

Translate this page into:
In-silico screening based on molecular simulations of 3,4-disubstituted pyrrolidine sulfonamides as selective and competitive GlyT1 inhibitors
⁎Corresponding author. Mohamed.elfadili@usmba.ac.ma (Mohamed El fadili),
⁎⁎Corresponding author. Mohammed.kara@usmba.ac.ma (Mohammed Kara)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
A systematic in-silico study based on molecular modeling techniques was conducted on thirty 3,4-disubstituted pyrrolidine sulfonamides derivatives to identify the drug candidate for treating schizophrenia and impairments associated with NMDA receptor hypofunction, through selective and competitive inhibition of GlyT1. QSAR analysis demonstrates that geometric and constitutional descriptors have a key function in human GlyT1 activity. The in-silico study concluded that the most active ligand labeled C19 was predicted to be a non-toxic inhibitor, with a desired ADME-Toxicity profile and a significant probability to penetrate the central nervous system (CNS). Molecular docking simulations confirmed that the C19 compound was docked to the active sites of drosophila melanogaster dopamine transporter (DAT) protein, creating a variety of chemical bonds towards TYR 124, ASP 475, GLU 480, ALA 479, and VAL 120 amino acids residues. The molecular dynamic (MD) technique combined with the MMGBSA approach confirmed that produced intermolecular interactions for the (DAT protein–C19 ligand) complex remain so stable during 100 ns of MD simulation time. Consequently, the C19 ligand is highly recommended for the treatment of schizophrenia and other disabilities linked to the hypofunction of glutaminergic NMDA receptors.
Keywords
NMDA
GlyT1
ADME-Toxicity
CNS
MD

1 Introduction
Over the last decade, the glycine transporter type 1 (GlyT1) localized at glutamatergic synapses has gained great attention as one of the major neurotransmitters that control memory and learning functions, and a key element of glycine metabolism in the central nervous system (CNS) (Eulenburg et al., 2005; Hudson et al., 2020; Lowe et al., 2009; Santora et al., 2018). Many pharmaceutical companies have focused on developing selective and competitive GluT1 inhibitors, to provide potential treatments for neurodegenerative disorders associated with glutamine N-methyl-D-aspartate (NMDA) receptor hypofunction, including schizophrenia (Atkinson et al., 2001; Ouellet et al., 2011; Wolkenberg and Sur, 2010; Wolkenberg et al., 2009). A number of these GlyT1 inhibitors have been repeatedly proven to be effective and successful in animal models of schizophrenia and clinically assessed for several symptom domains (Blackaby et al., 2010; Pinard et al., 2010a; Santora et al., 2018). A few years ago, Roche succeeded in developing a noncompetitive GlyT1 inhibitor as the most advanced compound, namely bitopertin (RG1678), which attained positive results to treat negative symptoms of schizophrenia patients. However, it did not successfully reduce these negative symptoms in the later stages of clinical trials, due to its very limited efficacy (Alberati et al., 2012; Pinard et al., 2010a). For this reason, the discovery of selective and competitive glycine transporter type 1 inhibitors continues to be of great interest to chemists in a wide range of literature (Amberg et al., 2018; Blackaby et al., 2010; Hudson et al., 2020; Łątka and Bajda, 2022; Pinard et al., 2010b; Rosenbrock et al., 2023; Santora et al., 2018). Recently, a structural family of 3,4-disubstituted pyrrolidine sulfonamides, was synthesized and experimentally evaluated through in vivo and in vitro assays to selectively and competitively inhibit the glycine transporter type 1. In this regard, our in-silico investigations were carried out on thirty derivatives of 3,4-disubstituted pyrrolidine sulfonamides which were detected thanks to their inhibitory activity against the type 1 of glycine transporter (Wang et al., 2018). In the beginning, we applied the quantitative structure–activity relationship (QSAR), which gained an advanced position in modern chemistry (Vilar et al., 2008), by developing two validated QSAR models which will be able to predict human GluT1 activity for 3,4-disubstituted pyrrolidine sulfonamides family, using multiple linear regression (MLR) and multiple nonlinear regression (MNLR) techniques (Mostoufi and Constantinides, 2023). Thereafter, we predicted the physicochemical and pharmacokinetic properties of candidate inhibitors able to cross the blood–brain barrier (BBB), penetrating the central nervous system (CNS). Afterwards, the most active inhibitor having an excellent absorption, distribution, metabolism, excretion, and toxicity (ADME-Tox) profile and meeting all drug-likeliness conditions (Tian et al., 2015), was chosen for molecular docking simulation as a technology widely applied in the field of drug discovery (Serrano et al., 2020), indicating the intermolecular interactions produced towards the dopamine transporter membrane protein encoded as 4 M48.pdb https://www.rcsb.org/structure/4M48 (Bank, n.d.). Moreover, the process of molecular docking for the (candidate ligand-targeted protein) complex was successfully validated using the docking validation protocol (El fadili et al., 2022a). The molecular stability of the studied complex was examined with the help of the molecular dynamics (MD) technique during 100 ns of MD simulation time, studying the conformational changes behaviors of the targeted protein towards the candidate drug with detailed temporal resolution and highest atomic precision (El fadili et al., 2022b; Hollingsworth and Dror, 2018; Hu et al., 2023; Karplus and McCammon, 2002). Finally, the molecular mechanics with generalized born and surface area (MMGBSA) solvation was equally conducted to calculate the binding free energies of the studied complex, to check again the molecular stability of the candidate ligand in complex with the protein target (Faisal et al., 2022; Ononamadu et al., 2021). Thus, the present study aims to discover a highly effective drug among thirty selective and competitive GluT1 inhibitors, predicted with a good ADME-Toxicity profile, and defined with a good level of molecular stability towards the targeted protein, which we can strongly recommend for the treatment of schizophrenia and other impairments associated with NMDA receptor hypofunction.
2 Materials and methods
2.1 Experimental database
The present work aims to study thirty of 3,4-disubstituted pyrrolidine sulfonamides shown in Table S1 (Wang et al., 2018), whose the human GlyT1 activities of Ki order were converted to pKi scale (pKi = - log10 Ki), and the experimental database was divided on training and test sets which contain 80 % and 20 % of active molecules, respectively.
2.2 Molecular descriptors calculation
To develop the quantitative structure–activity relationships (QSAR), a number of molecular descriptors were calculated, as resulted in Table S2. The constitutional descriptors were executed using ACD/chemsketch software (Österberg and Norinder, 2001), while the geometric, physicochemical, thermodynamic, and topological descriptors were obtained using ChemBio3D software (Milne, 2010).
2.3 Statistical methods
Initially, the principal component analysis (PCA) was applied as a multivariate statistical method, to reduce the dimension of calculated descriptors based on the correlation matrix, in which the non-correlated descriptors were kept well, while one of the two highly correlated descriptors (r greater than 0.9) was removed (Groth et al., 2013; Guerra Tort et al., 2020; Maćkiewicz and Ratajczak, 1993). Secondly, the linear and nonlinear relationships between human GluT1 activity and uncorrelated descriptors were studied using multiple linear regression (MLR) and multiple nonlinear regression (MNLR) techniques, respectively (Mostoufi and Constantinides, 2023). So, MLR and MNLR QSAR models, which were developed for training set of 24 molecules, were externally validated for test set on 6 novel molecules. Moreover, the mathematical models were internally validated with the help of cross validation with the leave one out procedure (CV-LOO) (Er-Rajy et al., 2022).
2.4 In-silico pharmacokinetics, drug-likeliness, and ADMET depiction exploration
Before proceeding with any clinical investigation, it is necessary to carry out an assessment of absorption, distribution, metabolism, excretion and toxicity (ADME-Tox) properties in the human body, checking the five rules of Lipinski (Lipinski et al., 1997), and respecting the violations number of Veber, Ghose, Egan, and Muegge (Egan et al., 2000; Egan and Lauri, 2002; Muegge et al., 2001; Veber et al., 2002) for the drug candidates, which were predicted as central nervous system (CNS) agents according to Boild-Eggs model. To achieve this objective, we examined the physicochemical and in-silico pharmacokinetics properties of the candidate’s compounds, with the help of pkCSM (Pires et al., 2015), and SwissADME (“SwissADME,” n.d.) servers.
