

Translate this page into:
In search of novel inhibitors of anti-cancer drug target fibroblast growth factor receptors: Insights from virtual screening, molecular docking, and molecular dynamics
⁎Corresponding authors. arif.khan@bge.uoda.edu.bd (Md. Arif Khan), firoz19701016@gmail.com (Firoz Ahmed) firoz@nstu.edu.bd (Firoz Ahmed)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract

Abstract
Fibroblast growth factor receptors (FGFR) are an essential player in oncogenesis and tumor progression. LY2874455 was identified as a pan-FGFR inhibitor and has gone through phase I clinical trial. In the current study, virtual screening was conducted against the PubChem database using a pharmacophore model generated from the crystal structure of FGFR4 inhibited by LY2874455. PubChem 137300327 was identified as the most suitable compound from this screening. Later, molecular docking and molecular dynamics studies conducted with FGFRs corroborated the initial finding. Analysis of ADMET properties disclosed that LY2874455 and PubChem 137300327 share alike properties. Our study suggests that PubChem 137300327 is a potential pan-FGFR inhibitor and can be exploited to treat different cancers following validation in proper wet-lab experiments and study in animal cancer models. This compound also follows Lipinski’s rules and can be used as a lead compound to synthesize more effective anticancer compounds.
Keywords
Fibroblast growth factor receptor
FGFR
Anti-cancer drug
Chemotherapy

1 Introduction
The FGFR (Fibroblast Growth Factor Receptor) protein family has five members- four strictly conserved transmembrane receptor tyrosine kinases, including FGFR1, FGFR2, FGFR3, FGFR4, and fibroblast growth factor receptor-like 1 protein (FGFRL1, also known as FGFR5). They originate from exclusive genes (FGFR1-4 and FGFRL1). Eighteen types of FGFs can bind to them, with heparin being a co-factor (Sarabipour & Hristova, 2016). Members of the FGFR family have critical roles in developing embryos, homeostasis, endocrine functions, metabolism, angiogenesis, and wound healing. However, in malignancies, FGFR mediated signaling becomes deregulated. In addition, they take part in cancer progression through their roles in multiplication and survival of cancer cells as well as formation of new blood vessels in cancer tissues (Itoh N, Ornitz, 2004; Babina & Turner, 2017; Luca et al., 2017; Ghedini et al., 2018). FGFR1-4 are cell surface receptors consisting of 800 amino acids and the following domains- i) three different extracellular immunoglobulin-like domains (D1-3) held together by malleable linker sequences, ii) one transmembrane helix domain, and iii) one intracellular tyrosine kinase domain for conveying signal into the cell interior.
In FGFR5, the tyrosine kinase domain is replaced by an intracellular histidine-rich motif. FGFR5 has roles in early testis development (Cotton et al., 2008) and interacts with extracellular signal-regulated kinase 1/2 (ERK1/2) mediated functions. FGFR5 is a newly discovered member of the FGFR family whose activities deserve further study (Itoh N, Ornitz, 2004; Babina & Turner, 2017; Luca et al., 2017; Zhou et al., 2016). D2-3 is the leading ligand-binding site of FGFRs and can efficiently maintain specificity in ligand binding. Linker sequence between D1 and D2 has a serine-rich region known as ‘acid box’ which has a role in interaction with ligands other than FGF and FGFR auto-inhibition process (Babina & Turner, 2017; Luca et al., 2017; Zhou et al., 2016). The first half of the D2 fragment houses a binding domain for heparan sulfate proteoglycan (HSGP). The binding of FGF to HSPG protects it from denaturation and degradation, and a ternary complex of FGF/FGFR/HSPG ensures adequate FGF–FGFR interactions (Babina & Turner, 2017; Zhou et al., 2016; Ghedini et al., 2018). Alternative splicing manifested in the second half of the D3 fragment of FGFR1-3 is the reason for seven FGFR isoforms (FGFR1b-3b, FGFR1c-3c, and FGFR4). These isoforms have a different propensity for expression in different cells. Usually, epithelial cells express b isoforms and c isoforms are abundant in mesenchymal cells. These isoforms are responsible for ensuring specificities in ligand binding and are differentially expressed in various tumors (Zhou et al., 2016; Perez-Garcia et al., 2018). Overexpression of FGF ligands and FGFR are observed in numerous cancers, such as astrocytoma, melanoma, ovarian, pancreatic, prostate, colorectal, gastric cancers, etc. (Koziczak et al., 2004; Dariya et al., 2018). FGFR gene amplification (66%), activating mutations (26%) that dimerize the FGFR in a ligand-independent manner or constitutively activate the intracellular kinase domain, oncogenic fusions due to chromosomal translocation (8%), and autocrine and paracrine signal mediated hyper-stimulation of FGFRs are some reasons behind the loss of balance in FGFR signaling in malignant tissues. These imbalances in FGFR signaling are directly responsible for uncontrolled angiogenesis, epithelial to mesenchymal transition, prohibition of apoptosis, cellular proliferation and migration, and drug resistance in cancer tissues (Zhou et al., 2016; Pearson et al., 2016; Helsten et al., 2016; Babina & Turner, 2017; Ghedini et al., 2018). These events demonstrate a significant correlation between tumor growth as well as survival and deregulated FGFR signaling pathways (Babina & Turner, 2017; Krook et al., 2021; Chae et al., 2017; Porta et al., 2017). Almost 7% of all cancers bear FGFR aberration in any form (Helsten et al., 2016). Malignancies where FGFR signaling is commonly deregulated include squamous cell carcinoma of the lung (approximately 13%), ovarian cancer (approximately 9%), breast cancer (approximately 18%), endometrial cell cancer (approximately 13%), and urothelial cancer (approximately 32%) (Luca et al., 2017). Various studies in miscellaneous tumor models in vitro and in vivo have shown that an arrest in cancer cell proliferation and apoptosis induction can be achieved by impeding FGFR signaling with the help of small molecule inhibitors and some such FGFR inhibitors are under various phases of clinical trial (Koziczak et al., 2004; Zhou et al., 2016; Kuriwaki et al., 2020; Porta et al., 2017; Zhang et al., 2017; Zhang et al., 2016; Wang et al., 2019; Fumarola et al., 2019; Zhang & Yu, 2020; Wu et al., 2021). Considering the impact and promise of blocking FGFR mediated signaling in cancer treatment, this study was designed to identify novel potent FGFR inhibitors with more desirable properties.
