

Intermediate-based virtual screening of c-Kit kinase inhibitors as potential anti-tumor agents via ab inito folding, molecular dynamics simulation, and molecular docking
⁎Corresponding authors. xyliu0427@163.com (Xueying Liu), 961638888@163.com (Xinlei Zhang)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Uncontrolled activation of c-Kit is closely related to the pathogenesis and progression of leukemia, gastrointestinal cancer, and other malignant diseases. Although there are several inhibitors available, due to the limitation of selectivity and the unfavorable side effects, designing and discovering highly selective inhibitors targeting c-Kit kinase, especially the gain of function mutation (for example c-Kit D816V), is still necessary. To identify novel c-Kit inhibitors, a metastable state-based virtual screening approach, which was successfully implemented in other kinase inhibitors, was employed in the current study. The results from our current study demonstrated the residues adjacent to the DFG motif within the activation loop could fold into short α-helices aside from the random coil, which was commonly found in the crystal structure. By expanding the conformation pool of the activation loop via PyRosetta-based ab initio folding protocol, we constructed a series of structural models of the c-Kit kinase intermediate between the inactive and active states. After evaluation of the thermal stability of the metastable state with molecular dynamics simulation, one structural model showed higher stability of α-helix, and the activation loop was retained. Considering the wild-type and D816V mutated KIT kinase shared similar metastable states during the kinase activation process, we developed a hypothesis that the identified intermediate might hold the potential to identify inhibitors targeting D816V mutations from the compound database. As expected, the intermediate structure showed higher selectivity to KIT D816V selective inhibitors, such as bezuclastinib, avapritinib, BLU-263, and elenestinib, than imatinib or masitinib. The virtual screening of the available KIT kinase inhibitor database further identified vorolanib, semaxanib, henatinib, and pexmetinib may possess potential inhibitory effects against wild type, as well as the mutated c-Kit kinase. The results from our current study not only proposed a novel structural model that could be used for the identification of selective c-Kit D816V inhibitors but also identified several potential inhibitors from available kinase inhibitors, which might shed new light on the design of new therapeutic approaches for c-Kit mutation-driven malignant diseases.
Keywords
c-Kit inhibitor
Intermediate-based virtual screening
Ab inito folding
Molecular dynamics simulation

1 Introduction
c-Kit belongs to the type III receptor tyrosine kinase family and plays a crucial role in cell differentiation and proliferation. Physiologically, the activation of c-Kit occurs via binding to its ligand, Stem cell factor, followed by the homodimerization of the receptor, and abolishing the auto-inhibitory interaction(Pathania et al., 2021). However, due to the overexpression and the occurrence of gain of function mutations, the dysregulation of c-Kit is commonly observed in the progression, invasion, and metastasis of various tumors, including leukemia, gastrointestinal stromal tumor (GIST), melanoma, and unilateral ovarian dysgerminoma(Abbaspour Babaei et al., 2016). The majority of patients with GISTs possess primary activating mutations of the KIT kinase gene, which frequently leads to ligand-independent activation of this receptor(Abdel-Magid 2021). Statistically, the analysis demonstrated the most common mutations are located in the kinase domain, including K642E, D820Y, D816V, N822K, or Y823D, and accounting for around 70 % of total cases(Rassner et al., 2023). Within all identified mutations, the D816V was significantly associated with poor prognosis in patients, contrasting with other types of mutations in KIT. Further studies showed the molecular mechanism was closely related to its unique kinetic properties that KIT D816V could accelerate autoactivate 586-fold faster than native c-Kit(Sheikh et al., 2022). Thus, blocking the catalytic activity of c-Kit kinase, especially the mutated c-Kit kinase is still one of the most attractive strategies for cancer therapy.
Similar to other tyrosine kinases, the activation of c-Kit was characterized by the conformational alteration of the activation loop (A-loop) within its kinase domain and the flipping of the DFG motif(Thomas and Roux 2021). In the auto-inhibitory state, the kinase domain adopted a DFG-out conformation, while it switches to DFG-in conformation when activated. Accordingly, the inhibitors targeting the kinase domain of c-Kit can be subdivided into two main categories. Type II inhibitors of KIT, also known as DFG-out inhibitors target the auto-inhibitory states, which contribute to stabilizing the KIT kinases in its auto-inhibitory states; while the Type I inhibitors target the DFG-in conformation of c-Kit, and function as competitive inhibitors of ATP. As one of the most famous DFG-out inhibitors, imatinib showed higher selectivity and binding affinity towards wild-type c-Kit. However, the typical DFG-out inhibitors, such as imatinib or masitinib, are less effective against activated c-Kit kinase(Castells and Akin 2021). In contrast, DFG-in inhibitors could inhibit the kinase activity of activated KIT or KIT D816V with efficacy comparable to wild-type KIT(Chen et al., 2022). However, due to the limitation of selectivity and the unfavorable side effects, discovering highly selective inhibitors targeting c-Kit kinase is still urgently needed.
Accumulating evidence demonstrating the inhibitors targeting the intermediate state showed more promising selectivity and prolonged residence time than typical type I and type II inhibitors(Tsai et al., 2019, Umezawa and Kii 2021), which soon became an attractive design strategy, and successfully used in the design of EGFR, MET, and ERK1/ERK2 inhibitors. The intermediate-based inhibitors are also known as type 1.5 inhibitors. Unlike type 1 or type 2 inhibitors, the binding of type 1.5 inhibitors could evoke spontaneous conformational alterations towards the DFG motif, αC helices, and P-loop, which resulted in the highly selectivity. For example, M. M. Sultan et al. identified an intermediate of SRC kinase with a potentially druggable pocket next to the ATP binding site, which formed by an unstructured A loop, an outward rotated αC helix via millisecond molecular dynamics simulation, and the intermediate state and pocket identified was considered drugable(Sultan et al., 2018). Similarly, Shukla et al. identified a structural intermediate of c-SRC kinase with a novel allosteric site that could be used for drug design (Shukla et al., 2014). However, due to the relatively high-energy and short-lived properties of the intermediate, the detailed structure information about the intermediate state or metastable state was limited. Thus, molecular simulation is still one of the most commonly used to strategy to capture and investigate the intermediate within the activation process of kinase.
Although KIT D816V possessed significantly increased flexibility of activation loop (A-loop), displayed a much shorter time required for activation, and a differed communication pattern between A-loop and kinase domain, the mutated KIT shared similar metastable states with wild-type KIT kinase(Laine et al., 2011, Laine et al., 2012, Ledoux et al., 2023). The findings indicated the identified metastable state could be used for the design of inhibitors towards D816V mutations.
