

Translate this page into:
Influence of end-capped engineering on 3-dimenional star-shaped triphenylamine-based donor materials for efficient organic solar cells
⁎Corresponding authors. adnan@cnu.ac.kr (Muhammad Adnan), riazhussain@uo.edu.pk (Riaz Hussain), jungyup.yang@kunsan.ac.kr (Jung Yup Yang), jclim@cnu.ac.kr (Jongchul Lim)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Star-shaped- triphenylamine-based (TPA) hole transporting materials (HTMs) are considered an up-and-coming candidate for developing efficient organic solar cells (OSCs). Therefore, we have designed partially oxygen bridged eight new novels (STRO1 – STRO8) and three-armed star-shaped HTMs with TPA core for the future development of efficient OSCs devices. Their photovoltaic, optical, and charge transport (CT) characteristics were studied and compared with the reference molecule (R). These designed materials have been characterized theoretically using various density functional theory (DFT) and time-dependent (TD-DFT) calculations. These planar configurated star-shaped molecules exhibited a red-shifted absorption, deeper HOMO levels, and improved extinction coefficients, enabling them to offer good phase separation morphology during blend formation. Furthermore, the distribution behavior of frontier molecular orbitals (FMOs), optical properties, open-circuit voltages, the density of states (DOS), transition density matrix (TDM), and reorganization energies of holes and electrons of the designed star-shaped molecules have been investigated. Besides, the complex study of STRO/PC61BM revealed the charge shifting process at the donor–acceptor interface. Therefore, our projected approach is a prerequisite in designing small molecule (SM)-based desirable photovoltaic materials for efficient OSCs, and light-emitting diodes (LEDs), and afterwards, these materials are suggested to the experimentalist for synthesis and to fabricate efficient photovoltaic devices.
Keywords
Star-shaped materials
Triphenylamine
Solar cells
Density functional theory

1 Introduction
Organic photovoltaic materials have a great hidden potential to fulfill future energy needs (Xiong et al., 2020) therefore, scientists are continuously struggling to synthesize such efficient photovoltaic materials, i.e., hole-transport or electron-transport materials (HTMs or ETMs) (Ma et al., 2020). Such molecules prove useful while device fabrication either organic or perovskite solar cells (OSCs or PSCs) to achieve stable and improved device performances. Therefore, until now, the advancement in these molecules has led the power conversion efficiency (PCE) to over 18 % in OSCs and over 25 % in PSCs (Cui et al., 2020; Gi-Hwan and Suk-Kim, 2021; Irshad et al., 2020; Adnan et al., 2020; Adnan and Lee, 2020).
Amongst, the HTM plays a pivotal role in improving and stabilizing the performances of the devices due to their ability to extract the photogenerated holes from the active layer and transfer these toward the metal electrode. Moreover, this is also acting a barrier between the active layer and the metal electrode. This process helps minimize the threat of recombination and improves the device's performance (Chang et al., 2012; Qin et al., 2019). Also, the HTMs have tremendous importance in minimizing the threat of device degradation by blocking the air moisture and the diffusion of the metal electrode into the active layer. The chemical structure of any HTM is essential and is considered a backbone for fabricating efficient and stable SCs. Therefore, various HTMs have been reported so far with numerous structures, including; organic, inorganic, polymers, and small-molecule (SM) based materials (Meng et al., 2018; Miao et al., 2020; Adnan et al., 2017; Adnan and Lee, 2018; Irshad et al., 2022; Irshad et al., 2022).
Among these, triphenylamines (TPAs) based HTMs are the most attractive and widely used materials in the solar cells field. To date, the best performance HTM is the SpiroOMeTAD and PTAA, widely used to prepare highly-efficient PSCs (Adnan et al., 2017). Though, some of the reports show that these materials degrade the SC device and limit the device stability as well (Yao et al., 2016). On the other hand, TPA is considered a better electron donor unit due to its ability to transform positive charges efficiently via radical cations and its capability to oxidize nitrogen centers (Bilal et al., 2020; Lv et al., 2020). Therefore, these TPA-based molecules are essential in preparing efficient SM-based organic and polymers materials (Adnan et al., 2021; Riaz et al., 2022; Alvina et al., 2022).
Star-shaped molecules are a group of SM-based organic materials having a central aromatic core attached to various functional groups (Liao et al., 2022). These materials exhibited a unique structure and are widely considered to have a characteristic of small organic and polymer molecules. Moreover, these materials have good solubility, better film formation properties, improved thermal stability, and can merge numerous structures to improve their physical and chemical characteristics (Adnan et al., 2017; Liao et al., 2022). Remarkably, these materials have been used widely in organic light-emitting diode (OLED) (Pathak et al., 2022), organic field effect transistors (OFET) (Aimi et al., 2022), and OSCs. Due to this reason, many researchers are interested in preparing such efficient HTMs having a heteroatom with fused aromatic rings because of their rigidity and coplanarity along with maximum overlap of π-orbitals for heads on π-stacking supporting intermolecular charge transport (Yang et al., 2016; Lu et al., 2022; Li et al., 2021; Zhang et al., 2022; Cui et al., 2022; Zhang et al., 2022; Zhang et al., 2016; Peng et al., 2022; Lei et al., 2022; He et al., 2022; Yu et al., 2020; Ren et al., 2022). Therefore, due to its ability to accumulate the features of SM-based organic materials and polymers, extended investigations of the star-shaped HTMs are an attractive area for research.
Herein, we designed and theoretically characterized ten new three-armed star-shaped donors (STRO1-STRO8), having a benzodithiophene (BDT) as the central-core unit, and modified end-capped units with multiple efficient functional groups. These star-shaped molecules have been designed to improve the materials' electrical, photovoltaic, and optoelectronic characteristics to ensure their best use in future SCs devices. For this purpose, we have modified the reference molecule (R) with different efficient end-capped units (Ma et al., 2021). The full names of the incorporated end-capped and bridged core units can be found in the supplementary section. In addition, the schematic device architecture and energy level diagram of the designed (STRO1-STRO8) series have been shown in Fig. 1.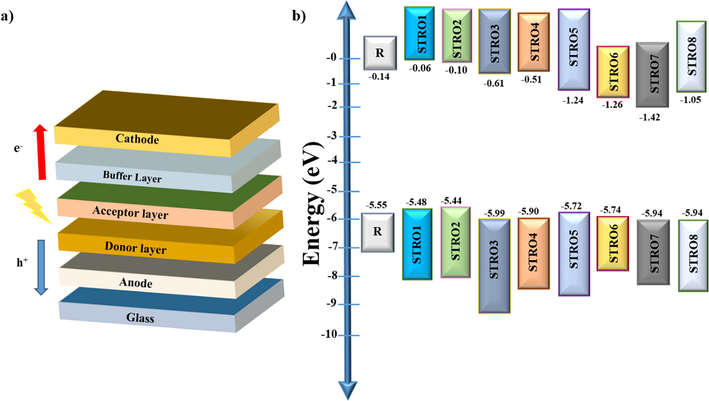
A) schematic device architecture, and b) energy level alignment of r and the designed star-shaped materials stro1-stro8.
