

Translate this page into:
Investigating disturbances of the core material system in the lung-gut axis of COPD based on the transcriptomics-metabolomics-microbiomics integration strategy
⁎Corresponding authors. linsong0228@163.com (Song Lin), yanlinqqhr@aliyun.com (Yan Lin)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
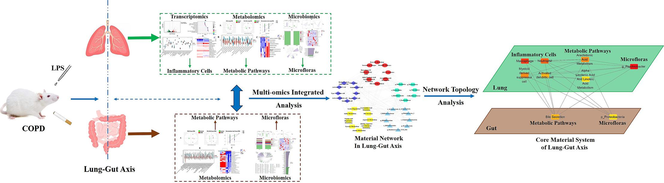
The COPDpathogenesis was explored based on the lung-gut axis about the interactions between the host and the microbes. The core material system in the lung-gut axis was analyzed by integrating transcriptomics-metabolomics-microbiomics. The inflammatory cells, including neutrophil etc., have undergone significant changes in COPD rats. The disorders of pulmonary arachidonic acid metabolism and fecal bile acid metabolism might markedly contribute to COPD. The altered of microbiota communities for COPD involved f_Pasteurellaceae and g_Ruminococcus_2, etc. in lung and gut
Abstract
Background
Although still the significance of the lung-gut axis for COPD is increasingly highlighted, it’s urgent to ulteriorly comprehend the sophisticated disturbance of the core material system along the lung-gut axis, which is of great importance for the accurate precaution and prognosis of COPD efficiently.
Aim of the study
The purpose of this study was to analyze the information connections of the lung-gut axis, thus supporting the effective treatment of COPD.
Materials and methods
An integrated multi-omics approach was applied to explore the lung-gut axis in COPD rats. Firstly, based on transcriptomics, the ssGSEA algorithm was used to evaluate changes in pulmonary inflammatory cells. Then, the disturbances of metabolic pathways in lung and feces were revealed by the Lilikoi algorithm using LC-MS and 1H NMR metabolomics. Next, the composition and function of microbial communities in lung and feces were analyzed by 16 s rRNA sequencing. Finally, the association analysis was employed to explore the possible crosstalk between the lung and gut. Furthermore, the core material system in the lung-gut axis was described based on network topology analysis.
Result
Firstly, 1652 differential expression genes (involving in immune response-regulating signaling pathway, etc.) and 15 types of inflammatory cells (including neutrophil, etc.) were identified related to COPD. 135 pulmonary differential metabolites (involving in arachidonic acid metabolism, etc.) and 105 fecal differential metabolites (involving in alanine metabolism, etc.) were revealed by metabolomics. The f_Pasteurellaceae, etc. and g_Ruminococcus_2, etc. were identified associated with COPD in lung and gut. Finally, disturbances of the core material system, composed of macrophage, neutrophil, activated dendritic cell, myeloid derived suppressor cell, arachidonic acid metabolism, alpha linolenic acid & linoleic acid metabolism, g_Psychrobacter in lung and bile secretion, p_Proteobacteria in gut, were obtained to analyze the possible information flow of the lung-gut axis.
Conclusion
The core material system for the lung-gut axis have been revealed, which might contribute to the illustration of the pathogenesis of COPD. In the future, more researches are required on the impact of the core material system in the lung-gut axis on the onset and recovery process of COPD, suggesting more precise identifying effective treatments for the disease.
Keywords
Transcriptomics
Metabolomics
Microbiomics
COPD
Lung-gut axis

1 Introduction
Chronic obstructive pulmonary disease (COPD) was the fourth leading cause of death globally, and it had influenced the health and life of 400 million people, defined as a common chronic and heterogeneous disease accompanied by persistent inflammation of the lung and airway remodeling (Christenson et al., 2022, Song et al., 2023). The characteristic pathological features of COPD were pulmonary interstitial edema, numerous inflammatory cell infiltration, pulmonary bullae and so on. And the pathogenesis of COPD was very complex and speculated to be the result of a combination of internal (such as inheritance and airway hyperresponsiveness) and external (such as air pollutants and smoking) factors (Christenson et al., 2022, Jiao et al., 2022). The commonly used anti-COPD drugs, for instance glucocorticoid and bronchodilator, could not effectively improve the downward trend of lung function. Furthermore, these drugs often led to serious complications and adverse reactions (Rabe and Watz 2017). In order to ameliorate current management strategies, the pathogenesis of COPD urgently needed to be deeply explored for developing effective prevention and treatment theories.
Recently, the critical role of the lung-gut axis, representing the interplay between the lung and the gut indicated by an increasing number of studies, in the pathology of COPD, is gradually becoming more and more spotlighted (Budden et al., 2016, Du et al., 2023). Although the lung and gut had different biological environments and physiological functions, they had similarities in mucosal structure and embryonic origin, and were the important parts of common mucosal immune system (CMIS) (Budden et al., 2016). Furthermore, the material network composed of inflammatory cells, microbiota and metabolic pathways was considered to have significant contributions to the information streams for the lung-gut axis (Wang et al., 2023). The inflammatory cells of CMIS could migrate between the lung and intestines, thereby affecting tissue homeostasis. Besides, some inflammatory cells participated in the mucosal barrier, which had a significant impact in resisting pathogens and repairing tissues (Huang et al., 2018, Wang et al., 2023). Moreover, during COPD, the disruption of mucosal environment and dysfunction of immune barrier could be caused by microbiota dysbiosis in both lung and gut, ultimately making a negative impact on the whole system (Budden et al., 2016, Song et al., 2023). And microbial translocation between lung and intestine seemed to play a positive role in restoring the gut-lung axis of COPD patients (Raftery et al., 2020, Wang et al., 2023). Meanwhile, it was noteworthy that some metabolites did have the ability to affect the activation, recruitment, and migration of inflammatory cells, thus ameliorating clinical symptoms. Besides, metabolites might also regulate the lung-gut axis by influencing the function of the microbiota (Budden et al., 2016, Corrêa et al., 2022). Although there had been some studies about the effects of microfloras, metabolic pathways or inflammatory cells on COPD, the complicated disturbances of the material network, especially the core material system, of the lung-gut axis in COPD, caused by the interactions between the host and the microbes, need to be systematically elucidated in depth, contributing to further understand the pathological mechanisms of COPD.
Hence, in this study, a systematic paradigm with the application of transcriptomics, metabolomics (by LC/MS and 1H NMR) and microbiomics along the lung-gut axis was established to investigate the pathogenesis of COPD. First, the significant biomarkers at the gene/metabolite/microflora level on the lung-gut axis were revealed by transcriptomics/metabolomics/microbiomics. Next, the changes of inflammatory cells, metabolic pathways and microbiota compositions were further evaluated. Then, the material network, composed of inflammatory cells, metabolic pathways and microfloras, along the lung-gut axis was explored based on multiple omics correlation analysis. Finally, topology analysis algorithm was adopted to analyze disturbances of the core material system in the lung-gut axis of COPD. All in all, our work attempted to give some light on the explanation of the impact of host − microbe interactions on the COPD pathogenesis, so that assist in the effective prevention and treatment of the disease.
2 Materials and methods
2.1 Reagents and chemical
HPLC (High Performance Liquid Chromatography) grade acetonitrile were offered by Fisher Scientific (Los Angeles, CA, USA). Distilled water was obtained from Watsons (Hangzhou, China). HPLC grade formic acid and methanol were from Shandong Yuwang Industrial Co., Ltd. (Yucheng, China). Deuterium oxide (D2O, 99.9 %) and LPS (lipopolysaccharide, Escherichia coli 055:B5) were provided by Sigma-Aldrich (Dorset, U.K.). Hongmei cigarettes (10 mg of tar and 0.8 mg of nicotine per cigarette) were supplied by Hongta Tobacco Group Company Limited (Yuxi, China). H&E Stain Kit was offered by Solarbio (Beijing, China). All oxidative stress indicator testing kits were obtained from the Nanjing Jiancheng Bioengineering Institute (Jiangsu, China). Enzyme-linked immunosorbent assay (ELISA) kits were offered by Jianglai Industrial Limited By Share Ltd. (Shanghai, China).
