

Translate this page into:
Integration of virtual screening and computational simulation identifies photodynamic therapeutics against human Protoporphyrinogen Oxidase IX (hPPO)
⁎Corresponding authors. psjgenesis@hanmail.net (Seok Ju Park), kwlee@gnu.ac.kr (Keun Woo Lee)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Photodynamic therapy (PDT) is a rapidly evolving area of cancer management against solid tumors. PDT is either administrated by injecting photosensitizer (porphyrins) or by accumulation of intracellular protoporphyrin IX via the inhibition of human Protoporphyrinogen Oxidase IX (hPPO). In this study, novel inhibitors of hPPO have been investigated by integrating virtual screening, molecular docking, and molecular dynamics (MD) simulation. A ligand-based pharmacophore was generated from a training set of 22 inhibitors of hPPO. The selected pharmacophore had four chemical features including three hydrogen bond acceptors and one hydrophobic. The pharmacophore was characterized by highest correlation coefficient of 0.96, cost difference of 53.20, and lowest root mean square deviation of 0.73. The resultant pharmacophore was validated by Fischer’s Randomization and Test Set Validation methods. The validated pharmacophore was used as a 3D query to screen chemical databases including NCI, Asinex, Chembridge, and Maybridge. The screening of chemical databases and the subsequent application of Lipinski’s Rule of Five, and ADMET Assessment Test, retrieved 1176 drug-like compounds. The drug-like compounds were subjected to molecular docking studies in the active site of hPPO to eliminate false positive hits and to elucidate their true binding orientation. Top three candidate molecules with high docking scores and hydrogen bond interactions with catalytic active residues were selected as best candidate inhibitors against hPPO. The binding stability of selected candidate inhibitors was evaluated by MD simulation. The MD simulation of hits portrayed strong hydrogen bonds and key hydrophobic interactions with catalytic active residues of hPPO including R59, R97, G159, G332 and flavin moiety of FAD (coenzyme of hPPO). Our study predicts three hit compounds against hPPO, which could possibly accumulate high concentration of protoporphyrinogen-IX, and thereby acting as an intracellular photosensitizer against tumor cells through photodynamic therapy.
Keywords
Photodynamic therapy (PDT)
hPPO Inhibition
Virtual screening
Pharmacophore modeling
Molecular docking simulation
Molecular dynamics (MD) simulation

- hPPO
-
human Protoporphyrinogen Oxidase IX
- PDT
-
photodynamic therapy
- MD
-
molecular dynamics
- 3D
-
3 dimensional
- ADMET
-
adsorption, distribution, metabolism, excretion and toxicity
- NCI
-
National institute of cancer
- FAD
-
Flavin adenine dinucleotide
- PPO
-
protoporphyrinogen oxidase IX
- Ki
-
inhibitory constant
- EC
-
enzyme commission
- DS
-
Discovery Studio v4.5
- HBA
-
hydrogen bond acceptor
- RMSD
-
root mean square deviation
- GOLD
-
genetic optimization of ligand docking
- PLP
-
Pearson linear potential
- ASP
-
Astex statistical potential
- TIP3P
-
transferable intermolecular potential 3P
- QSAR
-
Quantitative structure activity relationship
- HBD
-
hydrogen bond donor
- HYP
-
hydrophobic
- RA
-
ring aromatic
- NVT
-
Number of constant volume and temperature
- NPT
-
number of constant pressure and temperature
- PME
-
Particle Mesh Ewald
- vdW
-
van der Waal
- LINCS
-
linear constraint solver
- fs
-
femtosecond
- ps
-
picosecond
- VMD
-
visualization of molecular dynamics
- K
-
kelvin
Abbreviations
1 Introduction
Photodynamic therapy (PTD) is an emerging modality of cancer treatment and management, with special focus on solid tumors (Dougherty et al., 1998; Robertson et al., 2009). The theory behind this immense strategy involves the utilization of a photosensitizer, e.g. Protoporphyrinogen-IX, as the precursor of singlet oxygen. The photosensitizer is either injected to tumor localization, and is subsequently excited by light, or it is accumulated endogenously in tumor cells. Protoporphyrinogen-IX is a best photosensitizer in tumor therapy, where it is converted spontaneously to protogen (oxidized protoporphyrinogen-IX) in the presence of light and release highly reactive singlet oxygen (1O2) (Weishaupt et al., 1976; Foote, 1990; Fingar et al., 1997). Subsequently, this singlet oxygen causes cellular damage, involving cell apoptosis and cell disruption (Arnould and Camadro, 1998; Kato et al., 2010; Matringe et al., 1989).
Modifications of heme biosynthetic pathway by targeting protoporphyrinogen oxidase IX (PPO) remains a promising strategy in photodynamic therapy (Peng et al., 1997; Henderson et al., 1995). Protoporphyrinogen oxidase IX (EC 1.3.3.4) is an essential rate limiting enzyme of heme and/or chlorophyll biosynthetic pathways in prokaryotes as well as in eukaryotes (Poulson and Polglase, 1975). PPO catalyzes the photo-dependent oxidation of protoporphyrinogen-IX to protoporphyrin-IX (Poulson, 1976; Brenner and Bloomer, 1980). Accumulation of protoporphyrinogen-IX occurs as a result of either PPO inhibition or loss of its catalytic potential and is exported to cytoplasm, where it can spontaneously be oxidized to protogen by molecular oxygen upon light exposure (Masoumi et al., 2008). Being a strong photosensitizer, protogen accelerates the production of singlet oxygen that induces lipid peroxidation and cell apoptosis (Arnould and Camadro, 1998). Thus, PPO is considered as a dynamic target against several inhibitors like herbicides, bactericides and fungicides (Kato et al., 2010; Matringe et al., 1989). Furthermore, several studies have been recommended that PPO inhibitors may also be employed in cancer management via photodynamic therapy (Moghissi et al., 2009; Robertson et al., 2009; Halling et al., 1994; Rebeiz et al., 1992). Two fundamental studies have shown that the induction of endogenous accumulation of protoporphyrinogen-IX in tumor cells destroyed cancerous cells in the presence of light (Halling et al., 1994; Rebeiz et al., 1992). It has also been investigated that 5-aminolevulinic acid, being a precursor of porphyrin via porphobilinogen formation, induces porphyrin synthesis in murine tumor (Henderson et al., 1995). In another study, Peng et al. (1997) has been evaluated the therapeutic potential of 5-aminolevulinic acid by photodynamic therapy approach. Nevertheless, some of the small molecules have already been investigated as PPO inhibitors either in vitro and/or in vivo, but there is no practicing drug to date. Therefore, our study has emphasized on integration of virtual screening, molecular docking and molecular dynamic simulation for the identification of novel hPPO inhibitors.