2 Materials and methods
2.1 Ligand-based pharmacophore modeling and virtual Screening:
Standard crystal structures, from which the parameters necessary for effective ligand interactions are derived, are obligatory for constituting a pharmacophore model. Therefore, a ligand-based pharmacophore model for identifying novel inhibitors of human FGFRs was created using the crystal structure of LY2874455 bound FGFR4. This experimental structure was downloaded from the RCSB Protein Data Bank (PDB ID: 5XFF). LY2874455 is a pan-FGFR inhibitor. The pharmacophore model was obtained employing the Pharmit server (
https://pharmit.csb.pitt.edu/) (Sunseri & Koes, 2016). Features of the pharmacophore model are tabulated in Table 1 and shown in Fig. 1. PubChem database was screened against the pharmacophore model. ‘Inclusive’ and ‘Exclusive shape constraints' with tolerance level 1 were applied to include more matching compounds during the similarity search. 'Inclusive shape constraint' ensures that a minimum of one heavy atom of the compounds being screened falls within the pharmacophore aligned pose. 'Exclusive shape constraint' confirms that hits that possess heavy atom centers in their pharmacophore-aligned poses within the 'Exclusive shape' are discarded. Other following filters were also applied to identify more 'druglike' hits: ≤ 10 rotatable bonds, polar surface area ≤ 140 Å2, octanol–water partition coefficient log P value in the range of −0.4 to + 5.6, number of hydrogen bond acceptor ≤ 10, and number of hydrogen bond donor ≤ 5. The values of these parameters were chosen according to drug-likeness rules proposed by Lipinski (Lipinski et al., 1997), Ghose (Ghose et al., 1999), and Veber (Veber et al., 2002). The identified hits were next arranged serially according to ‘Vina scoring function’ to identify energetically favorable hits. Finally, only the hits having a maximum Vina score ≤ -8, minimized RMSD value from the original ligand (LY2874455) pose ≤ 2 Å, and single conformer were extracted.
Pharmacophore Description
Co-ordinates of Center
Radius (Å)
X
Y
Z
Hydrogen bond Acceptor
72.67
43.98
563.02
0.5
Hydrogen bond Acceptor
73.60
44.92
562.96
0.5
Hydrogen bond Acceptor
76.66
50.26
560.48
0.5
Hydrogen bond Acceptor
71.98
46.70
555.57
0.5
Hydrophobic Interactions
70.73
44.85
561.29
1.0
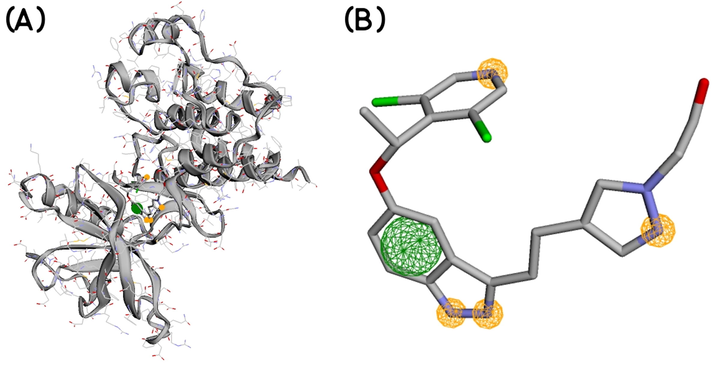
Features of the generated pharmacophore model; the yellow circles indicate hydrogen bond acceptors and the green circle indicates hydrophobic interaction site.
2.2 Molecular Docking
-
Molecular docking between PubChem 137300327 and V550LFGFR4 gatekeeper mutant:
To measure binding affinity between the receptor and the top-scoring compound (PubChem Compound ID-137300327), molecular docking was executed utilizing Autodock Vina (Trott & Olson, 2009). The 3D conformer of the top-scoring compound was downloaded in SDF (Structure Data File) format from the PubChem database ( https://pubchem.ncbi.nlm.nih.gov/compound/137300327) and converted to PDB format with the help of Open Babel (O’Boyle et al., 2011). Cleaned crystal structure of the receptor and the compound were transformed into PDBQT format with the help of AutoDock Tools 4.2 (Morris et al., 2009), a step essential for docking with AutoDock Vina. Before converting to the PDBQT format, polar hydrogens were added to the receptor, and the ligand bonds were made rotatable. A grid box centered on X: 72.500, Y: 48.000, and Z: 559.000 with 28 × 28 × 28 Å measurement was designed for site-specific docking. This grid box contained all the experiment-derived binding residues. Docking was conducted with an exhaustiveness level of 20. After execution of the docking process, nine binding poses were calculated along with binding energy in kcal/mol. The docking pose with the least binding energy, which also showed the least RMSD, was considered the most accurate prediction. An additional molecular docking tool, iGEMDOCK (Hsu et al., 2011), was also used for the docking study. A more exhaustive ‘accurate docking’ option was chosen with the following parameters: population size as 800, generations as 80, and numbers of solutions as 10. Other options were kept default.
The docked poses, as calculated by AutoDock Vina and iGEMDOCK, were superimposed on the native protein structure (PDB ID: 5XFF) and visualized by PyMol (The PyMOL Molecular Graphics System, Open-source PyMOL, Schrödinger, LLC.) (Fig. 1).