Total RNA extraction of rat lung tissues, reverse transcription of cDNA and qRT-PCR (Quantitative real-time PCR) were performed by TransZol Up reagent, EasyScript® One-Step gDNA Removal & cDNA Synthesis SuperMix kit and PerfectStart® Fast Green qPCR SuperMix (TransGen Biotech, Beijing, China).
2.2 Animals
A total of 16 male Wistar rats (220 to 250 g) were normally raised with temperature and humidity at 22 ± 2°C and 45 ± 15 % by Qiqihar Medical University Experimental Animal Center (Qiqihar, China), in a specialized pathogen free standard manner, under a natural light–dark cycle. The authorization of all animal operations was granted by the Ethics Committee of Qiqihar Medical University (QMU-AECC-2022–57). All rats were acclimatized for 7 days prior to experiment. Then, 16 rats were randomly divided into 2 groups, with eight animals each. Referring to previous research, COPD rats model was established (Weng et al., 2019). In brief, except for the 1st and 14th day, COPD group (8 rats) was exposed to the smoke of 8 cigarettes for 6 weeks (1 h each time, twice a day) in a self-produced fumigating box for smoking, and the concentration of smog was about 18 % (v/v) in the box. In addition, COPD group was treated with 200 μg/rat LPS intratracheally in the 1st and 14th day. Control group (8 rats) was not given fumigation. Finally, all rats were sacrificed on 12 h after the last administration to collect their lungs, BALF and intestinal contents. A schematic with the study design and other details are presented in Supplemental material.
2.3 Pathological evaluation of the lung
Based on our previous research, H&E staining, weight/dry (W/D) ratio, white blood cell (WBC) count and protein concentration in bronchoalveolar lavage fluid (BALF), as well as, the levels IL-1β, IL-6 and TNF-α and the activity of GSH, MDA, SOD in lung and BALF were adopted for pathological evaluation of the lung (Wang et al., 2020). Briefly, after washed with cold PBS, the upper or middle lobe of the right lung was used for H&E staining or W/D ratio. Supernatants/pelleted cells of BALF were used for the kit testing/Wright-Giemsa staining.
2.4 Transcriptomic research
Transcriptomic analysis was entrusted to Shanghai Bioprofile Technology CO., Ltd (Shanghai, China) for implementation. In brief, after RNA extraction and library construction, Agilent 2100 Bioanalyzer (Agilent Technologies Co. Ltd., USA) and Illumina Novaseq 6000 platform (Illumina, Inc., USA) were implemented on RNA quality assessment and sequencing. Gene expression profiles were analyzed by the limma package of R platform (Ritchie et al., 2015), and differentially expressed genes (DEGs) were recommended based on adjusted p (Benjamini-Hochberg false discovery rate adjusted p) < 0.05 and |log2FC| > 2. Then, Metascape (https://metascape.org) was used to perform enrichment analysis (Zhou et al., 2019). Based on 28 inflammatory cell subpopulations obtained from previous studies, the inflammatory cell situation of lungs was evaluated by ssGSEA which could be used to calculate the relative abundance of different inflammatory cells in each sample by comparing the gene expression data of each sample with a specific set of inflammatory cell genes (Barbie et al., 2009, Charoentong et al., 2017).
2.5 Metabolomic research
According to our previous research, sample preparation, sample analysis, data processing and metabolites identification were performed (Wang et al., 2022, Lin et al., 2023). In brief, precipitation protein operation was used for LC-MS metabolomics. TripleTOF™ 4600 (Sciex, Foster City, CA, USA) was used to analyze lung/BALF/feces samples in both negative and positive ionization modes. XCMS package of R platform and SIMCA-P (version14.0, Umetrics, Umea, Sweden) were employed for processing and analysing LC-MS metabolomic data (Benton et al., 2008). On the other hand, 1H NMR metabolomic data were collected by Bruker 500 MHz spectrometer (Bruker BioSpin, Karlsruhe, Germany), and data processing and metabolites identification were implemented by ASICS package of R platform (Lefort et al., 2021). The changes of metabolic pathways were evaluated by Lilikoi package (Fang et al., 2021). The remaining parameters were shown in Supplemental material.
2.6 Microbiomic research
Intestinal contents were transferred into sterile centrifuge tubes, rapidly stored in liquid nitrogen, and then transferred to −80 °C. Then, 16S rRNA sequencing analysis was executed by Shanghai Bioprofile Technology CO., Ltd (Shanghai, China). Briefly, microbial DNA was extracted from samples of BALF or intestinal contents following the manufacturer’s instructions. The NanoDrop 2000 (Thermo Scientific, USA) was used to detect final DNA concentration. V3–V4 regions of the 16S rRNA gene were amplified with degenerate PCR primers, 338F (5′-ACTCCTACGGGAGGCAGCA-3′) and 806 R (5′-GGACTACHVGGGTWTCTAAT-3′). The validated libraries were used to sequence on the Illumina NovaSeqPE250 platform (San Diego, USA), and QIIME 2 (version 2019.4) was used to analyze data.
2.7 RT-qPCR
RNA expression was measured by Quant Studio 3 PCR System (Thermo Fisher, USA). Then, using the comparative 2−ΔΔCT method, the relative expression level of the target genes was calculated. GADPH gene transcripts (Sangon Biotech, Shanghai, China) were used as the reference. The relevant primer sequences and the details of western blotting were shown in the Supplemental material.
2.8 Statistical analysis
Correlation analysis was used to explore the correlation among multiomics data. Cytoscape was used to visualize and analyze the material network of the lung-gut axis (Otasek et al., 2019). Statistical significances were calculated using Student’s t test. The p < 0.05 was adopted as the statistical significance threshold.