Considering the advantages of an intermediate-based drug design strategy during the design of kinase inhibitors, in this study, we implemented this approach in the discovery of c-Kit kinase inhibitors. To identify the potential metastable intermediate, secondary structure prediction, ab inito folding, and molecular dynamics simulation were employed in this study. The potential drugability of the identified intermediate was tested by molecular docking with a small dataset. Finally, potential c-Kit inhibitors were obtained by structural-based virtual screening.
2 Material and methods
2.1 Secondary structure profiling
The secondary structure profile of the activation loop was generated with PEP-FOLD 4.0 application(Rey et al., 2023) via RPBS Web Portal (https://mobyle.rpbs.univ-paris-diderot.fr/cgi-bin/portal.py#forms::PEP-FOLD4). The sequence of the activation loop of KIT kinase was used for secondary structure profile prediction, and the detailed sequence used for this experiment was “THGRITKICDFGLARDIKNDSNYVVKG”.
2.2 Ab inito folding of kinase domain within c-Kit kinase
The crystal structure describing the autoinhibitory conformation (PDBID:1T46) and active comconformation (PDBID:1PKG) of the tyrosine kinase domain within c-Kit was retrieved from Protein Data Bank. After removing water and co-crystallized ligand, the structural model was imported into the PyRosetta. The folding protocol was constructed according to the literature with adjustments. Briefly, a loop region between Cys 809 to Asn 831 was defined, and the centroid conformation of the defined loop region was generated with FragmentMover and CCDLoopClosureMover. After recovering the side chain conformation with PackRotamersMover, the full atom structure was subjected to the final refinement with LoopMover_Refine_CCD. The “score3” and “talaris2013” energy score function from PyRosetta was employed to evaluate the potential energy of the centroid and full atom structural model, respectively. For each initial structure, 5000 conformations (decoys) were generated. The root-mean-square deviations (RMSD) of the A-loop compared to the two reference structures were calculated with PyMOL, and the K-means clustering was performed by the scikit-learn package(Pedregosa et al., 2011) with Python. The three-dimensional structure of the generated decoys was visualized by PyMOL 2.5 (Open-source version).
2.3 Molecular dynamics simulation
The molecular dynamics (MD) simulations were performed to obtain the stable intermediate conformation. The hydrogen atoms were removed, and the protein structures were parameterized with the Amber 14SB force field(Maier et al., 2015). Then, the protein was embedded into a cubic box and dissolved within the TIP3P water model. The total atom number of the prepared systems was around 35,600, and the initial dimension was around 70.6 Å × 78.7 Å × 73.5 Å.
To remove the defects in structural models and to resolve the collision between hydrogen atoms, a three-staged energy minimization protocol was employed as a previous study. Briefly, the complex was subjected to an all-heavy atom position-restrained energy minimization (lasted for 500 steps), backbone atoms position-restrained energy minimization (for 500 steps), and an energy-minimized without restraints for another 1,000 steps. The energy-minimized system was gradually heated to 310 K from 0 K in 100 ps with all heavy atoms restrained. Finally, the restraints were gradually removed in a constant pressure environment (NPT) for a total of 2.5 ns. The simulation time step was set to 2 fs, and the van der Waals interactions were calculated at every step. The long-range electrostatic interactions were calculated every 5 steps by the particle mesh Ewald (PME) method(Fukuda et al., 2014) with a grid size of about 1 Å and a tolerance of 10−6. The cut-off for the van der Waals interactions was set to 9 Å. The temperature and pressure were controlled using Langevin dynamics and the Langevin piston barostat method. The SHAKE method was used to restrain hydrogen atoms and the tolerance was set to 10−8. Atomic coordinates of all atoms were saved every 1 ps. The production NPT run was performed at 310 K and 1 atm for at least 100 ns for each intermediate conformation by NAMD 2(Phillips et al., 2020). After obtaining the trajectory of the NPT production run, the RMSD, root-mean-square-fluctuations (RMSF) corresponding to the reference structure, as well as the secondary structure profile were calculated with Gromacs. The secondary structure of the protein was calculated by DSSP (Joosten et al., 2011) and visualized with do_dssp in Gromacs.
2.4 Preparation of compounds dataset and database
To evaluate the descriptive power of the structural model, a small dataset containing 6 known KIT inhibitors, including imatinib, masitinib, bezuclastinib, avapritinib, BLU-263, and elenestinib was constructed according to literature(Akin et al., 2022). To identify the potential inhibitors against KIT with D816V mutation, compounds from the public database of available kinase inhibitors currently in clinical trials (from phase I to IV), PKIDB(Carles et al., 2018, Bournez et al., 2020), were retrieved for virtual screening. Then, the three-dimensional structure of compounds in the test dataset and PKIDB were generated with the Ligprep application in Schroginder Suit. The bond angles and bond ordering were assigned, and the minimization using the OPLS-2005 force field with default settings.
2.5 Molecular docking
The protein structure of c-Kit obtained from molecular dynamics simulation was processed with the Protein Preparation Wizard application of Schrödinger Suite (Schrödinger 2020). The ionization state of the protein was calculated by PROPKA application(Olsson et al., 2011), and the hydrogen atoms were added to optimize the hydrogen bonds. Finally, the optimized structure was parameterized with the OPLS-2005 (Optimized Potentials for Liquid Simulations) force field and the energy minimization was performed with default settings(Sastry et al., 2013). The Glide application of the Schrodinger suit was employed for molecular docking. The grid which centered on the DFG motif within the activation loop was generated with a default value was generated using the default parameter, and the binding free energy was predicted by SP (standard precision) mode in the Glide application.
2.6 In vitro cytotoxicity test
The HMC1.1 cells containing V560G in the juxtamembrane domain of KIT and imatinib-resistant HMC-1.2 cells containing V560G and D816V dual mutations were maintained in Iscove's modified Dulbecco's medium (Gibico, Guangzhou, China) supplemented with 10 % fetal calf serum (Gibico, Guangzhou, China) at 37 °C, 5 % CO2. After treatment with Vorolanib of the indicated concentration for 24 h, the cell viability was measured with the Cell Counting Kit − 8 assay according to the literature(Yang et al., 2021).
2.7 Statistical analysis
Pooled data were analyzed by GraphPad prism software and visualized as the mean ± SEM. Differences between each group were analyzed using one-way ANOVA followed by Tukey post-hoc test. Probabilities (p) less than 0.05 were considered to be significant.