Molecular docking between PubChem 137300327 and other gatekeeper mutant proteins:
Docking between PubChem 137300327 and other FGFR proteins with gatekeeper mutation was also studied. Currently known FGFR mutations are FGFR1 (V561M), FGFR2 (V564F, V564I, and V564M), FGFR3 (V555L & V555M), and FGFR4 (V550L & V550M) (Abdel-Magid, 2019). Crystal structures of FGFR1-4 were downloaded from the RCSB PDB. Gatekeeper mutant structures of V561MFGFR1 (PDB ID: 4RWI), V564FFGFR2 (PDB ID: 7KIE), and V550MFGFR4 (PDB ID: 5XFJ) are available from PDB. V564IFGFR2 and V564MFGFR2 structures were derived by mutating 7KIE structure, and V555LFGFR3 & V555MFGFR3 structures were derived by mutating FGFR3 PDB structure (PDB ID: 6PNX) with the help of Discovery Studio Visualizer (Dassault Systèmes BIOVIA, Discovery Studio Visualizer v 20.1.0.19295, San Diego: Dassault Systèmes, 2020). These structures were later optimized by applying the CHARMM36 force field (Huang et al., 2013). ‘A’ chains of these proteins were used for docking with PubChem 137300327 by AutoDock Vina. Docking grid boxes were designed to contain the gatekeeper mutations (Supplementary Table 2). Other parameters were kept similar to 2.2 (a).
Complex
Receptor-ligand Interactions
Binding Energy (kcal/mol)
FGFR1 (V561M) - PubChem 137300327
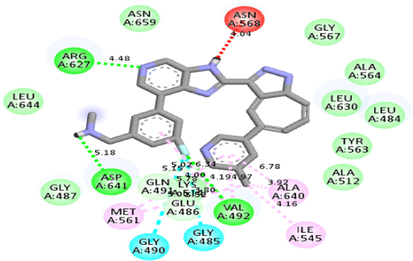
−10.3
FGFR2 (V564F) – PubChem 137300327
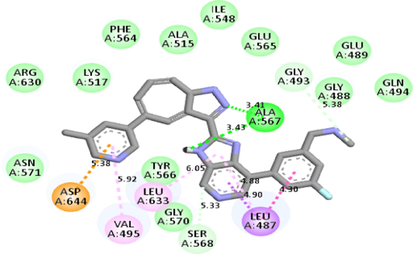
−11.3
FGFR2 (V564I) - PubChem 137300327
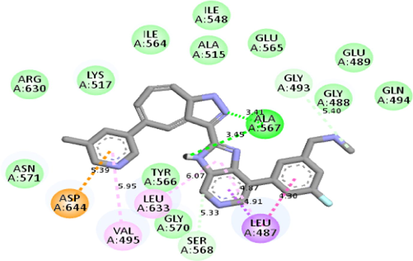
−11.2
FGFR2 (V564M) - PubChem 137300327
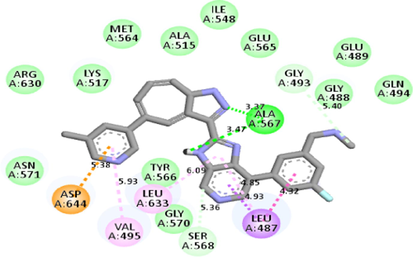
−11.0
FGFR3 (V555L) - PubChem 137300327

−10.5
FGFR3 (V555M) - PubChem 137300327
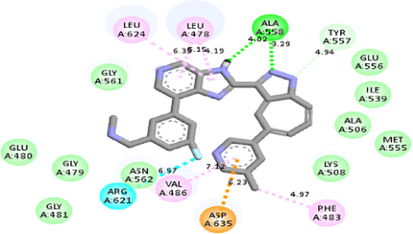
−9.6
FGFR4 (V550L) - PubChem 137300327
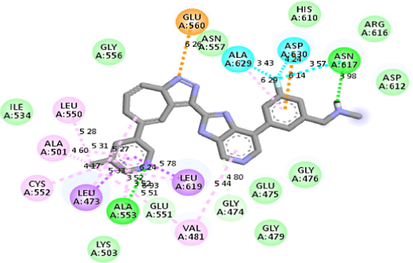
−9.9
FGFR4 (V550M) - PubChem 137300327
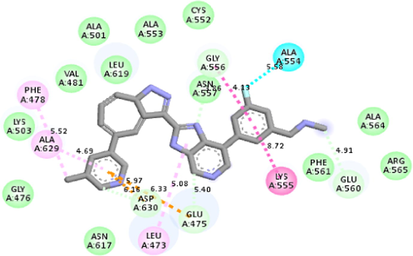
−10.1
Indicator

2.3 Molecular dynamics Simulation:
Since Kinase domains of FGFR1-4 share mostly identical residues (Wu et al., 2016), only one complex, PubChem 137300327 - V550LFGFR4 complex, was chosen for molecular dynamics study. It is logical that interactions of PubChem 137300327 with other FGFRs can be extrapolated from this MD simulation. The molecular dynamics study was performed in YASARA dynamics by applying AMBER14 force field. The receptor-ligand complex was initially cleaned, optimized, and the hydrogen network was oriented. TIP3P solvation model was used for solvating the complex and a virtual cubic cell was chosen. The complex was placed in the simulation box where each side of the box maintained a 20 Å distance from the complex. The physiological conditions were maintained in the simulation cell by applying pH 7.4, 310 K temperature, and 0.9% NaCl. Initial energy minimization was achieved by employing a simulated annealing method with steepest gradient algorithm (5000 cycles). The long-range electrostatic interactions were calculated using the Particle Mesh Ewalds (PME) method with a cut-off distance of 8.0 Å. By maintaining a constant pressure and the Berendsen thermostat, MD simulation was continued for 100 ns. The time step of the simulated system was fixed to 2.0 fs. The simulation trajectories were saved every 100 ps and used to calculate root mean square deviation (RMSD), root means square fluctuation (RMSF), solvent accessible surface area (SASA), radius of gyration, and number of hydrogen bonds.
2.4 In silico prediction of IC50:
Half-maximal inhibitory concentration value (IC50 value) is a measurement of efficacy of a given compound in inhibiting a desired biochemical/ biological process. An acceptable IC50 value greenlights the continuation of downstream processes in drug development. IC50 values of PubChem 137300327 and the reference inhibitor, LY2874455 were calculated using Cell-Line Cytotoxicity Predictor (https://way2drug.com/Cell-line/). It is a dedicated tool based on experimental data for evaluation of cytotoxicity against cancer cell lines. The compounds were submitted in SMILES format and the Pa value was set to 0.5.