2.5 Molecular docking simulation
To discover the chemical bonds produced between a drug candidate and a target protein, we performed molecular docking, an effective drug discovery technique (Bassani et al., 2022). The dopamine transport protein, was uploaded from proteins data bank (PDB), which was discovered by the X-ray diffraction technique with a resolution of 2.96 Å (Bank, n.d.; Kouranov, 2006). The targeted protein coded as 4 M48.pdb was prepared using Discovery Studio 2021 (BIOVIA) software package (Systèmes, 2020), removing the water molecules (H2O), chloride and sodium ions (Na+, Cl-) and all suspended ligands like as cholesterol (C27H46O) and nortriptyline (C19H21N). The gasteiger charges and polar hydrogens were added. Then, the prepared protein was docked to the candidate drug using AutoDock 4.2 software (Norgan et al., 2011), converting the complex to PDBQT format. The sizes of grid box were centralized on (−42.424 Å, −0.573 Å, 55.066 Å), putting the 3D- structure dimensions on (120, 120, 120) with a spacing of 0.375 Å. Finally, the results of intermolecular interactions produced for the most stable complex were visualized by mean of Discovery Studio 2021 (BIOVIA) software.
2.6 Molecular dynamic
To investigate the stability of intermolecular interactions produced between the candidate drug and targeted protein, we performed the molecular dynamics for studied complex during 100 ns of simulation time, using Desmond software, a package of Schrödinger LLC (Pyrhönen et al., 2022). The (candidate drug-targeted protein) complex was prepared with the assistance of Protein Preparation Maestro. Then, the pre-treated complex was optimized in the System Builder, working on a solvent model of TIP3P orthorhombic box, with application of OPLS force field (Kaminski et al., 2001). Additionally, the selected model was neutralized by the reproduction of physiological conditions, adding water molecules (H2O) and counter ions ([Na+, Cl-] equal to 0.15 M) with a temperature of 300 K and a pressure of 1 atm.
3 Results and discussion
3.1 Quantitative structure-activity relationships (QSARs)
3.1.1 Principal components analysis
The quantitative structure–activity relationship technique is intended to develop a predictive QSAR model between biological activity and uncorrelated descriptors. For this goal, we applied the principal component analysis (PCA) as a statistical method widely applied to minimize the size of molecular descriptors in a reduced number of factorial axes (or principal components) based on the correlation matrix (Bastianoni et al., 2021), as resulted in Table S2, which indicates thirty-two strong correlations (correlation coefficients of Pearson superior than 0.9). So, the development of QSAR model was focused only on weakly correlated descriptors.
3.1.2 Multiple linear regression
To study the linear relationship between the structural descriptors and human GluT1 activity, we applied the multiple linear regression (MLR) technique (Uyanık and Güler, 2013) on 24 molecules of training set, based on stepwise selection for hundreds of randomizations. Therefore, the best MLR QSAR model was obtained in equation (1), considering the best values of determination, adjustment, and correlation coefficients (R2, R2 adjusted, and R) and minimal root mean square deviation (RMSD).
The first predictive QSAR model developed in equation (1), demonstrates that human Glut1 activity of pKi order is strongly correlated with constitutional descriptors (in particular %H, %N, %O and D) and geometric descriptors (like as T E, and VDW E), with minimal root mean square deviation (RMSD of 0.546) and good values of determination, correlation, and adjustment coefficients (R2 = 0.746, R2 adjusted = 0.657, and R = 0.864, respectively). The six selected descriptors (Table S3) have significant impacts on human GluT1 activity, because the slope of each one is defined by a probability<0.05 (5%) for 95% of confidence interval, as displayed in Table 1.
Source
Value
Standard deviation
t
Pr > |t|
Lower terminal (95%)
Higher terminal (95%)
Constante
−60.528
12.047
−5.024
0.000
−85.945
−35.111
D
43.061
7.133
6.037
< 0.0001
28.011
58.110
%H
3.569
0.710
5.025
0.000
2.071
5.068
VDW E
0.321
0.101
3.162
0.006
0.107
0.534
T E
−0.087
0.026
−3.366
0.004
−0.142
−0.033
%N
−0.642
0,129
−4.969
0.000
−0.915
−0.369
%O
−0.539
0.117
−4.628
0.000
−0.785
−0.293
The developed MLR model shows that electronic density (D), mass percentage of hydrogen (%H), and van der Waals energy (VDW E) have a positive effect on human GluT1 activity, but mass percentages of nitrogen and oxygen (%N, %O), and torsion energy (T E) affect it negatively, as displayed in Fig. 1.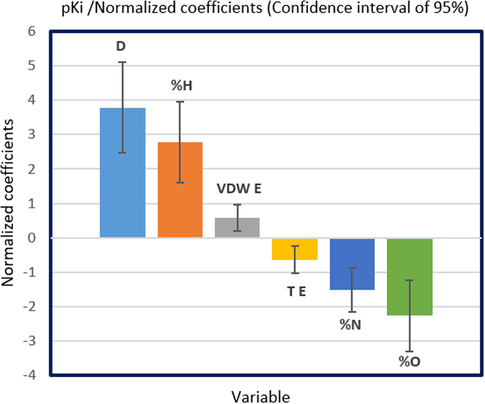
The impact of selected descriptors on human GluT1 activity.
The analysis of variance (Anova test) shows that calculated value of Fisher (F = 8.332) is superior to its critical value (F (30, 6) = 2.42, p < 5%), which indicates that null hypothesis (H0) was rejected, so there is an homogenous variance between the inhibitory activity and six selected descriptors as displayed in Table 2.
Source
DDL
Total square
Mean square
F
Pr > F
Model
6
14.923
2.487
8.332
0.000
Error
17
5.075
0.299
Adjusted total
23
19.998
3.2 Multiple non-linear regression
To model the nonlinear regression between the six uncorrelated descriptors, signaled by MLR method and GlyT1 activity, we performed the multiple non-linear regression (MNLR) method (Kravić et al., 2022), for twenty-four inhibitors (training set) based on following programmed function:
(El fadili et al., 2022b) Y: predicted activity by MNLR QSAR model. a0: constant of predictive model. Xi: selected descriptor. ai, bi: slopes of each descriptor for one and two degrees, respectively.
The second QSAR model, developed by the pre-programmed function of multiple non-linear regression confirms also that human GluT1 activity of pki order is affected by the six selected descriptors as expressed in equation (2). This mathematical model is done by a low deviation (RMSD equal to 0.636) and good values of determination and correlation coefficients (R2 = 0.777 and R = 0.882, respectively).
3.3 Validation of predictive QSAR models
3.3.1 External validation
External validation is an essential and critical step to examine the reliability of MLR and MNLR QSAR models, ensuring their application to new molecules included in the test set (Rezende et al., 2019). Based on developed models in equations (1) and (2) for twenty-four molecules (training set), we tested six novel molecules (2, 10, 11, 13, 19, 24) which are part of test set, and we obtained an external correlation coefficient of R2ext = 0.648 and R2ext’=0.872 for MLR and MNLR models, respectively. According to Golbreikh and Tropsha's study, the linear and non-linear models are externally validated because the external correlation coefficients are superior to 0.6. The Table 3, shows the predicted activities for six novel molecules by MLR and MNLR models, where the quantitative correlations are translated in Figs. 2 and 3, successively. * Indicates the test set molecules.
Compounds number
Molecules nomenclature
Observed pKi
Predicted pKi (MLR)
Predicted pKi (MNLR)
2*
(3R,4S)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)-N,4-diphenylpyrrolidin-3-amine.