3 Results and discussion
3.1 Secondary structure prediction revealed the presence of short α-helices adjacent to the DFG motif was one of the characteristics of the intermediate state
By comparing the crystal structure describing the kinase domain of c-Kit in autoinhibitory and activated states, we found a remarkable conformational switch of the A-loop. In its autoinhibited state, the A-loop adopted a “closed and folded” state, which characterized the presence of a short helix between C810 and G812, as well as a beta strand between D820 and L831(Mol et al., 2004). In the active state, the activation loop and DFG motif adopted an extended conformation, and the helix and strands were replaced by random coil(Mol et al., 2003) (As shown in Fig. 1A and Fig. 1B). As accumulating evidence demonstrated the activation of c-Kit kinase, as well as the member of SRC kinases required secondary structure changes of the activation loop(Ozkirimli and Post 2006), we hypothesize the metastable state of the c-Kit kinase domain might adopt a different secondary structure profile from identified crystal structures.
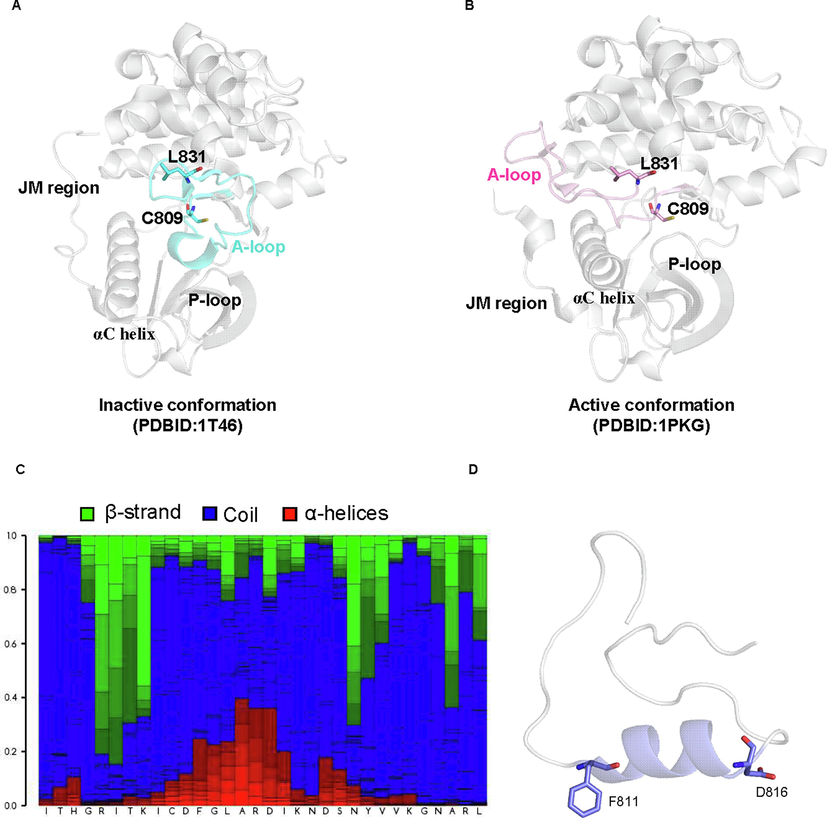
-
Secondary structure prediction revealed the presence of short α-helices adjacent to the DFG motif was one of the characteristics of the intermediate state. The crystal structure describing the KIT kinase in auto-inhibitory and activated conformation was shown in A and B, respectively. C. The results from the secondary structure prediction of residues in the activation loop, the presence of a-helices, b-strands, and the coil were colored red, blue, and green, respectively, while the representative structure was illustrated in D.
To obtain more information about the secondary structure profile about the possible metastable state, we performed a secondary structure prediction towards the activation loop. The results identified the presence of a β-sheet-containing structure within the D820 to L831, as well as a short β-strand located close to the beginning of the DFG motif within the kinase domain (Fig. 1C), which was in line with the available crystal structures. Interestingly, the secondary structure prediction revealed the residues within F811 to I817 adopted α-helices (colored in red) and random coil (colored in blue) with similar possibilities. However, the presence of short α-helices ranging from F811 to I817 (shown in Fig. 1D) was not observed in all available crystal structures. One the other hand, the presence of short α-helices was observed in the metastases found in many protein kinases, such as the anaplastic lymphoma kinase(Patil et al., 2021), Bruton tyrosine kinase(Sultan et al., 2017), Src kinase(Shukla et al., 2014) and Abl kinase(Paul et al., 2020). As a result, we consider the short helix within A-loop might be also a characteristics of metastable state in c-Kit enzyme, and we used it as a criterion for the identification of possible metastable states.
3.2 Ab initio folding and K-means clustering identified nine potential metastable conformations of c-Kit kinase, which showed remarkably different secondary structure profiles
Several computation methods were employed to study the dynamic properties of the kinase domain within c-Kit and identify the possible metastable state of c-Kit, and the results demonstrated the conformational alterations of residues within JM domain, P-loop, as well as the random coil located in N-lobe was closely related to the conformational switch between inactive and active state(Yang et al., 2009, Laine et al., 2011, Shukla et al., 2014, Paul et al., 2020). However, the detailed conformational information about the A-loop, which played an essential role in the activation process, was still limited. One of the possible reasons was the insufficient conformational sampling made by molecular dynamics simulation and enhanced sampling method(Del Alamo et al., 2022, Zhu et al., 2023). As a result, the PyRosetta package, which incorporated the Monte Carlo sampling technique was employed. Rosetta and PyRosetta were designed for de novo protein structure prediction, assembling fragments, which provided a promising platform to accomplish an extensive and sufficient sampling in the protein conformational space(Heilmann et al., 2020). Accordingly, Rosetta and PyRosetta were already widely used for the modeling and design of anti-bodies(Schoeder et al., 2021), enzymes(Bhattacharya and Cheng 2015), and other macromolecules(Leman et al., 2020).
To expand the conformational space of the activation loop within the kinase domain, and identify the structural mode of the intermediate, an ab inito folding-based structure predicting process was employed. As illustrated in Fig. 2A, the crystal structure describing the autoinhibitory and active state were employed as starting conformation, and subjected to ab initio folding with PyRosetta. After loop closure, side-chain packing, and energy minimization, the predicted conformation (decoys) was generated. To maximize the conformation pool, 5000 decoys for each starting structure were generated. Then, the RMSD of each decoy compared to the starting structure was calculated and plotted in Fig. 2B. Based on the result from the Silhouette analysis, K-means clustering of RMSD scatter identified nine clusters based on the RMSD plot, and the decoys belonging to each cluster were ranked based on their predicted energy. The top-scored poses bearing the lowest potential energy were selected as representatives, and visualized in Fig. 1C. Based on the conformation of the DFG motif, the identified metastable conformation could be subdivided into two different groups. In the first group (conformation a to d), the A-loop adopted a closed and folded structure. The β-sheet ranging from N822 to L831 was preserved, and the A-loop resided in the cleft between αC helices. Similar to the KIT kinase in its auto-inhibitory form, the DFG motif preserved its DFG-out conformation. While in conformation e to i, the DFG motif adopted a DFG-in conformation, which was similar to active KIT kinase. Besides, we noticed a remarkably different secondary structure profile within the predicted metastable intermediates. Based on the results from secondary structure prediction, we found that conformation b and conformation e, which possess the short α-helix within the A-loop were the structural model of potential metastable conformation.