2.5 Prediction of ADMET properties:
After a drug is taken per os, it undergoes the following stages sequentially: absorption (A), distribution (D), metabolism (M), and excretion (E). If a drug is administrated through intravenous or rarely, intra-arterial way, the absorption stage can be bypassed. This route is usually followed when a rapid onset of action and more bioavailability is desired. In drug designing, the study of ADME properties is crucial to get an idea of the properties of the drug being developed inside our bodies. Besides ADME properties, the study of toxicity (T) profile also deserves profound importance because sometimes intermediate metabolites can be significantly toxic to the body (Opo et al., 2021). Drug development is a long, costly, and tiresome process. Early detection of unsuitable ADMET properties can save money, time, and physical labor. In silico tools can be of great help in this regard and in some circumstances, are an alternative to animal testing in predicting these properties (Wu et al., 2020; Kar & Leszczynski, 2020; Brogi et al., 2020; Madden et al., 2020).
ADMET properties of the chosen compound, PubChem 137300327 were predicted by pkCSM ( https://biosig.unimelb.edu.au/pkcsm/) (Pires et al., 2015). In addition, similar properties of the native ligand of the pharmacophore model (LY2874455) were also calculated and compared with PubChem 137300327.
3 Results
3.1 Ligand-based pharmacophore modeling and virtual Screening:
450,708,705 conformers of 93,067,404 molecules deposited in the PubChem database were screened according to the pharmacophore model to obtain 576 small molecule hits. Finally, 22 compounds were procured (Supplementary Table 1) using the following filters: maximum binding score as −8 kcal/mol and maximum mRMSD (minimized root mean square deviation) as 2 Å. Among the 22 potential small molecules, the top-ranked hit molecule (PubChem CID: 137300327) having a −10.05 kcal/mol binding score together with 1.422 Å mRMSD was selected for further computational analyses.
3.2 Molecular Docking:
3.2 (a) Molecular docking between PubChem 137300327 and V550LFGFR4 gatekeeper mutant:
After scrutinizing the nine distinct binding modes calculated by Autodock Vina, the binding affinity of the most favorable mode was found −9.9 kcal/mol. iGEMDOCK calculated ‘Total Estimated Fitness Score’ was −128.449. This score was a summed-up contribution of Van Der Waals energy (-98.9901), hydrogen bonding energy (-29.4586), and electro statistic energy (0). iGEMDOCK does not provide binding energy; instead, it calculates an empirical pharmacophore-based score.
Autodock Vina and iGEMDOCK calculated docking poses superimposed on the 5XFF PDB structure are shown in Fig. 2. Also, the receptor-ligand interactions (Autodock Vina calculated) can be visualized in Table 2.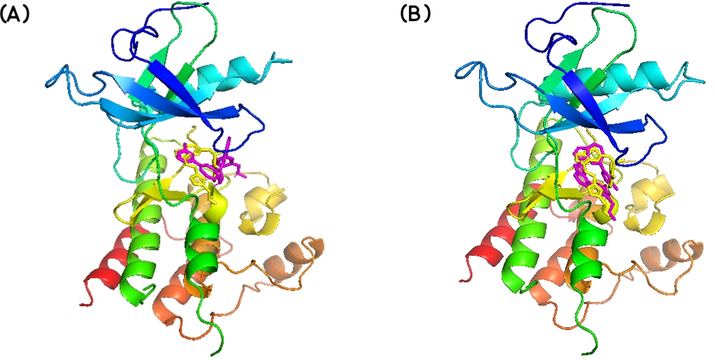
Best docked poses of the ligand to V550LFGFR4, as calculated by (A) AutoDock Vina and (B) iGEMDOCK, and superimposed on the native protein structure (PDB ID: 5XFF). LY2874455 is shown in yellow and PubChem- 137300327 is shown in pink.
3.2 (b) Molecular docking between PubChem 137300327 and other gatekeeper mutant proteins:
Molecular docking studies between other FGFR gatekeeper mutant proteins and PubChem 137300327 revealed that this ligand can also efficiently bind with all of these mutants. It bound to the mutant proteins with −9.6 to −11.3 kcal/mol binding energy. More interestingly, it also showed binding interactions with the mutated gatekeeper residues. Binding energy and interactions with different proteins can be found in Table 2.
3.3 Molecular dynamics (MD) Simulation:
MD simulation study was carried on to explore structural stability of the PubChem 137300327-V550LFGFR4 complex. RMSD of the C-alpha atoms was calculated to understand the flexibility status of the complex across the simulation trajectories. Fig. 3 (a) depicts that the complex experienced a relatively stable RMSD from the beginning to the end of the simulation. This complex exhibited a higher RMSD profile after 40 ns (but the deviation remained within 1 Å). However, the upper RMSD trend of this complex was stopped after 50 ns and a stable RMSD profile was maintained later on. This RMSD trend indicates the complex was stable and rigid throughout the simulation period.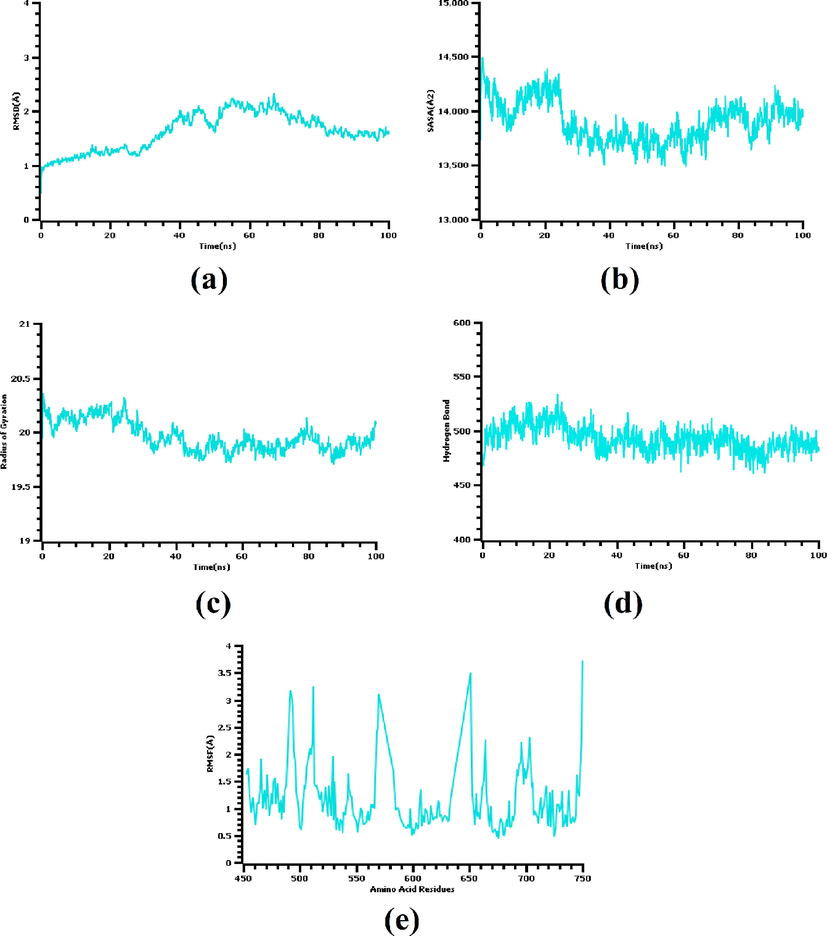
(a) Root mean square deviation, (b) solvent accessible surface area, (c) radius of gyration, (d) number of hydrogen bonds, and (e) root mean square fluctuation calculated from the molecular dynamics simulation of the V550LFGFR4-PubChem 137300327 complex.