6.701
5.447
5.606
10*
N-((3R,4S)-4-(4-fluorophenyl)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)pyrrolidin-3-yl)-5-(trifluoromethyl)pyridin-3-amine.
5.936
7.218
7.088
11*
(3R,4S)-N-(3-chlorophenyl)-4-(3-fluorophenyl)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)pyrrolidin-3-amine.
7.377
8.286
8.257
13*
N-((3R,4S)-4-(4-fluorophenyl)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)pyrrolidin-3-yl)-2-(trifluoromethyl)pyridin-4-amine.
6.928
7.986
7.870
19*
(3R,4S)-N-((R)-2,3-dihydro-1H-inden-1-yl)-4-(4-fluorophenyl)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)pyrrolidin-3-amine.
9.000
10.644
9.518
24*
4-(((3R,4S)-3-benzyl-4-phenylpyrrolidin-1-yl)sulfonyl)-1-methyl-1H-imidazole.
7.032
6.653
7.845
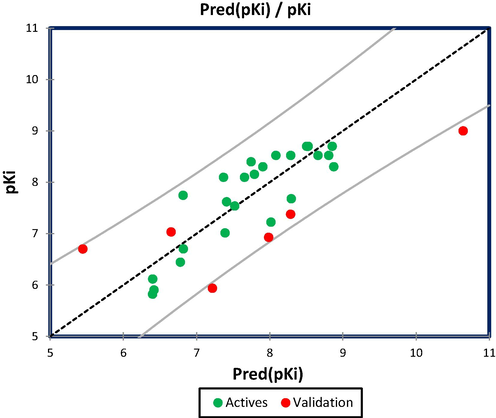
Quantitative correlation between observed and predicted GluT1 activity using MLR technique.
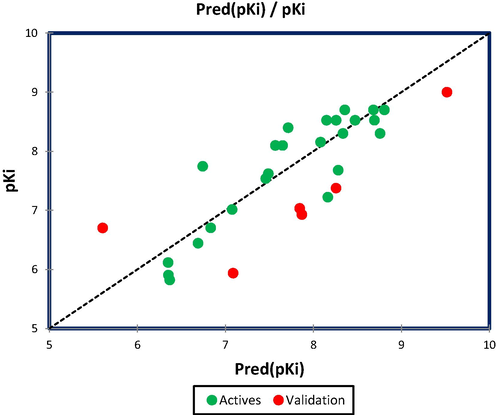
Quantitative correlation between observed and predicted GluT1 activity using MNLR technique.
3.3.2 Internal validation
A cross-validation (CV) technique was performed using the leave-one-out (LOO) procedure, for internal validation of the established QSAR models, testing each chemical compound once, modeling a new mathematical model on twenty-three molecules each time (N-1 = 23) and predicting human GluT1 activity for each tested molecule (Rafało, 2022). The process was repeated for all molecules in the training set, as resulted in Table 4. Afterwards, the quadratic coefficient (Q2 cv) of cross-validation, was calculated based on the following equation:
Molecules number
Molecules nomenclature
Observed pKi
Predicted pKi (MLR)
Predicted pKi (MNLR)
Predicted pKi (CVLOO)
1
2-chloro-N-((3R,4S)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)-4-phenylpyrrolidin-3-yl)-3-(trifluoromethyl)benzamide.
6.703
6.820
6.834
7.131
3
(3R,4S)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)-4-phenyl-N-(o-tolyl)pyrrolidin-3-amine.
5.821
6.399
6.367
6.648
4
(3R,4S)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)-4-phenyl-N-(m-tolyl)pyrrolidin-3-amine.
7.745
6.817
6,742
6.417
5
(3R,4S)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)-4-phenyl-N-(p-tolyl)pyrrolidin-3-amine.
5.903
6.419
6.353
6.634
6
(3R,4S)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)-4-phenyl-N-(3-(trifluoromethyl)phenyl)pyrrolidin-3-amine.
8.097
7.370
7.651
7.292
7
(3R,4S)-4-(4-fluorophenyl)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)-N-(3-(trifluoromethyl)phenyl)pyrrolidin-3-amine.
8.523
8.085
8.150
8.018
8
(3R,4S)-N-(3-chlorophenyl)-4-(4-fluorophenyl)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)pyrrolidin-3-amine.
8.523
8.288
8.259
8.247
9
(3R,4S)-N-(4-fluoro-3-(trifluoromethyl)phenyl)-4-(4-fluorophenyl)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)pyrrolidin-3-amine.
8.301
8.876
8.758
9.085
12
(3R,4S)-N-(3-chlorophenyl)-4-(2-fluorophenyl)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)pyrrolidin-3-amine.
7.678
8.296
8.282
8.394
14
(3R,4S)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)-4-(pyridin-3-yl)-N-(3-(trifluoromethyl)phenyl)pyrrolidin-3-amine.
6.116
6.401
6.350
6.494
15
(3R,4S)-4-(4-fluorophenyl)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)-N-(3-(trifluoromethoxy)phenyl)pyrrolidin-3-amine.
8.398
7.745
7.714
7.639
16
N-((3R,4S)-4-(4-fluorophenyl)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)pyrrolidin-3-yl)-4-(trifluoromethyl)pyridin-2-amine.
8.097
7.656
7.568
7.576
17
N-((3R,4S)-4-(4-fluorophenyl)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)pyrrolidin-3-yl)-6-(trifluoromethyl)pyrimidin-4-amine.
8.301
7.906
8.337
7.629
18
(3R,4S)-N-(4-fluoro-3-(trifluoromethoxy)phenyl)-4-(4-fluorophenyl)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)pyrrolidin-3-amine.
8.699
8.504
8.357
8.477
20
(3R,4S)-N-(4-fluoro-3-(trifluoromethyl)phenyl)-4-(4-fluorophenyl)-1-((1-methyl-1H-1,2,3-triazol-4-yl)sulfonyl)pyrrolidin-3-amine.
8.523
8.806
8.694
8.896
21
4-(((3S,4R)-3-(4-fluorophenyl)-4-((3-(trifluoromethoxy)phenoxy)methyl)pyrrolidin-1-yl)sulfonyl)-1-methyl-1H-imidazole.
8.155
7.790
8.082
7.649
22
((3R,4S)-4-(4-fluorophenyl)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)pyrrolidin-3-yl)(3-(trifluoromethoxy)phenyl)methanone.
7.538
7.522
7.459
7.515
23
N-(((3S,4S)-4-(4-fluorophenyl)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)pyrrolidin-3-yl)methyl)-3-(trifluoromethoxy)aniline.
7.222
8.017
8.164
8.108
25
4-(((3R,4S)-4-(4-fluorophenyl)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)pyrrolidin-3-yl)oxy)-6-(trifluoromethyl)pyrimidine.
7.013
7.390
7.077
7.583
26
(3R,4S)-4-(4-fluorophenyl)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)-N-(3-(trifluoromethoxy)phenyl)pyrrolidine-3-carboxamide.
6.444
6.779
6.690
7.137
27
(3R,4R)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)-4-((R)-tetrahydro-2H-pyran-2-yl)-N-(3-(trifluoromethoxy)phenyl)pyrrolidine-3-carboxamide.
8.523
8.660
8.474
8.714
28
(3R,4R)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)-4-((S)-tetrahydrofuran-2-yl)-N-(3-(trifluoromethoxy)phenyl)pyrrolidine-3-carboxamide.
7.620
7.411
7.489
6.921
29
(3R,4R)-4-(1,3-dioxepan-2-yl)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)-N-(3-(trifluoromethoxy)phenyl)pyrrolidine-3-carboxamide.
8.699
8.854
8.806
8.968
30
(3R,4R)-4-(1,3-dioxan-2-yl)-1-((1-methyl-1H-imidazol-4-yl)sulfonyl)-N-(3-(trifluoromethoxy)phenyl)pyrrolidine-3-carboxamide.
8.699
8.527
8.682
8.445
As: Yob: the observed activity, Ypr: the predicted activity, and Ymean: the mean of observed activities.