To accomplish the objective, we employed ligand-based pharmacophore modeling approach to generate a suitable 3D query which represents the essential features of the ligand complementing the hot spots of the active site of hPPO. The pharmacophpore model was validated by Fischer’s Randomization and Test Set Validation methods to assist its specificity and efficiency towards hPPO inhibitors. The validated model was used as a 3D query to screen four chemical databases including NCI, Asinex, Maybridge, and Chembridge, and the best fitted compounds were retrieved. The retrieved compounds were evaluated for their drug-like properties by Lipinski’s Rule of Five and ADMET Assessment Test methods. The initial binding poses for candidate compounds into the active site of hPPO were predicted through molecular docking. The best docked candidate molecules were subjected to molecular dynamics (MD) simulation to affirm the conformational space and binding stability of the final candidate molecules. Finally, this in silico approach for the identification of potential inhibitors of hPPO may contribute to a new avenue in photo-induced cancer therapy.
2 Results and discussion
2.1 Formulation of training set and pharmacophore modeling
To accomplish our goal, a training set of 22 structurally diverse inhibitors of hPPO was formulated and their respective 2D structures were drawn in Accelrys Draw v4.2 (Fig. 1).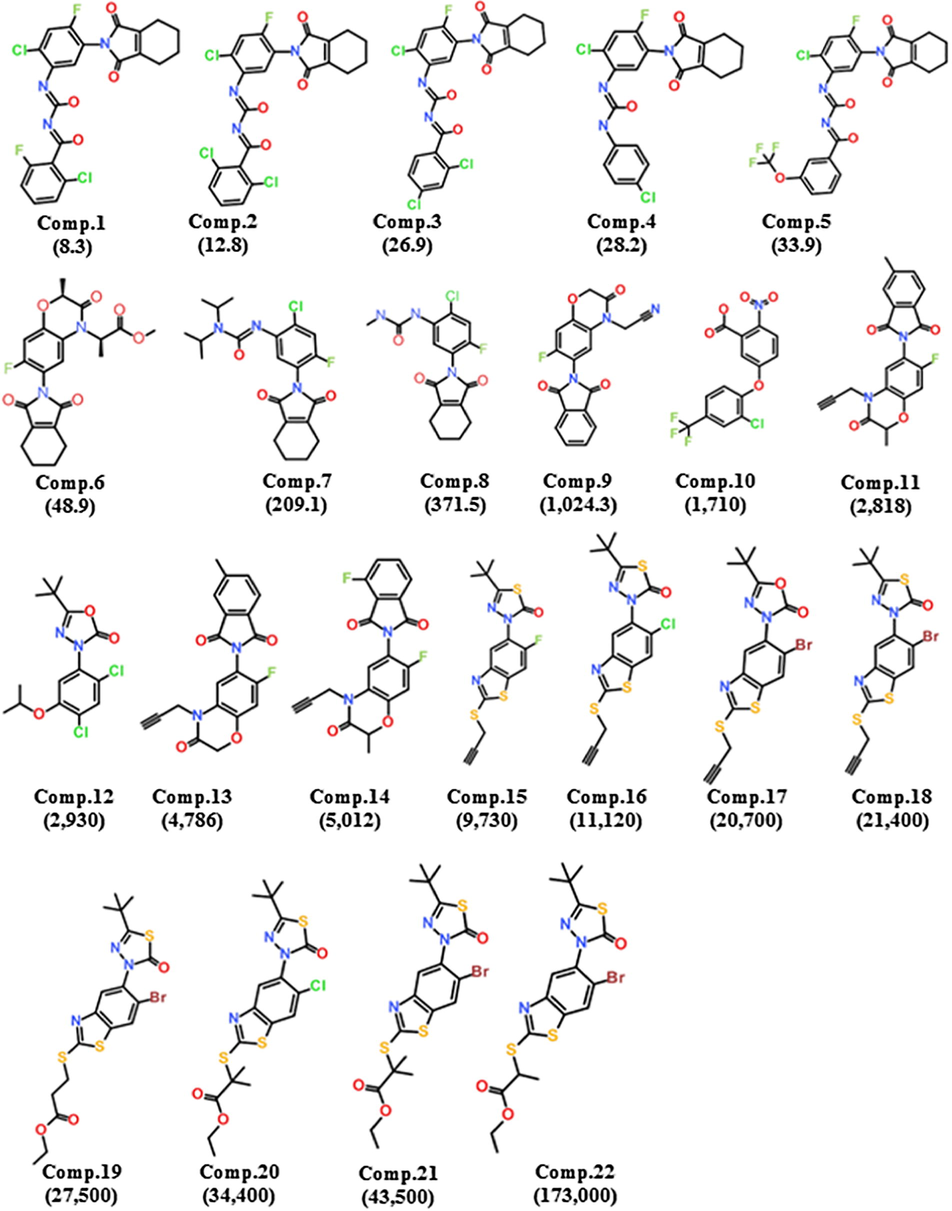
Training set compounds: 2D chemical structures and Ki (nmol/L) values of 22 training set compounds for hypotheses generation.
Based on experimental Ki value, the entire training set was classified into three classes: most active (+++, Ki < 1000 nmol/L), moderate active (1000 nmol/L < Ki ≤ 5000 nmol/L, ++), and least active (Ki > 5000 nmol/L, +) as depicted in Table 1.
Compound no.
Fit value
Exp. Ki (nmol/L)
Pred. Ki (nmol/L)
Errora
Exp. scaleb
Pred. scaleb
1
9.35
8.3
23
+2.8
+++
+++
2
9.28
12.9
28
+2.2
+++
+++
3
9.17
26.9
36
+1.3
+++
+++
4
9.13
12.2
39
+1.4
+++
+++
5
9.32
33.9
25
−1.3
+++
+++
6
9.23
48.9
31
−1.6
+++
+++
7
8.35
229.1
240
+1.0
++
++
8
8.31
371.5
260
−1.4
+++
+++
9
8.22
1023.1
320
−3.2
++
+++
10
7.01
1710.0
5100
+3.0
++
++
11
7.06
2818.4
4600
+1.6
++
++
12
6.53
2930.0
16,000
+5.4
++
+
13
7.07
4786.3
4500
−1.1
++
++
14
7.07
5011.9
4500
−1.1
+
++
15
6.57
9730.0
14,000
+1.5
+
+
16
6.61
11120.0
13,000
+1.2
+
+
17
6.06
20700.0
46,000
+2.2
+
+
18
6.58
21400.0
14,000
−1.5
+
+
19
6.61
27500.0
13,000
−2.1
+
+
20
6.52
34400.0
16,000
−2.2
+
+
21
6.40
43500.0
21,000
−2.1
+
+
22
6.33
173000.0
25,000
−6.9
+
+
Accordingly, ten hypotheses with statistical parameters were generated using the training set compounds. Several cost values including fixed cost, total cost, and null cost were employed to select the best hypothesis (Table 2).