Solvent accessible surface area (SASA) of the docked complex was also explored. A higher than initial SASA value indicates an expansion in the protein surface area (usually due to unfolding) (Savojardo et al., 2021). The complex under study had a higher SASA profile from the beginning to 20 ns [Fig. 3 (b)] which demonstrates an initial expansion in the surface area of the receptor. After that, the receptor became stable, the SASA profile lowered, and reached its steady-state by 25 ns. This steady trend was maintained till the completion of the simulation time.
A complex's low radius of gyration (Rg) profile defines its compact nature, whereas a higher Rg profile suggests flexible nature. The Rg profile of the complex under investigation was nearly similar till the end of the simulation period which indicates the complex maintained a stable and rigid status during whole simulation [Fig. 3 (c)]. The number of hydrogen bonds in this complex was found near uniform in the trajectories which also implies stable ligand binding [Fig. 3 (d)]. Besides, maximum amino acid residues had an RMSF value less than 2.5 Å, which further assures stability of the complex [Fig. 3 (e)].
3.4 In silico prediction of IC50:
From IC50 predictions, it was found that our identified compound PubChem 137300327 and the reference inhibitor, LY2874455 share similar IC50 values (Table 3) which further supports the prospect of this compound. Here, Pa is the probability of belonging to the sub-class of active compounds used in the training set and Pi is the probability of belonging to the sub-class of inactive compounds used during training.
Compound
Pa
Pi
Cell-line
PubChem 137300327
0.576
0.019
HCT-116 (Colon carcinoma)
LY2874455
0.576
0.009
MDA-MB-468 (Breast adenocarcinoma)
LY2874455
0.611
0.009
DMS-114 (Lung carcinoma)
3.5 Prediction of ADMET properties:
ADMET properties of both PubChem 137300327 and LY2874455 were calculated and compared. Details of the predicted properties are available in Table 4. pkCSM calculated a total of 7 absorption parameters, four distribution parameters, seven metabolism parameters, two excretion parameters, and ten toxicity parameters. Comparison among these parameters revealed that there are minor differences in various properties between the two compounds.
Property
Model Name
Unit
Predicted Value
PubChem 137300327
LY2874455
Absorption
Water solubility
Numeric (log mol/L)
−3.009
−3.866
Caco2 permeability
Numeric (log Papp in 10^-6 cm/s)
1.385
0.77
Intestinal absorption (human)
Numeric (% Absorbed)
89.268
89.521
Skin Permeability
Numeric (log Kp)
−2.735
−2.737
P-glycoprotein substrate
Categorical (Yes/No)
Yes
Yes
P-glycoprotein I inhibitor
Categorical (Yes/No)
Yes
Yes
P-glycoprotein II inhibitor
Categorical (Yes/No)
Yes
Yes
Distribution
VDss (human)
Numeric (log L/kg)
0.198
0.204
Fraction unbound (human)
Numeric (Fu)
0.098
0.007
BBB permeability
Numeric (log BB)
−1.298
−1.671
CNS permeability
Numeric (log PS)
−2.359
−2.394
Metabolism
CYP2D6 substrate
Categorical (Yes/No)
No
No
CYP3A4 substrate
Categorical (Yes/No)
Yes
Yes
CYP1A2 inhibitor
Categorical (Yes/No)
Yes
Yes
CYP2C19 inhibitor
Categorical (Yes/No)
Yes
Yes
CYP2D6 inhibitor
Categorical (Yes/No)
No
No
CYP2D6 inhibitor
Categorical (Yes/No)
No
No
Excretion
Total Clearance
Numeric (log ml/min/kg)
0.78
0.551
Renal OCT2 substrate
Categorical (Yes/No)
Yes
No
Toxicity
AMES toxicity
Categorical (Yes/No)
Yes
No
Max. tolerated dose (human)
Numeric (log mg/kg/day)
0.304
0.163
hERG I inhibitor
Categorical (Yes/No)
No
No
Oral Rat Acute Toxicity (LD50)
Numeric (mol/kg)
2.281
2.87
Oral Rat Chronic Toxicity (LOAEL)
Numeric (log mg/kg_bw/day)
1.477
1.535
Skin Sensitization
Categorical (Yes/No)
No
No
T.Pyriformis toxicity
Numeric (log ug/L)
0.285
0.313
Minnow toxicity
Numeric (log mM)
0.402
−1.344
4 Discussion
FGFRs act as receptor tyrosine kinases (RTK) and FGFR targeted anti-cancer therapy is a well-researched topic. However, one particular obstacle faced here is mutation, especially, gatekeeper mutations that lead to chemotherapeutic resistance (Sohl et al., 2015; Azam et al., 2008; Dai et al., 2019). The gatekeeper residue is present at the first part of the hinge region that connects the N-terminal and C-terminal lobes of the kinase domain. The gatekeeper amino acid residue in kinases can be threonine, leucine, phenylalanine, or valine. Nevertheless, in wild-type FGFRs, it is always a valine. RTKs transfer the phosphate group from ATP to their intracellular protein substrate's tyrosine residue. Small molecule inhibitors of RTKs compete with ATP for binding to the active site. For effective binding, they need to accommodate themselves in the hydrophobic pocket behind the active site. Moreover, these inhibitors also attach to the gatekeeper residue via critical hydrogen bonds. When cancer cells are exposed to small molecule inhibitor drugs, their self-protection strategy involves mutation of the relatively smaller hydrophobic gatekeeper residue to a larger hydrophobic residue, for instance, isoleucine, leucine, methionine. This results in a steric clash between the inhibitor and the mutant residue, and elimination of critical hydrogen bonds. As a result, the inhibitor losses its efficacy (Zhou et al., 2020; Ryan et al., 2019; Dai et al., 2019; Wu et al., 2018; Yoza ET AL., 2016; Sohl et al., 2015; Bunney et al., 2015; O’Hare et al., 2009; Blaukat et al., 2007).