According to Golbreikh and Tropsha's study, although the result of quadratic coefficient (Q2cv = 0.546) exceeds 0.5, so the developed QSAR model is internally validated with a good reliability and strong predictivity.
3.3.3 Validation using Y-randomisation test
According to Golbreikh and Tropsha's study, the highest value of quadratic coefficient generated by cross validation technique with the leave one out procedure (CVLOO) is probable to be overestimated and may refers to a lucky correlation, so the internal validation is necessary but still not enough sufficient. For this reason, the Y-Randomization test is so required (Golbraikh and Tropsha, 2002; Halder and Jha, 2010). Using java Platform SE binary, we evaluated the predictive accuracy of developed QSAR model through one hundred randomizations as resulted in Table S4. The value of cR2p criteria was 0.621, which exceeds 0.5 threshold. Moreover, the resulted correlation, determination, and cross-validation with the leave-one-out procedure coefficients of the original model are so higher than the marked values for all 100 randomizations. So, the predicted GlyT1 activity by the originally developed model are not due to random chance.
3.3.4 Statistical criterias of Golbreikh and Tropsha's study
Golbreikh and Tropsha's statistical study emphasizes that a certain number of postulated conditions must be verified before the quantitative structure–activity relationship model can be applied to other new molecules. In this part, we noted that the generated model by MLR technique satisfy with success all postulated threshold conditions of Tropsha and Golbraikh's theory, as presented in Table 5. Consequently, the developed model is qualified by an accurate and precise predictivity, which affirms again that human GlyT1 activity is really affected by the six selected descriptors.
Statistical parameters
Developed Equations
Model scores
Threshold
Comment
0.746
> 0.6
Accepted
0.657
> 0.6
Accepted
0.648
> 0.6
Accepted
0.529
> 0.5
Accepted
R2rand
The average of 100 R2rand (i)
0.243
< R2
Accepted
‘LOO’ rand
The average for 100
‘LOO‘ rand (i)
−0.660
< Q2cv
Accepted
cR2p
cR2p = R*
0.621
> 0.5
Accepted
3.4 In-silico pharmacokinetics ADMET prediction
We initiated the in-silico screening of thirty human GlyT1 inhibitors, using the BOILED-Egg model as a widely applied predictive technique in the field of medicinal chemistry, to discover drug candidates that could penetrate the central nervous system (CNS), as resulted in Fig. 4. We noticed that most of the chemical compounds are part of white Egan Eggs, so they are predicted to be absorbed by the gastrointestinal tract, except the molecules marked as 2, 3, 4, 5, and 19, which are part of the yellow part of Egan-egg, thus they are predicted to be effluated from the central nervous system (CNS) by the P-glycoprotein, and predicted as potent inhibitors with an important ability to cross the blood–brain barrier (BBB). However, molecule 24 colored in red was predicted to not be effluated from the central nervous system, but it’s capable to pass through the blood–brain barrier (BBB) (Daina and Zoete, 2016). Therefore, we conclude that just five glycine transporter type 1 inhibitors (C2, C3, C4, C5, and C19) which are predicted as central nervous system agents with the highest probability to cross the blood–brain barrier.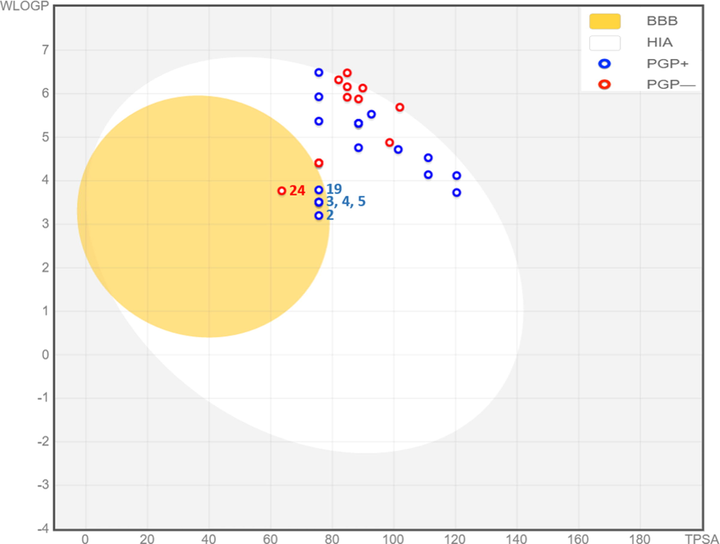
The predictive model of BOILED-Egg for 3,4-disubstituted pyrrolidine.
Subsequently, we studied the physicochemical and pharmacokinetic properties of all central nervous system agents, as shown in Table 6 and Table 7, respectively. Based on the properties of nortriptyline as a positive control molecule co-crystallized with the DAT protein, we noticed that all predicted inhibitors are in good agreement with nortriptyline, where they respect all five rules of Lipinski (MW ≤ 500 g/mol, 40 ≤ MR ≤ 130, HBD < 5, Log P (Octanol/Water) < 5, HBA ≤ 10) (Er-rajy et al., 2022; Er-rajy et al., 2023a; Er-rajy et al., 2023b), and satisfy the violations number of Veber, Ghose, Egan, and Muegge (Jia et al., 2020). Moreover, the pharmacokinetics properties reveal a good ADME-Toxicity profile, where all predicted molecules are defined by a good human intestinal absorption (HIA superior than 94 %), and the values of CNC and BBB permeabilities are included in (-2 to −3) Log PS, and higher than −1 Log BB, respectively. Thus, they have a good permeabilities to the central nervous system (CNS) and the blood–brain barrier (BBB). In addition, they are all predicted as substrates of 3A4 cytochrome. C4 and C24 compounds and C19 as the most active ligand, are predicted as potent inhibitors for 2C9 and 3A4 cytochromes. Also, they are defined by an excretion of a total clearance inferior than 1 Log ml/min/kg. According to the AMES toxicity test, all chemical compounds are predicted as non-toxic inhibitors of the human body, without any skin sensitization, but they have a known positive hepatotoxicity.