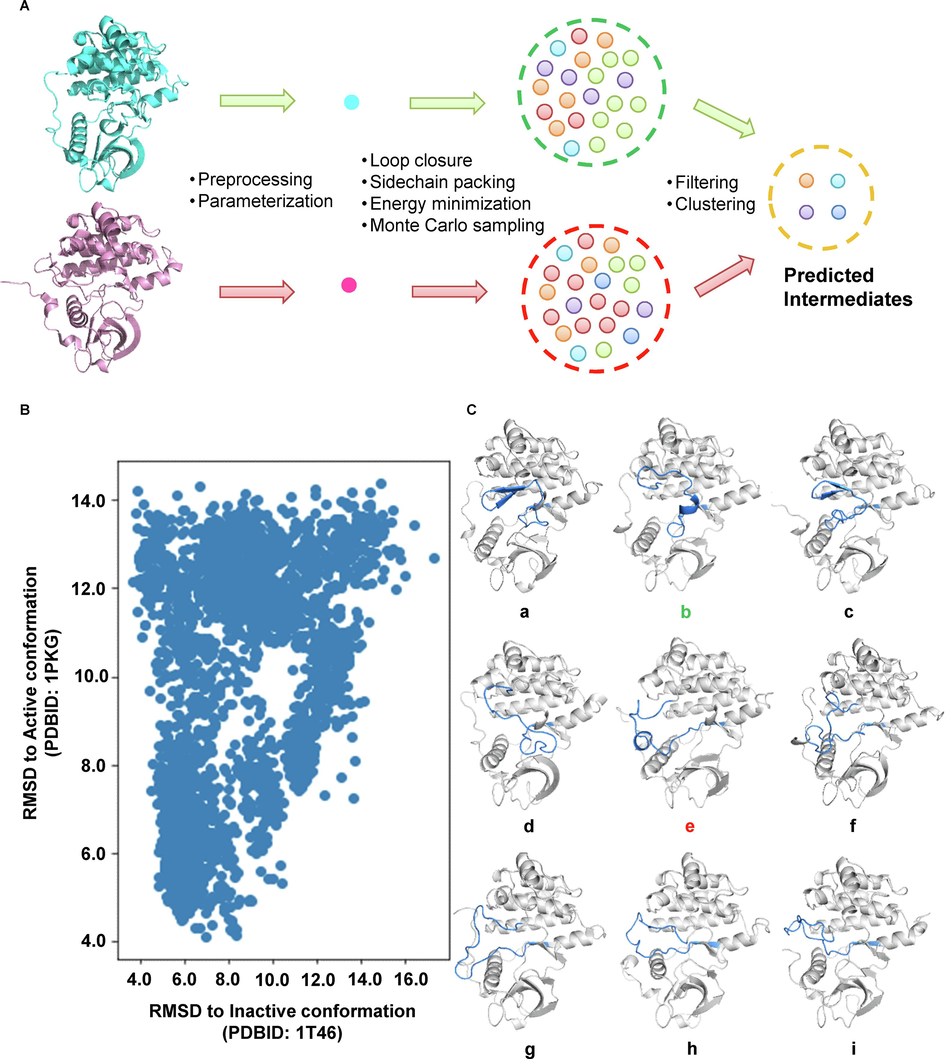
-
Ab initio folding and K-means clustering identified nine potential metastable conformations of c-Kit kinase, which showed remarkably different secondary structure profiles. A. The illustration of the intermediate generation process. The crystal structure of the kinase domain of c-Kit in the autoinhibitory state (PDBID: 1 T46, colored cyan) and active state (PDBID: 1PKG, colored pink) were used as the initial structure for ab inito folding. For each initial structure, 5000 decoys were generated. The RMSD of each decoy related to the initial structures was plotted in B, and the representative structure of each cluster was visualized with a cartoon in C..
A careful analysis of the identified metastable state revealed the unfolding of the A-loop happened before DFG-flip, indicating the unfolding of the A-loop was one of the pre-requests for DFG-flip and kinase activation. Besides the identified metastable states b and e which may possess higher stability, we also found four intermediate conformation f to the conformation i, which was similar to the “DFG-up”, “DFG-in” and “DFG-down” states identified in a metadynamics simulation(Jiang et al., 2022).
3.3 Molecular dynamic simulation identified one possible metastable state with promising thermal stability
In vitro and in silico studies demonstrated there were a variety of intermediates that existed between the active and inactive states, however, their thermal stability differed significantly(Laine et al., 2011). Accumulating evidence revealed that only the stable intermediate along the transition pathway may play an essential role in regulating the catalytic activity or molecular function(Kannan and Zacharias 2014, Ghosh and Ranjan 2020). The formation of stable intermediate was accompanied by the exposure of buried druggable pockets, which made them an attractive target in the discovery of novel kinase inhibitors (Umezawa and Kii 2021). Accordingly, the thermal stability of two potential intermediates was investigated with molecular dynamics simulations, and the secondary structure profile, as well as the RMSD of the A-loop referring to the initial structure, was measured. The higher stability of the secondary structure profile and the lower RMSD value indicated the increased thermal stability of the simulation system.
As shown in Fig. 3, the molecular dynamics simulation did not alter the overall secondary structure of KIT kinase, however, the α-helical structure of A-loop within the conformation b was gradually replaced with 3-helix (Fig. 3A, middle panel), indicating the helix was less stable. While, the α-helix within the conformation e was preserved during the simulation, which indicated the higher stability compared with conformation b (Fig. 3B, middle panel). On the other hand, we observed less variation in the RMSD plot at 80 to 100 ns within conformation e, further indicating the increased thermal stability of α-helix within the activation loop (Fig. 3A and B, lower panel).