1.1 Computational details
Gaussian 09 W software was used for calculations, and for graphical studies Gauss View 5.0 program was used (Frisch et al., 2009; Atiq et al., 2023). We initially optimized the reference molecule R with various DFT functionals before starting the theoretical characterization of the designed star-shaped series (STRO1-STRO8). The DFT functionals used in this study are; B3LYP (Civalleri et al., 2008), cam-B3LYP (Adamo and Barone, 1998), ωB97XD, (Yanai et al., 2004) and MPW1PW91, (Chai and Head-Gordon, 2008). For absorption properties, we have employed TD-DFT for B3LYP, cam-B3LYP, ωB97XD, and MPW1PW91 along with 6-31G(d,p) basis sets in the solvent phase (CHCl3) (Parr, 1980). The UV–vis absorption spectra and λmax values of the designed star-shaped series (STRO1-STRO8) and R (Ma et al., 2021) were compared, showing close agreement at the CAM-B3LYP level of theory. Thus, the CAM-B3LYP level of theory was used for further characterizations of the designed star-shaped series (STRO1-STRO8) and R. Moreover, other vital characterizations such as; FMO analysis, open-circuit voltage (Voc), excitation energies (Ex), reorganization energies, oscillator strength (f), DOS, and TDM analysis of the designed star-shaped series (STRO1-STRO8) and R molecule were calculated at the CAM-B3LYP /6-31G(d,p) level of theory. For data dissemination from Gaussian, origin 8.1 programs were used, and for DOS analysis, PyMOlyze program. Numerous computations relating to hole/electron mobility, Voc, and fill factor (FF) have been made to estimate the materials' PCE. The exact DFT functionals have been used to estimate reorganization energy, i.e., internal (λint) and external (λext) energy. Moreover, because of the expected minor role in managing the outer atmosphere, λext is being overlooked. The reorganization energy is estimated with the following Eqs. (1) and (2) (Adnan et al., 2017; Riaz et al., 2022)
Hence, neutral material optimization's cationic and anionic energies are and, respectively. Moreover, are the anionic and cationic energies of cation and anion geometries optimization, respectively. Moreover, define the energies of the neutral molecules optimized at anionic and cationic states. E0 is the neutral molecule energy at the ground state geometry. The UV–vis spectrum was taken with Origin 8.1 software and the HOMO-LUMO with Multiwfn 3.7.
2 Results and discussion
Fig. 2 shows the molecular structure of the R molecule used in this study along with the used efficient end-capped units to modify this molecule further to investigate its potential for efficient OSCs devices. As shown in Fig. 3, initially, we did optimization of the R molecule with four various DFT functionals to find the best one for further characterizations of the designed materials. These functional includes; B3LYP, CAM-B3LYP, MPW1PW91, ωB97XD, and at 6-31G(d,p). Subsequently, we carried out optical measurements of the R to check its UV–vis response both in the gas and solvent phase. Remarkably, the R molecule presented good values of absorption and is highly red-shifted among all of the employed DFT functionals, such as; 485.20 nm (B3LYP), 388.97 nm (CAMB3LYP), 462.33 (MPW1PW91), and 379.01 nm (ωB97XD), as shown in Fig. 3.
Chemical structure of STRO (R) along-with eight newly proposed star-shaped molecules (STRO1-STRO8).
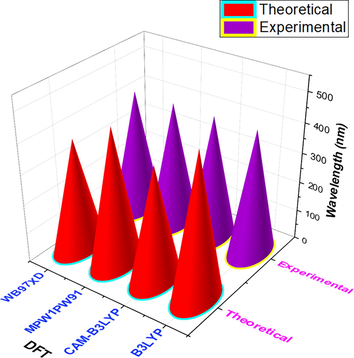
The optimization of R molecule at four different DFT functionals.
The experimentally achieved UV–vis value of R is 419.00 nm and is relatively nearer to the theoretically calculated value of 388.97 at CAM-B3LYP/6-31G(d,p). Thus, we have selected B3LYP/6-31G(d,p) functional for the following measurements of this designed three-armed star-shaped series (STRO1-STRO8) along with the R molecule. The chemical structure of R and the used efficient end-capped units are shown in Fig. 2, and the designed star-shaped series (STRO1-STRO8) structure can be found in Figure S1. The relatively optimized structures of the designed star-shaped series (STRO1-STRO8) and R can be seen in Fig. 4.
The optimized chemical structures of the designed STRO1-STRO8 and R molecules.
2.1 Frontier molecular orbitals (FMOs) analysis
The FMO analysis of R and the designed three-armed star-shaped series (STRO1-STRO8) are investigated intensively. In these calculations, we checked the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) at CAM-B3LYP/6-31G(d,p), as shown in Fig. 5. Both the HOMO and LUMO levels formations show their specific electronic distribution patterns, positions, and energy levels alignments which are pretty different from each other just because of the presence of different end-capped units attached over the base-core parts. These various energy level values also significantly influenced the bandgaps (Eg) and the optical and photovoltaic parameters of the designed star-shaped series (STRO1-STRO8). The HOMO-LUMO values of R and the designed star-shaped series (STRO1-STRO8) have been shown in Fig. 9b, and the corresponding values are tabulated in Table 1.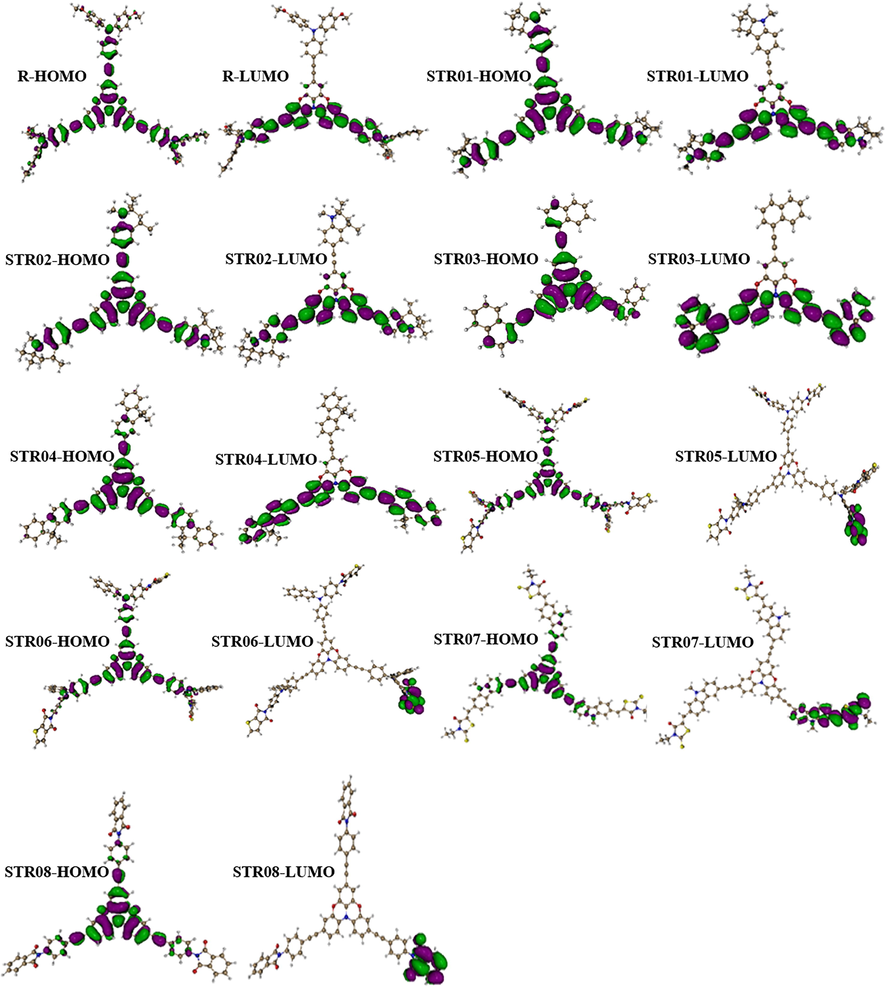
The formation of HOMO and LUMO of the designed star-shaped series (STRO1-STRO8) and R.