Compounds number
Physical and chemical properties
Lipinski violations
Veber violations
Egan violations
Ghose violations
Muegge violations
Molecular weight (g/mol)
Molar refractive index
Rotatable bonds Number
Log P (Octanol/ Water)
H-bond Acceptors Number
H-bond donors Number
Categorical (Yes/No)
Threshold
≤ 500
40 ≤ MR ≤ 130
< 10
< 5
≤ 10
< 5
C2
382.48
109.01
5
3.03
4
1
Yes
Yes
Yes
Yes
Yes
C3
396.51
113.98
5
2.65
4
1
Yes
Yes
Yes
Yes
Yes
C4
396.51
113.98
5
3.24
4
1
Yes
Yes
Yes
Yes
Yes
C5
396.51
113.98
5
3.00
4
1
Yes
Yes
Yes
Yes
Yes
C19
440.53
119.90
5
3.37
6
1
Yes
Yes
Yes
Yes
Yes
C24
381.49
109.48
5
2.55
4
0
Yes
Yes
Yes
Yes
Yes
Nortriptylene
263.38
86.06
3
3.22
1
1
Yes
Yes
Yes
Yes
No
Compounds number
Absorption
Distribution
Metabolism
Excretion
Toxicity
Intestinal Absorption (human)
BBB permeability
CNS permeability
Substrate
Inhibitor
Total Clearance
AMES Toxicity
Hepatotoxicity
Skin Sensitization
Cytochromes
2D6
3A4
1A2
2C19
2C9
2D6
3A4
Numeric (% Absorbed)
Numeric (Log BB)
Numeric (Log PS)
Categorical (Yes/No)
Numeric (Log ml/min/kg)
Categorical (Yes/No)
C2
94.986
0.175
−2.334
No
Yes
No
Yes
Yes
No
No
0.27
Not toxic
Yes
No
C3
96.126
0.219
−2.264
No
Yes
No
Yes
No
No
Yes
0.269
Not toxic
Yes
No
C4
95.608
0.142
−2.254
No
Yes
No
Yes
Yes
No
Yes
0.264
Not toxic
Yes
No
C5
95.522
0.125
−2.26
No
Yes
No
Yes
No
No
Yes
0.272
Not toxic
Yes
No
C19
94.049
0.151
−2.313
No
Yes
No
No
Yes
No
Yes
0.41
Not toxic
Yes
No
C24
99.251
0.066
−2.216
No
Yes
Yes
Yes
Yes
No
Yes
0.114
Not toxic
Yes
No
Nortriptylene
97.482
0.808
−1.196
No
Yes
Yes
No
No
Yes
No
0.929
Not toxic
Yes
Yes
3.5 Molecular docking
The results of molecular docking simulation exposed in the Fig. 5, demonstrate the intermolecular interactions produced between the dopamine transporter (DAT) protein as a key therapeutic strategy to treat clinical depression and neuropathic pain (Adamski et al., 2017; Belhassan et al., 2019), and the compound C19 as the most active ligand with a good ADME-Toxicity profile. 2D and 3D dimensional visualizations confirm that C19 ligand forms two hydrogen bonds towards ASP 475 and GLU 480 amino acids residues of DAT protein in A chain, with nuclear distances of 3.90 Å and 3.07 Å respectively, taking into account that the chemical bonds of the hydrogen bonding type make the (ligand–protein) complex so stable. Additionally, C19 reacts to VAL 120 and ALA 479 amino acids forming two chemical bonds of Pi-Alkyl type at 6.08 Å and 3.95 Å, respectively. And Pi-Pi T- shaped bond with TYR 124 amino acid at 5.39 Å. Consequently, ASP 475, GLU 487, VAL 120, ALA 479, and TYR 124 are the main sites of glycine transporter type 1 inhibition for the candidate drug. The resulted chemical bonds include intermolecular interactions similar to those detected towards Tyr 124, Ala 479, and Val 120 amino acid residues, which have been produced by two selective GluT1 inhibitors, after being bound to the same membrane protein of drosophila melanogaster dopamine transporter (DAT), which were also designed to improve memory performance while treating depression and the cognitive symptoms associated with schizophrenia (El fadili et al., 2023; Hudson et al., 2020; Santora et al., 2018).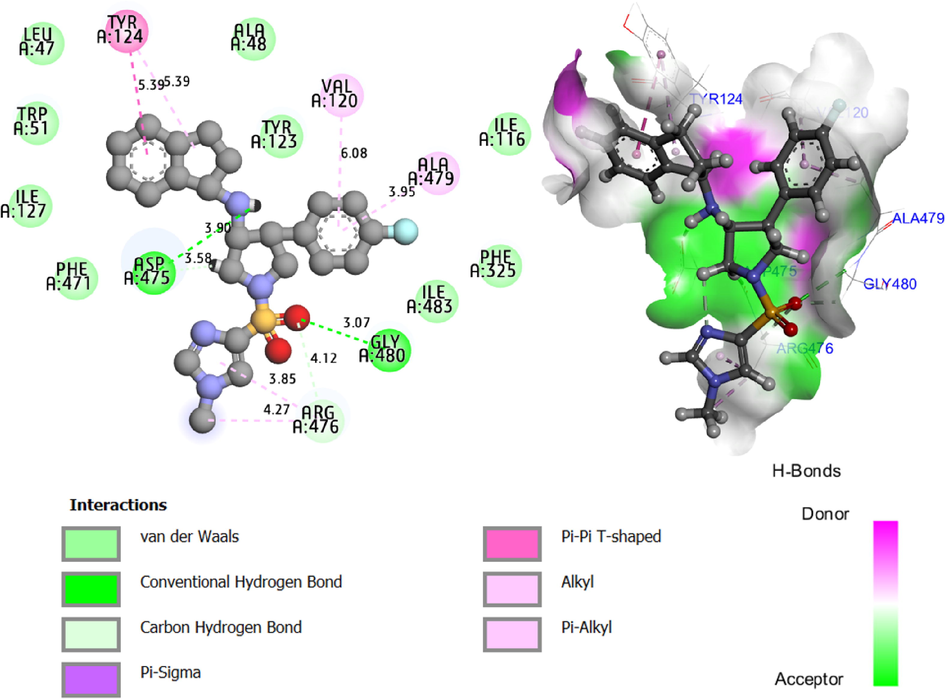
2D (left) and 3D (right) intermolecular interactions, produced for (C19 ligand - DAT protein) complex with binding energy of −8.96 Kcal/mol.
3.6 Docking validation protocol
The process of molecular docking simulations was examined using docking validation protocol or re-docking methodology for the (C19 ligand – DAT protein) complex. In the first time, we have compared the intermolecular interactions produced for the studied complex with the active sites of nortriptyline as co-crystallized ligand bound to the same chain of targeted protein. Where, the active sites of nortriptyline are the residues of TYR 124, PHE 325, and PHE 43 amino acids, which are experimentally extracted using the ProteinsPlus online server (“Zentrum für Bioinformatik: Universität Hamburg - Proteins Plus Server,” n.d.), as resulted in Fig. 6A, and TYR 124, PHE 325, PHE 43, VAL 120, and ALA 479 amino acids, which are theoretically produced using Autodock tools, as presented in Fig. 6B. Therefore, we noticed that TYR 124, VAL 120, and ALA 479 amino acids are the same amino acids, which were obtained for (C19 - DAT protein) complex. In second time, we have used the superposition of co-crystallized and docked nortriptyline, and we have obtained a good overlay pose with a minimal root mean square deviation (RMSD = 0.128) which is<2 Å as shown in Fig. 6C. Therefore, we conclude that the candidate drug was docked in the active sites of targeted protein, and the process of molecular docking is successfully validated (Abdullahi et al., 2020).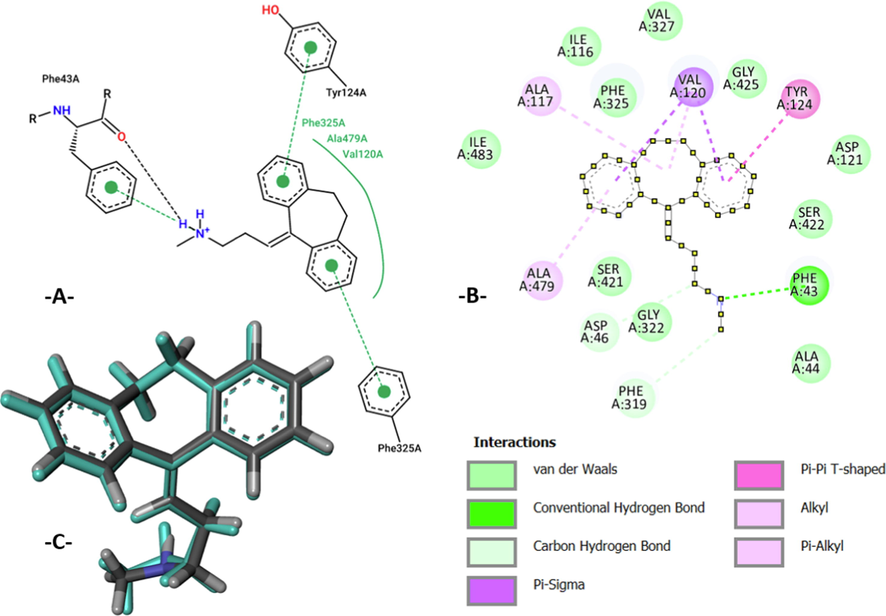
The active sites of co-crystallized ligand of DAT protein in A-chain. B: 2D intermolecular interactions produced for the (docked nortriptyline - DAT protein) complex with a binding energy of − 9.02 kcal/mol. C: Re-docking pose with a RMSD value of 0.128 Å (co-crystallized nortriptyline colored in cyan superposed on docked nortriptyline).