3 Results
3.1 Histopathological changes in COPD rats
H&E staining result indicated that, compared to the control group, inflammatory cell infiltration and airway wall thickness were significantly deteriorated in COPD group, further more capillary congestion and interstitial edema were obviously observed in COPD group, which suggested the evident morphological alterations in the lung tissues of COPD group (Fig. 1A). Similarly, the counts of WBC, neutrophils (NEU), lymphocyte (LY), monocytes (MON), protein concentration in BALF and W/D ratio of COPD group were obviously higher than those of control group (Fig. 1B). Besides, significant differences in inflammatory cytokines and oxidative stress indicators in lung and BALF, such as IL-1β, IL-6, TNF-α and SOD, MDA, GSH (Fig. 1C and D), were also observed in COPD group, showing abnormal changes of pulmonary permeability and inflammation in COPD rats. The above results indicated that there were significant histopathological changes in the COPD group, expressing COPD model had been successfully replicated.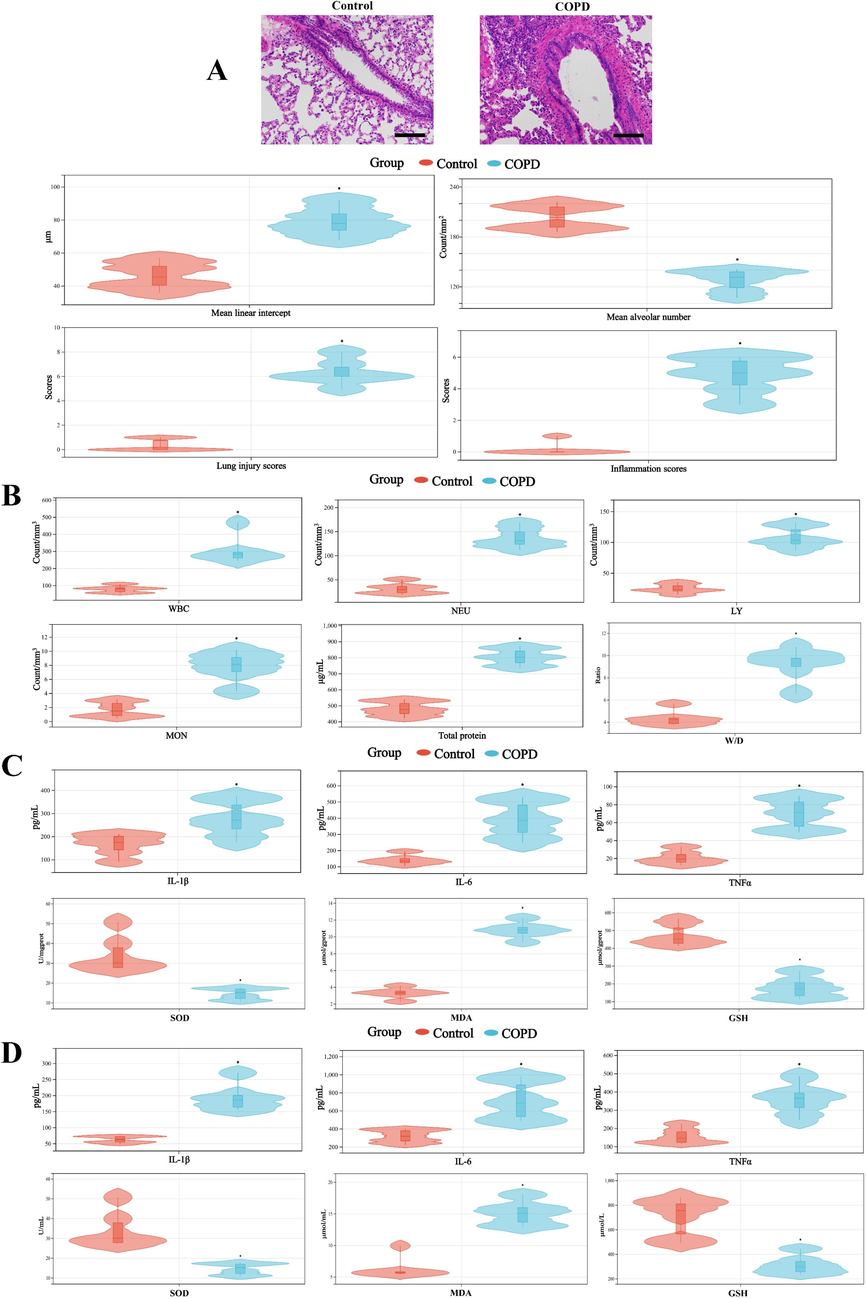
A, Morphological changes of lung (100 × magnification) and the quantitative analysis of the lung pathological staining result. B, The counts of WBC, NEU, LY, MON, protein concentration in BALF and W/D ratio. The levels of IL-1β, IL-6, TNFα and the activity of SOD, MDA, GSH in lung (C) and BALF (D). *, p < 0.05, compared with control group (n = 6).
3.2 The results of pulmonary transcriptomic research
Based on the aforementioned threshold, 1652 DEGs were screened out (Fig. 2A and Table S1). Moreover, RT-qPCR result also illustrated the abnormal expression of representative DEGs, such as IL-1β, PTGS2, NLRP3 and so on (Fig. S1). Then, enrichment analysis was adopted to explore the biological significance of the above DEGs, and top 10 terms were drawn in Fig. 2B. The result showed that some terms concern with immune response, such as immune response-regulating signaling pathway, leukocyte migration, leukocyte activation and so on, were closely related to COPD, which indicated a close correlation between the immune microenvironment and the pathogenesis of COPD. Accordingly, to further evaluate the changes in the pulmonary immune microenvironment of COPD, 28 types of inflammatory cells situation were investigated by ssGSEA (Fig. 2C). Furthermore, the results confirmed the significant changes in the COPD pulmonary 15 types of inflammatory cells, including neutrophil, Type 17 T helper cell, macrophage, ect. (Fig. 2D).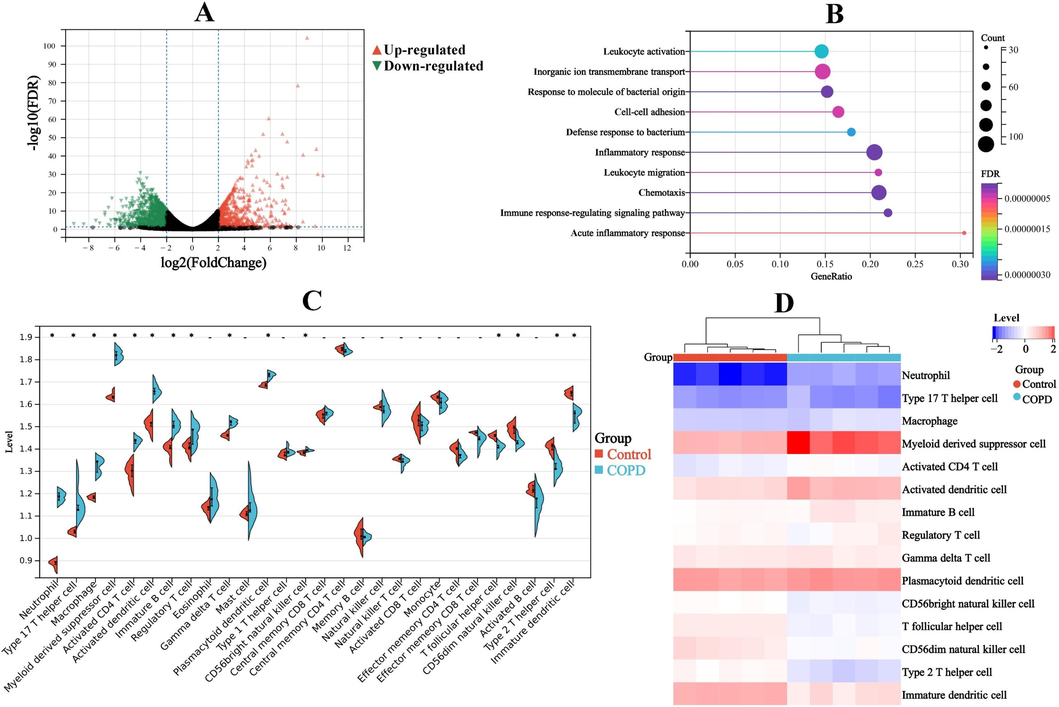
A, Volcano plot. B, Enrichment analysis of top 10 terms. C, Expression levels of 28 types of inflammatory cells. D, 15 types of inflammatory cells with significant changes. *, p < 0.05, compared with control group.