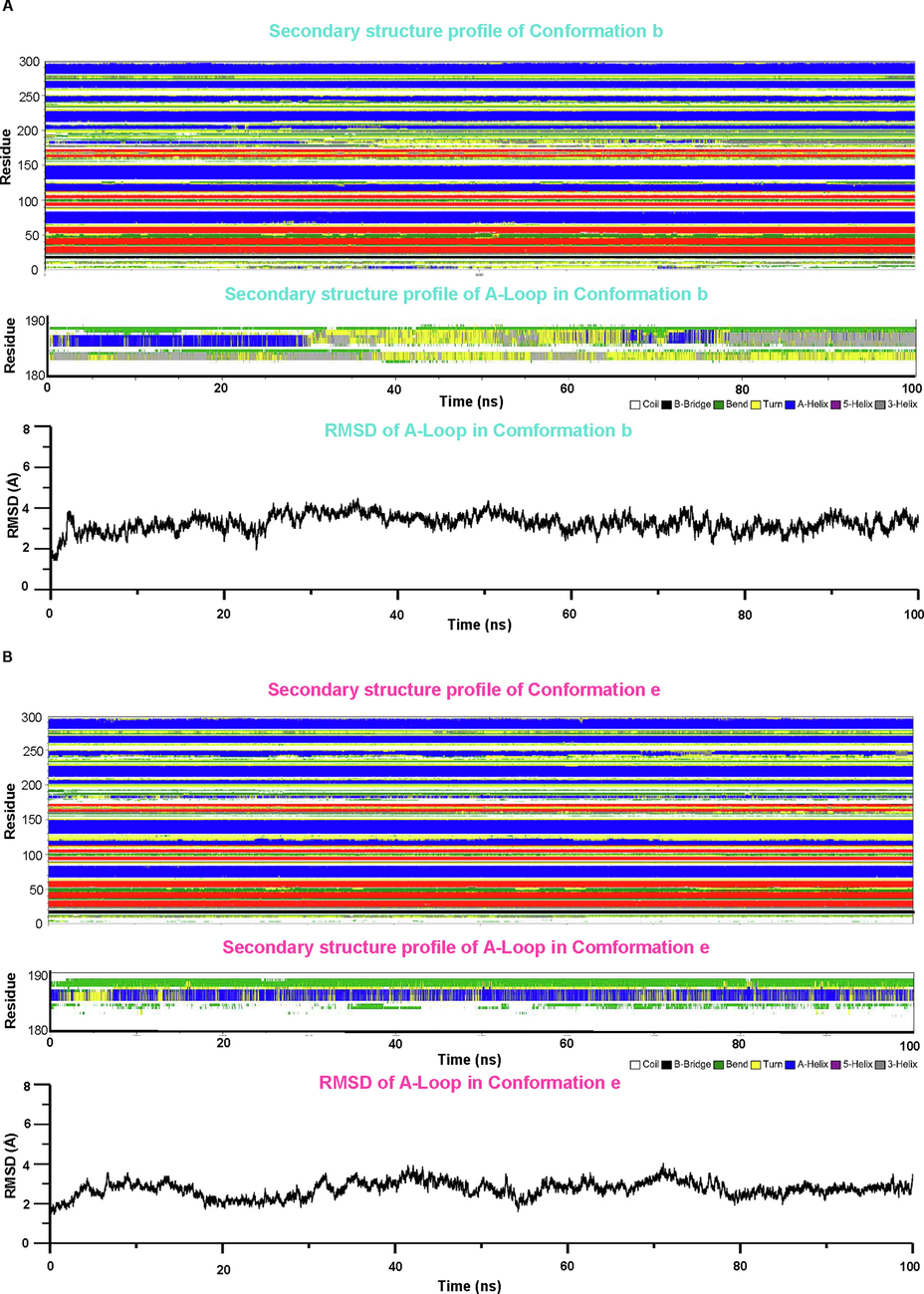
-
Molecular dynamic simulation identified one possible metastable state with promising thermal stability. The secondary structure profile of conformation b and conformation e was obtained from molecular dynamics simulation and was shown in the upper panels of A and B. The secondary structure of the A-loop (residues between 180 and 190) was plotted in the middle panel of A and B. The RMSD of the A-loop along with the simulation was shown in the lower panel of A and B.
In addition, the RMSF of two structural models was calculated. Comparing the RMSF profile of two structural model, we observed the overall RMSF, as well as the RMSF of A-loop (high-lighted in red frame), was smaller within confirmation e than conformation e (As shown in Fig. 4). Consequently, the conformation e was selected as metastable conformation for the following experiments.
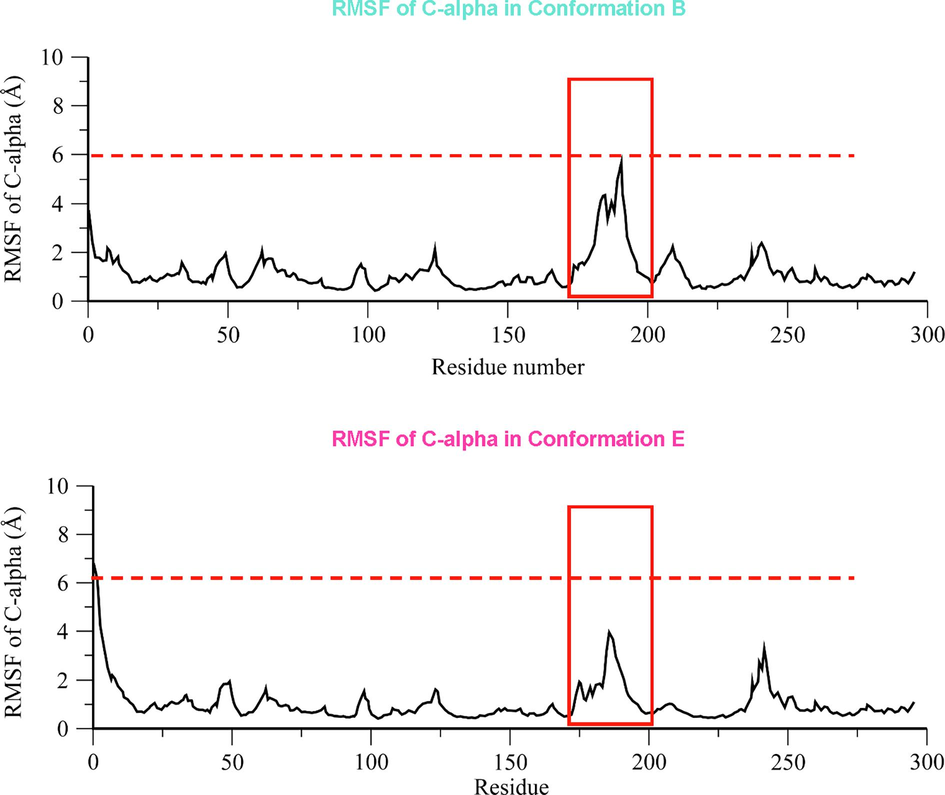
-
The RMSF profile of C-alpha in conformation b and conformation e. The RMSF profile of conformation b and conformation e was plotted in the upper and lower panel, respectively. The RMSF of C-alpha atom in A-loop was highlighted in red square.