Molecules
HOMO (EHOMO)
(eV)
LUMO (ELUMO)
(eV)
Eg = ELUMO - EHOMO
(eV)
Reference
−5.55
−0.14
5.41
STRO1
−5.48
−0.06
5.41
STRO2
−5.44
−0.10
5.34
STRO3
−5.99
−0.61
5.37
STRO4
−5.90
−0.51
5.39
STRO5
−5.72
−1.24
4.49
STRO6
−5.74
−1.26
4.49
STRO7
−5.94
−1.42
4.52
STRO8
−5.94
−1.05
4.88
The calculated HOMO and LUMO energies for R is −5.55 and −0.14, whereas, these values for the designed star-shaped series (STRO1-STRO8) are; −5.48, −5.44, −5.99, −5.90, −5.72, −5.74, −5.94, and − 5.94 eV for HOMO, and − 0.06, −0.10, −0.61, −0.51, −1.24, −1.26, −1.42, and − 1.05 eV for LUMO, respectively. These FMO analyses revealed that the designed star-shaped series (STRO1-STRO8) have a distinctive distribution pattern and their energy levels values are quite comparable with the R. On the other hand, the obtained Eg for the designed star-shaped series (STRO1-STRO8) is much lower than the R molecules, making them more appropriate for photovoltaic applications because the lower Eg materials are highly desirable for photovoltaic applications. Furthermore, these lower Eg values were obtained due to the specific positioning of the efficient end-capped units, various good conversions of the charges between the two orbitals, and the existence of the different donor–acceptor (D-A) units that dynamically took part during this charge transformation phenomenon.
Smaller Eg materials are highly desirable for efficient solar cells, and interestingly, our all-designed star-shaped series shows much narrower Eg values than the R molecule. As shown in the Fig. 9, the Eg of R is 5.41 eV, whereas the Eg values for the designed star-shaped series are; 5.41 eV (STRO1), 5.34 eV (STRO2), 5.37 eV (STRO3), 5.39 eV (STRO4), 4.49 eV (STRO5), 4.49 eV (STRO6), 4.52 eV (STRO7), and 4.88 eV (STRO8) respectively. Among all designed star-shaped series, the STRO8 shows a much lower Eg than the other molecules due to its efficient end-capped alterations, whereas the Eg values for other molecules (STRO1-STRO7) are also very close to each other and much comparable with the R, indicating our efficient designing strategy. The formulated decreasing Eg trend for all molecules is; R > STRO1 > STRO4 > STRO3 > STRO2 > STRO8 > STRO7 > STRO5 > STRO6.
2.2 Optical properties
The optical parameters of the designed star-shaped series (STRO1-STRO8) and R have been estimated by performing UV–vis calculations at B3LYP/6-31G(d,p), both in the gas and in the solvent phase. Moreover, we also calculated the excitation energy (Ex), oscillator strength (f), and molecular orbital assignments of the designed star-shaped series (STRO1-STRO8) and R, as shown in Table 2. The UV–vis (experimental) of R is 419.00 nm, which is a little bit higher than the calculated UV–vis value of 388.97 nm.
Molecule
DFT Calculated λmax (nm)
Experimental λmax (nm)
Ex
(eV)
fos
Major MO Assignment
Reference
388.97
419
3.19
2.47
HOMO->LUMO (69 %)
STRO1
371.34
3.34
1.90
HOMO->LUMO (79 %)
STRO2
371.61
3.34
1.93
HOMO->LUMO (78 %)
STRO3
376.30
3.29
1.60
HOMO->LUMO (75 %)
STRO4
375.33
3.30
1.93
HOMO->LUMO (76 %)
STRO5
377.57
3.28
2.38
HOMO->LUMO (73 %)
STRO6
378.22
3.28
2.28
HOMO->LUMO (71 %)
STRO7
380.18
3.26
2.95
HOMO->LUMO (72 %)
STRO8
370.59
3.35
1.70
HOMO->LUMO (77 %)
Interestingly, it was found that both the experimental and calculated UV–vis value is quite close to each other, ensuring our efficient strategy to build and characterize the materials with appropriate methods. This also ensures the suitability of any new photovoltaic material to estimate its inner potential before synthesizing it. The related UV–vis absorption spectrum of the designed star-shaped series (STRO1-STRO8) and R is shown in Fig. 6. Previously, it was reported that the molecules with higher optical properties are necessary to prepare an efficient SCs device ().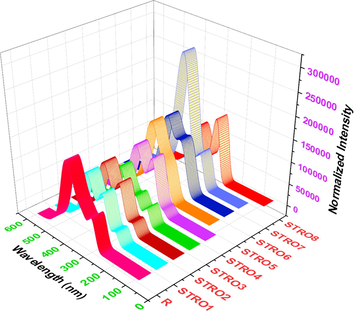
The UV–Visible absorption of the designed star-shaped series (STRO1-STRO8) and R.
Thus, our all-designed star-shaped series (STRO1-STRO8) was found to be very red-shifted in absorption spectrum than R. The calculated UV–vis of the designed star-shaped series (STRO1-STRO8) is; 371.34 nm for STRO1, 371.61 nm for STRO2, 376.30 nm for STRO3, 375.33 nm for STRO4, 377.57 nm for STRO5, 378.22 nm for STRO6, 380.18 nm for STRO7, and 370.59 nm for STRO8. These investigations show that our all-designed star-shaped series (STRO1-STRO8) displayed a better absorption phenomenon than R. Among this designed star-shaped series (STRO1-STRO8), the highest UV–vis value was seen for STRO7, which is quite comparable with R. All other designed star-shaped materials also presented an excellent UV value. All are very near to R. Therefore, after these results, we can predict that these good UV values are just because of efficient end-capped engineering of these molecules with some efficient functional groups. Furthermore, the decreased calculated absorption behavior of the designed star-shaped series (STRO1-STRO8), and R is; R > STRO7 > STRO6 > STRO5 > STRO3 > STRO4 > STRO2 > STRO1 > STRO8. This shows that our designed star-shaped series (STRO1-STRO8) is suitable for efficient HTMs in OSCs.
The excitation energy (Ex) is also considered a key factor in estimating the hidden parameters of molecules used for SCs. Therefore, it was reported that the lower values of Ex are good for the effective charge transformation from lower HOMO to LUMO levels (Adnan et al., 2017; Zhang et al., 2017; Li et al., 2017). This process is quite helpful in enhancing the efficiency of the molecules during device formation (Bilal et al., 2020). So, these Ex calculations have been performed with the same method at TD-DFT- CAM-B3LYP/6-31G(d,p). The derived Ex value of R is 3.19 eV, while, for the designed star-shaped series (STRO1-STRO8) are; 3.34 eV for STRO1, 3.34 eV for STRO2, 3.29 eV for STRO3, 3.30 eV for STRO4, 3.28 eV for STRO5, 3.28 eV for STRO6, 3.26 eV for STRO7, and 3.35 eV for STRO8. In particular, the designed star-shaped series (STRO1-STRO8) showed quite lower Ex than R. These lower energies are due to the incorporation of efficient end-capped functional units on the bridged core.
With the incorporation of these functional units, we can achieve desirable characteristics from any molecule, and we can design a molecule according to our requirements. Among this designed star-shaped series (STRO1-STRO8), STRO7 shows a much lower value of Ex than R and the other designed star-shaped series (Table 3). While, the other designed materials also presented suitable and comparable values of Ex than R. Therefore, with these analyses, it is revealed that our all-designed star-shaped series (STRO1-STRO8) has an excellent potential to use in efficient SCs devices.
Molecules
EHOMO-LUMO
Eopt
Eb
Reference
5.41
3.19
2.22
STRO1
5.41
3.34
2.07
STRO2
5.34
3.34
2.00
STRO3
5.37
3.29
2.08
STRO4
5.39
3.30
2.09
STRO5
4.49
3.28
1.21
STRO6
4.49
3.28
1.21
STRO7
4.52
3.26
1.26
STRO8
4.88
3.35
1.53
2.3 Density of state analysis (DOS)
The DOS analyses were performed for R and the designed star-shaped series (STRO1-STRO8) to measure the working potential of the designed materials (STRO1-STRO8), i.e., their bandgap alignments and the charge transfer properties (Adnan et al., 2017; Adnan et al., 2021). For this, the partial and total DOS (PDOS and TDOS) analyses have been carried out to estimate the FMOs distribution alignments along with their percentage contribution for R and the designed star-shaped series (STRO1-STRO8). This DOS analysis provides critical information regarding various energy levels per unit increment. In particular, for any specific energy level, the possibility of states estimated relative to their larger DOS values and vice versa (Adnan et al., 2021).