3.3 The results of pulmonary and fecal metabolomic research
Based on LC-MS and 1H NMR analytical platforms, the metabolic profiles of lung and intestinal contents were considered. On the one hand, as shown in Fig. 3A and Fig. S2A, the PCA and PLS-DA of pulmonary metabolomic data performed apparent separation between control and COPD groups, and the permutation test suggested a low risk of overfitting, implying COPD group was noticeable different from control group in terms of metabolic pattern. Then, potential candidates were screened based on the threshold (VIP > 1, p < 0.05 and fold change > 2). Finally, 135 differential metabolites (DMs) were obtained in pulmonary metabolomics (Table S6). Heatmap analysis of 135 pulmonary DMs were drawn, and the results indicated that remarkable differences in 135 pulmonary DMs between control and COPD group (Fig. S2A). Next, pathway analysis showed that 135 pulmonary DMs mainly involved 17 metabolic pathways, including lipid metabolism (such as arachidonic acid metabolism, glycerophospholipid metabolism, phospholipid biosynthesis, etc.), ABC transporters and so on (Fig. 3B). In addition, further analysis suggested that 11 of the above metabolic pathways had undergone significant changes (Fig. 3C). On the other hand, 105 DMs were obtained in fecal metabolomic research (Table S6, Fig. 4A and Fig. S2B). And, 38 pathways (significant changes in 33 pathways), such as amino acid metabolism (alanine metabolism, tyrosine metabolism, etc.) and bile acid metabolism (bile secretion, bile acid biosynthesis, etc.), were calculated by pathway analysis of 105 fecal DMs (Fig. 4B and C).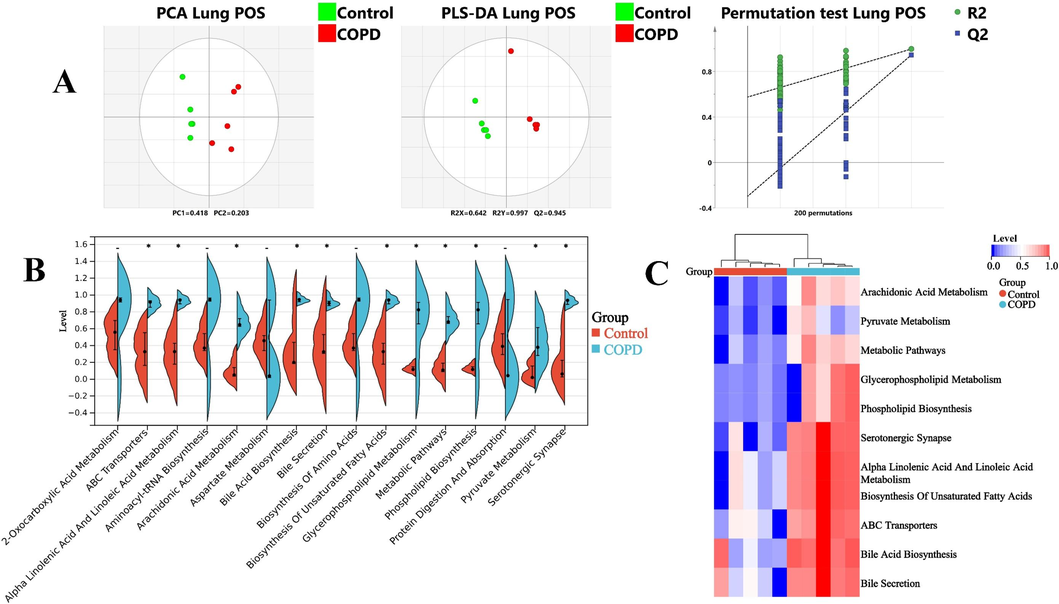
A, LC-MS metabolomic analysis of lung in positive mode: PCA, PLS-DA and permutation test. B, Expression levels of 17 pathways in lung. C, 11 pathways with significant changes in lung. *, p < 0.05, compared with control group.
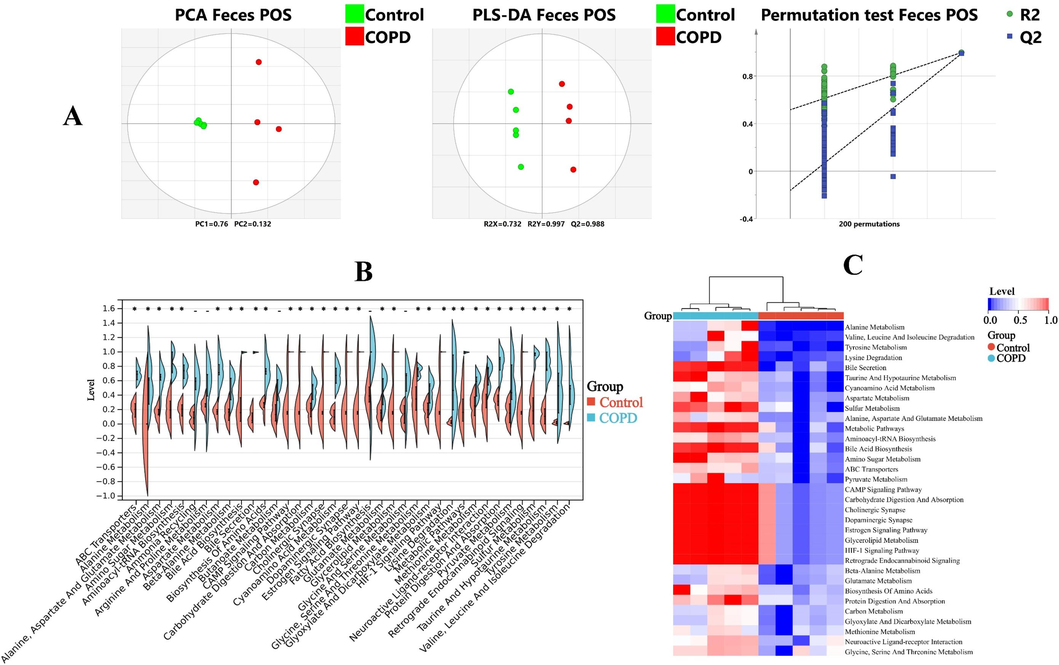
A, LC-MS metabolomic analysis of feces in positive mode: PCA, PLS-DA and permutation test. B, Expression levels of 38 pathways in feces. C, 33 pathways with significant changes in feces. *, p < 0.05, compared with control group.