3.7 Molecular dynamic simulation
High-performance molecular dynamics simulation was performed to study the stability of intermolecular interactions produced for the (C19 ligand - DAT protein) complex during 100 ns (El fadili et al., 2022b; Er-rajy et al., 2023). The results of the conformational changes presented in Fig. 7 show that the root mean square deviation (RMSD) of the candidate ligand oscillates in parallel with the RMSD values of the protein target during the first thirty nanoseconds, then destabilizes a little, and stabilizes again during the last thirty nanoseconds due to the effect of heavy atoms as oxygen and nitrogen which make the ligand–protein complex so stable. In addition, there are no deviations greater than 3 Å, which demonstrates that the simulation remains in equilibrium during the molecular dynamics simulation time without any noticeable deviation. Secondly, the root mean square fluctuation (RMSF) values show only two remarkable fluctuations for the residuals 470 and 530, which exceeded 3 Å, as shown in Fig. 7. So, we conclude that the ligand does not diffuse far from its primary binding site.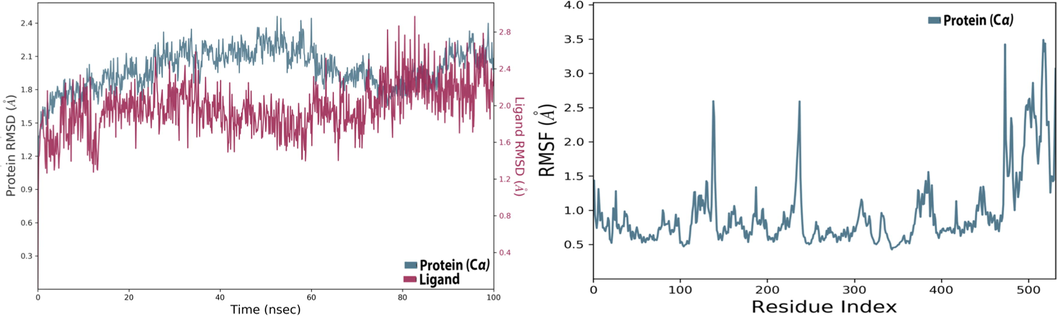
The changes of Root Mean Square Deviation (RMSD) and Root Mean Square Fluctuation (RMSF) during 100 ns, for the dopamine transporter protein complexed with C19 ligand.
In addition, we recorded negligible fluctuations for ligand properties, such as radius of gyration (r Gyr), molecular surface area (MolSA), solvent accessible surface area (SASA), and polar surface area (PSA), which all oscillate in a very small range over 75 ns, making just one remarkable fluctuation at the same time, due to the influence of nitrogen, oxygen, sulfur, and fluorine, as typical heteroatoms characterizing this organic molecule (AlAmiery, 2023; TANWER and Shukla, 2023). However, they stabilize again until the rest of the molecular dynamics simulation time as displayed in Fig. 8, which demonstrates minimal changes in the compactness of the candidate ligand. Consequently, the targeted protein is very flexible after being bound to the candidate ligand.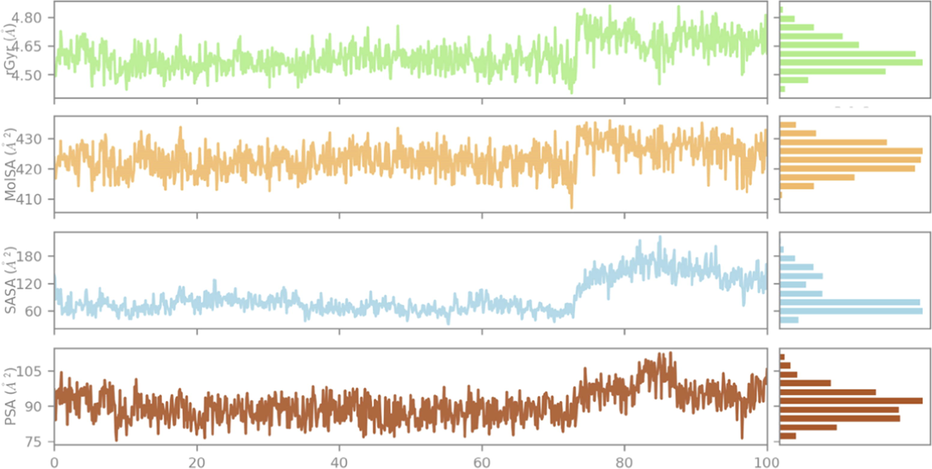
The changes in radius of gyration (r Gyr), molecular surface area (MolSA), solvent accessible surface area (SASA), and polar surface area (PSA), over 100 ns of molecular dynamics simulation time.
Furthermore, the upper panel shown in Fig. 9, reflects the specified number of contacts produced by the candidate ligand towards the targeted protein throughout molecular dynamics simulation time, while the lower panel displays the residues of amino acids, which reacted with the C19 compound along the trajectory. The scale on the right-hand side of the graph shows that the strongest connection is one that corresponds to a dark orange residue that has entered into more than one chemical reaction with the candidate ligand (Ebenezer et al., 2022). For this, ASP 475 amino acid residue has the main function in the stability of (protein–ligand) complex throughout 100 ns of MD simulation time, which presents an important interaction fraction of H-bonds as displayed on the left side of Fig. 9.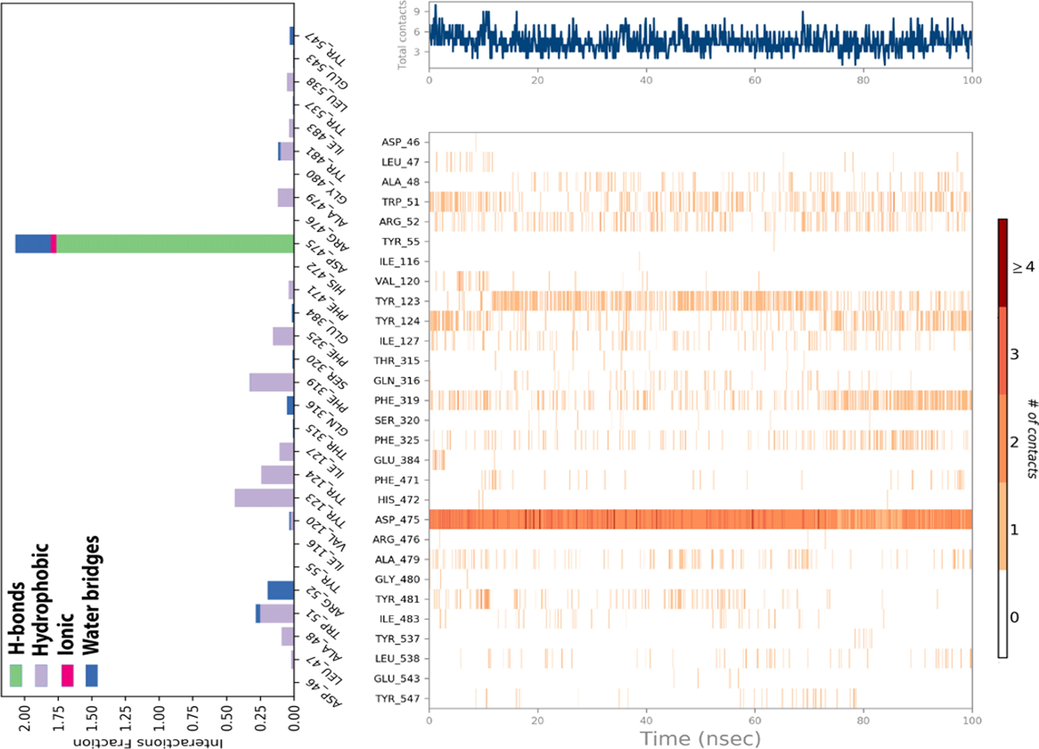
The histogram of interaction fractions between DAT protein and C19 ligand (left), and the timeline depiction of intermolecular interactions and chemical contacts of candidate ligand in the active sites of targeted protein (right).