Hypo no.
Total cost
Cost differencea
RMSDb
Correlation (r)
Max fit
Featuresc
Hypo1
99.68
53.20
0.73
0.96
9.65
3HBA, 1HYP
Hypo2
99.76
53.12
0.76
0.95
9.27
3HBA, 1HYP
Hypo3
101.06
51.82
0.78
0.95
10.07
2HBA, 1HYP, 1RA
Hypo4
101.78
51.10
0.88
0.94
9.14
3HBA, 1HYP
Hypo5
102.36
50.52
0.92
0.93
8.79
3HBA, 1HYP
Hypo6
102.90
49.98
0.93
0.93
9.09
3HBA, 1HYP
Hypo7
103.80
49.08
0.99
0.92
8.60
2HBA, 1HYP, 1RA
Hypo8
103.94
48.94
1.00
0.92
8.78
2HBA, 1HYP, 1RA
Hypo9
104.60
48.28
0.98
0.92
9.78
3HBA, 1HYP
Hypo10
104.68
48.20
103
0.92
8.56
2HBA, 1HYPA, 1RA
The distribution of cost values remain different, where fixed cost is the lowest possible cost and represents a simplest model that fits all the data perfectly. Conversely, null cost is equal to maximum error cost. A pharmacophore is considered to be statistically significant if the difference between the fixed cost and null cost remains greater. Several studies have shown that the possibility of correlating experimental and estimated activities enhances to 75–90%, if the cost difference ranges between 40 and 60 bits (Gupta et al., 2010; Kim et al., 2008). In this study, difference between the fixed cost and null cost was 53.20 bits, which reveals that the quality of the generated pharmacophore is significant to use as a 3D query for screening of chemical databases. Moreover, a best pharmacophore must have total cost closed to fixed cost and far from the null cost (Nayana et al., 2009; Debnath, 2002). Our results also observed that all the ten hypotheses had total cost closed to fixed cost, e.g. 92.79 and far from null cost i-e. 152.89; which in turn agrees the criteria of best model quality (Table 2). Apart from that, other statistically significant factors including cost values, correlation coefficient (r), pharmacophore features, and root mean square deviation (RMSD) of the entire generated hypotheses are depicted in Table 2. The RMSD value evaluates the quality of pharmacophore in terms of prediction of the training set compounds. The RMSD of ten hypotheses ranged between 0.73 and 1.03, where the Hypo1 showed a lowest RMSD score of 0.73 (Table 2). Correlation coefficient (r) enumerates geometric fit index and its value should be greater than 0.90 (Debnath 2002). Our results showed high correlation coefficient value of 0.96 for Hypo1, which represents consensus correlation by linear regression. Collectively, the entire results confirmed that Hypo1 comprised of three hydrogen bond acceptors (HBA) and one hydrophobic (HYP) features, is the best ranked pharmacophore model among the generated hypotheses (Fig. 2).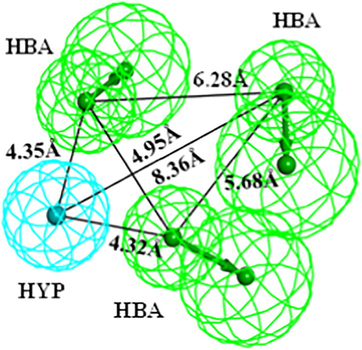
Chemical features of the selected pharmacophore ‘Hypo1’ with inter-feature distance constraints. Hypo1 consists of three HBA (green) and one HYP (cyan) features. The inter-feature distance constraints have been shown in angstrom (Å).
To evaluate the predictive capability of the Hypo1, an activity-based prediction of the training set compounds was conducted. The regression analysis revealed that all the highly active compounds were predicted in the same scale, whereas among the two moderate active compounds, one was overestimated as highly active, and the other was underestimated as least active. Additionally, one least active was overestimated as moderate active among the entire training set compounds (Table 1). These results suggested that the corresponding pharmacophore is capable to predict the Ki values in the same order of magnitude as the experimental values with high accuracy. Our results also confirmed that all the features in Hypo1 were perfectly mapped to highly active compound with a fit value of 9.65 (Fig. S1A). In contrast, the least active compound mapped onto only 3 features of the selected pharmacophore (Fig. S1B). These results recommended that the selected pharmacophore is efficient to discriminate highly active from least active compounds against hPPO.
2.2 Pharmacophore validation
The representative pharmacophore model was validated by two methods. First, Hypo1 was validated by Fischer’s Randomization at 95% confidence level. Hypo1 achieved the lowest total cost value of 99.68 among all the 19 spreadsheets generated, which advocates the statistical significance of the pharmacophore (Fig. 3A).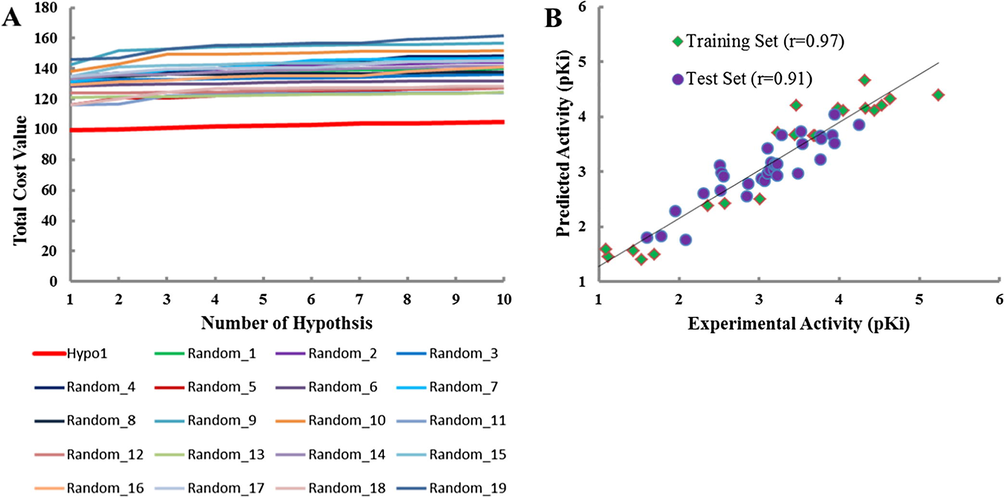
Pharmacophore validation. (A) Fischer’s Randomization. The total cost of Hypo1 remains lower to the total costs of the 19 scrambled runs generated during the Fischer’s Randomization run; (B) Test Set Validation. Correlation plot between the Hypo1 predicted inhibitory activities and experimental activities of 22 training set compounds as well as 34 test set compounds.
