

Translate this page into:
LC-MS/MS quantification of vitamin K1 after simple precipitation of protein from low volume of serum
⁎Corresponding author. shaotingw@whu.edu.cn (Shao-Ting Wang)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Quantification of vitamin K1 in serum is still quite a challenge for most analytical laboratories. For long, protein precipitation (PP) coupled with liquid–liquid extraction (LLE) or solid phase extraction (SPE) is the only choice for sample preparation before LC-MS/MS analysis. The objective of this study is to establish a novel method for the simple and rapid quantification of K1. To this end, three issues are illuminated in the first place: (i) the release efficiency of K1 is influenced by the type of precipitator and the volume ratio of serum to precipitator during PP; (ii) the absolute recovery of K1 will be compromised by the partition effect during extraction; (iii) the volume of injected serum per run should be proper rather than high to prevent the potential ion suppression effect. Benefitting from these findings, a single-step PP method is developed for K1 quantification for the first time. For pretreatment, isopropanol (100 μL), instead of the frequently used methanol, ethanol and acetonitrile, is utilized to release K1 from serum (25 μL), which realize the excellent release efficiency (>90%) and dilution effect (1:4) simultaneously. After validation, the method can provide favorable accuracy (91.5–112.2%) and precision (less than13.5%) in the wide concentration range (0.1–2.5 ng/mL). As a result, such method may provide a feasible and high-throughput alternative for evaluating K1 status in practice.
Keywords
Phylloquinone
Vitamin K
LC-MS/MS
Recovery
Matrix effect

1 Introduction
Vitamins K are a group of well-known fat-soluble vitamins, which share a 2-methyl-1,4-naphthoquinone structure with various unsaturated side chains. These compounds play significant roles in blood coagulation, cardiovascular health and bone metabolism (Fusaro et al., 2020). For long, vitamin K1 (K1, also named as phylloquinone) is regarded as one of the most frequently used biomarkers to access vitamin K status in vivo (Thane et al., 2006; Harrington et al., 2008; Shea et al., 2012). However, ascribing to its sub-stoichiometric abundance in serum (two orders of magnitude lower than the concentration of 25-hydroxyvitamin D3) and ultra-hydrophobic nature (more hydrophobic than α-tocopherol), the measurement is still quite a challenge (Fusaro et al., 2017; Zhang et al., 2019; Card et al., 2020).
In the past twenty years, numerous mass spectrometry-based methods, mainly liquid chromatography-tandem mass spectrometry (LC-MS/MS), have been developed to analyze K1 in blood matrix (Suhara et al., 2005; Ducros et al., 2010; Gentili et al., 2014; Konieczna et al., 2016; Riphagen et al., 2016; Nannapaneni et al., 2017; Sandvik et al., 2017; Hu et al., 2018; Andersen et al., 2019; Dunovska et al., 2019; Zhang et al., 2020; Arachchige et al., 2021; Card et al., 2022; Meinitzer et al., 2022). As shown in Table 1, two steps were carried out successively for sample preparation in all cases: protein precipitation (PP) and target extraction (Suhara et al., 2005; Ducros et al., 2010; Gentili et al., 2014; Konieczna et al., 2016; Riphagen et al., 2016; Nannapaneni et al., 2017; Sandvik et al., 2017; Hu et al., 2018; Andersen et al., 2019; Dunovska et al., 2019; Zhang et al., 2020; Arachchige et al., 2021; Card et al., 2022). In the first step, hitherto, no consensus has been reached for either the solvent selection or the sample/solvent proportion. Among the published researches, methanol, ethanol, isopropanol and acetonitrile have all been utilized with the sample to solvent ratio ranging from 2/1 to 1/4. Actually, this “releasing” step is the prerequisite to achieve satisfying recovery, because K1 is originally integrated with triacylglycerol-rich lipoproteins in blood. Nonetheless, limited focus has been dedicated on this aspect, which left a blind spot for method evaluation (as discussed in Section 2.4). In the second step, solid phase extraction (SPE) or liquid–liquid extraction (LLE) coupling with nitrogen evaporation was utilized to concentrate the supernatant after PP. In practice, such processes probably suffer from drawbacks like lengthy, chemical-polluting or high-cost. Additionally, as K1 is light-sensitive and prone to emerge non-specific adsorption to containers, the more intricate the experimental procedures are, the lower the recovery and reproducibility can be. In result, there are a number of controversies among previous methods, for example whether there were matrix effect after LLE (Gentili et al., 2014; Hu et al., 2018; Andersen et al., 2019; Zhang et al., 2020; Arachchige et al., 2021); whether the precipitation should be removed before LLE (Konieczna et al., 2016; Dunovska et al., 2019; Zhang et al., 2020); whether the tube type would affect K1 quantification (Andersen et al., 2019; Arachchige et al., 2021). In practice, such immaturity on methodology has, at least, partly contributed to the misty phenomena in vitamin K related clinical and nutritional trails (Shea et al., 2021; Kaesler et al., 2021).
NO.