Fig. 7 shows that the DOS analysis primarily checks donor participation: acceptor units for forming HOMO-LUMO distributions and the molecules Eg. In Fig. 4, the designed spaces between the two high peaks (black lines) in the middle of the spectrum represent the Eg of the material. While these black lines correspond to the HOMO and LUMO levels of the molecule. Furthermore, it is seen that the HOMO density open-out over the core units of the molecule, whereas the LUMO density spreads on the acceptor region part of the molecule. Because of the different charge density distributions, both HOMO and LUMO densities get detached from each other and show an individual distribution pattern related to the specific characteristic of the molecule. Interestingly, a similar distribution behavior has been seen for the designed star-shaped series (STRO1-STRO8), which is also very comparable with R. This shows our efficient HTMs designing approach for the efficient SCs devices.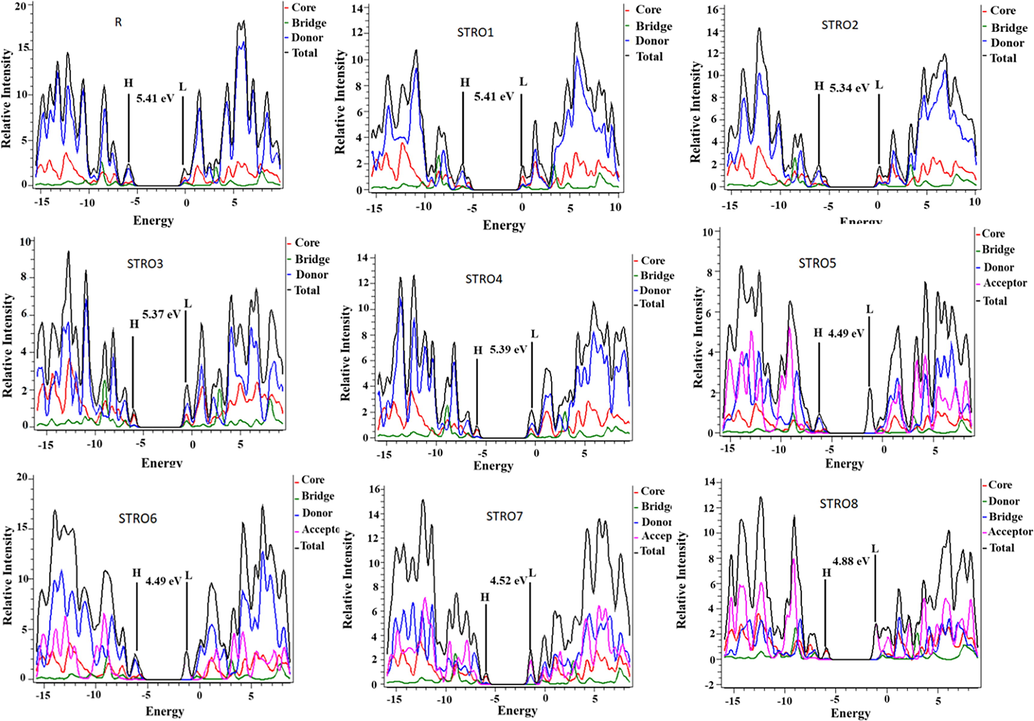
The density of states graphs of the designed STRO1-STRO8 and R molecules.
2.4 Reorganization energy
The reorganization energy analysis has been performed to identify the explicit nature of the designed star-shaped series (STRO1-STRO8) along with R. These calculations have great importance for any optical material to identify its nature, whether it would be an electron or a hole transport material (Adnan et al., 2017; Adnan et al., 2021). These analyses also confirm the selection of materials and the corresponding device architecture regarding the nature of the molecule. We got two types of data from these analyses, i.e., the energy of electrons and holes. Both of these energies have their role in determining the performance of any materials and have various charge transfer rates. In particular, the smaller the reorganization energy, the higher the charge transfer rate for that particular molecule. During these analyses, we observe different charge mobility values for the designed star-shaped series (STRO1-STRO8) and for R, which significantly affects the performance of these materials when these are employed to prepare SCs devices. Thus, scientists divide the reorganization energy into two various types; external (λext) and internal (λint) reorganization energy. The λint provides information regarding internal molecular changes due to the polarization factor, while λext is associated with the instant external transformation of the molecules (Adnan et al., 2017). The estimated RE values for hole and electrons are shown in Table 4.
Molecules
telectron
thole
Reference
0.0055
0.0097
0.04
0.17
STRO1
0.0052
0.0059
0.05
0.30
STRO2
0.0049
0.0062
0.05
0.30
STRO3
0.0034
0.0048
0.03
0.44
STRO4
0.0034
0.0049
0.38
0.43
STRO5
0.0066
0.0057
0.00
0.22
STRO6
0.0064
0.0074
0.00
0.20
STRO7
0,0039
0.0049
0.00
0.42
STRO8
0.0040
0.0060
0.00
0.56
Importantly, in these calculations, there is no λext role observed; thus, we have ignored this factor. While we dealt extensively with the molecules' internal charge transformation, we have characterized our all-designed star-shaped series (STRO1-STRO8) and R at CAM-B3LYP/6-31G(d,p) to calculate the relative energy of electrons and holes. Fig. 8 shows the graphical representation of the obtained reorganization energy values; the corresponding obtained energy values have been summarized in Table 4.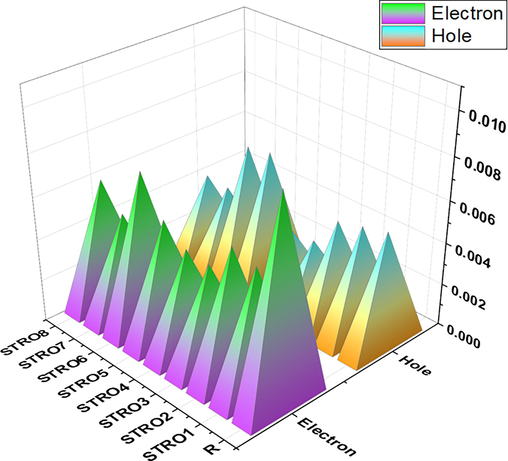
The reorganization energy of holes and electron for the designed STRO1-STRO8 and R molecules.
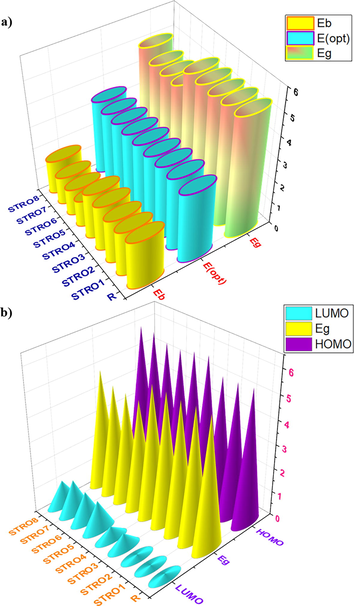
a) The calculated binding energy, first-principle excitation energy, and HOMO-LUMO Eg, and b) the HOMO, LUMO and the Eg for the designed STRO1-STRO8 and R molecule.
2.5 Open circuit voltages
The performance of any photovoltaic device is calculated with current density (Jsc), Voc, and a fill factor. Therefore, the Voc have great importance and role in determining the performance of any photovoltaic material when they are used to prepare a device (Adnan et al., 2021; Riaz et al., 2022). To this end, we have calculated the Voc for the designed star-shaped series (STRO1-STRO8) and R at CAM-B3LYP/6-31G(d,p). The Voc is considered to have the value of the total current extracted from the device. It is also influenced by the saturated as well as by the photo-generated current of the device (Adnan et al., 2021). The most crucial factor in obtaining high voltage values from the device is to have the good energy levels alignments of the used materials (Riaz et al., 2022).