Second, the pharmacophore was validated by external test set which was comprised of 34 structurally diverse compounds other than training set compounds (Fig. S2). Herein, we have obtained an acceptable correlation coefficient value of 0.91 by linear regression analysis between the Hypo1 predicted and experimental activities of test set compounds (Fig. 3B). Therefore, our results corroborated that the Hypo1 is able to discriminate between the active and moderate active compounds.
2.3 Screening of chemical databases
The validated pharmacophore model was employed to filter the best-mapped compounds from chemical databases including NCI, Asinex, Maybridge, and Chembridge. The number of compounds in each database was 238,819; 231,262; 59,652; and 50,000 for NCI, Asinex, Maybridge, and Chembridge, respectively. Hypo1 mapped a total of 217,913 compounds from all the databases. Since, the fit value of mapping of the selected pharmacophore to highly active compound of the training set was used as cut-off value (9.35); hence the candidate molecules with fit value greater than 9.35 were filtered. The successful compounds were further subjected to additional filtrations like Lipinski’s Rule of Five and ADMET Descriptors to characterize and subsequently retrieve the drug-like compounds. Both these approaches evaluate compounds in terms of their drug-like properties, for instance, number of hydrogen bond donor and/or acceptor, logP value, molecular weight, blood-brain barrier, solubility, and toxicity. A total of 1176 candidate hits could successfully pass the drug-like properties (Table S1).
2.4 Molecular docking simulation
Molecular docking was performed to predict the binding mode of hit compounds. To date, a single crystal structure of hPPO has been resolved (PDB ID: 3NKS) (Qin et al., 2011). The candidate hits as well as the training set compounds were docked into the active site of hPPO. The catalytic active pocket of hPPO is shaped by R59, R62, R97, F169, F331, G332, I419 and the flavin moiety of FAD (Flavin adenine dinucleotide) (Qin et al., 2011). The ChemPLP score of 74.16 and ASP score of 28.32 obtained by highly active compound of the training set (hereafter Reference) was preferred as cut-off value for the evaluation of candidate hits (Table 3).
Systems
ChemPLP score
ASP score
hPPO + Hit1
76.58
34.76
hPPO + Hit2
77.94
24.28
hPPO + Hit3
78.72
30.50
hPPO + Reference
74.16
28.32
The candidate hits with high ChemPLP score and high ASP scores than cut-off value and interactions with catalytic active residues of hPPO were selected. Since, the ChemPLP score was used as decision making parameter during the docking analysis, therefore, Hit2 was also considered as best candidate compound despite of its low ASP score than the Reference (Table 3). Our docking results illustrated that all the three candidate hits had strong hydrogen bond as well as hydrophobic interactions with catalytic active residues of hPPO. For instance, Hit1 showed hydrogen bond with catalytic active residues including R62, R97, G169 and FAD. Moreover, Hit1 also showed strong hydrophobic interactions with other important residues of hPPO e.g. π-π interaction with F331 and L344. Hit2 established hydrogen bond interactions with active residues R59 and R97 of hPPO. Hit2 also formed several hydrophobic interactions with catalytic site residues of hPPO. The docking simulation of Hit3 showed hydrogen bond interactions with R97, F98 and G169 of hPPO. Additionally, Hit3 also established van der Waals, alkyl, π-alkyl and π-cation interactions with residues shaping the catalytic site of hPPO. Finally, the top three hits were selected as final candidates. The selected candidate hits mapped well onto all the four features of Hypo1 (Fig. 4).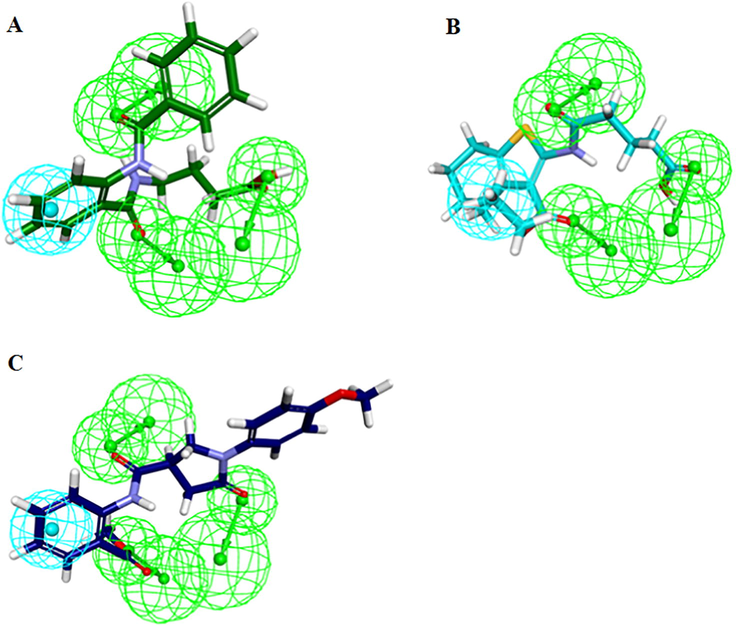
Hypo1 mapped onto the hit molecules. (A) Hit1, (B) Hit2, (C) Hit3. The HBA and HYP features are colored as green and cyan respectively.
2.5 Molecular dynamics simulation
In order to affirm the binding stability and pose orientation of each candidate hit in the active site of hPPO, molecular dynamics simulation of 30-ns was performed. The docked pose of each candidate hit was taken as input data and a total of four different systems (Reference and the three candidate hits) were designed. Our simulation results showed that all the systems have converged well and established a stable root mean square deviation (RMSD) which ranges between 2 and 3 Å throughout the simulation period (Fig. 5).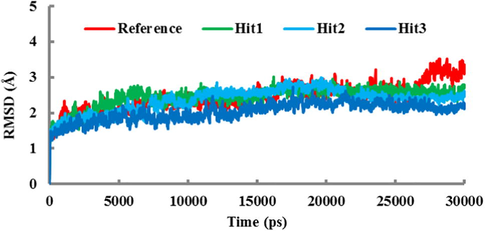
The RMSD plot of four systems. The RMSD profile for the Cα atoms of the protein during the 30-ns simulation. Colors are assigned as red (Reference compound), green (Hit1), cyan (Hit2), and blue (Hit3).