Sample
consumption
Protein
precipitation
Extraction
Injected serum
per run
Sensitivity
(LLOQ)
Reference
1a
500 μL
Ethanol (2 mL)
LLE with hexane (3 mL); SPE; evaporation of hexane/diethyl ether (97:3, 5 mL)
∼250 μL
0.04 ng/mL
(8)
2a
500 μL
Ethanol (2 mL)
LLE with cyclohexane (5 mL); evaporation
∼100 μL
0.04 ng/mL
(9)
3a
500 μL
Ethanol (1.5 mL)
LLE for three times with hexane (1.5 mL); evaporation; cold-induced lipid elimination
∼200 μL
0.1 ng/mL
(10)
4b
200 μL
Methanol (0.8 mL)
LLE for two times with hexane (0.8 mL); evaporation
∼20 μL
0.33 ng/mL
(11)
5b
350 μL
Ethanol (0.175 mL)
SPE; evaporation of methanol/isopropanol/hexane (2:1:1, 2 mL)
∼60 μL
0.06 ng/mL
(12)
6a
300 μL
Methanol (1 mL)
LLE with hexane (2.5 mL); evaporation
∼30 μL
0.1 ng/mL
(13)
7b
250 μL
Methanol/ethanol
(1:1, 0.8 mL)SPE
∼8 μL
0.1 ng/mL
(14)
8a
200 μL
Ethanol (0.6 mL)
LLE with cyclohexane (1 mL); evaporation
∼130 μL
0.1 ng/mL
(15)
9b
160 μL
Isopropanol/ZnSO4 (1 mol/L) /NH3 (25%) in water (94/1/5, 0.4 mL)
On-line SPE
∼20 μL
0.03 ng/mL
(16)
10b
500 μL
Ethanol (2 mL)
LLE for two times with hexane (4 mL); evaporation; SPE; evaporation of hexane/diethyl ether (97:3, 9 mL)
∼30 μL
0.03 ng/mL
(17)
11b
250 μL
Methanol (1 mL)
LLE for two times with hexane (2.5 mL); evaporation
∼30 μL
0.01 ng/mL
(18)
12a
45 μL
Acetonitrile
(0.2 mL)LLE with hexane (0.4 mL); re-LLE with hexane (0.3 mL); evaporation; SPE
∼ 7 μL
0.7 ng/mL
(19)
13a
250 μL
Ethanol (0.6 mL)
LLE with hexane (3 mL); evaporation; SPE; evaporation
∼250 μL
< 0.36 ng/mL
(20)
14
25 μL
Isopropanol
(0.1 mL)–
∼ 2 μL
0.1 ng/mL
This work
Actually, our group has been trying to quantify K1 since our primary attempt on detecting fat-soluble vitamins in 2017 (Yuan et al., 2017; Le et al., 2018). During the past five years, we kept collecting the experience from the existed methods and trying to improve the feasibility and reliability for K1 quantification. In this work, we illuminated the previous predicament in K1 analysis from two aspects: (i) the incomplete recovery of K1 by the suboptimum sample preparation protocol; (ii) the potential ion suppression effect by injecting excessive sample matrix. Thereafter, a one-step PP-based LC-MS/MS method was developed and validated, which provided the first alternative to analyze serum K1 without LLE or SPE.
2 Materials and methods
2.1 Chemicals and resources
Standards of K1 (V-030–1 mL) and vitamin K1-d7 (5,6,7,8-d4, 2-methyl-d3) (K1-d7, 705470-1MG) were bought from Sigma-Aldrich (Beijing, China). Methanol, ethanol, isopropanol, acetonitrile, n-hexane, ammonium acetate and formic acid at HPLC-grade were all derived from Fisher-Scientific (New Jersey, USA). All the standards and chemical reagents were provided by manufacturers with guaranteed composition and purity. Thus, no specific treatment and characterization was carried out before use. The water used throughout the study was purified by a Milli-Q apparatus (Millipore, Bedford, MA).
The human serum samples used throughout the study were derived from Department of Clinical Laboratory of Renmin Hospital of Wuhan University (Wuhan, China). These samples were obtained from the excess samples after routine biochemical detection services. The whole study was supervised under the Ethics Committee of Renmin Hospital of Wuhan University (WDRY2021-K041) and abided by the Declaration of Helsinki principles.
2.2 Apparatus and parameters
The LC separation was carried out on a Shimadzu LC-30AD-CL system by using Kinetex 2.6 μm C18 100 Å (100 × 3 mm) column. Formic acid in methanol (0.2% v/v) with ammonium acetate (10 mmol/L) was used as mobile phase. The flow program was set as Supplemental Table S1. The column temperature was maintained at 65 °C to decrease back-pressure and shorten retention time. No noticeable column deterioration was observed with regular use up to 5 months. The retention time of K1 and K1-d7 was 2.2 min. To diminish contamination toward ion source, LC fluid from 0 to 1.7 min and 2.7–6.0 min was discarded by using an integrated diverter valve. A SIL-30AC-MP-CL auto-sampler was used for injection. The injecting volume was 10 μL for all the performed experiments. After injection, both the inner and outer walls of sampling syringe were auto-washed with methanol (200 μL).
The MS detection was performed on a Shimadzu MS-8050-CL spectrometer with electrospray ionization source under positive ion mode. The multiple reaction monitoring parameters are listed in Supplemental Table S2. The ESI parameters were as follows: DL temperature (250 °C), heat block temperature (400 °C), nebulizing gas (2 L/min), drying gas (10 L/min), heating gas (10 L/min) and interface temperature (350 °C). Data acquisition and processing were performed with Lab Solution 5.53 SP2 software.
2.3 Calibrators and quality controls
Both K1 and K1-d7 were dissolved and diluted with methanol to prepare stock solutions (100 ng/mL). Internal standard (IS) solution was prepared by further diluting stock solution of K1-d7 with methanol to 5 ng/mL.