Therefore, we have estimated the Voc values from the energy level alignments after establishing the donor:acceptor complex. These analyses revealed that the well-matched energy levels could produce the maximum voltage value we can obtain from the device. Therefore, a well-matched energy levels alignment of the used materials for fabricating a device is considered the backbone for higher device performance.
Moreover, based on the nature of our designed star-shaped series (STRO1-STRO8) and R, we have selected a well-known polymer acceptor material (PC61BM) for establishing D:A blend to calculate the voltage values of the designed star-shaped series (STRO1-STRO8) and R, respectively. Fig. 11 shows the arrangement of the D:A for blend formation to extract their Voc.(see Fig. 10)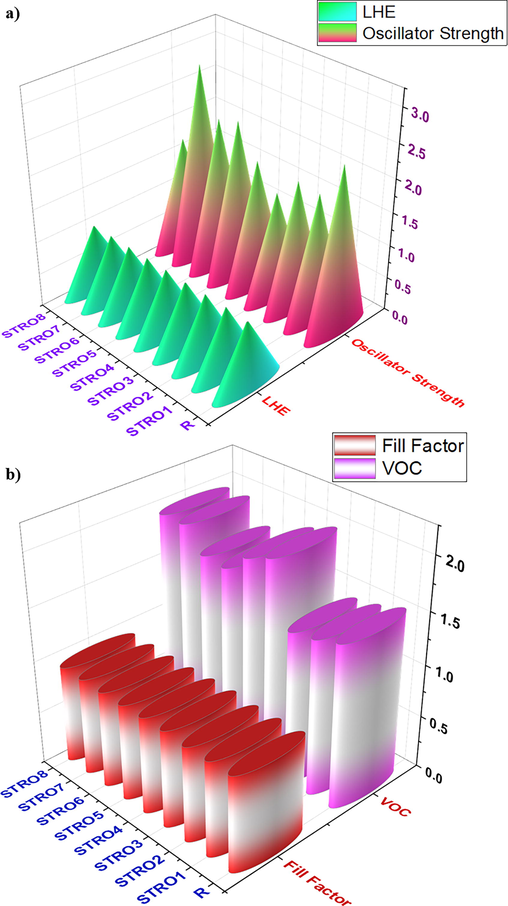
A) the calculated light harvesting efficiency and oscillator strength, while, b) the estimated open-circuit voltages and fill factor for the designed stro1-stro8 and r molecule.
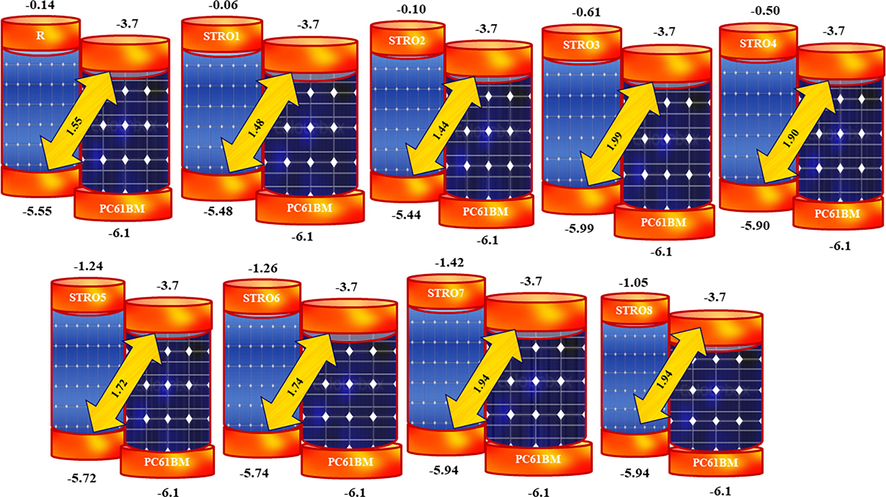
The optimized open-circuit voltages of the designed STRO1-STRO8 and R molecules.
From these analyses, the theoretically estimated Voc for R is 1.85 V, while for the designed star-shaped series (STRO1-STRO8), these are; 1.78 V, 1.74 V, 2.29 V, 2.2 V, 2.02 V, 2.04 V, 2.24 V, and 2.24 V, respectively. Among this designed star-shaped series (STRO1-STRO8), STRO7 presented the highest value of Voc of 2.24 eV, and the lowest noted value for this designed material is for STRO2, which 1.74 eV, while this value for R is 1.85 eV, respectively. Remarkably, it was revealed that all of our designed materials (STRO1-STRO8) showed a good value of Voc. Among them, some of the molecules, such as STRO1 (1.78 eV) and STRO2 (1.74 eV), presented very comparable values with R, whereas STRO3 to STRO8 presented higher Voc values than R. And these values are in the range of 2.29 eV, 2.2 eV, 2.02 eV, 2.04 eV, 2.24 eV, and 2.21 eV, respectively. Hence, these analyses also revealed our efficient design for building efficient photovoltaic materials for the next-generation solar cells devices.
2.6 Transition density matrix (TDM)
The TDM is a conventional concept in quantum chemistry and a non-local visualization tool to estimate the electron-hole pair coherences and excitonic effects of molecular excitations (Riaz et al., 2022; Adnan et al., 2021). It could be defined as the density matrix linking the ground state (GS) molecular excitations with a specifically excited state excitation (Adnan et al., 2017; Riaz et al., 2022; Adnan et al., 2021). Therefore, we have estimated TDM analyses at B3LYP/6-31G(d,p). These TDM maps have estimated the particular position of electrons, holes, and electron-holes pair coupling within the molecule. Furthermore, these TDM analyses also provide information regarding different excited state transitions (Adnan et al., 2021). To get better insights from the TDM plots, we have split the TDM maps into various parts per the nature of that particular molecule, i.e., acceptor, donor, and bridge core unit (Alvina et al., 2022; Adnan et al., 2021). Among these analyses, there is also a possibility of hydrogen effects, so we have passed over this effect throughout all our analyses due to its non-contributing charge transitions. The obtained TDM maps are shown in Fig. 12.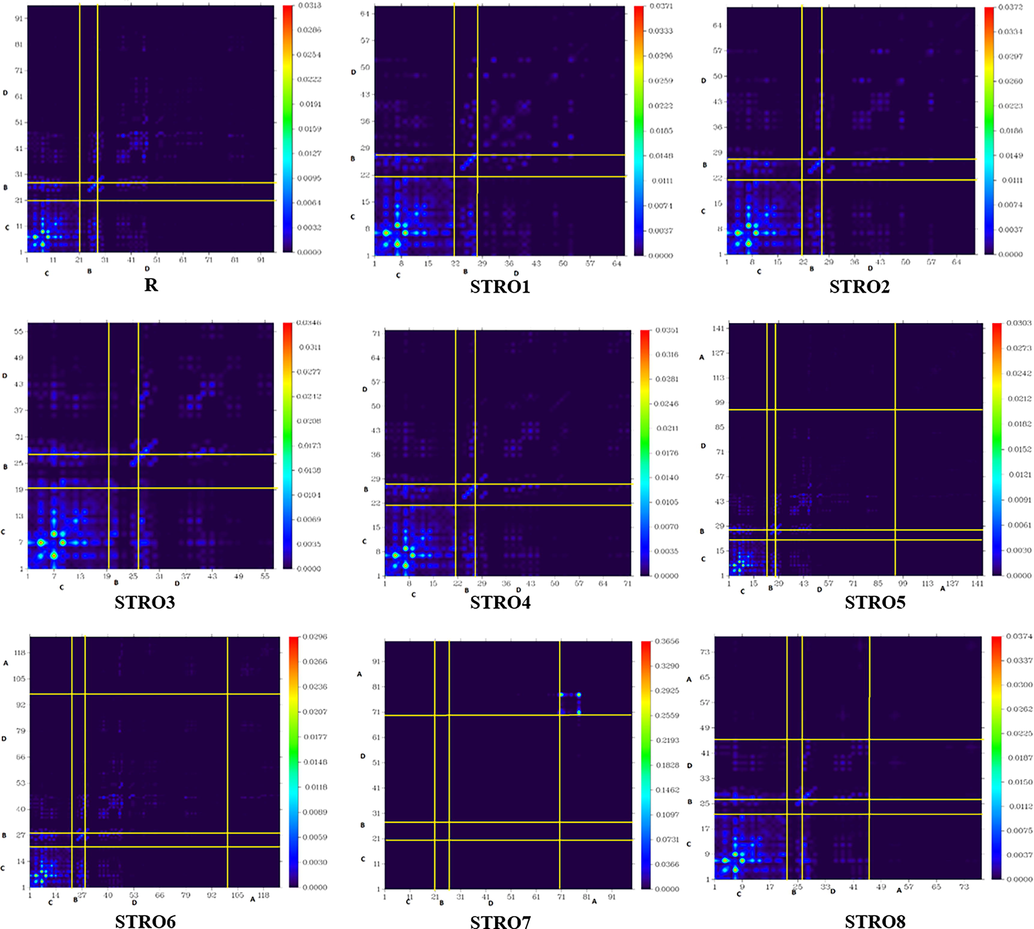
The transition density matrix spectrums of the designed STRO1-STRO8 and R molecules.