3.4 The identified metastable structural model showed enhanced efficacy targeting TKIs which was effective against D816V
A careful examination of the identified metastable conformation revealed it possesses the structural characteristics of both autoinhibitory c-Kit kinase and active c-Kit kinase. Although the A-loop adopted a partially extended conformation, the DFG motif preserved an in conformation, indicating it may represented a semi-active conformations, which may existed in the mutated c-Kit. Accordingly, a small dataset containing inhibitors targeting wild-type KIT (imatinib and masitinib) and KIT D186V (bezuclastinib, avapritinib, BLU-263, and elenestinib) was constructed, and their binding affinity of identified metastable conformation was predicted by molecular docking in standard precision mode. Interestingly, imatinib and masitinib, two inhibitors that were not effective on D816V mutations showed higher binding affinity towards conformation b than conformation e, while the selective KIT D816V inhibitors showed higher binding affinities towards conformation e(Fig. 5A). The detailed analysis of the binding pocket and binding poses revealed that the partial unfolding of the A-loop evoked the conformational alterations in the sidechain of V603, L595, F811, L799, and Y672, and led to the formation of a hydrophobic pocket adjacent to the DFG motif. At the same time, the rearrangement of the A-loop also evoked the conformational change of charged residues, such as D677, D816, K623, and R796. On the one hand, the newly formed hydrophobic pocket could stabilize the hydrophobic scaffold of inhibitors targeting KIT D816 mutations. On the other hand, the alteration of positively charged K263 and R796 could impair the repulse between KIT and inhibitors, and further stabilize the selective KIT D816V inhibitors within the binding pocket (Fig. 5 B and C).
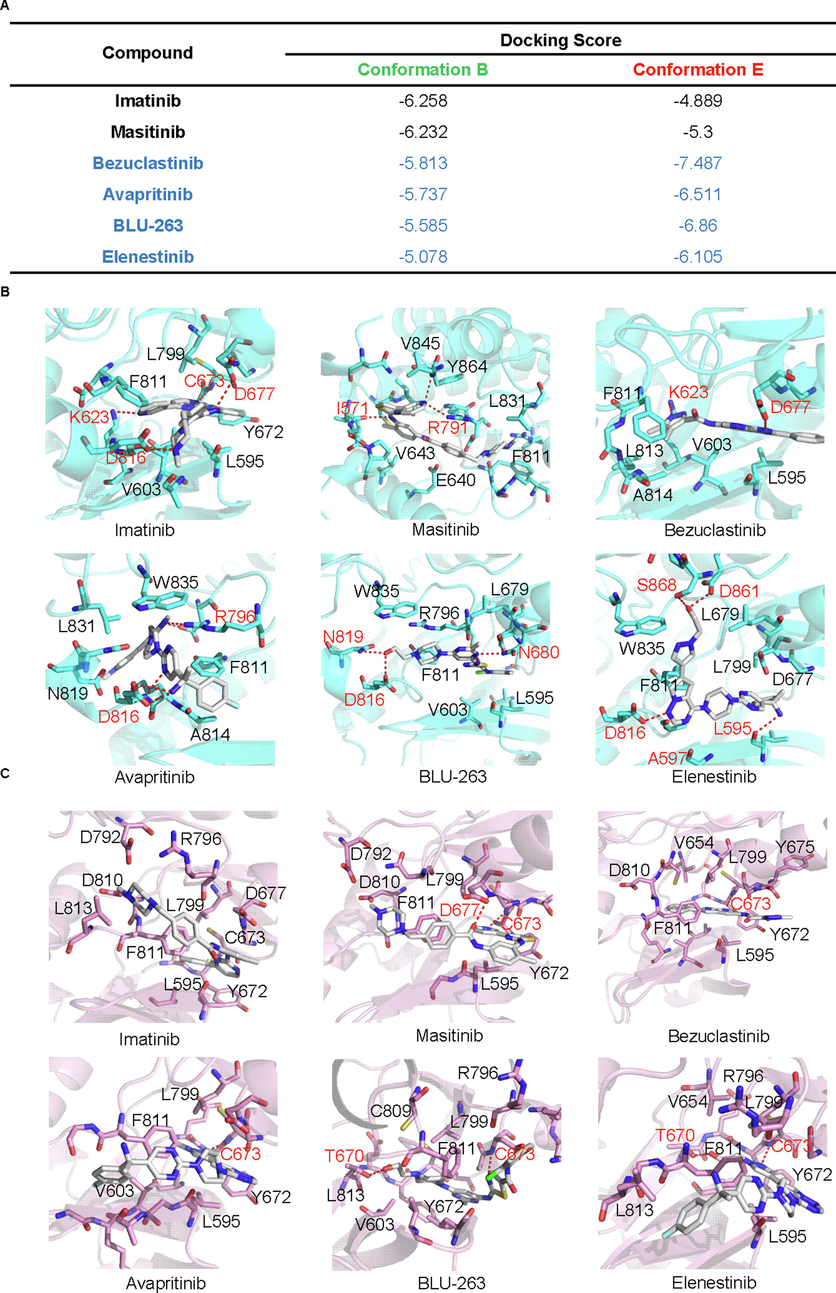
-
The identified metastable structural model showed enhanced efficacy in targeting TKIs which was effective against D816V. The predicted binding free energy of identified metastable states and selected TKIs was obtained from molecular docking (Glide in standard precision mode). The docking score of each TKI was shown in A, and the three-dimensional structure of the complex was shown in B and C. The carbon atoms in the ligand were colored in white. The carbon atoms within conformation b and conformation e were colored cyan and pink, respectively. The nitrogen, oxygen, and chloride atom was colored blue, red, and green, respectively.