LY2874455 (2-[4-[(E)-2-[5-[(1R)-1-(3,5-dichloropyridin-4-yl)ethoxy]-1H-indazol-3yl]ethenyl]pyrazol-1-yl]ethanol) is a pan-FGFR inhibitor, having the ability to selectively inhibit FGFR1-4 with the following IC50 values, respectively: 2.8 nM, 2.6 nM, 6.4 nM and 6 nM. This compound has also undergone a phase I clinical trial consisting of patients with advanced-stage cancers (Zhao et al., 2011; Michael et al., 2017; Hanes et al.,2019). In the current study, a pharmacophore model was generated to identify inhibitors of human FGFRs using an experimental structure of LY2874455 bound to FGFR4 (PDB ID: 5XFF). The rationale for selecting this experimental structure was containment of LY2874455 in FGFR4 with a gatekeeper residue mutation (V550L) and good crystal quality (resolution: 2.70 Å). Screening of the PubChem database to satisfy the pharmacophore model revealed PubChem 137300327 as the most suitable compound. Later, molecular docking and dynamics studies supported this finding. The docking analysis found that the identified ligand is well accommodated in the hydrophobic pocket and does not face any steric clash with the gatekeeper mutant (V550LFGFR4).
Interestingly, this residue showed alkyl interaction with the ligand (Table 2). Docking studies with other gatekeeper mutant FGFR proteins supported that PubChem 137300327 can bind to all of them with equal efficiency and thus, is a potential pan-FGFR inhibitor like LY2874455. Moreover, this compound also inhibits WNT signaling pathway ( https://pubchem.ncbi.nlm.nih.gov/patent/EP-3284744-A1), another crucial pathway involved in cancer (Zhan et al., 2017; Jung & Park, 2020; Mo et al., 2019). So, this compound can provide synergistic Inhibition in different malignancies. From ADMET analyses of the identified ligand and LY2874455, it was found that both of them share near similar properties.
Similarities in structural and ADMET properties with LY2874455, and binding affinities with the FGFR gatekeeper mutant proteins suggest PubChem 137300327 is also a pan-FGFR inhibitor. Additionally, this compound also obeys Lipinski’s ‘rule of five’ (Lipinski et al., 1997; Hefti, 2008): molecular weight- 477.5 Da, octanol–water partition coefficient (log P)- 4, hydrogen bond donor count-3, and hydrogen bond acceptor count-7 (data provided by PubChem). So, this compound can also be used as a lead to derive other more effective FGFR inhibitors.
Like LY2874455, several other pan-FGFR inhibitors have also recently been reported. These include FIIN-2, JNJ-42756493, ponatinib (Katoh, 2016), rogaratinib (Grünewald et al., 2019), DW14383 (Dai et al., 2021), erdafitinib, pemigatinib (Weaver and Bossaer, 2021), futibatinib, infigratinib, derazantinib, ASP5878, etc. Deatailed information about currently identified FGFR inhibitors can be found here (https://www.selleckchem.com/subunits/FGFR1_FGFR_selpan.html). Among these compounds, erdafitinib has achieved FDA approval for treating metastatic urothelial carcinoma with FGFR2 and FGFR3 alterations, and pemigatinib for unresectable cholangiocarcinoma with FGFR2 alterations (Weaver and Bossaer, 2021). FDA has approved derazantinib for treating intrahepatic cholangiocarcinoma and futibatinib (Goyal et al., 2020, Rizzo et al., 2021) has pocketed ‘Breakthrough Therapy Designation’ from FDA. Infigratinib (Javle et al., 2021) was approved by FDA for treating adults who were previously treated but currently suffer from unresectable locally advanced or metastatic cholangiocarcinoma with a fibroblast growth factor receptor 2 (FGFR2) fusion or other rearrangements. Several other pan-FGFR inhibitors are currently under various phases of clinical trial.
Computational techniques can ease the initial processes of drug target identification and drug discovery (Mahfuz et al., 2021; Mahfuz et al., 2020; Mahfuz et al., 2021). Considering the promises hold by pan-FGFR inhibitors, in vivo and clinical studies of LY2874455, and the results obtained from this thorough computational study, PubChem 137300327 can be regarded as a suitable agent for inhibiting all FGFRs. The limitation of this study is lack of in vitro experiments. Further studies on cancer cell lines, animal models of malignancies, and clinical trials can prove the promise hold by this compound.
CRediT authorship contribution statement
A M U B Mahfuz: Conceptualization, Data curation, Formal analysis, Methodology, Visualization, Writing - original draft. Md. Arif Khan: Conceptualization, Project administration, Resources, Supervision, Writing - review & editing. Suvro Biswas: Writing - original draft. Shamima Afrose: Writing - original draft. Shafi Mahmud: Formal analysis, Writing - original draft. Newaz Mohammed Bahadur: Writing - review & editing. Firoz Ahmed: Supervision, Writing - review & editing.
References
- Second-Generation FGFR Inhibitors for the Treatment of Cancers Harboring Mutated FGFRs. ACS Med. Chem. Lett.. 2019;10:1374-1375.