After these analyses, we noticed that the electronic charge density specifically covers the bridge-core and end-capped functional units. Moreover, it was also investigated that all of our designed star-shaped series (STRO1-STRO8) involved efficient charge transportation from the D to A region, and the bridging units in between them facilitate this process without interruption. This process of an efficient charge transformation was realized because of the modulation of the molecule by attaching various efficient functional end-capped units on the R molecule and not disturbing the bridged-core unit. The used end-capped units tend to extract the electronic charge density, and the bridged part facilitates this charge density distribution process. Therefore, the charge density transformation process continues in the molecules, ensuring improved optical and electronic characteristics. Hence, this active intra-molecular charge transformation coherence among R and the designed star-shaped series (STRO1-STRO8) showed our efficient modeling strategy to build up a required characteristic photo-voltaic molecule. The hole/electrons overlap heat maps of STRO (R) along-with eight newly proposed star-shaped molecules (STRO1-STRO8) are shown in Figure S2.
2.7 Electrostatic potential (ESP)
The ESP analyses were carried out to investigate the R's three-dimensionally charge distribution (x,y,z) and the designed star-shaped series (STRO1-STRO8). These analyses gave us some valuable information regarding charge distribution visualization and the other charge-related characteristics of the materials (Adnan et al., 2021). Moreover, they also provide information related to the shape and size of the molecules. Therefore, these analyses are considered highly desirable for predicting the behavior of organic molecules (Riaz et al., 2022). In our study, we used these analyses to investigate the presence of different active charging sites and their specific distribution within the molecules. Usually, the molecules have various types of charging sites that present molecules and every specific site is related to a specific fundamental property of that molecule and considerably influences the performance of the materials. Fig. 13 shows the representation of ESP plots calculated at CAM-B3LYP/6-31G(d,p) for R and the designed star-shaped series (STRO1-STRO8). These ESP plots presented the different color patterns within the molecule, which are considered to have a specific property for that molecule for their electrostatic potential. For instance, the soft green color for the neutral region, the blue color represents the electropositive region, and the red color is for the negative region of that particular molecule.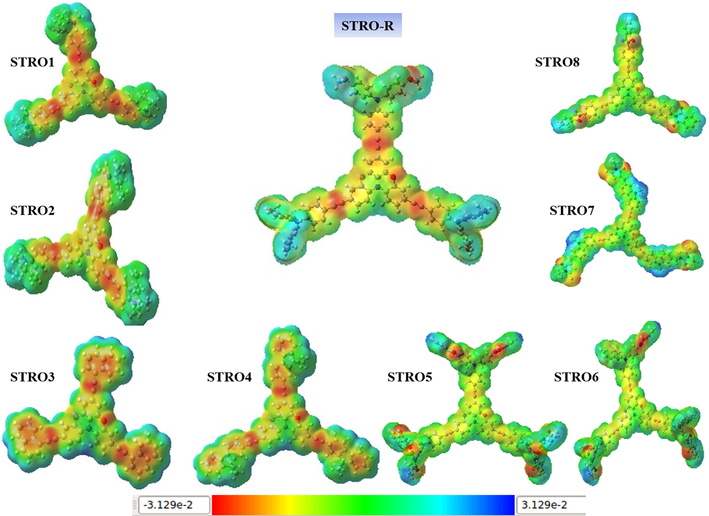
The MEP plots for the designed STRO1-STRO8 and R molecule.
Remarkably, our designed star-shaped series (STRO1-STRO8) exhibited a comparable color pattern to the R molecule, ensuring our efficient modeling strategy. Furthermore, due to their possible employment in the next-generation photovoltaic device, the molecules are made highly valuable by the exitance of such a combination of features. Therefore, these ESP plots greatly help estimate the inner hidden potential of any organic material for SCs devices and can save our time and cost if we use this technique before synthesizing and particulate photovoltaic material. Furthermore, after these ESP analyses, we successfully estimated the electrostatic characteristics for our designed star-shaped series (STRO1-STRO8) and R molecule and found that these designed star-shaped series (STRO1-STRO8) have a similar electrostatic potential as R, and thus, ensuring our efficient modeling approach to build up improved characteristics photovoltaic materials.
2.8 Light harvesting efficiency (LHE)
The LHE is the specific capability of any molecule to generate charge carriers after exposure to light (Alvina et al., 2022). Furthermore, it is also highly related to the Jsc production from any photovoltaic material during the fabrication process, and it is estimated by the following Eq. (3).
Where ɸinj represents the efficacy of electronic injection, and ηcollect shows the specific efficiency of charge collection. The LHE process has been estimated with Eq. (4), and obtained values are tabulated in Table 5.
Molecules
Voc
(V)
Oscillator Strength
Fill Factor
LHE
Reference
1.55
2.47
0.9171
0.9666
STRO1
1.48
1.90
0.9140
0.9874
STRO2
1.44
1.93
0.9122
0.9883
STRO3
1.99
1.60
0.9320
0.9749
STRO4
1.90
1.93
0.9294
0.9883
STRO5
1.72
2.38
0.9236
0.9958
STRO6
1.74
2.28
0.9243
0.9948
STRO7
1.94
2.95
0.9306
0.9989
STRO8
1.94
1.70
0.9306
0.9800
Here, f is the oscillator strength, and its corresponding values are shown in Table 5 for R and the designed star-shaped series (STRO1-STRO8). These (f’) values have been calculated in the solvent phase and are used to determine the LHE. Moreover, we can use gaseous phase values using the same form for this measurement. Remarkably, all of our newly designed star-shaped series (STRO1-STRO8) showed a higher value of LHE than R (Table 5), ensuring the higher ability to generate higher Jsc values among SCs devices. Furthermore, the designed molecule STRO5 presented the highest possible value of LHE (99.58 %), which is also an indication of higher Jsc for these molecules when employed as next-generation photovoltaic devices. While all other designed star-shaped molecules presented a good value of LHE, and all are very close to each other and higher than the R molecule. Therefore, these results also support our efficient design strategy to build efficient photovoltaic materials.
2.9 Fill factor (FF)
The working potential of any photovoltaic device is estimated mainly by the three key factors; Voc, Jsc, and FF. To this end, we have also estimated Voc and FF for the designed star-shaped series (STRO1-STRO8) and for R. The representative FF values can be seen in Table 5. Equation (6) was used to estimate the FF of the designed star-shaped series (STRO1-STRO8) and for R.
Here,
signifies the stabilized Voc and “e” is the elementary charge that is fix at 1. The Voc of the molecules is assessed using equation (5), KB is Boltzmann constant 8.61733034x10-5 electron volts/kelvin, and “T” is the temperature set at 300 K. (Parr, 1980; Lu et al., 2022) The estimated Voc and FF have been shown in Table 5. Among these, the modified STRO3 to STRO8 molecules presented a much-improved value of FF in contrast to other modified molecules (STRO1 and STRO2), and the FF percentage is quite comparable with the R. The calculated FF of STRO7 and STRO8 shows a theoretical FF value of 0.9306, at the same time, the R has 0.917, whereas the observed FF for STRO3 to STRO6 are also improved than R molecule, whereas SRO1 and STRO2 have very closer FF values than R. These great values of FF for our all designed molecules (STRO1-STRO8), showed our efficient designing approach to design efficient photovoltaic molecules for SCs applications. Furthermore, based on the afor-mentioned calculated Voc and FF values, a decent approximation of PCE could be invented for all the newly designed three-armed star-shaped series (STRO1-STRO8), by using Eq. (6).
Herein, Pinput represents the power generated from the light source. It is familiar that the Jsc mainly depends on the charge mobility rates, bandgap, and LHE of materials. Thus, we have calculated all these properties theoretically and found that all newly modeled three-armed star-shaped materials (STRO1-STRO8) showed an improved value than R. Furthermore, they also have pretty comparable FF and Voc values, which encourage us to present these materials (STRO1-STRO8) for the future development of efficient SCs devices. Among these, STRO3-STRO8 presented much higher Voc and FF values in contrast to R, whereas STRO1 and STRO2 showed much closer Voc and FF values relative to R, ensuring their capability to be used for the next-generation high-performance SCs devices.
2.10 Charge transfer analysis (CTA)
The complex study (D:A) has been performed to investigate the CT process between the designed three-armed star-shaped donor and a well-known acceptor martial (PC61BM). To do so, we have used the best working designed material, STRO6 among all, to realize the CT process of this complex, as shown in Fig. 14. The STRO was chosen because of its capacity to show red-shifting absorption, desirable hole-electron reorganization energy, lower Eg, Ex and Eb, Voc, and a good value of Voc and FF. It was seen that the CT is mainly affected by LUMO of the acceptor and HOMO of the donor, and the lower LUMO are valuable because of their ability to induce better charge shifting, helping to boost the optoelectronic properties of the molecules. While the obtained Eg difference directly affects the PCE values of the fabricated devices.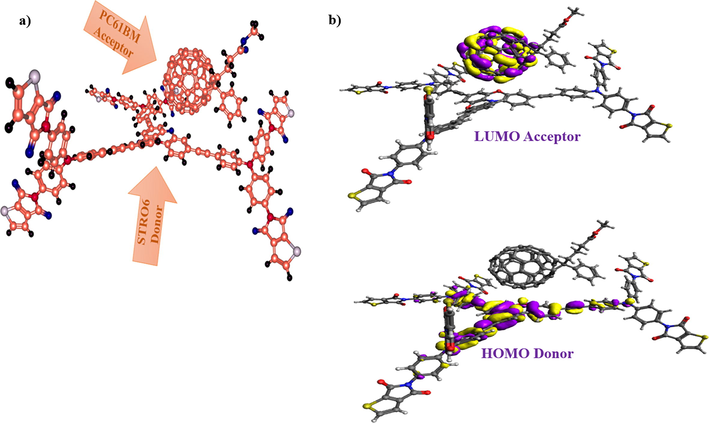
A) the optimized structures of the complex (donor stro6 and polymer acceptor pc61BM), and b) the formed LUMO and HOMO patterns of donor:acceptor complex.
Moreover, the formed orientation of the D:A complex also has a vital role in tuning the electronic transitions of the molecules. Therefore, here both the D and the A molecules formed the parallel geometry to ensure an efficient charge transport phenomenon between them. Among this complex, we have realized that the HOMO density covers the core region of the molecule, whereas the LUMO density is on the acceptor units only (Fig. 14b). Therefore, this phenomenon ensures the shifting of charges from D to A parts. Moreover, the devised MO diagram confirms the designated charge transfer symmetry of D and A materials to assure the formation of an efficient active layer. Moreover, the corresponding estimated values for the possible energy loss during charge generation process as well as energy loss while charge recombination are shown in Table 6.
Molecules
ELL
(LDonor - LAcceptor)
ECT
(LAcceptor - HDonor)
Energy loss incurred during charge generation
Energy loss incurred during charge recombination
Reference
1.95
3.46
1.95
1.91
STRO1
2.03
3.39
2.03
1.91
STRO2
1.99
3.35
1.99
1.91
STRO3
1.48
3.90
1.48
1.91
STRO4
1.59
3.81
1.59
1.91
STRO5
0.85
3.63
0.85
1.91
STRO6
0.83
3.65
0.83
1.91
STRO7
0.67
3.85
0.67
1.91
STRO8
1.04
3.85
1.04
1.91
3 Conclusion
In conclusion, we have reported 1eoght novel three-armed star-shaped SMs (STRO1-STRO8) for O/PSCs. The core structure has partially oxygen bridged TPA in the periphery. This newly designed three-armed star-shaped (STRO1-STRO8) series presented excellent photovoltaic, optoelectronic, and charge transport characteristics for applications in OSCs. Overall, all of the designed star-shaped (STRO1-STRO8) series presented good values of FF (ranges until 93 %) and Voc (ranges until 1.99 V) as compared with R, which could be attributed to the improved and stable amorphous character of the materials. Furthermore, our designed star-shaped materials (STRO1-STRO8) bear a lower symmetry and thus may play a vital role in the formation and stabilization of amorphous films, and their improved planarity could maintain a better hole-transport character. Moreover, the designed series (STRO1-STRO8) showed a lower Eg and Ex and improved dipole moment than R. Interestingly, the designed star-shaped series (STRO1-STRO8) presented superior photo-physical and optoelectronic properties in-contrast to R. Amongst, STRO5 and STRO6 showed lowest bandgap (4.49 eV) than others and also with R (5.41 eV). Moreover, the R molecule presented the observed UV–vis absorption of 388.97 nm, while our designed STRO1-STRO8 molecules also presented a much closer absorption phenomenon, and the values are in the range of 371.34 nm to 380.18 nm, respectively. Moreover, all the designed star-shaped series (STRO1-STRO8) also showed a much lower Ex than R. Afterward, the studied properties were successfully compared with R, and a central coherence was found with improved characteristics. Hereafter, the designed star-shaped materials (STRO1-STRO8) are highly proposed to the experimentalists for the next-generation, efficient, and high-performance OSCs devices.
Acknowledgement
This research was supported by Nano Material Technology Development Program through the National Research Foundation of Korea (NRF) funded by Ministry of Science and ICT (grant number-2021M3H4A1A02057007). This work was also financially supported by research funds of Chungnam National University, Republic of Korea. The authors are highly thankful to the COMSAT University, Abbottabad, Pakistan for providing their computational facility support.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The m PW and m PW1PW models. J. Chem. Phys.. 1998;108:664-675.
- [Google Scholar]
- Efficient synthesis and characterization of solvatochromic fluorophore. Bull. Korean Chem. Soc.. 2017;38:1052-1057.
- [Google Scholar]
- Fine tuning the optoelectronic properties of triphenylamine based donor molecules for organic solar cells. Zeitschrift für Physikalische Chemie.. 2017;1:1127-1139.
- [Google Scholar]
- All sequential dip-coating processed perovskite layers from an aqueous lead precursor for high efficiency perovskite solar cells. Sci. Rep.. 2018;8:2168.
- [Google Scholar]
- Facile all-dip-coating deposition of highly efficient (CH3)3NPbI3-x Cl x perovskite materials from aqueous non-halide lead precursor. RSC Adv.. 2020;10:29010-29017.
- [Google Scholar]
- Highly efficient planar heterojunction perovskite solar cells with sequentially dip-coated deposited perovskite layers from a non-halide aqueous lead precursor. RSC Adv.. 2020;10:5454-5461.
- [Google Scholar]
- Banana-shaped nonfullerene acceptor molecules for highly stable and efficient organic solar cells. Energy Fuels. 2021;35:11496-11506.
- [Google Scholar]
- In silico designing of efficient C-shape non-fullerene acceptor molecules having quinoid structure with remarkable photovoltaic properties for high-performance organic solar cells. Optik. 2021;241:166839
- [Google Scholar]
- Fabrication of solution-processable OFET memory using a nano-floating gate based on a phthalocyanine-cored star-shaped polymer. Mater. Adv.. 2022;3:3128-3134.
- [Google Scholar]
- Synergistic engineering of end-capped acceptor and bridge on arylborane-arylamine macrocycles to boost the photovoltaic properties of organic solar cells. Optical Mater.. 2022;123:111907
- [Google Scholar]
- Fused ring pyrrolo [3, 2-b] pyrrole-based tilde-shaped acceptor molecules for highly efficient organic solar cells. J. Phys. Chem. Solids 2023:111228.
- [Google Scholar]
- Designing triphenylamine-configured donor materials with promising photovoltaic properties for highly efficient organic solar cells. ChemistrySelect. 2020;5:7358-7369.
- [Google Scholar]
- Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys.. 2008;10:6615-6620.
- [Google Scholar]
- Synthesis, molecular and photovoltaic properties of donor–acceptor conjugated polymers incorporating a new heptacylic indacenodithieno [3, 2-b] thiophene arene. Macromolecules. 2012;45:9282-9291.
- [Google Scholar]
- B3LYP augmented with an empirical dispersion term (B3LYP-D*) as applied to molecular crystals. CrstEngComm. 2008;10:405-410.
- [Google Scholar]
- Thermal performance of modified melamine foam/graphene/paraffin wax composite phase change materials for solar-thermal energy conversion and storage. J. Cleaner Production. 2022;367:133031
- [Google Scholar]
- Single-junction organic photovoltaic cells with approaching 18% efficiency. Adv. Mater.. 2020;32:1908205.
- [Google Scholar]
- Gaussian 09, Revision A.02. Wallingford, CT: Gaussian, Inc.; 2009.
- Development of perovskite solar cells with > 25% conversion efficiency. Joule. 2021;5:1033-1035.
- [Google Scholar]
- A vanadium-based fluoroxide cathode material for lithium-ion storage with high energy density. Adv. Sustainable Syst.. 2022;6:2200122.
- [Google Scholar]
- Efficient planar heterojunction inverted perovskite solar cells with perovskite materials deposited using an aqueous non-halide lead precursor. Bull. Korean Chem. Soc.. 2020;41:937-942.
- [Google Scholar]
- Simple preparation of highly efficient MAxFA1− xPbI3 perovskite films from an aqueous halide-free lead precursor by all dip-coating approach and application in high-performance perovskite solar cells. J. Mater. Sci.. 2022;57:1936-1946.
- [Google Scholar]
- Sequentially dip-coated processed MAyFA1− yPbI3− xBrx perovskite layers from an aqueous halide-free lead precursor for efficient perovskite solar cells. J. Mater. Sci.: Mater. Electron. 2022:1-11.
- [Google Scholar]
- Rational design strategy of novel energy storage systems: toward high-performance rechargeable magnesium batteries. Small. 2022;18:2200418.
- [Google Scholar]
- Effects of π-conjugation on electrochemical properties within hole-transporting materials for perovskite solar cells from first principle and molecular dynamics. Organic Electron.. 2017;43:96-104.
- [Google Scholar]
- Carbon-based materials for fast charging lithium-ion batteries. Carbon. 2021;183:721-734.
- [Google Scholar]
- Inhibiting excessive molecular aggregation to achieve highly efficient and stabilized organic solar cells by introducing a star-shaped nitrogen heterocyclic-ring acceptor. Energy Environ. Sci.. 2022;15:384-394.
- [Google Scholar]
- An asymmetric encoder–decoder model for Zn-ion battery lifetime prediction. Energy Rep.. 2022;8:33-50.
- [Google Scholar]
- Influences of quinoid structures on stability and photovoltaic performance of nonfullerene acceptors. Solar RRL. 2020;4:2000286.
- [Google Scholar]
- Improving open-circuit voltage by a chlorinated polymer donor endows binary organic solar cells efficiencies over 17%, Science China. Chemistry. 2020;63:325-330.
- [Google Scholar]
- Pyridine-triphenylamine hole transport material for inverted perovskite solar cells. J. Energy Chem.. 2021;54:395-402.
- [Google Scholar]
- Organic and solution-processed tandem solar cells with 17.3% efficiency. Science. 2018;361:1094-1098.
- [Google Scholar]
- Organic solar cells based on small molecule donors and polymer acceptors operating at 150 °C. J. Mater. Chem. A. 2020;8:10983-10988.
- [Google Scholar]
- Parr, R.G. Density functional theory of atoms and molecules. In Horizons of Quantum Chemistry: Proceedings of the Third International Congress of Quantum Chemistry Held at Kyoto, Japan, October 29-November 3, (1980) 5-15.
- Triazatruxene based star-shaped thermally activated delayed fluorescence emitters: modulating the performance of solution-processed non-doped OLEDs via side-group engineering. J. Mater. Chem. C. 2022;9:7363-7373.
- [Google Scholar]
- Alloy-Type anodes for high-performance rechargeable batteries. Angewandte Chemie Int. Ed.. 2022;61:e202206770.
- [Google Scholar]
- Tuning terminal aromatics of electron acceptors to achieve high-efficiency organic solar cells. J. Mater. Chem. A. 2019;7:27632-27639.
- [Google Scholar]
- Triggering ambient polymer-based Li-O2 battery via photo-electro-thermal synergy. Nano Energy. 2022;98:107248
- [Google Scholar]
- Ab Initio Study of Two-Dimensional Cross-Shaped Non-Fullerene Acceptors for Efficient Organic Solar Cells. ACS omega. 2022;7:10638-10648.
- [Google Scholar]
- Fused-ring bislactone building blocks for polymer donors. Sci. Bull.. 2020;65:1792.
- [Google Scholar]
- A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP) Chem. Phys. Lett.. 2004;393:51-57.
- [Google Scholar]
- Screening π-conjugated bridges of organic dyes for dye-sensitized solar cells with panchromatic visible light harvesting. Nanotechnology. 2016;27:265701
- [Google Scholar]
- Molecular design of benzodithiophene-based organic photovoltaic materials. Chem. Rev.. 2016;116:7397-7457.
- [Google Scholar]
- Fast rate and long life potassium-ion based dual-ion battery through 3D porous organic negative electrode. Adv. Functional Mater.. 2020;30:2001440.
- [Google Scholar]
- Mo 6+–P 5+ co-doped Li 2 ZnTi 3 O 8 anode for Li-storage in a wide temperature range and applications in LiNi 0.5 Mn 1.5 O 4/Li 2 ZnTi 3 O 8 full cells. Inorganic Chem. Front.. 2022;9:35-43.
- [Google Scholar]
- Design new hole transport materials for efficient perovskite solar cells by suitable combination of donor and core groups. Organic Electron.. 2017;49:255-261.
- [Google Scholar]
- Constructing advanced electrode materials for low-temperature lithium-ion batteries: a review. Energy Rep.. 2022;8:4525-4534.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.104709.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