3.5 Strucutre-based virtual screening identified vorolanib, semaxanib, henatinib, and pexmetinib may possess promising inhibitory effects on KIT D816V mutation
To identify the potential KIT D816V inhibitors from kinase inhibitors, a structure-based virtual screening process was performed, and for the compounds from PKIDB, a collection of kinase inhibitors was employed. The threshold of predicted binding free energy was set to −6.50 Kcal/mol, and we identified 114 inhibitors out of a total of 369 compounds. As shown in Supplementary Table 1, the majority of hit compounds belonged to the Tyr kinase family. Besides, we found our docking-based virtual screening protocol identified the majority of selective D816V inhibitors, such as toceranib, amuvatinib, avapritinib (predicted binding free energy: −7.537 Kcal/mol), and nilotinib (predicted binding free energy: 7.329 Kcal/mol) from available tyrosine kinase inhibitors. The ten top-scored compounds were listed in Table 1, and we found two out of ten selected compounds were selectively targeting D816V mutations. Consequently, we identified vorolanib, semaxanib, henatinib, and pexmetinib as potential inhibitors that target the D816V mutations.
| Compound | Identified Targets | Kinase families | docking score | Used as a KIT inhibitor | Sensitive to D816V |
|---|---|---|---|---|---|
| Vorolanib | Unknown | −9.647 | No | Unknown | |
| Toceranib | KIT | KIT | −9.606 | Yes | Yes(Liao et al., 2002) |
| Famitinib | KIT; KDR; FLT4; PDGFRA; PDGFRB; FLT1; FLT3 | Tyr | −9.344 | Yes | Unknown |
| Semaxanib | KDR | Tyr | −9.341 | Yes | Unknown |
| Orantinib | KDR; PDGFRB; FGFR1 | Tyr | −9.221 | No | Unknown |
| Henatinib | KDR | Tyr | −9.017 | No | Unknown |
| Pexmetinib | MAPK14; TEK | CMGC; Tyr | −8.749 | No | Unknown |
| Tanuxiciclib | Unknown | −8.321 | No | Unknown | |
| Amuvatinib | KIT; MET; RET; FLT3; PDGFRB | Tyr | −8.304 | Yes | Yes(Abdellateif et al., 2023) |
| Entospletinib | SYK | Tyr | −8.111 | No | Unknown |
3.6 In vitro validation of virtual screening
To evaluate the potential therapeutic effects of identified inhibitors, cell viability of the HMC1.1, and HMC1.2 cell lines, which possess D816V single mutation and D816V, V560G dual mutations was measured. As shown in Fig. 6, Vorolanib, the top-scored compound showed promising cytotoxicity against not only the HMC 1.1 cells, but also the HMC 1.2 cells. The results from the in vitro experiment further confirmed our prediction.
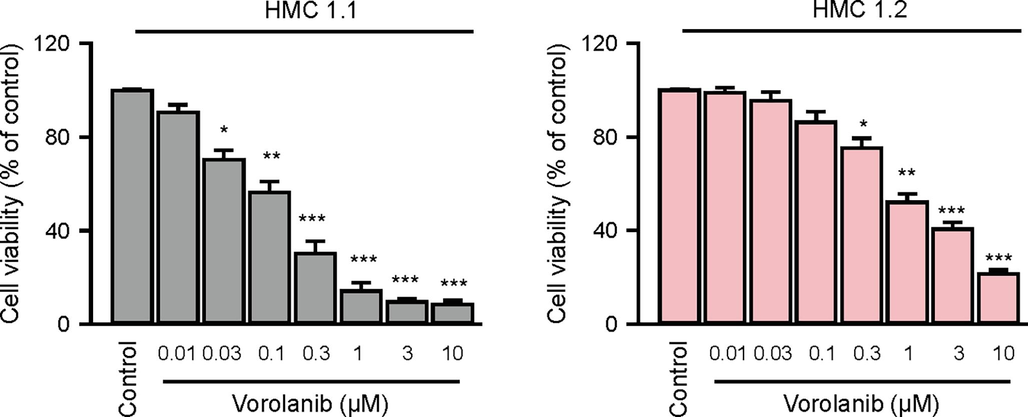
-
In vitro validation of virtual screening. The cytotoxic effects of effects of Vorolanib, the top-scored compound, on HMC1.1 and HMC1.2 cell line was measured by CCK-8 assay. The pooled data was visualized as mean ± S.E.M of four independent experiments. The one-way ANOVA and Tukey post-hoc test were employed for statistical analysis. *: <0.05, **: <0.01, ***: <0.001.
4 Conclusion
In summary, by employing secondary structure prediction and ab initio folding, we expanded the conformational space of the kinase domain of the c-Kit kinase, especially the conformational space of the activation loop. Besides, we identified a novel metastable conformation between the inactive and active state of the c-Kit kinase with high possibility, which was characterized by the presence of short α-helices within the activation loop. The molecular dynamics simulation and molecular docking studies demonstrated the metastable state identified from ab inito folding showed promising thermal stability and potent selectivity towards selective inhibitors targeting D816V mutations. The structure-based virtual screening identified vorolanib, semaxanib, henatinib, and pexmetinib, four kinase inhibitors that may possess promising inhibitory activity against c-Kit D816V mutations. Our study not only proposed a novel structure model for screening selective inhibitors against c-Kit D816V mutations but also identified four tyrosine kinase inhibitors that held the potential for the development of c-Kit inhibitors.
Funding
This study was supported by the National Natural Science Foundation of China (No. 82304804), the Guangdong Basic and Applied Basic Research Foundation (No. 2022A1515110734), and Fundamental Research Funds for the Central Universities, Sun Yat-sen University (No. 23ptpy38).
CRediT authorship contribution statement
Lu Jin: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Writing – original draft. Chunguo Qian: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. Zhao Wei: Data curation, Formal analysis, Methodology, Validation. Dongxu Zhang: Methodology, Validation, Visualization. Jiayue Xi: Data curation, Formal analysis, Investigation, Methodology, Validation. Dingkang Sun: Data curation, Formal analysis. Linke Fu: Data curation, Formal analysis, Methodology, Validation. Xueying Liu: Conceptualization, Data curation, Formal analysis, Software, Writing – review & editing. Xinlei Zhang: Conceptualization, Funding acquisition, Project administration, Validation, Writing – original draft, Writing – review & editing.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Receptor tyrosine kinase (c-Kit) inhibitors: A potential therapeutic target in cancer cells. Drug Des. Devel. Ther.. 2016;10:2443-2459.
- [CrossRef] [Google Scholar]
- c-Kit receptors as a therapeutic target in cancer: Current insights. Onco Targets Ther.. 2023;16:785-799.
- [CrossRef] [Google Scholar]
- The potential of c-KIT kinase inhibitors in cancer treatment. ACS Med. Chem. Lett.. 2021;12:1191-1192.
- [CrossRef] [Google Scholar]
- Tyrosine kinase inhibitors for the treatment of indolent systemic mastocytosis: Are we there yet? J. Allergy Clin. Immunol.. 2022;149:1912-1918.