The individual RMSD value for each system further affirmed that all the three candidate hits had low RMSD values than the Reference compound in last 5-ns trajectory (Fig. 5).
To further get insight into the stability of protein-ligand complex, the potential energy for each system was calculated. The potential energy of all the four systems remained stable during the entire simulation period and no abnormal behavior was observed (Fig. S2). To trace the binding orientation, the representative structures of all the protein-ligand complexes were taken from the last 5-ns trajectory and superimposed. It was observed that all the candidate hits oriented in the same pattern and occupied the same catalytic pocket as the Reference compound (Fig. S3). Furthermore, the molecular interactions of each ligand with catalytic active residues of hPPO were inferred. As mentioned earlier that the active site of hPPO is shaped by R59, Arg62, R97, G169, F331, G332, I419 and the flavin moiety of the FAD. Our computational analysis observed that the binding mechanism of Reference compound was established by hydrogen bond formation with the catalytic site residues Arg97 and Gly345 of hPPO (Fig. 6A).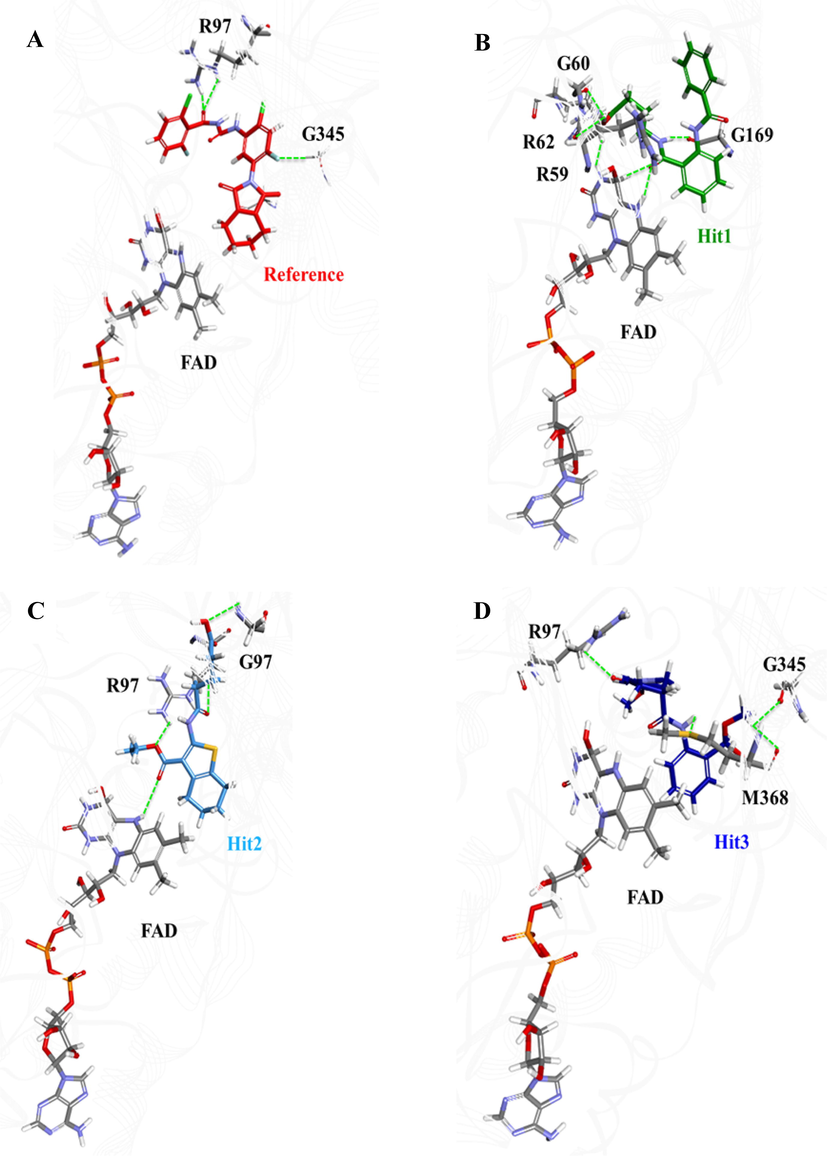
Illustration of pose orientation and hydrogen bond formation of the Reference compound and the three hit molecules in the active site of hPPO. (A) Reference: red, (B) Hit1: green, (C) Hit2: cyan, and (D) Hit3: blue. Hydrogen bonds between hPPO and ligands are shown as green dotted lines.
Several other hydrophobic interactions were also observed between the Reference compound and the residues in catalytic pocket of hPPO (Table 4).
Systems
Hydrogen bond (<3.0 Å)
Hydrophobic interactions
van der Waals interactions
hPPO + Reference
Arg97, G169, G345
Arg62, Ala172, Val314, Leu334, Val347, Met368
Gly60, Arg168, Val170, Gln226, Gly332, His333, Leu344, Ile346, Ile419, FAD
hPPO + Hit1
Gly60, Arg59, Arg62, Gly169, FAD
Val314, Leu334, Val347, Met368
Pro58, Ile61, Arg62, Arg168, Val170, Phe171, Ala172, Gln226, Gly332, His333, Leu344, Gly345, Ile346, Ile419
hPPO + Hit2
Arg97, Gly332, FAD
Leu344, Met368, FAD
Arg59, Arg62, Phe98, Arg168, Gly169, Ala172, Val170, Val314, Phe331, His333, Leu334, Gly345, Ile346, Val347, Tyr348, Ile419
hPPO + Hit3
Arg62, Gly345, Met368
Ala172, Met368, FAD
Arg59, Gly60, Leu166, Arg168, Gly169, Val170, Leu334, Leu344, Gly345, Ile346, Val347, Leu369, Ile419
Our computational analysis showed that Hit1 has hydrogen bond interactions with R59, G60, R62, G169 and FAD as shown in Fig. 6B. Our analysis observed that the hydroxyl group of acidic moiety of Hit1 has hydrogen bond with carbonyl oxygen of G60 and R59, and amino group of isoalloxazine ring of FAD (Fig. 6B). Additionally, hydrogen bond formation was observed between the oxygen of amino-terminus of benzamidobenzoyl moiety of Hit1 and amino group of isoalloxazine ring of FAD. Apart from hydrogen bonding, Hit1 has also showed several hydrophobic interactions including π-alkyl stacking between the benzene ring of benzoyl group and side chains of V314 and M368, and benzene ring of benzamide group and side chains of V347 and L334 (Table 4). Our analysis observed that benzoamidobenzoyl moiety of Hit1 is oriented towards the hydrophobic subpocket of hPPO by making van der Waals interactions with A172, I419 and G345. All these findings are in strong agreement of the interaction pattern established by aciflurine (Qin et al., 2011).