The Molecular Mechanics with Generalized Born and Surface Area (MMGBSA) solvation was equally conducted to calculate the binding free energies of the C19 ligand in complex with DAT protein, as resulted in Table 8 (Faisal et al., 2022; Ononamadu et al., 2021). The MMGBSA approach shows that the binding energies are negatively very large, indicating that the candidate drug have reacted with the protein target with low energies (minimal ΔG Bind scores), which makes the complex so stable with optimal energies during the molecular dynamic’s simulation time.
Studied complex
MMGBSA dG Bind
MMGBSA dG Bind Coulomb
MMGBSA dG Bind Covalent
MMGBSA dG Bind Hbond
MMGBSA dG Bind Lipo
MMGBSA dG Bind Solv GB
MMGBSA dG Bind vdW
(C19 – protein) complex
−51.417899
−56.518147
1.557139
−0.820922
–23.877425
73.360626
−45.119169
4 Conclusion
In summary, quantitative structure–activity relationships reveal that constitutional and geometric descriptors have a key function in the human GluT1 activity of the 3,4-disubstituted pyrrolidine sulfonamides family. In-silico screening indicates that C2, C3, C4, C5, and C19 were predicted as non-toxic inhibitors of the human body, and functioning as central nervous system agents with the highest probability to cross the blood–brain barrier, respecting Lipinski, Veber, Egan, Muegge, and Rhose conditions. The compound C19 having the highest activity and predicted with a good ADME-Tox profile, was chosen for molecular docking and molecular dynamics simulations, which indicate that this candidate ligand produced stable intermolecular interactions with ASP 475, GLU 480, VAL 120, ALA 479, and TYR 124 amino acids residues of dopamine transporter protein, because the studied properties of (C19 ligand – DAT protein) complex remained perfectly stable until 100 ns of molecular dynamic simulation time with lowest binding energies of MMGBSA solvation. Therefore, C19 ligand is strongly recommended as candidate drug to treat schizophrenia and similar disabilities in relation with glutaminergic N-methyl-D-aspartate receptor hypofunction.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Funding
This research was funded by Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2023R141), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Acknowledgment
The authors extend their appreciation to Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2023R141), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
References
- In silico QSAR and molecular docking simulation of some novel aryl sulfonamide derivatives as inhibitors of H5N1 influenza A virus subtype. Beni-Suef Univ. J. Basic Appl. Sci.. 2020;9:2.
- [CrossRef] [Google Scholar]
- Novel family of fused tricyclic [1,4]diazepines: Design, synthesis, crystal structures and molecular docking studies. Tetrahedron. 2017;73:3377-3386.
- [CrossRef] [Google Scholar]
- AlAmiery, A., 2023. Gravimetric and Density Functional Theory Investigations on 4-Amioantipyrin Schiff base as an inhibitor for mild steel in HCl solution. Prog. Color Colorants Coat. https://doi.org/10.30509/pccc.2023.167077.1196
- Glycine reuptake inhibitor RG1678: A pharmacologic characterization of an investigational agent for the treatment of schizophrenia. Neuropharmacology. 2012;62:1152-1161.
- [CrossRef] [Google Scholar]
- Discovery of novel aminotetralines and aminochromanes as selective and competitive glycine transporter 1 (GlyT1) inhibitors. J. Med. Chem.. 2018;61:7503-7524.
- [CrossRef] [Google Scholar]
- ALX 5407: A potent, selective inhibitor of the hGlyT1 glycine transporter. Mol. Pharmacol.. 2001;60:1414-1420.
- [CrossRef] [Google Scholar]
- Bank, R.P.D., n.d. RCSB PDB - 4M48: X-ray structure of dopamine transporter elucidates antidepressant mechanism [WWW Document]. URL https://www.rcsb.org/structure/4M48 (accessed 1.6.23a).
- Re-exploring the ability of common docking programs to correctly reproduce the binding modes of non-covalent inhibitors of SARS-CoV-2 Protease Mpro. Pharmaceuticals. 2022;15:180.
- [CrossRef] [Google Scholar]
- Multivariate analysis applied to aquifer hydrogeochemical evaluation: A case study in the coastal significant subterranean water body between “Cecina River and San Vincenzo”, Tuscany (Italy) Appl. Sci.. 2021;11:7595.
- [CrossRef] [Google Scholar]
- Study of novel triazolo-benzodiazepine analogues as antidepressants targeting by molecular docking and ADMET properties prediction. Heliyon. 2019;5:e02446.
- [Google Scholar]
- Identification of an orally bioavailable, potent, and selective inhibitor of GlyT1. ACS Med. Chem. Lett.. 2010;1:350-354.
- [CrossRef] [Google Scholar]
- A BOILED-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem. 2016;11:1117-1121.
- [CrossRef] [Google Scholar]
- Unveiling of pyrimidindinones as potential anti-norovirus agents—A pharmacoinformatic-based approach. Molecules. 2022;27:380.
- [CrossRef] [Google Scholar]
- Prediction of intestinal permeability. Adv. Drug Delivery Rev.. 2002;54:273-289.
- [CrossRef] [Google Scholar]
- Prediction of drug absorption using multivariate statistics. J. Med. Chem.. 2000;43:3867-3877.
- [CrossRef] [Google Scholar]
- El fadili, M., Er-rajy, M., Imtara, H., Kara, M., Zarougui, S., Altwaijry, N., Al kamaly, O., Al Sfouk, A., Elhallaoui, M., 2022a. 3D-QSAR, ADME-Tox In Silico Prediction and Molecular Docking Studies for Modeling the Analgesic Activity against Neuropathic Pain of Novel NR2B-Selective NMDA Receptor Antagonists. Processes 10, 1462. https://doi.org/10.3390/pr10081462.
- El fadili, M., Er-Rajy, M., Kara, M., Assouguem, A., Belhassan, A., Alotaibi, A., Mrabti, N.N., Fidan, H., Ullah, R., Ercisli, S., Zarougui, S., Elhallaoui, M., 2022b. QSAR, ADMET In Silico Pharmacokinetics, Molecular Docking and Molecular Dynamics Studies of Novel Bicyclo (Aryl Methyl) Benzamides as Potent GlyT1 Inhibitors for the Treatment of Schizophrenia. Pharmaceuticals 15, 670. https://doi.org/10.3390/ph15060670.
- El fadili, M., Er-rajy, M., Imtara, H., Noman, O.M., Mothana, R.A., Abdullah, S., Zerougui, S., Elhallaoui, M., 2023. QSAR, ADME-Tox, molecular docking and molecular dynamics simulations of novel selective glycine transporter type 1 inhibitors with memory enhancing properties. Heliyon e13706. https://doi.org/10.1016/j.heliyon.2023.e13706.
- Er-rajy, M., El fadili, M., Imtara, H., Saeed, A., Ur Rehman, A., Zarougui, S., Abdullah, S.A., Alahdab, A., Parvez, M.K., Elhallaoui, M., 2023. 3D-QSAR Studies, Molecular Docking, Molecular Dynamic Simulation, and ADMET Proprieties of Novel Pteridinone Derivatives as PLK1 Inhibitors for the Treatment of Prostate Cancer. Life 13, 127. https://doi.org/10.3390/life13010127.
- 2D-QSAR modeling, drug-likeness studies, ADMET prediction, and molecular docking for anti-lung cancer activity of 3-substituted-5-(phenylamino) indolone derivatives. Struct. Chem.. 2022;33:973-986.
- [CrossRef] [Google Scholar]
- QSAR, molecular docking, ADMET properties in silico studies for a series of 7-propanamide benzoxaboroles as potent anti-cancer agents. Chinese J. Anal. Chem.. 2022;50:100163
- [CrossRef] [Google Scholar]
- Design of novel anti-cancer drugs targeting TRKs inhibitors based 3D QSAR, molecular docking and molecular dynamics simulation. Journal of Biomolecular Structure and Dynamics. 2023;0:1-14.