- [CrossRef] [Google Scholar]
- Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat. Struct. Mol. Biol.. 2008;15:1109-1118.
- [CrossRef] [Google Scholar]
- Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer. 2017;17:318-332.
- [CrossRef] [Google Scholar]
- Blaukat A. Tyrosine Kinases. xPharm: The Comprehensive Pharmacology Reference. Elsevier; 2007. pp. 1–4. doi:10.1016/B978-008055232-3.62957-5.
- Editorial: In silico Methods for Drug Design and Discovery. Front. Chem.. 2020;8:612.
- [CrossRef] [Google Scholar]
- The Effect of Mutations on Drug Sensitivity and Kinase Activity of Fibroblast Growth Factor Receptors: A Combined Experimental and Theoretical Study. EBioMedicine.. 2015;2:194-204.
- [CrossRef] [Google Scholar]
- Inhibition of the fibroblast growth factor receptor (FGFR) pathway: the current landscape and barriers to clinical application. Oncotarget.. 2017;8:16052-16074.
- [CrossRef] [Google Scholar]
- Cellular signaling by fibroblast growth factors (Fgfs) and their receptors (Fgfrs) in male reproduction. Endocr. Rev.. 2008;29(2):193-216.
- [Google Scholar]
- Fibroblast Growth Factor Receptors (FGFRs): Structures and Small Molecule Inhibitors. Cells.. 2019;8:614.
- [CrossRef] [Google Scholar]
- DW14383 is an irreversible pan-FGFR inhibitor that suppresses FGFR-dependent tumor growth in vitro and in vivo. Acta Pharmacol. Sin.. 2021;42:1498-1506.
- [CrossRef] [Google Scholar]
- EGFR and FGFR in Growth and Metastasis of Colorectal Cancer. In: Nagaraju G.P., ed. Role of Tyrosine Kinases in Gastrointestinal Malignancies. Singapore: Springer Singapore; 2018. p. :141-170.
- [CrossRef] [Google Scholar]
- Expanding the Arsenal of FGFR Inhibitors: A Novel Chloroacetamide Derivative as a New Irreversible Agent With Anti-proliferative Activity Against FGFR1-Amplified Lung Cancer Cell Lines. Front. Oncol.. 2019;9:179.
- [CrossRef] [Google Scholar]
- Future applications of FGF/FGFR inhibitors in cancer. Expert Rev. Anticancer Ther.. 2018;18:861-872.
- [CrossRef] [Google Scholar]
- A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem.. 1999;1:55-68.
- [CrossRef] [Google Scholar]
- FOENIX-CCA2: A phase II, open-label, multicenter study of futibatinib in patients (pts) with intrahepatic cholangiocarcinoma (iCCA) harboring FGFR2 gene fusions or other rearrangements. JCO.. 2020;38:108.
- [CrossRef] [Google Scholar]
- Rogaratinib: A potent and selective pan-FGFR inhibitor with broad antitumor activity in FGFR-overexpressing preclinical cancer models. Int. J. Cancer. 2019;145:1346-1357.
- [CrossRef] [Google Scholar]
- Preclinical Evaluation of the Pan-FGFR Inhibitor LY2874455 in FRS2-Amplified Liposarcoma. Cells.. 2019;8:189.
- [CrossRef] [Google Scholar]
- Requirements for a lead compound to become a clinical candidate. BMC Neurosci.. 2008;9:S7.
- [CrossRef] [Google Scholar]
- The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clin. Cancer Res.. 2016;22:259-267.
- [CrossRef] [Google Scholar]
- iGEMDOCK: a graphical environment of enhancing GEMDOCK using pharmacological interactions and post-screening analysis. BMC Bioinf.. 2011;12:S33.
- [CrossRef] [Google Scholar]
- CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem.. 2013;34:2135-2145.
- [CrossRef] [Google Scholar]
- Evolution of the Fgf and Fgfr gene families. Trends Genet.. 2004;20:563-569.
- [CrossRef] [Google Scholar]
- Infigratinib (BGJ398) in previously treated patients with advanced or metastatic cholangiocarcinoma with FGFR2 fusions or rearrangements: mature results from a multicentre, open-label, single-arm, phase 2 study. Lancet Gastroenterol. Hepatol.. 2021;6:803-815.
- [CrossRef] [Google Scholar]
- Wnt signaling in cancer: therapeutic targeting of Wnt signaling beyond β-catenin and the destruction complex. Exp. Mol. Med.. 2020;52:183-191.
- [CrossRef] [Google Scholar]
- Open access in silico tools to predict the ADMET profiling of drug candidates. Expert Opin. Drug Discov.. 2020;15:1473-1487.
- [CrossRef] [Google Scholar]
- FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review) Int. J. Mol. Med.. 2016;38:3-15.
- [CrossRef] [Google Scholar]
- Blocking of FGFR signaling inhibits breast cancer cell proliferation through downregulation of D-type cyclins. Oncogene. 2004;23:3501-3508.
- [CrossRef] [Google Scholar]
- Fibroblast growth factor receptors in cancer: genetic alterations, diagnostics, therapeutic targets and mechanisms of resistance. Br. J. Cancer. 2021;124:880-892.
- [CrossRef] [Google Scholar]
- Structure-based drug design of 1,3,5-triazine and pyrimidine derivatives as novel FGFR3 inhibitors with high selectivity over VEGFR2. Bioorg. Med. Chem.. 2020;115453
- [CrossRef] [Google Scholar]
- Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev.. 1997;23:3-25.
- [CrossRef] [Google Scholar]
- FGFR-targeted therapeutics for the treatment of breast cancer. Expert Opin. Invest. Drugs. 2017;26:303-311.
- [CrossRef] [Google Scholar]
- A Review of In Silico Tools as Alternatives to Animal Testing: Principles Resources and Applications. Altern Lab Anim.. 2020;48:146-172.
- [CrossRef] [Google Scholar]
- Characterization of ribonucleotide reductases of emerging pathogens Elizabethkingia anophelis and Elizabethkingia meningoseptica and streptonigrin as their inhibitor: a computational study. Journal of Biomolecular Structure and Dynamics 2021:1-13.
