50:1214-1243. https://link.springer.com/article/10.1007/s10593-014-1585-1
- [CrossRef] [Google Scholar]
- Reactions of pyrazole-3(5)-diazonium salts with 4-hydroxy-2H-chromen-2-one and isochroman-1,3-dione. Chem. Heterocycl. Compd.. 2015;51:734-737. https://link.springer.com/article/10.1007/s10593-015-1766-6
- [CrossRef] [Google Scholar]
- Maiorova, O.A., Babkina, N.V., Egorova, A.Yu., 2015. Studies of stereochemistry of 3-(arylhydrazono)furan-2(3H)-ones, synthesis of 4-(arylhydrazono)pyridazin-3(1H)-ones. Chem. Heterocycl. Compd. 51, 514–517. doi: 10.1007/s10593-015-1730-5, URL https://link.springer.com/article/10.1007/s10593-015-1730-5.
- Crystal structure of 3-(2-(2-nitrophenyl)hydrazono)-5-phenyl-3H-furan-2-one. J. Struct. Chem.. 2015;56:803-805. https://link.springer.com/article/10.1134/S0022476615040320
- [CrossRef] [Google Scholar]
- Synthesis of pyrazoles and condensed pyrazoles. J. Heterocycl. Chem.. 1999;36:321-332. https://onlinelibrary.wiley.com/doi/abs/10.1002/jhet.5570360202
- [CrossRef] [Google Scholar]
- Acid- and base-catalyzed modifications of 3-[aryl(hetaryl)hydrazinylidene]-3H-furan-2-ones. Russ. J. Org. Chem.. 2015;51:1305-1307. https://link.springer.com/article/10.1134/S107042801509016X
- [CrossRef] [Google Scholar]
- 13C and 1H NMR study of azo coupling products from diazonium salts and furan-2(3H)-ones. Magn. Reson. Chem.. 2015;53(10):853-856. https://onlinelibrary.wiley.com/doi/abs/10.1002/mrc.4270
- [CrossRef] [Google Scholar]
- Reactions of 3H-furan-2-ones and 2H-chromen-2-ones with pyrazole-3(5)-diazonium salts. Heterocycl. Commun.. 2018;24(4):183-185. https://www.degruyter.com/view/journals/hc/24/4/article-p183.xml
- [CrossRef] [Google Scholar]
- Darstellung, E/Z-Isomerie und gehinderte Rotation an N-substituierten Aminomethylen-chromandionen,-pyrandionen und -pyridindionen. Monatsh. Chem.. 1975;106:963-971. https://link.springer.com/article/10.1007/BF00900875
- [CrossRef] [Google Scholar]
- Osipov, A.K., Anis’kov, A.A., Grinev, V.S., Yegorova, A.Yu., 2017. Study of E/Z isomerization of (arylamino)methylidenefuran‐2(3H)‐ones by 1H, 13C, 15N spectroscopy and DFT calculations in different solvents. Magn. Reson. Chem. 55, 730–737. doi: 10.1002/mrc.4583, URL https://onlinelibrary.wiley.com/doi/abs/10.1002/mrc.4583.
- Linked drug-drug conjugates based on triterpene and phenol structures. Rational synthesis, molecular properties, toxicity and bioactivity prediction. Arabian J. Chem.. 2020;13(2):8793-8806. URL https://www.sciencedirect.com/science/article/pii/S1878535220304044
- [CrossRef] [Google Scholar]