- [Google Scholar]
- QSAR, molecular docking, and molecular dynamics simulation–based design of novel anti-cancer drugs targeting thioredoxin reductase enzyme. Struct Chem 2023
- [Google Scholar]
- Glycine transporters: essential regulators of neurotransmission. Trends Biochem. Sci.. 2005;30:325-333.
- [CrossRef] [Google Scholar]
- Identification and inhibition of the druggable allosteric site of SARS-CoV-2 NSP10/NSP16 methyltransferase through computational approaches. Molecules. 2022;27:5241.
- [CrossRef] [Google Scholar]
- Guerra Tort, C., Aguiar Pulido, V., Suárez Ulloa, V., Docampo Boedo, F., López Gestal, J.M., Pereira Loureiro, J., 2020. Electronic Health Records Exploitation Using Artificial Intelligence Techniques, in: 3rd XoveTIC Conference. Presented at the XoveTIC Conference, MDPI, p. 60. https://doi.org/10.3390/proceedings2020054060
- Validated predictive QSAR modeling of N-aryl-oxazolidinone-5-carboxamides for anti-HIV protease activity. Bioorg. Med. Chem. Lett.. 2010;20:6082-6087.
- [CrossRef] [Google Scholar]
- Molecular dynamics simulation of the interaction of food proteins with small molecules. Food Chem.. 2023;405:134824
- [CrossRef] [Google Scholar]
- Azetidine-based selective glycine transporter-1 (GlyT1) inhibitors with memory enhancing properties. Bioorg. Med. Chem. Lett.. 2020;30:127214
- [CrossRef] [Google Scholar]
- A drug-likeness toolbox facilitates ADMET study in drug discovery. Drug Discov. Today. 2020;25:248-258.
- [CrossRef] [Google Scholar]
- Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B. 2001;105:6474-6487.
- [CrossRef] [Google Scholar]
- Molecular dynamics simulations of biomolecules. Nat. Struct. Biol.. 2002;9:646-652.
- [CrossRef] [Google Scholar]
- The RCSB PDB information portal for structural genomics. Nucleic Acids Res.. 2006;34:D302-D305.
- [CrossRef] [Google Scholar]
- Nonlinear multivariate regression algorithms for improving precision of multisensor potentiometry in analysis of spent nuclear fuel reprocessing solutions. Chemosensors. 2022;10:90.
- [CrossRef] [Google Scholar]
- Analysis of binding determinants for different classes of competitive and noncompetitive inhibitors of glycine transporters. IJMS. 2022;23:8050.
- [CrossRef] [Google Scholar]
- Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev.. 1997;23:3-25.
- [CrossRef] [Google Scholar]
- The discovery of a structurally novel class of inhibitors of the type 1 glycine transporter. Bioorg. Med. Chem. Lett.. 2009;19:2974-2976.
- [CrossRef] [Google Scholar]
- Milne, G.W.A., 2010. Software Review of ChemBioDraw 12.0. J. Chem. Inf. Model. 50, 2053–2053. https://doi.org/10.1021/ci100385n.
- Mostoufi, N., Constantinides, A., 2023. Linear and nonlinear regression analysis, in: Applied Numerical Methods for Chemical Engineers. Elsevier, pp. 403–476. https://doi.org/10.1016/B978-0-12-822961-3.00008-X.
- Simple selection criteria for drug-like chemical matter. J. Med. Chem.. 2001;44:1841-1846.
- [CrossRef] [Google Scholar]
- In silico identification and study of potential anti-mosquito juvenile hormone binding protein (MJHBP) compounds as candidates for dengue virus - Vector insecticides. Biochem. Biophys. Rep.. 2021;28:101178
- [CrossRef] [Google Scholar]
- Prediction of drug transport processes using simple parameters and PLS statistics The use of ACD/logP and ACD/ChemSketch descriptors. Eur. J. Pharm. Sci.. 2001;12:327-337.
- [CrossRef] [Google Scholar]
- First-time-in-human study with GSK1018921, a selective GlyT1 inhibitor: Relationship between exposure and dizziness. Clin. Pharmacol. Ther.. 2011;90:597-604.
- [CrossRef] [Google Scholar]
- Selective GlyT1 inhibitors: Discovery of [4-(3-Fluoro-5-trifluoromethylpyridin-2-yl)piperazin-1-yl][5-methanesulfonyl-2-((S)-2,2,2-trifluoro-1-methylethoxy)phenyl]methanone (RG1678), a promising novel medicine to treat schizophrenia. J. Med. Chem.. 2010;53:4603-4614.
- [CrossRef] [Google Scholar]
- pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem.. 2015;58:4066-4072.
- [CrossRef] [Google Scholar]
- Molecular dynamics prediction verified by experimental evaluation of the solubility of different drugs in poly(decalactone) for the fabrication of polymeric nanoemulsions. Adv. NanoBiomed. Res.. 2022;2:2100072.
- [CrossRef] [Google Scholar]
- Cross validation methods: Analysis based on diagnostics of thyroid cancer metastasis. ICT Express. 2022;8:183-188.
- [CrossRef] [Google Scholar]
- Rezende, K.B. de C., Cunha, A.J.L.A. da, Amim Junior, J., Bornia, R.G., 2019. External validation of the Fetal Medicine Foundation algorithm for the prediction of preeclampsia in a Brazilian population. Pregnancy Hypertension 17, 64–68. https://doi.org/10.1016/j.preghy.2019.05.006.
- Development of the novel GlyT1 inhibitor, iclepertin (BI 425809), for the treatment of cognitive impairment associated with schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 2023
- [CrossRef] [Google Scholar]
- Design and synthesis of novel and selective glycine transporter-1 (GlyT1) inhibitors with memory enhancing properties. J. Med. Chem.. 2018;61:6018-6033.
- [CrossRef] [Google Scholar]
- QN-Docking: An innovative molecular docking methodology based on Q-Networks. Appl. Soft Comput.. 2020;96:106678
- [CrossRef] [Google Scholar]
- SwissADME [WWW Document], n.d. URL http://www.swissadme.ch/ (accessed 1.7.23).
- Systèmes, D., 2020. Free Download: BIOVIA Discovery Studio Visualizer [WWW Document]. Dassault Systèmes. URL https://discover.3ds.com/discovery-studio-visualizer-download (accessed 1.7.23).
- TANWER, S., Shukla, S., 2023. Cefuroxime: A Potential Corrosion Inhibitor for Mild Steel in Sulphuric Acid Medium. Prog. Color Colorants Coat. 16. https://doi.org/10.30509/pccc.2022.166974.1165.
- The application of in silico drug-likeness predictions in pharmaceutical research. Adv. Drug Deliv. Rev.. 2015;86:2-10.
- [CrossRef] [Google Scholar]
- A study on multiple linear regression analysis. Procedia. Soc. Behav. Sci.. 2013;106:234-240.
- [CrossRef] [Google Scholar]
- Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem.. 2002;45:2615-2623.
- [CrossRef] [Google Scholar]
- Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. CTMC. 2008;8:1555-1572.
- [CrossRef] [Google Scholar]
- De Novo design, synthesis, and biological evaluation of 3,4-disubstituted pyrrolidine sulfonamides as potent and selective glycine transporter 1 competitive inhibitors. J. Med. Chem.. 2018;61:7486-7502.
- [CrossRef] [Google Scholar]
- Recent progress in the discovery of non-sarcosine based GlyT1 inhibitors. CTMC. 2010;10:170-186.
- [CrossRef] [Google Scholar]
- Discovery of GlyT1 inhibitors with improved pharmacokinetic properties. Bioorg. Med. Chem. Lett.. 2009;19:1492-1495.
- [CrossRef] [Google Scholar]
- Zentrum für Bioinformatik: Universität Hamburg - Proteins Plus Server [WWW Document], n.d. URL https://proteins.plus/ (accessed 1.15.23).
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.105105.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1
Supplementary data 2
Supplementary data 2