- [CrossRef] [Google Scholar]
- Carfilzomib as a potential inhibitor of NADH-dependent enoyl-acyl carrier protein reductases of Klebsiella pneumoniae and Mycobacterium tuberculosis as a drug target enzyme: insights from molecular docking and molecular dynamics. Journal of Biomolecular Structure and Dynamics 2020:1-17.
- [CrossRef] [Google Scholar]
- A network-biology approach for identification of key genes and pathways involved in malignant peritoneal mesothelioma. Genomics Inform. 2021;19
- [CrossRef] [Google Scholar]
- A Phase 1 Study of LY2874455, an Oral Selective pan-FGFR Inhibitor, in Patients with Advanced Cancer. Targ Oncol.. 2017;12:463-474.
- [CrossRef] [Google Scholar]
- The role of Wnt signaling pathway in tumor metabolic reprogramming. J Cancer.. 2019;10:3789-3797.
- [CrossRef] [Google Scholar]
- AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem.. 2009;30:2785-2791.
- [CrossRef] [Google Scholar]
- AP24534, a Pan-BCR-ABL Inhibitor for Chronic Myeloid Leukemia, Potently Inhibits the T315I Mutant and Overcomes Mutation-Based Resistance. Cancer Cell. 2009;16:401-412.
- [CrossRef] [Google Scholar]
- Structure based pharmacophore modeling, virtual screening, molecular docking and ADMET approaches for identification of natural anti-cancer agents targeting XIAP protein. Sci. Rep.. 2021;11:4049.
- [CrossRef] [Google Scholar]
- High-Level Clonal FGFR Amplification and Response to FGFR Inhibition in a Translational Clinical Trial. Cancer Discov.. 2016;6:838-851.
- [CrossRef] [Google Scholar]
- pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem.. 2015;58:4066-4072.
- [CrossRef] [Google Scholar]
- FGFR a promising druggable target in cancer: Molecular biology and new drugs. Crit. Rev. Oncol./Hematol.. 2017;113:256-267.
- [CrossRef] [Google Scholar]
- Futibatinib, an investigational agent for the treatment of intrahepatic cholangiocarcinoma: evidence to date and future perspectives. Expert Opin. Invest. Drugs. 2021;30:317-324.
- [CrossRef] [Google Scholar]
- The FGFR1 V561M Gatekeeper Mutation Drives AZD4547 Resistance through STAT3 Activation and EMT. Mol. Cancer Res.. 2019;17:532-543.
- [CrossRef] [Google Scholar]
- Mechanism of FGF receptor dimerization and activation. Nat. Commun.. 2016;7:10262.
- [CrossRef] [Google Scholar]
- Solvent Accessibility of Residues Undergoing Pathogenic Variations in Humans: From Protein Structures to Protein Sequences. Front Mol. Biosci.. 2021;7:626363
- [CrossRef] [Google Scholar]
- Illuminating the molecular mechanisms of tyrosine kinase inhibitor resistance for the FGFR1 gatekeeper mutation: the Achilles’ heel of targeted therapy. ACS Chem. Biol.. 2015;10:1319-1329.
- [CrossRef] [Google Scholar]
- Pharmit: interactive exploration of chemical space. Nucleic Acids Res.. 2016;44:W442-W448.
- [CrossRef] [Google Scholar]
- Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2009; NA-NA. doi:10.1002/jcc.21334.
- Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem.. 2002;45:2615-2623.
- [CrossRef] [Google Scholar]
- The Discovery and Development of a Series of Pyrazolo[3,4-d]pyridazinone Compounds as Novel Covalent Fibroblast Growth Factor Receptor (FGFR) Inhibitors by Rational Drug Design. J. Med. Chem. 2019
- [CrossRef] [Google Scholar]
- Crystal Structure of the FGFR4/LY2874455 Complex Reveals Insights into the Pan-FGFR Selectivity of LY2874455. Wlodawer A, editor. PLoS ONE. 2016;11:e0162491
- [CrossRef] [Google Scholar]
- LY2874455 potently inhibits FGFR gatekeeper mutants and overcomes mutation-based resistance. Chem. Commun.. 2018;54:12089-12092.
- [CrossRef] [Google Scholar]
- Computational Approaches in Preclinical Studies on Drug Discovery and Development. Front. Chem.. 2020;8:726.
- [CrossRef] [Google Scholar]
- Design, synthesis and biological evaluation of pyrazolo[3,4-d]pyridazinone derivatives as covalent FGFR inhibitors. Acta Pharm. Sinica B. 2021;11(3):781-794.
- [Google Scholar]
- Biophysical characterization of drug-resistant mutants of fibroblast growth factor receptor 1. Genes Cells. 2016;21:1049-1058.
- [CrossRef] [Google Scholar]
- Discovery and biological evaluation of a series of pyrrolo[2,3-b]pyrazines as novel fgfr inhibitors. Molecules. 2017;22(4):583.
- [Google Scholar]
- Design, Synthesis and Biological Evaluation: 5-amino-1H-pyrazole-1- carbonyl derivatives as FGFR Inhibitors. Lett. Drug Des. Discovery. 2020;17(11):1330-1341.
- [Google Scholar]
- Design, synthesis and biological evaluation of 6-(2,6-Dichloro-3,5-dimethoxyphenyl)-4-substituted-1h-indazoles as potent fibroblast growth factor receptor inhibitors. Molecules. 2016;21(10):1407.
- [Google Scholar]
- A Novel, Selective Inhibitor of Fibroblast Growth Factor Receptors That Shows a Potent Broad Spectrum of Antitumor Activity in Several Tumor Xenograft Models. Mol. Cancer Ther.. 2011;10:2200-2210.
- [CrossRef] [Google Scholar]
- Characterization of FGFR signaling pathway as therapeutic targets for sarcoma patients. Cancer Biol. Med.. 2016;13:260-268.
- [CrossRef] [Google Scholar]
- FGF/FGFR signaling pathway involved resistance in various cancer types. J. Cancer.. 2020;11:2000-2007.
- [CrossRef] [Google Scholar]
- Fibroblast growth factor receptor (FGFR) inhibitors: A review of a novel therapeutic class. J. Oncol. Pharm. Pract.. 2021;27:702-710.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2022.103882.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