Our data showed that Hit2 has formed four hydrogen bonds with hPPO including residues R97, G332 and the flavin moiety of FAD (Fig. 6C). It was observed that the oxygen atoms of ethoxy and oxopentanoic acid groups of Hit2 have formed hydrogen bonds with side chain of R97. Hit2 has also been interacted by its terminal oxygen of acetate group with another catalytic active residue G332. Our results follow Qin et al. (2011) findings that aciflurine has hydrogen bond interaction with G332. Moreover, the carboxyl oxygen of oxopentanoic acid moiety of Hit2 formed stable hydrogen bond interaction with FAD (Fig. 6C). Based on these observations, we suggest that Hit2 may have same mode of hPPO inhibition as the Reference compound but with high affinity. We support our suggestion of high affinity of Hit2 towards hPPO by the formation of strong hydrogen bonds with Arg97, Gly332 and FAD. Hit2 was also found to establish several hydrophobic and van der Waals interactions with active site residues of hPPO (Table 4). Benzene ring of oxopentanoic acid moiety has strong hydrophobic center which formed alkyl interactions with positively charged scars of M368, L344, and FAD. Oxopenanoic acid group of Hit2 also formed π-sulfur interaction with sulfur containing M368. Since, R97 has been shown as the essential residue participating in catalytic reaction driven by hPPO, therefore, our rational approach suggested that the formation of hydrogen bonds of Hit2 with R97, G332 and FAD might be strong enough to prevent the binding of substrate (protoporphyrinogen-IX) in the active site of hPPO in competitive inhibition.
The third candidate compound, Hit3, was also evaluated by MD simulation to infer its binding mode and conformational stability in hPPO. Our analysis showed that polar hydrogen of the amino group of benzoate formed hydrogen bond with sulfur atom of M368. Other hydrogen bonds were observed between the oxygen atom of oxo-pyrrolidine group of Hit3 and side chain of R62 as well as methyl group of methoxyphenyl and carboxyl oxygen of G345 (Fig. 6D). Moreover, Hit3 showed π-alkyl interactions between its aminobenzoate moiety and M368, A172 and FAD. All the hydrophobic and van der Waals interactions with the catalytic pocket residues of hPPO are depicted in Table 4.
To evaluate the binding affinity of selected candidate molecules in the active site of hPPO, the inter-molecular hydrogen bonds of the compounds were monitored during the entire simulation period (Fig. 7). Our computational analysis observed that the average number of hydrogen bonds for all the hit compounds remained higher than that of the Reference compound. Among the hit compounds, Hit1 showed highest number of hydrogen bonds (Fig. 7). The comparative analysis suggested that Hit2 and Hit3 also showed greater number of hydrogen bonds than the Reference compound.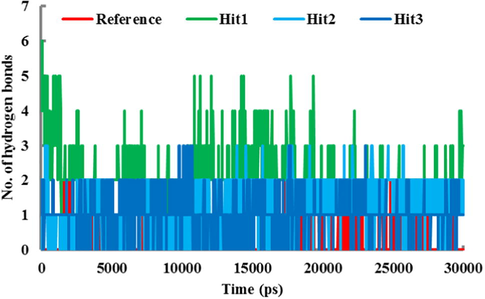
The number of intermolecular hydrogen bonds between hPPO and the compounds during 30-ns MD simulation. Red, green, cyan and blue represent reference, Hit1, Hit2, and Hit3 respectively.
Finally, PubChem Structure (Wang et al., 2014), which is an online tool, was used to verify that the selected hit compounds are not tested experimentally as PDT induced therapeutics and can be recommended as potent hPPO inhibitors. Based on these observations, we propose these three hit compounds as novel candidates for hPPO inhibition (Fig. 8).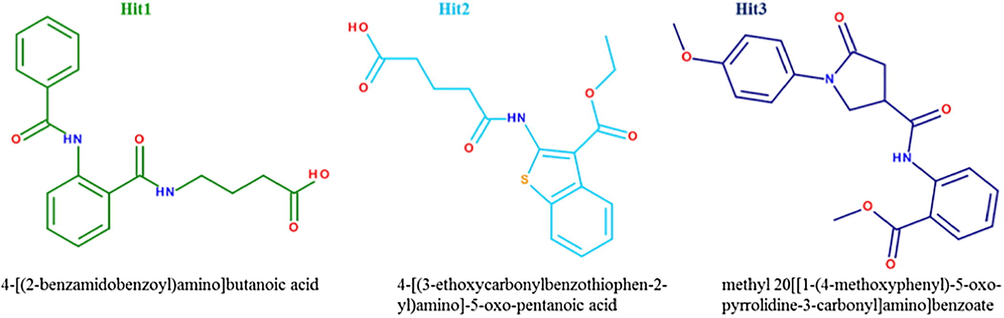
2D structures and IUPAC names of the three hit molecules.
3 Conclusion
Inhibition of heme biosynthetic pathway has been considered as a promising target in several cancer therapies. PPO is the last rate limiting enzyme of heme biosynthetic pathway and its inhibition results in high accumulation of protogen, which later on produces singlet oxygen in the presence of light and causes apoptosis and cellular destruction. In this study, we attempted to identify novel inhibitors against hPPO by ligand-based pharmacophore modeling. Herein, a pharmacophore model was generated from training set compounds. The best pharmacophore model had four chemical features including three HBA and one HYP. Hypo1 had the highest correlation coefficient (r) of 0.96, cost difference of 53.20, and low RMSD of 0.73. The pharmacophore was validated by Fischer’s Randomization and Test Set Validation method, which confirmed the ability of Hypo1 to retrieve active compounds against hPPO. Thereafter, Hypo1 was used as a 3D structural query to screen the selected chemical libraries and retrieved the best fitted compounds. Lipinski’s Rule of Five and ADMET Descriptors were used to eliminate the non-drug like compounds. Furthermore, the drug-like compounds were subjected to molecular docking to eliminate the false positive hit molecules. Finally, MD simulation was employed to affirm the stability of each ligand into the catalytic active site of hPPO. The newly identified hit molecules formed stable hydrogen bond interactions with catalytic active residues e.g. R59, R97, and G332 of hPPO. The MD simulation also confirmed that each hit molecule formed hydrophobic interactions with residues occupying the catalytic active site of hPPO. Finally, we proposed three hit molecules as fundamental virtual platforms for the development of photodynamic therapeutics against hPPO inhibition.
4 Materials and methods
4.1 Data set preparation
Already known inhibitors of protoporphyrinogen oxidase IX (PPO) were identified from literature survey (Zhang et al., 2009; Zuo et al., 2011) and binding database (http://www.bindingdb.org/bind/index.jsp). A total of 56 compounds were selected and classified into training set (22 compounds) and test set (34 compounds). Thereafter, the inhibitory constant expressed as Ki value was taken as selective parameter for the classification of compounds. Based on Ki values, all the training set compounds were classified into highly active, moderate active, and least active sets. The 2D structures of training set compounds were drawn in Accelrys Draw v4.2, and subsequently converted to their 3D format in Discovery Studio v4.5 (DS). The training set compounds were energy minimized by CHARMm forcefield with energy threshold value of 20 kcal/mol. Subsequently, a maximum of 255 conformers were generated for each compound and these conformers were used for hypotheses generation, fitting compounds into the hypotheses and estimating the activity of compounds.
4.2 Pharmacophore model generation
To identify the essential features of the training set compounds, they were subjected to Feature Mapping protocol, implanted in DS. The essential features of the training set compounds were employed to generate pharmacopohore models, while using 3D QSAR Generation protocol, implanted in DS. The essential pharmacophoric features were included hydrogen bond acceptor (HBA), hydrogen bond donor (HBD), ring aromatic (RA), hydrophobic (HYP), and hydrophobic aromatic (Hy-Ar). The uncertainty value was defined as 3 for all the training set compounds. Other parameters were selected as minimum number of features “1”, maximum number of features “5”, conformation generation “best” and the Fischer’s validation “95%”, while the rest of the parameters were used as default values. At the end, ten qualitative pharmacophore models were generated with corresponding statistical parameters like cost values, fit values, correlation coefficient values, and root mean square range.
4.3 Pharmacophore model evaluation
In order to confirm whether the resultant pharmacophore model was reliable enough to mimic the essential features of hPPO inhibitors, validation of the pharmacophore models was carried out. The Fischer’s Randomization method was employed to select the best pharmacophore model. Our protocol relied on 95% significance of the best hypothesis, where 19 random sets were derived from the original training set by shuffling randomly and 19 different pharmacophore models were generated. These randomly created pharmacophores were plotted against the selected hypothesis in terms of total cost, where the selected hypothesis must have lowest total cost (Sakkiah and Lee, 2012).
To further validate our pharmacophore model, structurally diverse inhibitors of hPPO, which were not included in training set, were taken and assigned as test set. The test set was used to predict correlation between the experimental and predicted activity values estimated by the selected pharmacophore.
4.4 Pharmacophore-based screening of chemical databases
The validated pharmacophore was used as a 3D query to retrieve the best fitted candidate molecules from chemical databases like NCI, Asinex, Chembridge, and Maybridge. The screening of chemical databases was carried out by pharmacophore using Ligand Pharmacophore Mapping module in DS with Best and Flexible parameters. The pharmacophore-fitted molecules were evaluated for their drug-like properties. The drug-like properties were analyzed by Lipinski’s Rule of Five and ADMET Descriptors module, implanted in DS. Finally, the drug-like hit molecules were subjected to molecular docking to elucidate the binding orientation in the active site of hPPO.
4.5 Molecular docking simulation
Molecular docking is an in silico approach to predict binding affinity, pose orientation, and molecular interactions between ligand and receptor. Herein, molecular docking studies were carried out by using Genetic Optimization of Ligand Docking (GOLD v5.2.2) package (Morris et al., 1998). GOLD package relies on ChemPLP (Pearson Linear Potential) as main scoring function, which accounts for energy from external hydrogen bonds, external van der Waals (vdW) and internal torsion. On the other hand, ASP (Astex Statistical Potential) score was used as rescoring function, which deals with atom-atom distance potential and Chemscore terms (Yan et al., 2014). For molecular docking simulation, a high resolution (1.9 Å) crystal structure of hPPO (PDB ID: 3NKS) was taken (Qin et al., 2011). The initial structure was cleaned by removing water molecules and hetero atoms. The missing residues were gap-filled by Loop Insertion protocol, implanted in DS. CHARMm forcefield was used to carry out energy minimization and hydrogen atoms were added. The well prepared hPPO structure was sued as protein input file for docking analysis. The docking coordinates were selected from active site residues e.g. R59, R97, F331, G332 and hPPO bound inhibitor (aciflurine). The drug-like molecules along with training set compounds were energy minimized and used as ligand input file for docking in the active site of hPPO.
4.6 Molecular dynamics (MD) simulation
All simulations were performed in GROMACS v5.1.2 (Abraham et al., 2015) with CHARMm36 forcefield (Huang et al., 2017) parameters. The topology and coordinates of all the ligands were parameterized by SWISSPARAM (Zoete et al., 2011). For each ligand, independent MD system was designed in a TIP3P water model in an octahedral simulation box (Jorgensen et al., 1983). Periodic boundary conditions were applied in all directions to imitate infinite system. During the simulation, Particle Mesh Ewald (PME) method using 10 Å cut-off distance for real-space Ewald interactions and vdW interaction was employed (Darden et al., 1993). Dispersion correction was used to accommodate energy and pressure terms caused by vdW truncation. Bond lengths were restrained by LINCS algorithm that endorsed a 2-fs time frame in all simulations (Hess et al., 1997). Prior to simulation, all the systems were neutralized by adding sodium (Na+) as counter ions. The energy minimization was carried out by steepest descent minimization with a maximum force of 10 kJ/mol/nm to remove any unfavorable contacts. To avoid configurational changes during equilibration, all the atoms were position restrained. Each system was equilibrated in two phases; first, systems were simulated for 100 ps at NVT ensemble and constant temperature of 300 K, which was controlled by V-rescale method (Bussi et al., 2007). Each NVT equilibrated system was subjected to NPT equilibration for 100 ps at an isotropic pressure of 1.0 bar, employing Parrinello-Rahman barostat (Parrinello and Rahman, 1981). Production MD was performed for 30 ns in flexible mode and data was collected after each 2 fs interval. During the production period, V-rescale thermostat and Parrinello-Rahman barostat were employed to maintain temperature (300 K) and pressure (1 bar). All the analyses of MD simulations were carried out by VMD and DS software.