3.4 The results of pulmonary and fecal microbiomic research
As shown in Fig. 5A, α diversity indexes of pulmonary microbiomics, for instance, Shannon and Simpson indices showed significant changes. Furthermore, NMDS and PCoA analysis also illustrated the significant differences of pulmonary microflora in control and COPD group, which declared the dissimilarity of β diversity (Fig. 5B). And further relative abundance analysis manifested that the microbiota compositions of COPD lung were significantly altered in various levels, for example, at genus level, Sphingomonas, Acinetobacter and so on were significantly increased in COPD group, however Rothia, Enhydrobacter and so on were markedly reduced in COPD group (Fig. 5C and D). Then, linear discriminant analysis (LDA) and LDA effect size (LEfSe) were used to evaluate the representative microflora of lung at different taxa level, and f_Pasteurellaceae (Pasteurellaceae family), o_Pasteurellales (Pasteurellaceae order), g_Rodentibacter (Rodentibacter genus) and so on exhibited the sensitivity to COPD (Fig. 5E). Similarly, in regard to fecal microbiomics, there were also differences of α and β diversity in control and COPD group (Fig. 6A and B). The significantly increasing microbiota in COPD group were g_Bifidobacterium (Bifidobacterium genus), g_Alloprevotella (Alloprevotella genus), etc., and the markedly reducing microbiota in COPD group were g_Turicibacter (Turicibacter genus), g_Romboutsia (Romboutsia genus), etc. (Fig. 6C and D). Additionally, g_Ruminococcus_2 (Ruminococcus_2 genus), p_Actinobacteria (Actinobacteria phylum), etc. were filtered as the microflora associated with COPD (Fig. 6E).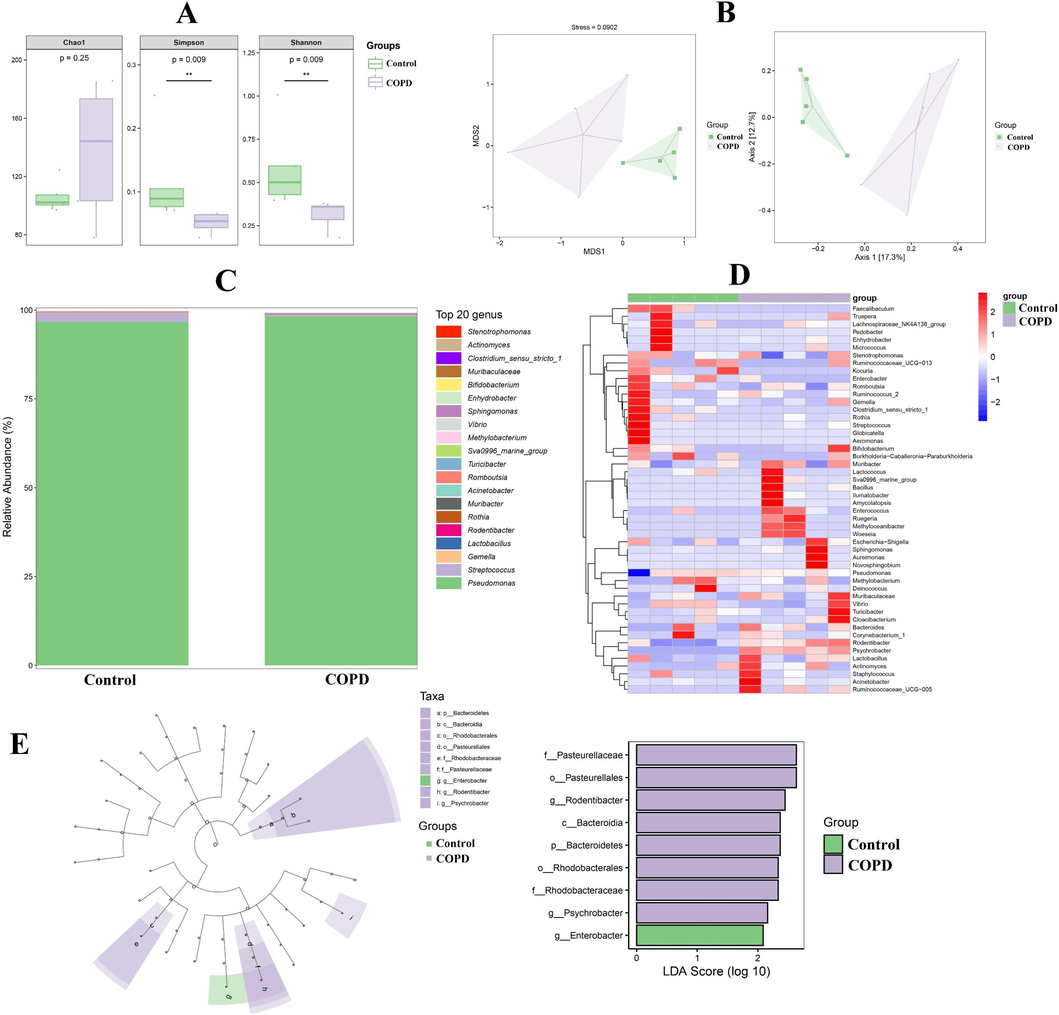
The results of pulmonary microbiomic research. A and B, Alpha and beta diversity analysis. C, Relative abundance of top 20 microfloras at genus level. D, Heatmap of microfloras at genus level. E, LDA and LEfSe.
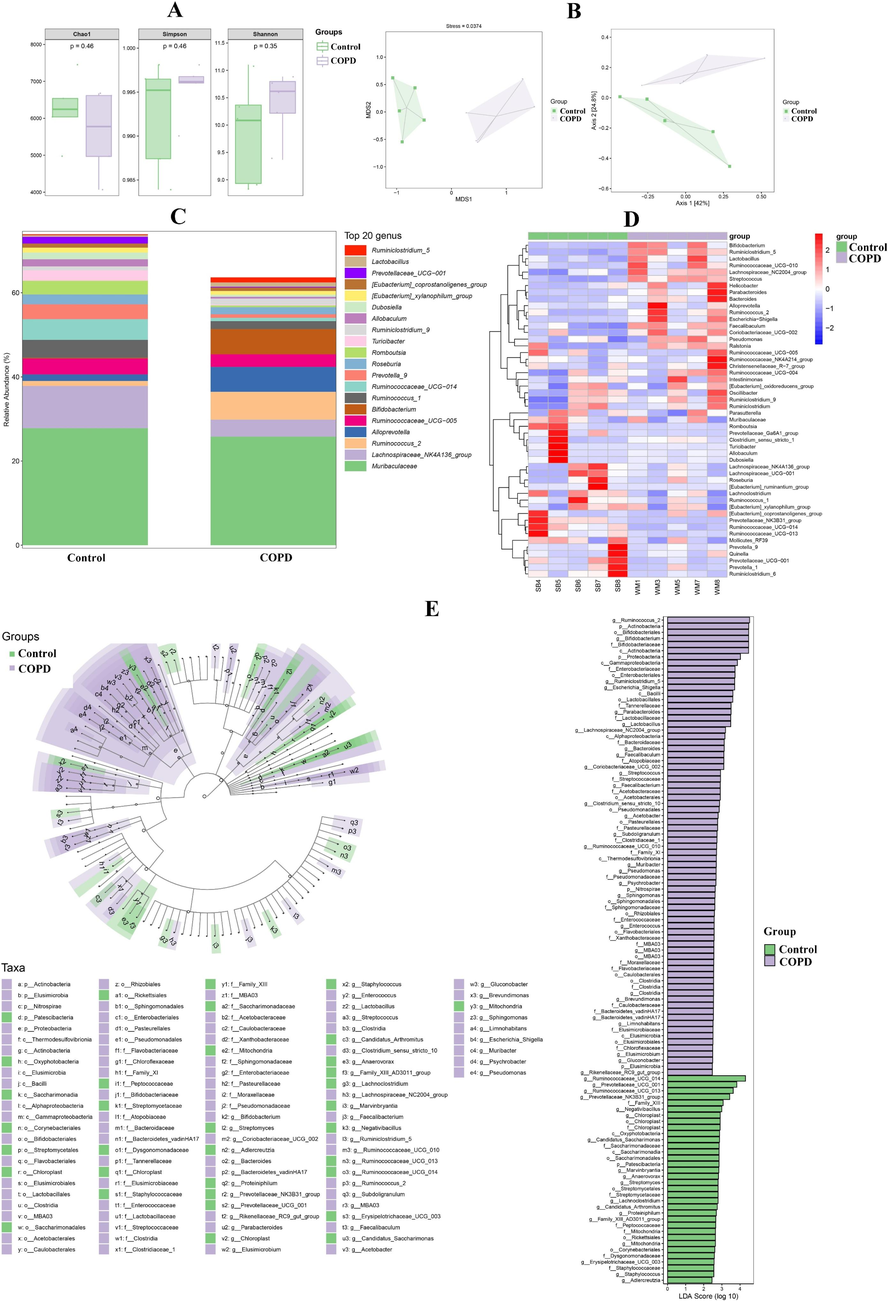
The results of fecal microbiomic research. A and B, Alpha and beta diversity analysis. C, Relative abundance of top 20 microfloras at genus level. D, Heatmap of microfloras at genus level. E, LDA and LEfSe.
3.5 Multiple omics correlation analysis
Correlation analysis was implemented to explore the relationship between multiple omics results and the possible crosstalk between the lung and gut. Firstly, in lung samples, the top 10 inflammatory cells and metabolic pathways in significance ranking, as well as top 9 microfloras in LDA score were used for correlation analysis (Fig. 7A–C). Neutrophil, myeloid derived suppressor cell and activated dendritic cell showed the best correlation with metabolic pathways/microfloras, attributing to the absolute value of correlation coefficients (|r|) between these cells and the five metabolic pathways/two microfloras being greater than or equal to 0.9. Similarly, for the pulmonary metabolic pathways, arachidonic acid metabolism, metabolic pathways, serotonergic synapse, alpha linolenic acid and linoleic acid metabolism as well as biosynthesis of unsaturated fatty acids represented the optimum connection with the inflammatory cells/microfloras. Besides, o_Pasteurellales, f_Pasteurellaceae, g_Rodentibacter and g_Psychrobacter (Psychrobacter genus) had the best correlation with the pulmonary inflammatory cells/metabolic pathways. Then, in the result of the link between fecal metabolomics and microbiomics, there was the optimal association between three metabolic pathways (Alanine metabolism, tyrosine metabolism and lysine degradation) and five microfloras (p_Actinobacteria, c_Actinobacteria (Actinobacteria class), f_Bifidobacteriaceae (Bifidobacteriaceae family), g_Bifidobacterium and o_Bifidobacteriales (Bifidobacteriales order)) with |r| ≥ 0.9 (Fig. S3A).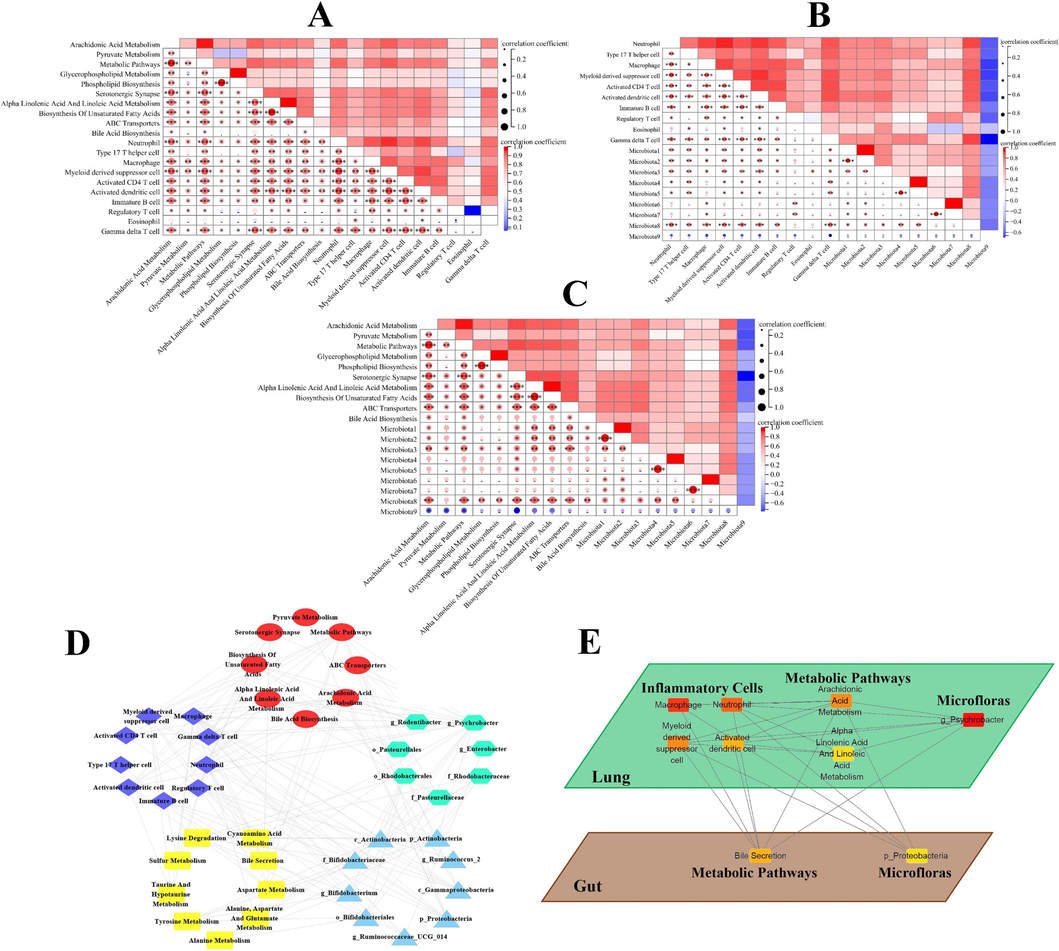
A, pulmonary correlation heatmap of inflammatory cells and metabolic pathways, B, inflammatory cells and microfloras, C, metabolic pathways and microfloras. (The information of microfloras were shown in supplementary material.) D, multiomics correlation network: purple diamond, pulmonary inflammatory cells; red circle, pulmonary metabolic pathways; green hexagon, pulmonary microfloras; yellow rectangle, fecal metabolic pathways; blue triangle, fecal microfloras. E, the core system, which was composed of 9 nodes with the highest degree values, of multiomics correlation network.
Next, in order to further reveal the material network of the lung-gut axis, the correlation coefficients between the individual omics results were calculated, and multiomics correlation network was established based on |r| ≥ 0.9 (except for the |r| between fecal microfloras and pulmonary inflammatory cells/metabolic pathways/microfloras being greater than or equal to 0.7) (Fig. S3B–G). And the material network composed of 9 pulmonary inflammatory cells, 8 pulmonary metabolic pathways, 7 pulmonary microfloras, 9 fecal metabolic pathways and 9 fecal microfloras preliminarily revealed the systematic correlation of the lung-gut axis in COPD (Fig. 7D).
Finally, cytohubba algorithm was adopted to unveil the core system in multiomics correlation network (Fig. 7E). The results showed that the pulmonary key inflammatory cells/metabolic pathways/bacteria closely related to COPD might be macrophage, neutrophil, myeloid derived suppressor cell, activated dendritic cell/arachidonic acid metabolism, alpha linolenic acid & linoleic acid metabolism/g_Psychrobacter. Similarly, bile secretion and p_Proteobacteria might be the key metabolic pathways and bacteria in feces, and played an important role in COPD.
4 Discussion
4.1 The transcriptomic analysis of lung
First, the immune response related gene sets were identified as closely related gene sets to COPD in lung transcriptomics (Fig. 2B). Abnormal immune status, especially the excessive activation of inflammation, was recognized as a significant characteristic of COPD, which could lead to the alteration in the structural cells of the airway and lung, accompanied by the activation and/or migration of inflammatory cells (Budden et al., 2016, Caramori et al., 2016). Our enrichment analysis results also showed the activation and migration of leukocyte, indicating that excessive activation of inflammation in COPD. Furthermore, the result of inflammatory cell analysis showed that most inflammatory cells in the COPD group were increased (Fig. 2C and D). Moreover, the studies showed the increase in the number of inflammatory cells seemed to be a core immunopathological feature of COPD (Caramori et al., 2016).
4.2 The metabolomic analysis of lung and gut
Then, as revealed by pathway analysis, lipid metabolism might play an important role on pulmonary metabolic disorder in COPD (Fig. 3). Accumulating investigations had significantly stated the understanding of the role of lipids as important participants in inflammation, which had a profound impact on COPD (Kotlyarov and Kotlyarova 2021). Additionally, previous researches had implicated that turbulence of lipid homeostasis might intervene in the course of COPD by influencing pulmonary surfactant production. Pulmonary surfactant was mainly composed of lipids, which was of great significance for maintaining the integrity of alveolar structure and function (Fan et al., 2023). On the other hand, amino acid metabolism and bile acid metabolism were main disordered metabolic pathways in fecal metabolomics (Fig. 4). Dysbolism of amino acid in intestinal contents had been described in COPD patients (Bowerman et al., 2020). Considering the significant impact of amino acid metabolism on intestinal health, regulating amino acid metabolism has become an effective attempt to improve COPD symptoms (Jiao et al., 2022). Similarly, the important regulatory effects of bile acids as gut hormones on intestinal homeostasis had also been explained in recent researches. The increasing researches suggested that bile acids could ensure physiological function of the intestine by affecting lipid metabolism homeostasis, inflammation and mucosal immunity (Bowerman et al., 2020).
4.3 The microbiomic analysis of lung and gut
Next, the microbiome of lung and gut were determined by 16S rRNA gene sequencing. As shown in Fig. 5, there was a significant change in the pulmonary microbiota (such as f_Pasteurellaceae, o_Pasteurellales and g_Rodentibacter) of COPD group, which indicated impaired lung function and abnormal inflammatory state probably. Previous studies indicated that abnormal changes in f_Pasteurellaceae and o_Pasteurellales occurred in patients with respiratory morbidity, therefore, significant changes of these microbiota detected in our results might reflect impaired lung function of COPD group (Bowerman et al., 2020). While the disorder of g_Rodentibacter manifested abnormalities in lung inflammation, as there was an obvious correlation between the abundance of g_Rodentibacter and inflammatory cytokine level in lung (Jin et al., 2021). In addition, in fecal 16S rRNA gene sequencing result, some respiratory disease-related microbiota (such as p_Actinobacteria and g_Ruminococcus_2) were identified (Fig. 6). Previous research of fecal microbiome profiles had revealed that p_Actinobacteria was increased in COPD patients, which was similar to our results (Wu et al., 2021). And the result of He et al. showed that adjusting the abundance of Ruminococcus_2 had potential effects on the treatment of respiratory disease (He et al., 2023).
4.4 Disturbances of the core material system in the lung-gut axis
Firstly, macrophage, neutrophil, activated dendritic cell and myeloid derived suppressor cell were recommended as important pulmonary inflammatory cells in the core systems of the lung-gut axis (Fig. 7E). The increased macrophages would cause a more pro-inflammatory state, which might lead to tissue damage and dysfunction of phagocytosis, contributing to the progression of COPD collectively (Yamasaki and Eeden 2018). Furthermore, the defective functions of macrophages could cause changes in pulmonary microbial community structure related to acute exacerbations during COPD (Naito et al., 2017, Yamasaki and Eeden 2018). The increase of neutrophil was the commonest inflammatory phenotype in COPD, which would drive mucus hypersecretion and airways damage, as well as, cause changes in bacterial abundance probably (Brightling and Greening 2019). In addition, activated dendritic cells, as significant orchestrators of immunity, which linked innate immune response with adaptive immune response, increased with the severity of COPD deteriorating (Freeman et al., 2009, Van Pottelberge et al., 2009). And activated dendritic cells had potential important effects on COPD’s local antigen presentation, induction of lymphocyte proliferation, pulmonary lymphogenesis and promotion of sustained inflammation (Van Pottelberge et al., 2009). Meanwhile, the accumulation of myeloid derived suppressor cells might provide a potential basis for the COPD blunted immune response (Kolahian et al., 2016).
Secondly, the arachidonic acid metabolism and alpha linolenic acid & linoleic acid metabolism displayed significant effects on the lung-gut axis for COPD in the aspect of pulmonary metabolomics (Fig. 7E). Most of the metabolites related to the above metabolic pathway were polyunsaturated fatty acids (PUFAs). PUFAs, as important bioactive mediators, had significant value in the development and elimination of inflammation (Kotlyarov and Kotlyarova 2021). The study had revealed differences in PUFAs metabolism between COPD and control groups, and this difference might be related to airway epithelial remodeling (van der Does et al., 2019).
Next, g_Psychrobacter was identified as an important pulmonary microbiota in the core systems (Fig. 7E). According to the potential relationship between g_Psychrobacter and cytokine production & inflammatory cell infiltration, it was speculated that g_Psychrobacter might affect the core system of the lung-gut axis by intervening in pulmonary inflammation (Liu et al., 2021).
Then, the pathways related to bile acid metabolism (bile secretion) had shown potential impacts on the core system of the lung-gut axis in terms of research on intestinal contents (Fig. 7E). There was the evidence to suggest that abnormal bile acid metabolism might be a trigger for activating macrophages and driving gut inflammation (Wang et al., 2020). Moreover, the interaction between bile acid metabolism and gut microbiota had a significant impact on the physiological function of the intestine, as well as the lung-gut axis (Cai et al., 2022, Jiao et al., 2022, Du et al., 2023).
Finally, as shown in Fig. 7E, p_Proteobacteria in feces was mined to demonstrate its importance as a participant in the lung-gut axis. The disorder of p_Proteobacteria had been observed in COPD patients, and researches showed that regulating p_Proteobacteria to intervene COPD might be an effective method for treating the disease (Wu et al., 2021, Mao et al., 2022). However, further research was needed to explore the impact of p_Proteobacteria on the COPD lung-gut axis.
In addition, current research still had some limitations, such as the need for further exploration of the process of information transmission within the lung-gut axis, as well as the need to expand the sample size to further confirm and develop our hypothesis. Meanwhile, abovementioned matters will be explored subsequently in our further study. Altogether, in this study, the core material system composed of inflammatory cells-metabolic pathways-microfloras along the lung-gut axis was established to investigate the pathogenesis of COPD, providing available assistance for the effective precaution and prognosis of disease.
5 Conclusions
In the present study, an integrated transcriptomics-metabolomics-microbiomics approach had been successfully applied to analyze the pathogenesis of COPD based on the lung-gut axis from the perspective of the interactions between the host and the microbes. First, in pulmonary transcriptomic research, immune response related terms, for example immune response-regulating signaling pathway, leukocyte migration, leukocyte activation, etc., involving 1652 DEGs (such as IL-1β, PTGS2, NLRP3 and so on) were considered as important contributors to lung abnormalities in COPD. The ssGSEA result further demonstrated the significant changes in 15 types of inflammatory cells, including neutrophil, Type 17 T helper cell, macrophage, ect., in COPD lung. Then, 135 pulmonary DMs involved 17 metabolic pathways, including arachidonic acid metabolism, 2-oxocarboxylic acid metabolism, ABC transporters, ect. and 105 fecal DMs involved 38 metabolic pathways, such as alanine metabolism, bile secretion and so on, were revealed by LC-MS and 1H NMR metabolomic platforms. Next, f_Pasteurellaceae, o_Pasteurellales, etc. and g_Ruminococcus_2, p_Actinobacteria, etc. were identified as the microflora associated with COPD in lung and gut. Finally, the material network was established by multi-omics correlation analysis to reveal potential connections in the lung-gut axis. Furthermore, the core system, composed of macrophage, neutrophil, activated dendritic cell, myeloid derived suppressor cell, arachidonic acid metabolism, alpha linolenic acid & linoleic acid metabolism, g_Psychrobacter in lung and bile secretion, p_Proteobacteria in gut, of the material network was deeply explored to analyze the possible information flow of the lung-gut axis, which might contribute to the illustration of the pathogenesis of COPD, providing new insights for the effective prevention and treatment of the disease. In addition, various techniques, such as single cell sequencing and targeted metabolomics, will be adopted to further validate the reliability and accuracy of our research results in the subsequent work. Next, we will focus on the core material of the lung gut axis to explore whether effective treatment of COPD can be achieved by regulating arachidonic acid metabolism, macrophage and so on. Furthermore, based on in-depth exploration of the pathological mechanism of COPD, effective therapeutic drugs and delivery routes will be developed. All in all, in the future, more researches are required on the further exploration for the impact of the core material of the lung-gut axis on the onset and recovery process of COPD, suggesting more precise identifying effective treatments for COPD with emphasis.
CRediT authorship contribution statement
Tianyang Wang: Writing – original draft, Methodology, Formal analysis. Fang Wang: Investigation. Ruinan Ren: Investigation. Yikun He: Investigation. Qi Yu: Validation, Data curation. Guoan Zhao: Software, Formal analysis. Jinling Zhang: Validation, Data curation. Qi Liu: Validation, Data curation. Ying Lyu: Visualization. Weiwei Jia: Visualization. Wenbao Wang: Validation, Data curation. Fanchen Meng: Visualization. Song Lin: Writing – review & editing, Supervision, Project administration, Funding acquisition, Conceptualization. Yan Lin: Supervision, Funding acquisition, Conceptualization.
Funding
This study was supported by Heilongjiang Provincial Natural Science Foundation of China (LH2022H110), National Natural Science Foundation of China (82405233, 82074148), Research Institute of Medicine & Pharmacy of Qiqihar Medical University (2023-ZDPY-001, QMSI2023E-01, QMSI2021B-03, 2022-ZDPY-004) and Science and Technology Bureau of Qiqihar (LHYD-2021011).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108-112.
- [CrossRef] [Google Scholar]
- XCMS2: Processing Tandem Mass Spectrometry Data for Metabolite Identification and Structural Characterization. Anal. Chem.. 2008;80:6382-6389.
- [CrossRef] [Google Scholar]
- Disease-associated gut microbiome and metabolome changes in patients with chronic obstructive pulmonary disease. Nat. Commun.. 2020;11
- [CrossRef] [Google Scholar]
- Airway inflammation in COPD: progress to precision medicine. Eur. Respir. J.. 2019;54
- [CrossRef] [Google Scholar]
- Emerging pathogenic links between microbiota and the gut–lung axis. Nature Reviews Microbiology.. 2016;15:55-63.
- [CrossRef] [Google Scholar]
- Gut microbiota-derived bile acids in intestinal immunity, inflammation, and tumorigenesis. Cell Host Microbe. 2022;30:289-300.
- [CrossRef] [Google Scholar]
- Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep.. 2017;18:248-262.
- [CrossRef] [Google Scholar]
- Butyrate: Connecting the gut-lung axis to the management of pulmonary disorders. Front. Nutr.. 2022;9
- [CrossRef] [Google Scholar]
- The lung-gut crosstalk in respiratory and inflammatory bowel disease. Front. Cell. Infect. Microbiol.. 2023;13
- [CrossRef] [Google Scholar]
- Alveolar type II epithelial cell FASN maintains lipid homeostasis in experimental COPD. JCI Insight. 2023;8
- [CrossRef] [Google Scholar]
- Lilikoi V2.0: a deep learning–enabled, personalized pathway-based R package for diagnosis and prognosis predictions using metabolomics data. GigaScience. 2021;10
- [CrossRef] [Google Scholar]
- Lung Dendritic Cell Expression of Maturation Molecules Increases with Worsening Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med.. 2009;180:1179-1188.
- [CrossRef] [Google Scholar]
- Shaoyao-Gancao-Tang regulates the T-helper-type 1/T-helper-type 2 ratio in the lung and gut and alters gut microbiota in rats with ovalbumin-induced asthma. Journal of Ethnopharmacology.. 2023;309
- [CrossRef] [Google Scholar]
- S1P-dependent interorgan trafficking of group 2 innate lymphoid cells supports host defense. Science. 2018;359:114-119.
- [CrossRef] [Google Scholar]
- The therapeutic effect of Xuanbai Chengqi Decoction on chronic obstructive pulmonary disease with excessive heat in the lung and fu-organs based on gut and lung microbiota as well as metabolic profiles. J. Chromatogr. B. 2022;1198
- [CrossRef] [Google Scholar]
- Single Treatment of Vitamin D3 Ameliorates LPS‐Induced Acute Lung Injury through Changing Lung Rodentibacter abundance. Mol. Nutr. Food Res.. 2021;66
- [CrossRef] [Google Scholar]
- The emerging role of myeloid-derived suppressor cells in lung diseases. Eur. Respir. J.. 2016;47:967-977.
- [CrossRef] [Google Scholar]
- Anti-Inflammatory Function of Fatty Acids and Involvement of Their Metabolites in the Resolution of Inflammation in Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci.. 2021;22
- [CrossRef] [Google Scholar]
- Joint Automatic Metabolite Identification and Quantification of a Set of 1H NMR Spectra. Anal. Chem.. 2021;93:2861-2870.
- [CrossRef] [Google Scholar]
- Exploring the key factors of schizophrenia relapse by integrating LC-MS/1H NMR metabolomics and weighted correlation network analysis. Clin. Chim. Acta. 2023;541
- [CrossRef] [Google Scholar]
- RS-5645 attenuates inflammatory cytokine storm induced by SARS-CoV-2 spike protein and LPS by modulating pulmonary microbiota. Int. J. Biol. Sci.. 2021;17:3305-3319.
- [CrossRef] [Google Scholar]
- The Bufei Jianpi Formula Improves Mucosal Immune Function by Remodeling Gut Microbiota Through the SCFAs/GPR43/NLRP3 Pathway in Chronic Obstructive Pulmonary Disease Rats. Int. J. Chron. Obstruct. Pulmon. Dis.. 2022;17:1285-1298.
- [CrossRef] [Google Scholar]
- Bacteriological incidence in pneumonia patients with pulmonary emphysema: a bacterial floral analysis using the 16S ribosomal RNA gene in bronchoalveolar lavage fluid. Int. J. Chron. Obstruct. Pulmon. Dis.. 2017;12:2111-2120.
- [CrossRef] [Google Scholar]
- Cytoscape Automation: empowering workflow-based network analysis. Genome Biol.. 2019;20
- [CrossRef] [Google Scholar]
- Links Between Inflammatory Bowel Disease and Chronic Obstructive Pulmonary Disease. Front. Immunol.. 2020;11
- [CrossRef] [Google Scholar]
- limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res.. 2015;43:e47-e.
- [CrossRef] [Google Scholar]
- Imbalance of gut microbiota is involved in the development of chronic obstructive pulmonary disease: A review. Biomed. Pharmacother.. 2023;165
- [CrossRef] [Google Scholar]
- Dynamic differences in dietary polyunsaturated fatty acid metabolism in sputum of COPD patients and controls. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of. Lipids. 2019;1864:224-233.
- [CrossRef] [Google Scholar]
- The Role of Dendritic Cells in the Pathogenesis of COPD: Liaison Officers in the Front Line. COPD: Journal of Chronic Obstructive Pulmonary Disease.. 2009;6:284-290.
- [CrossRef] [Google Scholar]
- Gut microbial bile acid metabolite skews macrophage polarization and contributes to high-fat diet-induced colonic inflammation. Gut Microbes. 2020;12
- [CrossRef] [Google Scholar]
- The Bidirectional Gut–Lung Axis in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med.. 2023;207:1145-1160.
- [CrossRef] [Google Scholar]
- Metabolomic profile perturbations of serum, lung, bronchoalveolar lavage fluid, spleen and feces in LPS-induced acute lung injury rats based on HPLC-ESI-QTOF-MS. Anal. Bioanal. Chem.. 2020;412:1215-1234.
- [CrossRef] [Google Scholar]
- An integrated pathological research for precise diagnosis of schizophrenia combining LC-MS/1H NMR metabolomics and transcriptomics. Clin. Chim. Acta. 2022;524:84-95.
- [CrossRef] [Google Scholar]
- Cathelicidin LL-37 restoring glucocorticoid function in smoking and lipopolysaccharide-induced airway inflammation in rats. Chin Med J (engl). 2019;132:569-576.
- [CrossRef] [Google Scholar]
- Variations in fecal microbial profiles of acute exacerbations and stable chronic obstructive pulmonary disease. Life Sci.. 2021;265
- [CrossRef] [Google Scholar]
- Yamasaki, K. and S. F. v. Eeden, 2018. Lung Macrophage Phenotypes and Functional Responses: Role in the Pathogenesis of COPD. International Journal of Molecular Sciences. 19, https://doi.org/10.3390/ijms19020582.
- Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun.. 2019;10
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2024.106056.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary Data 1
Supplementary Data 1