Three sets of samples, including the mixture of patients’ serum with the pre-detected concentration of K1 lower than 0.2 ng/mL (as BLANK MATRIX I), VD-DDC Mass Spect Gold plasma (Golden West Diagnostics, Temecula, CA; as BLANK MATRIX II) and the mixture of water and methanol (80%, v/v; as BLANK MATRIX III), were enrolled to prepare apparent blank matrix. These samples were kept at room temperature and allowed to stand for three full days under natural light before use. The calibration samples (C1 to C6) were prepared by spiking K1 into these candidate blank matrices (C0). The spiked concentrations were 0.1, 0.25, 0.5, 0.8, 1.6 and 2.5 ng/mL, respectively. Such designation was referred the previous studies to cover the physiological range of K1 (Fusaro et al., 2017; Zhang et al., 2019; Card et al., 2020). All these samples (C0 to C6) were analyzed by the established LC-MS/MS method. The effective peak area ratios of K1 to K1-d7 were calculated by subtracting the value of C0 from C1 to C6. The calibration was then built by the linear least squares regression model utilizing these effective peak area ratios versus the nominal spiked concentrations. The quality controls of four levels (High-, Medium-, Low-, and LLOQ-QC) were prepared by spiking K1 (2.5, 0.8, 0.25 and 0.1 ng/mL) into BLANK MATRIX II.
All the prepared stock solutions, IS solution, calibration samples and QC samples were wrapped in tin foil and kept at 4 °C for short-term storage (within 3 days) or −80 °C for long-term storage (up to 180 days). During these periods of time, no obvious degradation (less than 15%) was observed after investigating the relative recovery (as described in method validation).
2.4 The efficiency of different pretreatments
As summarized in Table 1, previous studies utilized very different pretreatment strategies for K1 extraction. Among these pioneering contributions, the recovery was always discussed after normalization with IS (as relative recovery). In contrast, the absolute recovery, which directly reflected the efficiency of K1 release, has not been systematically investigated yet. To lighten this blind spot, we carried out two sets of experiments, which investigated K1′s releasing-efficiency after PP (EXPERIMENT I) and PP coupled with LLE (EXPERIMENT II). The serum samples used in this section were derived from the same serum pool, which was prepared by mixing serum samples from random patients (n = 40). The concentration of K1 in such mixture was pre-analyzed by the established LC-MS/MS method (0.52 ng/mL, as CONCENTRATION 0).
For EXPERIMENT-I, certain volume of methanol, ethanol, isopropanol or acetonitrile (100, 150, 200, 400 or 800 μL, presented as VOLUME 1) was added into serum samples (50 μL, presented as VOLUME 0), creating the proportion of serum to precipitator from 1/2 to 1/16. After vortexing (1 min) and centrifuging (12,000 g at 4 °C for 2 min), the supernatant (100 μL) was collected and mixed with IS solution (5 μL) for LC-MS/MS analysis. The apparent concentration in this supernatant (the peak area ratio of K1 to IS, similarly hereinafter) was presented as CONCENTRATION 1. After removing all the supernatant, ethanol (200 μL, presented as VOLUME 2) was added to re-extract the residual K1 from the precipitate. After vortexing (1 min) and centrifuging (12,000 g at 4 °C for 2 min), the supernatant (100 μL) was also collected and mixed with IS solution (5 μL) for LC-MS/MS analysis. The apparent concentration in this supernatant was presented as CONCENTRATION 2. Accordingly, the releasing-efficiency of K1 by PP can be calculated by Eq. (1). And the releasing-efficiency of K1 by washing the precipitate can be calculated by Eq. (2).
2.5 The impact of sample consumption
Considering the low abundance of K1 in blood, the existed methods always imported large volume of serum/plasma (200–500 μL) to ensure their sensitivity. However, as other hydrophobic interference would be also condensed at the same time, the accumulation of ion suppression may potentially compromise the K1 response during MS detection. To investigate such impact, we carried out EXPERIMENT III and EXPERIMENT IV.
In EXPERIMENT III, serum (1 mL) was precipitated with ethanol (4 mL) and LLE with n-hexane (20 mL). Afterward, different volume of n-hexane (0.2, 0.4, 1, 2, 4 or 10 mL) was transferred into separate tubes for nitrogen drying (presented as GROUP 1–6). The dried samples were reconstituted with ethanol (100 μL) and IS solution (5 μL) for LC-MS/MS analysis. As a result, the volume of “injected serum per run” could be estimated as 1, 2, 5, 10, 20 or 50 μL for each sample group (as shown in Eq. (4). The ion suppression effect on K1 could be evaluated by comparing the detected peak area of K1 in GROUP 2–6 to its expected value calculated from GROUP (1) (as shown in Eq. (5). And the ion-suppression effect on IS could be evaluated by comparing the detected peak area of IS in GROUP 2–6 to the result from GROUP (1) (as shown in Eq. (6).
In EXPERIMENT IV, serum (50 μL) was precipitated by using isopropanol (200 μL). The supernatant (100 μL) was mixed with IS solution (5 μL) before subjecting to LC-MS/MS analysis (presented as GROUP 7). In this case, the volume of injected serum per run was 2 μL. The corresponding ion suppression effect on K1 and IS could be also evaluated by Eqs. (5) and (6).
2.6 Sample preparation
Serum sample (25 μL) was combined with IS solution (5 μL) and isopropanol (100 μL). After vortexing (1 min) and centrifuging (12,000 g at 4 °C for 2 min), the supernatant (100 μL) was transferred into a 96-well plate for LC-MS/MS analysis. As the whole process could be finished within 5 min, no specific light-prevention was carried out during sample pretreatment. But the sampled 96-well plate should be covered by tin foil (EasyPierce 20 μm Thin Foil from Thermo Scientifi-AB1720) to prevent long-time light exposure and solvent evaporation before injection.
2.7 Method validation
The method was validated according to the guidelines for bio-analytical method validation from Food and Drug Administration and document C62-A from Clinical and Laboratory Standards Institute. All the determinations were performed by triplicate unless expressly stated.
The calibration was built by the linear least squares regression model utilizing peak area ratios of K1 to IS versus the nominal spiked concentrations. All the three apparent blank matrices were compared for developing calibrations. Based on the established linear regression, the allowable bias should be within 15% for all calibrators (within 20% at the LLOQ). The method sensitivity was reflected by the lowest concentrations of the calibrators (LLOQ level).
The carry-over effect was studied by detecting BLANK MATRIX II after three successive analyses of upper limit of calibrators. The residual signal should be no>15% of LLOQ.
The IS-normalized matrix effect was evaluated by spiking 0.5 and 2.0 ng/mL of K1 into six random serum samples from different donors. The detected difference between spiked and initial serum was compared with the spiked concentration. The consistency should be within 85–115% for all the six samples.
Accuracy was determined as apparent recovery (dividing the measured concentrations to the spiked values) by analyzing all the four levels of QC samples in one day (n = 6, intra-day) and in consecutive days (n = 10, inter-day). The results should be within 80–120% for LLOQ-QC and 85–115% for others. And imprecision was calculated as CV of those measurements, which should be lower than 20% for LLOQ-QC and 15% for others.
Stability was investigated by using Medium-QC. Serum samples were wrapped in tin foil and kept at 25 °C for 1 day, 4 °C for 3 days or −80 °C for 180 days before pretreatment. The freeze–thaw test was performed for three cycles. And for treated samples, the supernatant was kept within the covered 96-well plate in auto-sampler for 24 h before injection. The apparent recovery should be within 85–115%.
2.8 Method application
To examine the applicability of this new method, we carried out K1 quantification in one hundred serum samples from donors for health screening. All the samples were collected among October to November 2021. Serum was separated and transferred into 1.5 mL Eppendorf tubes within 2 h after collection. All the obtained tubes were stored at −80 °C in darkness until further treatment. Before determination, the thawing process was conducted in darkness at 4 °C.
3 Results
3.1 Method development
Considering the discrepancy among previous pretreatment protocols for K1, we concluded there were three issues awaiting careful investigation before developing more advanced methods: (i) how to realize efficient dissociation of K1 from serum matrix; (ii) whether the enrichment effect from LLE/SPE was essential to preserve adequate detecting sensitivity; (iii) whether the high volume of “injected serum per run” would cause ion suppression for MS response. In accordance, four series of experiments (EXPERIEMTN I to IV) were conducted successively.
The results of EXPERIMENT I were summarized in Fig. 1 and Supplemental Table S3 & S4. Interestingly, acetonitrile, which was commonly used for PP in blood samples, exhibited weak capacity for K1 release. At all the tested volume ratios, the releasing-efficiency was less than 16.4%. And after PP with acetonitrile, K1 seemed to be tightly retained on the aggregated precipitate. Even after washing with ethanol,K1 cannot be re-released efficiently (4.3–8.9%). As for alcohols, the completeness of K1 release increased with the volume ratio. In general, isopropanol exhibited better performance than methanol and ethanol, especially at low volume ratio. Taking the case of 1:3 as a typical example, the releasing-efficiency after PP with isopropanol can reach 92.3%, which was about 13 times higher than methanol and 1.5 times higher than ethanol. Unlike acetonitrile, after PP with alcohols, the residual K1 in precipitate can be well regained by re-extracting with ethanol. These phenomena clearly indicated alcohols were more suitable for counteracting the adhesion between K1 and serum matrix than acetonitrile. Although the detailed mechanism was not fully understood yet, we speculated the precipitator’s polarity and hydrophobicity should be the main determinants.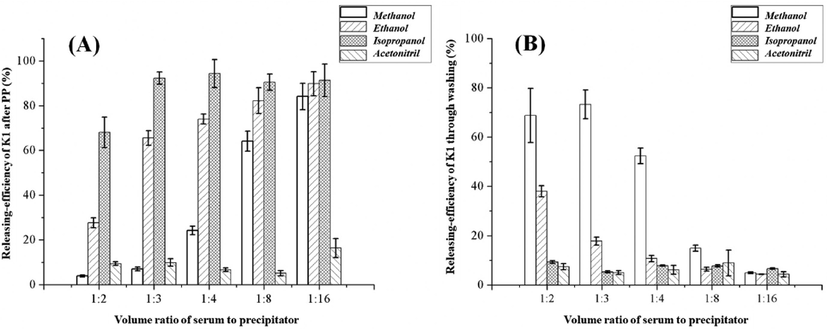
(A) The releasing-efficiency of K1 after protein precipitation by methanol, ethanol, isopropanol or acetonitrile. (B) The releasing-efficiency of K1 regaining from washing the residual precipitate (obtained from protein precipitation) with ethanol.
The results of EXPERIMENT II were summarized in Fig. 2 and Supplemental Table S5 & S6. In general, combining with EXPERIMENT I, these experiments showed that LLE treatment can accelerate K1 release during PP. Also taking the volume ratio of 1:3 as a typical example, profiting from LLE, the releasing-efficiency reached 50.7% for methanol and 70.5% for ethanol. Meanwhile, the residual K1 in precipitate decreased to 22.5% for methanol and 6.3% for ethanol. However, LLE possessed very limited improvements for acetonitrile groups, which still preserved only 7.4–15.0% as the releasing-efficiency. And the partition effect between precipitator and extractor would compromise the releasing-efficiency. This drawback gave rise to distinctive inflection points of releasing-efficiency in methanol group at 1:8 and ethanol group at 1:4. Even at best circumstance, over 25% of K1 would be lost due to such effect. Moreover, isopropanol, which exhibited best capacity for K1 release in EXPERIMENT I, was not well compatible with LLE treatment. After introducing hexane, isopropanol would be homogenized into the upper phase. Consequently, the time cost of the following nitrogen drying process would be obviously prolonged.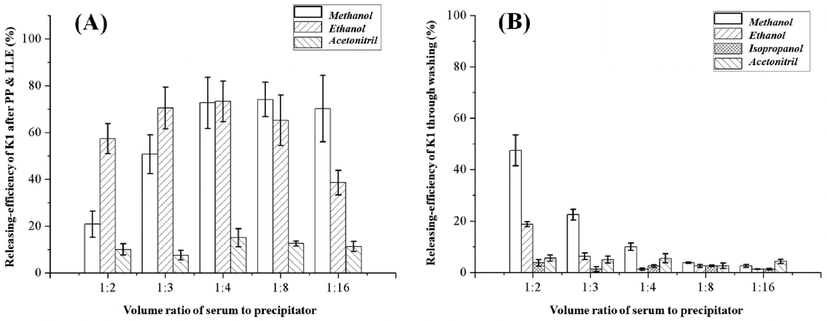
(A) The releasing-efficiency of K1 after protein precipitation by methanol, ethanol, isopropanol or acetonitrile coupled with LLE by hexane. (B) The releasing-efficiency of K1 regaining from washing the residual precipitate (obtained from protein precipitation coupled with LLE) with ethanol. Of note, as isopropanol is miscible with n-hexane, the obtained upper phase after LLE need too much time to dry under general circumstance. In result, this group of samples was not included in (A).
Additionally, we also evaluated the releasing-efficiency of IS (K1-d7). In this experiment, we added 1 ng/mL of IS into BLANK MATRIX II serving as the analytical sample. The sample underwent similar treatments as in EXPERIMENT I and II. Prior to injection, we introduced K1 standard solution (10 μL, 5 ng/mL) to serve as the internal standard of K1-d7. The final results were as expected, showing the very similar performance of K1-d7 with K1.
The results of EXPERIMENT III were summarized in Fig. 3 and Supplemental Table S7 & S8. Along with the increase of injected serum per run (from 1 to 50 μL), the ion suppression effect significantly aggravated for both K1 (up to 92.2%) and IS (up to 95.4%). Fortunately, both compounds shared similar changing trends (as shown in Fig. 3A), which indicated the accuracy of K1 quantification could be partly preserved from ion suppression by using IS normalization. Nonetheless, the detected absolute peak area of K1 was significantly affected by such ion suppression effect (as shown in Fig. 3B). For GROUP (1) to (3), the signal response was still comparable with the theoretical value calculated from GROUP (1). But for GROUP (4) to (6), the detected peak area of K1 did not grow proportionally at all. In fact, the signal of GROUP (6) even became lower than GROUP (5). When compared GROUP (6) to GROUP (3), ten times increase in injected serum per run only brought about 14% increase on signal intensity. In result, for PP&LLE based methods, the detecting sensitivity of K1 was not necessarily always correlated with the volume of injected serum per run (or initial sample consumption). “Proper” rather than “large” sample involvement should be applied to achieve satisfying analytical performance.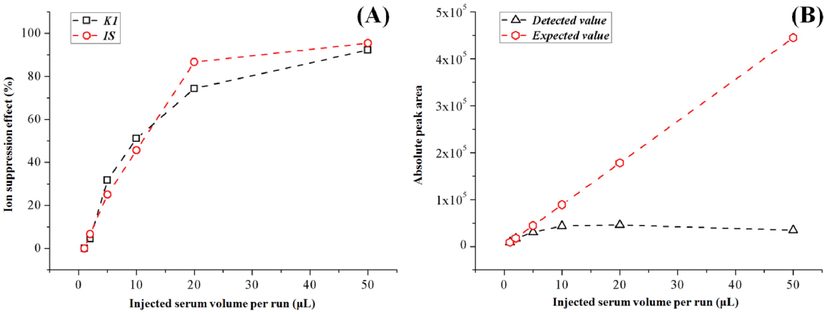
(A) The impact of different injected serum volume per run on ion suppression effect of K1 and IS (K1-d7). (B) The impact of different injected serum volume per run on detected peak area and expected peak area (calculated from GROUP (1) of K1.
Taking the results from EXPERIMENT I, II and III together, two points could be learned: (1) instead of the mainstream PP&LLE treatment, excellent releasing-efficiency of K1 could be simply realized by direct PP with isopropanol; (2) considering ion suppression, small volume of injected serum per run (2–5 μL) could achieve comparable detecting sensitivity with the conventionally used high volume of injected serum per run (50 μL or more). Therefore, EXPERIMENT IV was carried out to test whether K1 could be efficiently analyzed after single PP treatment with isopropanol. According to the results, the ion suppression effect of GROUP 7 was 1.7% for K1 and 4.9% for IS. Such data was at the same level with GROUP (2), which possessed identical volume of injected serum per run (2 μL). This phenomenon indicated that sample after PP would not cause higher ion suppression than PP&LLE. In addition, profiting from the better releasing-efficiency (94% vs 73%), the detected peak area of K1 in GROUP 7 was even slightly higher than GROUP (2) (20643 vs 17012). To sum up, under optimized condition, the simple PP strategy could be an attractive substitute for PP&LLE treatment in K1 analysis.
3.2 Method validation
The retention time of K1 and IS was 2.22 and 2.20 min. The linear range was prepared as 0.1–2.5 ng/mL so that the LLOQ was set at 0.1 ng/mL. The regressions obtained from all the three apparent blank matrices were shown in Supplemental Table S9, in which the R2 was all higher than 0.996. All calibrators from the established linear regression exhibited the bias less than 10%, except for C1 from BLANK MATRIX I (revealing 17.7%), which was also within the acceptable criteria. As the slopes and intercepts exhibited good consistency, the sample pretreatment strategy was considered possessing negligible matrix bias. Considering C0 from BLANK MATRIX I still preserved quantifiable K1 (0.08 ng/mL, as shown in Supplemental Figure S1), BLANK MATRIX II (in which K1 was nearly undistinguishable) was chosen to build the methodology.
No detectable K1 could be observed (the ratio of signal to noise less than 3) after successive injection of three calibrator samples at C6 level. It demonstrated the carryover effect was negligible for the proposed method.
To study the IS-normalized matrix effect, two concentrations of K1 (0.5 and 2.0 ng/mL) was spiked into six different real sample matrices. The background concentrations of K1 in these samples were detected as 0.13, 0.19, 0.23, 0.27, 0.44 and 0.45 ng/mL. After spiking 0.5 ng/mL, the detected concentrations were 0.69, 0.72, 0.73, 0.75, 0.91 and 1.02 ng/mL. And after spiking 2.0 ng/mL, the detected concentrations were 2.03, 2.15, 2.44, 2.35, 2.57 and 2.44 ng/mL. In result, the consistency was calculated as 93–113% with CV of 7%. In addition, we also performed the above spiking-experiments towards standard samples (possessing the same K1 concentrations with the serum samples). The obtained peak area ratio of K1 to IS was quite comparable to the serum group (with the difference less than 9.7%). All these results indicated the matrix effect (as well as the processing recovery) was acceptable after IS-normalization.
The results of accuracy and imprecision were summarized in Supplemental Table S10. In all cases, the accuracy was in the range of 91.5 to 112.2%. And the corresponding imprecision was observed less than 13.5%. These results exhibited the favorable reliability of the developed method on K1 quantification.
During stability test, no significant degradation was observed under all the tested condition (apparent recovery 89.4–106.6%), which demonstrated K1 was relatively stable in darkness. In contrast, when serum sample was uncovered during storage at 25 °C, the recovery revealed gradual decrease (92.3% for 12 h, 88.1% for 24 h, 70.7% for 48 h and 51.1% for 72 h). Such results indicated that to obtain convincing K1 status, long-time light exposure should be strictly forbidden before measurement.
3.3 Method application
By using the developed method, K1 was successfully quantified in all the enrolled samples (as shown in Supplemental Table S11). The concentration was determined as 0.07 to 2.94 ng/mL (0.57 ± 0.49 ng/mL). The typical chromatograms from one sample with the concentration (0.14 ng/mL) near LLOQ level were shown in Fig. 4. Such results exhibited the excellent applicability of this new method in practice.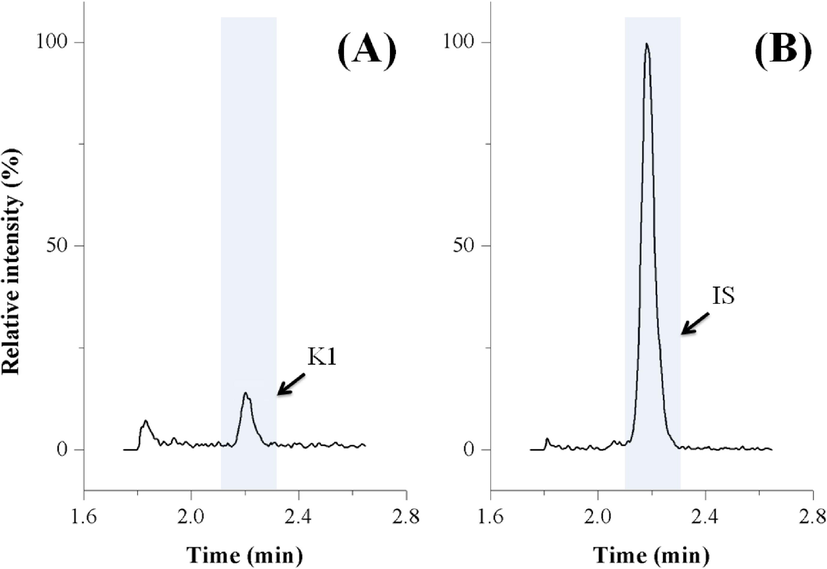
The typical LC-MS/MS chromatograms of K1 and IS (K1-d7) from a clinical sample with the K1 concentration of 0.14 ng/mL.
4 Discussion
In the past two decades, significant efforts have been dedicated to develop applicable methodology for K1 analysis in blood. Currently, it is known that precise quantification of K1 requires the utilization of PP to release it from the matrix, followed by concentration and purification through LLE or SPE (if necessary). However, what remains unclear is whether different PP methods exhibit varying release efficiencies, whether the extraction process will decrease the K1 recovery, and to what extent the sample matrix can weaken the detecting performance.
According to the pretreatment approach (as summarized in Table 1), the previous methods can be divided into three categories: (i) PP coupled with SPE (Riphagen et al., 2016; Sandvik et al., 2017; Andersen et al., 2019); (ii) PP coupled with LLE after excluding precipitate (Konieczna et al., 2016; Dunovska et al., 2019; Zhang et al., 2020); (iii) PP coupled with LLE with precipitate (Nannapaneni et al., 2017; Hu et al., 2018; Arachchige et al., 2021; Card et al., 2022; Suhara et al., 2005; Ducros et al., 2010; Gentili et al., 2014). However, unlike the case of other fat-soluble vitamins (including vitamin A/D/E), K1 assessment was still not popularized in routine clinical laboratories. The results from this study illustrated the following three main issues for K1 detection.
First, the releasing-efficiency of K1 can be encumbered by the imperfect PP strategies. According to the results from EXPERIMENT I, to achieve satisfactory releasement (>80%) (Yuan et al., 2016), the required volume ratio of serum to precipitator should be 1:16 for methanol and 1:8 for ethanol. However, in the methods of (i) PP coupled with SPE (Riphagen et al., 2016; Sandvik et al., 2017; Andersen et al., 2019) and (ii) PP coupled with LLE after excluding precipitate (Konieczna et al., 2016; Dunovska et al., 2019; Zhang et al., 2020), the applied volume ratio was only in the range of 2:1 to 1:4. This phenomenon indicated K1 may be partly lost (along with discarding the precipitate) before the SPE or LLE taking place. Apart from the frequently used methanol, ethanol and acetonitrile, in the present work, we found isopropanol was a superior releaser for K1 from serum matrix. Excellent releasing-efficiency (>90%) can be achieved with the volume ratio of 1:3. Such low dilution factor raised the possibility of direct analysis of K1 after PP without further condensation process. Noteworthily, there has been one study introducing isopropanol during PP (Andersen et al., 2019). Nonetheless, the irritant ammonium hydroxide was involved at the same time. And the method was built on an on-line SPE system, which somewhat compromised the accessibility for routine laboratories.
Second, during LLE, the partition effect between precipitator and extractor was prone to decrease the absolute recovery of K1. Comparing the results from EXPERIMENT I to EXPERIMENT II, when 400 μL and 800 μL ethanol was used for PP, 17.0% and 51.2% of K1 would be lost after performing LLE with n-hexane (1 mL). Thereupon, to eliminate such adverse effect, multi-cycle LLE or LLE with large volume of extractor was always required, which in turn lowered the throughput of the extracting and drying process. All the LLE methods (Nannapaneni et al., 2017; Hu et al., 2018; Suhara et al., 2005; Ducros et al., 2010; Gentili et al., 2014; Konieczna et al., 2016; Dunovska et al., 2019; Zhang et al., 2020; Arachchige et al., 2021; Card et al., 2022) could suffer from such issue more or less. On the other hand, one latest study imported acetonitrile-based PP coupled with LLE (Arachchige et al., 2021). According to the results from Fig. 2 and Supplemental Table S5, the releasing-efficiency was probably less than 20% through those treatments. We speculated such low recovery may be the main reason for the relatively high LLOQ (0.7 ng/mL) of that methodology.
Third, the detecting efficiency for K1 could be apparently influenced by the volume of injected serum per run. For long, there were inconsistent conclusions about whether LLE was adequate (Nannapaneni et al., 2017; Hu et al., 2018; Zhang et al., 2020) or inadequate (Ducros et al., 2010; Gentili et al., 2014; Dunovska et al., 2019; Arachchige et al., 2021) to exclude ion suppression for K1 analysis. According to the results from EXPERIMENT III, after condensation with LLE, the ion suppression effect on K1 and IS would reach over 75%, if the volume of injected serum per run was higher than 20 μL. Such outcome indicated that, without further purification by SPE or cold-induced lipid elimination (Gentili et al., 2014; Dunovska et al., 2019; Arachchige et al., 2021; Card et al., 2022), the methods depending on the crude LLE treatment would likely preserve certain matrix interference for K1 detection (especially if the volume of injected serum per run was high). In regard of the signal intensity (as shown in Fig. 3B and Supplemental Table S8), benefitting from the low ion suppression effect, the injected serum per run of 2 to 5 μL could realize comparable detecting sensitivity with the case of 50 μL or more.
Collecting all the above findings, we developed the isopropanol-based PP method for K1 quantification. The volume ratio of serum to isopropanol was set as 1:4 so that the excellent releasing-efficiency (>90%) and favorable dilution effect could be achieved simultaneously. Afterward, 10 μL of the supernatant was injected for LC-MS/MS analysis, which provided the volume of injected serum per run of 2 μL. To be noticed, even with this minimized sample involvement, we found a refresh step (2.7–6.0 min, as shown in Supplemental Table S1) was necessary to prevent matrix accumulation on column. If the LC program was terminated at 2.7 min, the signal intensity of K1 would sharply decrease>90% after successively analyzing three samples. Under these optimized conditions, K1 could be reliably determined in the range of 0.1–2.5 ng/mL by using only 25 μL of serum sample. After enrolling one hundred random samples to test the applicability, only four of them fell outside the range (as shown in Supplemental Table S11). Such results demonstrated the quantitative range could well fulfill the routine requests for clinical evaluation of K1 status in vivo.
5 Conclusion
In comparison to previous studies, the most significant advancement of this research lies in the successful quantification of endogenous vitamin K1 in serum using a direct PP approach. By circumventing multiple pretreatments such as LLE, SPE, and solvent evaporation typically found in existing methods, the new method exhibits high throughput, cost-effectiveness, and ease of operation. We believe these characteristics make it very suitable for widespread adoption in routine clinical laboratories.
CRediT authorship contribution statement
Hai-Bo Wang: Investigation, Validation. Rui Peng: Investigation. Juan Le: Methodology. Shao-Ting Wang: Conceptualization, Methodology, Validation, Writing – original draft, Writing – review & editing, Supervision.
Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities (grant number 2042020kf0067) and the Interdisciplinary Innovative Talents Foundation from Renmin Hospital of Wuhan University (grant number JCRCFZ-2022-001). And also thank Professor Yu-Qi Feng (Department of Chemistry, Wuhan University) for the kind academic counselling on method validation.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Quantitation of vitamin K1 in serum using online SPE-LC-MS/MS and the challenges of working with vitamin K. J. Chromatogr. B. 2019;1117:41-48.
- [CrossRef] [Google Scholar]
- Absolute quantification of eleven A, D, E and K vitamers in human plasma using automated extraction and UHPLC-Orbitrap MS. Anal. Chim. Acta. 2021;1181:338877
- [CrossRef] [Google Scholar]
- Characterization and traceability of two generations of standard reference material for the measurement of vitamin K1(phylloquinone) at endogenous concentrations in human plasma and serum. Biomed. Chromatogr.. 2022;36:e5378.
- [Google Scholar]
- Laboratory assessment of vitamin K status. J. Clin. Pathol.. 2020;73:70.
- [CrossRef] [Google Scholar]
- Quantitative determination of plasma vitamin K1 by high-performance liquid chromatography coupled to isotope dilution tandem mass spectrometry. Anal. Biochem.. 2010;401:7-14.
- [CrossRef] [Google Scholar]
- LC–MS/MS quantitative analysis of phylloquinone, menaquinone-4 and menaquinone-7 in the human serum of a healthy population. PeerJ. 2019;7:e7695.
- [CrossRef] [Google Scholar]
- Vitamin K plasma levels determination in human health. Clin. Chem. Lab. Med. (CCLM). 2017;55:789-799.
- [Google Scholar]
- Vitamin K effects in human health: new insights beyond bone and cardiovascular health. J. Nephrol.. 2020;33:239-249.
- [CrossRef] [Google Scholar]
- Rapid, high performance method for the determination of vitamin K1, menaquinone-4 and vitamin K1 2,3-epoxide in human serum and plasma using liquid chromatography-hybrid quadrupole linear ion trap mass spectrometry. J. Chromatogr. A. 2014;1338:102-110.
- [CrossRef] [Google Scholar]
- A study of the prevalence of vitamin K deficiency in patients with cancer referred to a hospital palliative care team and its association with abnormal haemostasis. J. Clin. Pathol.. 2008;61:537.
- [CrossRef] [Google Scholar]
- A simple, sensitive, and high-throughput LC-APCI-MS/MS method for simultaneous determination of vitamin K1, vitamin K1 2,3-epoxide in human plasma and its application to a clinical pharmacodynamic study of warfarin. J. Pharm. Biomed. Anal.. 2018;159:82-91.
- [CrossRef] [Google Scholar]
- Vitamin K and cardiovascular complications in chronic kidney disease patients. Kidney Int.. 2021;100:1023-1036.
- [CrossRef] [Google Scholar]
- The LC-MS method for the simultaneous analysis of selected fat-soluble vitamins and their metabolites in serum samples obtained from pediatric patients with cystic fibrosis. Journal Of Pharmaceutical And Biomedical Analysis. 2016;124:374-381.
- [CrossRef] [Google Scholar]
- New LC-MS/MS method with single-step pretreatment analyzes fat-soluble vitamins in plasma and amniotic fluid. J. Lipid Res.. 2018;59:1783-1790.
- [CrossRef] [Google Scholar]
- Development of a liquid chromatography mass spectrometry method for the determination of vitamin K1, menaquinone-4, menaquinone-7 and vitamin K1–2,3 epoxide in serum of individuals without vitamin K supplements. Clin. Chem. Lab. Med.. 2022;60:1011-1019.
- [CrossRef] [Google Scholar]
- A sensitive and rapid UFLC-APCI-MS/MS bioanalytical method for quantification of endogenous and exogenous Vitamin K1 isomers in human plasma: Development, validation and first application to a pharmacokinetic study. Talanta. 2017;164:233-243.
- [CrossRef] [Google Scholar]
- Measurement of plasma vitamin K1 (phylloquinone) and K2 (menaquinones-4 and -7) using HPLC-tandem mass spectrometry. Clin. Chem. Lab. Med. (CCLM). 2016;54:1201-1210.
- [CrossRef] [Google Scholar]
- Routine Supercritical Fluid Chromatography Tandem Mass Spectrometry Method for Determination of Vitamin K1 Extracted from Serum with a 96-Well Solid-Phase Extraction Method. The Journal of Applied Laboratory Medicine. 2017;1:637-648.
- [CrossRef] [Google Scholar]
- Circulating Phylloquinone Concentrations of Adults in the United States Differ According to Race and Ethnicity. J. Nutr.. 2012;142:1060-1066.
- [CrossRef] [Google Scholar]
- Perspective: Evidence before Enthusiasm-A Critical Review of the Potential Cardiovascular Benefits of Vitamin K. Adv. Nutr.. 2021;12:632-646.
- [CrossRef] [Google Scholar]
- Method for the Determination of Vitamin K Homologues in Human Plasma Using High-Performance Liquid Chromatography-Tandem Mass Spectrometry. Anal. Chem.. 2005;77:757-763.
- [CrossRef] [Google Scholar]
- Plasma phylloquinone (vitamin K1) concentration and its relationship to intake in British adults aged 19–64 years. Br. J. Nutr.. 2006;96:1116-1124.
- [CrossRef] [Google Scholar]
- Investigation of the “true” extraction recovery of analytes from multiple types of tissues and its impact on tissue bioanalysis using two model compounds. Anal. Chim. Acta. 2016;945:57-66.
- [CrossRef] [Google Scholar]
- Quantification of menadione from plasma and urine by a novel cysteamine-derivatization based UPLC–MS/MS method. J. Chromatogr. B. 2017;1063:107-111.
- [CrossRef] [Google Scholar]
- A concise review of quantification methods for determination of vitamin K in various biological matrices. J. Pharm. Biomed. Anal.. 2019;169:133-141.
- [CrossRef] [Google Scholar]
- Reversed phase UPLC/APCI-MS determination of Vitamin K1 and menaquinone-4 in human plasma: Application to a clinical study. J. Pharm. Biomed. Anal.. 2020;183:113147
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.105023.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