Acknowledgments
This research was supported by the Next-Generation BioGreen 21 Program (PJ01106202) from Rural Development Administration (RDA) of Republic of Korea. And also supported by the Pioneer Research Center Program (NRF-2015M3C1A3023028) through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning.
References
- GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19-25.
- [Google Scholar]
- The domain structure of protoporphyrinogen oxidase, the molecular target of diphenyl ether-type herbicides. Proc. Natl. Acad. Sci.. 1998;95:10553-10558.
- [Google Scholar]
- The enzymatic defect in variegate prophyria. Studies with human cultured skin fibroblasts. N. Engl. J. Med.. 1980;302:765-769.
- [Google Scholar]
- Particle Mesh Ewald: An N-log(N) method for Ewald sums in large systems. J. Chem. Phys.. 1993;98:10089-10092.
- [Google Scholar]
- Pharmacophore mapping of a series of 2,4-diamino-5-deazapteridine inhibitors of Mycobacterium avium complex dihydrofolate reductase. J. Med. Chem.. 2002;45:41-53.
- [Google Scholar]
- Photodynamic therapy using a protoporphyrinogen oxidase inhibitor. Cancer Res.. 1997;57:4551-4556.
- [Google Scholar]
- Foote, C.S., 1990. Chemical mechanism of photodynamic action. In: Gomer, C.J. (Ed.), Future Directions and Application in Photodynamic Therapy. vol. IS6. SPIE Optical Engineering Press, Bellingham, Washington, pp. 115–126.
- Pharmacophore modeling of substituted 1,2,4-Trioxanes for quantitative prediction of their antimalarial activity. J. Chem. Inf. Model.. 2010;50:1510-1520.
- [Google Scholar]
- Halling, B.P., Yuhas, D.A., Finger, V.F., Winkelmann, J.W., 1994. Protoporphyrinogen oxidase inhibitors for tumor therapy. In: Duke, S.O., Rebeiz, C.A. (Eds.), Porphyric Pesticides: Chemistry, Toxicology, and Pharmaceutical Applications. American Chemical Society (ASP) Symp, Washington, DC, USA, pp. 280–290.
- Photosensitization of murine tumor, vasculature and skin by 5-aminolevulinic acid-induced porphyrin. Photochem. Photobiol.. 1995;62:780-789.
- [Google Scholar]
- LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem.. 1997;18:1463-1472.
- [Google Scholar]
- CHARMM36m: an improved forcefield for folded and intrinsically disordered proteins. Nat. Methods.. 2017;14:71-73.
- [Google Scholar]
- Comparison of simple potential functions for simulating liquid water. J. Chem. Phys.. 1983;79:926-935.
- [Google Scholar]
- Identification of a gene essential for protoporphyrinogen IX oxidase activity in the cyanobacterium Synechocystis sp. PCC6803. Proc. Natl. Acad. Sci.. 2010;107:16649-16654.
- [Google Scholar]
- New serotonin 5-HT6 ligands from common feature pharmacophore hypotheses. J. Chem. Inf. Model.. 2008;48:197-206.
- [Google Scholar]
- Complex formation between protoporphyrinogen IX oxidase and ferrochelatase during haem biosynthesis in Thermosynechococcus elongatus. Microbiology. 2008;154:3707-3714.
- [Google Scholar]
- Protoporphyrinogen oxidase as a molecular target for diphenyl ether herbicides. Biochem. J.. 1989;260:231-235.
- [Google Scholar]
- Photofrin PDT for early stage oesophageal cancer: long term results in 40 patients and literature review. Photodiagn. Photodyn. Ther.. 2009;6:159-166.
- [Google Scholar]
- Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem.. 1998;19:1639-1662.
- [Google Scholar]
- Structural analysis of carboline derivatives as inhibitors of MAPKAP K2 using 3D QSAR and docking studies. J. Chem. Inf. Model.. 2009;49:53-67.
- [Google Scholar]
- Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys.. 1981;52:7182-7190.
- [Google Scholar]
- 5-Aminolevulinic acid-based photodynamic therapy: principles and experimental research. Photochem. Photobiol.. 1997;65:235-251.
- [Google Scholar]
- The enzymatic conversion of protoporphyringen IX to protoporphyrinogen IX in mammalian mitochondria. J. Biol. Chem.. 1976;251:3720-3733.
- [Google Scholar]
- The enzymic conversion of protoporphyrinogen IX to protoporphyrin IX. Protoporphyrinogen oxidase activity in mitochondrial extracts of Saccharomyces cerevisiae. J. Biol. Chem.. 1975;250:1269-1274.
- [Google Scholar]
- Structural insight into human variegate porphyria disease. FASEB J.. 2011;25:653-664.
- [Google Scholar]
- Photodestruction of tumor cells by induction of endogenous accumulation of protoporphyrin IX: enhancement by 1, 10-phenanthroline. Photochem. Photobiol.. 1992;55:431-435.
- [Google Scholar]
- Photodynamic therapy (PDT): a short review on cellular mechanisms and cancer research applications for PDT. J. Photochem. Photobiol. B, Biol.. 2009;96:1-8.
- [Google Scholar]
- Pharmacophore-based virtual screening and density functional theory approach to identify novel butyrylcholinesterase inhibitors. Acta Pharmacol. Sin.. 2012;33:964-978.
- [Google Scholar]
- PubChem BioAssay: 2014 update. Nucleic Acids Res.. 2014;42 (Database issue), D1075 –D1082
- [Google Scholar]
- Identification of singlet oxygen as the cytotoxic agent in photoinactivation of a murine tumor. Cancer Res.. 1976;36:2326-2329.
- [Google Scholar]
- Comparative assessment of scoring functions on an updated benchmark: 2. Evaluation methods and general results. J. Chem. Inf. Model.. 2014;54:1717-1736.
- [Google Scholar]
- Bioactive conformation analysis of cyclic imides as protoporphyrinogen oxidase inhibitor by combining DFT calculations, QSAR and molecular dynamic simulations. Bioorgan. Med. Chem.. 2009;17:4935-4942.
- [Google Scholar]
- SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem.. 2011;32:2359-2368.
- [Google Scholar]
- Quantitative structure-activity relationship of 1,3,4-thiadiazol-2(3H)-ones and 1,3,4-oxadiazol-2(3H)-ones as human protoporphyrinogen oxidase inhibitors. Bioorgan. Med. Chem.. 2011;20:296-304.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.arabjc.2018.04.009.
Appendix A
Supplementary material
Supplementary data 1
Supplementary data 1