- [CrossRef] [Google Scholar]
- De novo protein conformational sampling using a probabilistic graphical model. Sci. Rep.. 2015;5:16332.
- [CrossRef] [Google Scholar]
- Comparative assessment of protein kinase inhibitors in public databases and in PKIDB. Molecules. 2020;25
- [CrossRef] [Google Scholar]
- PKIDB: A curated, annotated and updated database of protein kinase inhibitors in clinical trials. Molecules. 2018;23
- [CrossRef] [Google Scholar]
- Finding the right KIT inhibitor for advanced systemic mastocytosis. Nat. Med.. 2021;27:2081-2082.
- [CrossRef] [Google Scholar]
- Current progress and open challenges for applying tyrosine kinase inhibitors in osteosarcoma. Cell Death Discov.. 2022;8:488.
- [CrossRef] [Google Scholar]
- Sampling alternative conformational states of transporters and receptors with AlphaFold2. Elife. 2022;11
- [CrossRef] [Google Scholar]
- The zero-multipole summation method for estimating electrostatic interactions in molecular dynamics: Analysis of the accuracy and application to liquid systems. J. Chem. Phys.. 2014;140:194307
- [CrossRef] [Google Scholar]
- Sampling of the conformational landscape of small proteins with Monte Carlo methods. Sci. Rep.. 2020;10:18211.
- [CrossRef] [Google Scholar]
- The conformational transition pathways and hidden intermediates in DFG-flip process of c-met kinase revealed by metadynamics simulations. J. Chem. Inf. Model.. 2022;62:3651-3663.
- [CrossRef] [Google Scholar]
- A series of PDB related databases for everyday needs. Nucleic Acids Res.. 2011;39:D411-D419.
- [CrossRef] [Google Scholar]
- Role of tryptophan side chain dynamics on the Trp-cage mini-protein folding studied by molecular dynamics simulations. PLoS One. 2014;9:e88383
- [CrossRef] [Google Scholar]
- Mutation D816V alters the internal structure and dynamics of c-KIT receptor cytoplasmic region: Implications for dimerization and activation mechanisms. PLoS Comput. Biol.. 2011;7:e1002068
- [CrossRef] [Google Scholar]
- Allosteric communication across the native and mutated KIT receptor tyrosine kinase. PLoS Comput. Biol.. 2012;8:e1002661
- [CrossRef] [Google Scholar]
- Receptor tyrosine kinase KIT: Mutation-induced conformational shift promotes alternative allosteric pockets. Kinases and Phosphatases. 2023;1:220-250.
- [CrossRef] [Google Scholar]
- Macromolecular modeling and design in Rosetta: Recent methods and frameworks. Nat. Methods. 2020;17:665-680.
- [CrossRef] [Google Scholar]
- Inhibition of constitutively active forms of mutant kit by multitargeted indolinone tyrosine kinase inhibitors. Blood. 2002;100:585-593.
- [CrossRef] [Google Scholar]
- ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput.. 2015;11:3696-3713.
- [CrossRef] [Google Scholar]
- Structure of a c-kit product complex reveals the basis for kinase transactivation. J. Biol. Chem.. 2003;278:31461-31464.
- [CrossRef] [Google Scholar]
- Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J. Biol. Chem.. 2004;279:31655-31663.
- [CrossRef] [Google Scholar]
- PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput.. 2011;7:525-537.
- [CrossRef] [Google Scholar]
- Src kinase activation: A switched electrostatic network. Protein Sci.. 2006;15:1051-1062.
- [CrossRef] [Google Scholar]
- A holistic view on c-Kit in cancer: Structure, signaling, pathophysiology and its inhibitors. Biochim. Biophys. Acta. 2021;1876:188631
- [CrossRef] [Google Scholar]
- Computational studies of anaplastic lymphoma kinase mutations reveal common mechanisms of oncogenic activation. PNAS. 2021;118
- [CrossRef] [Google Scholar]
- Diversity of long-lived intermediates along the binding pathway of imatinib to Abl kinase revealed by MD simulations. J. Chem. Theory Comput.. 2020;16:7852-7865.
- [CrossRef] [Google Scholar]
- Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys.. 2020;153:044130
- [CrossRef] [Google Scholar]
- Development of highly sensitive digital droplet PCR for detection of cKIT mutations in circulating free DNA that mediate resistance to TKI treatment for Gastrointestinal Stromal Tumor (GIST) Int. J. Mol. Sci.. 2023;24
- [CrossRef] [Google Scholar]
- PEP-FOLD4: a pH-dependent force field for peptide structure prediction in aqueous solution. Nucleic Acids Res.. 2023;51:W432-W437.
- [CrossRef] [Google Scholar]
- Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des.. 2013;27:221-234.
- [CrossRef] [Google Scholar]
- Modeling immunity with rosetta: Methods for antibody and antigen design. Biochemistry. 2021;60:825-846.
- [CrossRef] [Google Scholar]
- Role and significance of c-KIT receptor tyrosine kinase in cancer: A review. Bosn. J. Basic Med. Sci.. 2022;22:683-698.
- [CrossRef] [Google Scholar]
- Activation pathway of Src kinase reveals intermediate states as targets for drug design. Nat. Commun.. 2014;5:3397.
- [CrossRef] [Google Scholar]
- Millisecond dynamics of BTK reveal kinome-wide conformational plasticity within the apo kinase domain. Sci. Rep.. 2017;7:15604.
- [CrossRef] [Google Scholar]
- Towards simple kinetic models of functional dynamics for a kinase subfamily. Nat. Chem.. 2018;10:903-909.
- [CrossRef] [Google Scholar]
- Thomas, T. and B. Roux, 2021. Tyrosine kinases: complex molecular systems challenging computational methodologies. European Physical Journal B. 94, doi: ARTN 203 10.1140/epjb/s10051-021-00207-7.
- How electrostatic coupling enables conformational plasticity in a tyrosine kinase. J. Am. Chem. Soc.. 2019;141:15092-15101.
- [CrossRef] [Google Scholar]
- A simple colorimetric method for viable bacteria detection based on cell counting Kit-8. Anal. Methods. 2021;13:5211-5215.
- [CrossRef] [Google Scholar]
- Steered molecular dynamics simulations reveal the likelier dissociation pathway of imatinib from its targeting kinases c-Kit and Abl. PLoS One. 2009;4:e8470.
- [CrossRef] [Google Scholar]
- Enhancing conformational sampling for intrinsically disordered and ordered proteins by variational autoencoder. Int. J. Mol. Sci.. 2023;24
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2024.105979.
Appendix A
Supplementary material
The following are the Supplementary data to this article:




