

Translate this page into:
Microwave assisted synthesis of 2-amino-4-chloro-pyrimidine derivatives: Anticancer and computational study on potential inhibitory action against COVID-19
⁎Corresponding authors at: Deanship of Scientific Research, Imam Abdulrahman Bin Faisal University, P.0. Box 1982, Dammam 31441, Saudi Arabia (F. Qureshi); Department of Nano-Medicine Research, Institute for Research and Medical Consultations (IRMC), Imam Abdulrahman Bin Faisal University, P.O. Box 1982, Dammam 31441, Saudi Arabia (M. Nawaz) frqureshi@iau.edu.sa (Faiza Qureshi), mnnmuhammad@iau.edu.sa (Muhammad Nawaz),
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Pyrimidine derivatives have been synthesized and characterized by spectroscopic and analytical tools. Anticancer activity of synthesized derivatives (1–7) was tested against HCT116 and MCF7 cell line. Molecular docking was completed for derivatives (1 – 7) versus SARS-CoV-2 main protease. The derivative 6 was the best candidate with a binding energy score of – 8.12 kcal/mol and inhibition constant of 1.11 µM. Computational ADME, drug-likeness, pharmacokinetic attributes and toxicity of derivatives 1 – 7 were investigated.
Abstract
We report microwave synthesis of seven unique pyrimidine anchored derivatives (1–7) incorporating multifunctional amino derivatives along with their in vitro anticancer activity and their activity against COVID-19 in silico. 1–7 were characterized by different analytical and spectroscopic techniques. Cytotoxic activity of 1–7 was tested against HCT116 and MCF7 cell lines, whereby 6 exhibited highest anticancer activity on HCT116 and MCF7 with EC50 values of 89.24 ± 1.36 µM and 89.37 ± 1.17 µM, respectively.
Molecular docking was performed for derivatives (1–7) on main protease for SARS-CoV-2 (PDB ID: 6LU7). Results revealed that most of the derivatives had superior or equivalent affinity for the 3CLpro, as determined by docking and binding energy scores. 6 topped the rest with highest binding energy score of −8.12 kcal/mol with inhibition constant reported as 1.11 µM. ADME, drug-likeness, and pharmacokinetics properties of 1–7 were tested using Swiss ADME tool. Toxicity analysis was done with pkCSM online server.
All derivatives showed high GI absorption. Except 1 and 3, all derivatives showed blood brain barrier permeability. Most derivatives showed negative logKp values suggesting derivatives are less skin permeable and bioavailability score of all derivatives was 0.55. The toxicity analysis demonstrated that all derivatives have no skin sensitization properties. 6 and 7 showed maximum tolerated dose (Human) values of −0.03 and −0.018, respectively and absence of AMES toxicity.
Keywords
Chloropyrimidine
Microwave synthesis
Anticancer
Molecular docking
Coronavirus
SARS-CoV-2

1 Introduction
Regardless of the fast-track pervasive research and significant advancement, cancer remains one of the fatal diseases and still warrants unceasing research for newer anticancer molecules (Napiórkowska et al., 2019). The unresolved capacity of cancer cells developing resistance to remedies poses a profound barrier in chemotherapy making incessant efforts for identification of selective and less-toxic anti-cancer agents crucial (Spaczyńska et al., 2019).
Many anticancer potent derivatives of pyrimidine are reported till now. N-trisubstituted pyrimidine compounds for human tumor cell lines in vitro (Luo et al, 2014) and 1,2,3-triazole-pyrimidine derivatives were effective against quite a few cancer cell lines (Ma et al., 2014). Halogenated pyrimidine derivatives were tested in vitro versus HCT116, A549, K562 and U937 cell lines (Munikrishnappa et al., 2016). Nsubstituted pyrimidine reported with high potential against T-47D and MDA-MB-468 cell lines and greater inhibition versus several kinases (El-Deeb and Lee, 2010). Aminobenzazolyl pyrimidine demonstrated great anticancer potential when tested for leukaemia, renal and prostate cancers (Chikhale et al., 2018). Also, diarylurea moiety is reported for treatment of cancer and a related example is Sorafenib (Wilhelm et al., 2006).
Our research group has been working on multifunctional amino derivatives synthesis (Nawaz et al., 2020; Rahim et al., 2020; Taha et al., 2020; Gollapalli et al., 2018; Taha et al., 2018; Ibrahim et al., 2020; Nawaz et al., 2014) with antidiabetic and cytotoxic activity (Qureshi et al., 2020; Nawaz et al., 2022), and for this research we synthesized pyrimidine derivatives. Pyrmidine, a multi therapeutical nitrogen carrying heterocyclic ring, proven against carbohydrate digesting enzymes, bacteria and even cancer cell line (Taha et al., 2017; Slyusarenko et al., 1989; Nawaz et al., 2016; Soylem et al., 2017; Altaf et al., 2015; Krause et al., 2017; Salar et al., 2017) and also many other therapeutic benefits, was selected as parent molecule, to be treated with a variety of functional moieties and the resulting compounds were characterized as per protocol.
Coronavirus COVID-19 is an RNA virus that emerged and started its spread since December 2019 in China. Later, it was recognized that this virus is responsible for causing the transmittable infections between humans leading to severe infectious disease COVID-19. Resembling to the severe acute respiratory syndrome, the new pandemic triggering virus is labeled as SARS-CoV-2. A single chain positive RNA genome containing a spiral nucleotide. Corona virus replication depends upon RNA-dependent RNA polymerase (RDR) (Mei-Yue et al., 2020).
Pandemic borne efforts aimed on repurposing known drugs in hopes of finding fast tracked clinical solution, however, it did not work effectively and the search for a new drug in ongoing. Up till July 25, 2022, 576,023,223 cases were reported worldwide, including 6,405,351 deaths. The spread when charted show increasing trend peaking in January 2022 (Worldometer, 2022) with situation worse in some countries.
Till now, several drug candidates have been tested for the control and prevention of coronavirus. Since, one main enzyme is responsible for copying genetic material of coronavirus, any compound with the potential to inhibit RDR could be a promising therapeutic candidate against SARS-CoV-2 (Chhetri et al., 2021). Stating the obvious and following the trend with other human viruses like hepatitis and HIV, blocking or inhibiting the replication of the virus holds the key to the development of the effective antiviral or therapeutic agent against corona virus(es). Mpro (PDB ID: 6LU7) has been identified as the main protease (Zumla et al., 2016; Shirato et al., 2013) as it is vital for the coronavirus replicase polyprotein (Fehr and Perlman, 2015; Somboon et al., 2021) and has been the focus of rapid scientific studies around the globe with respect to corona virus(es). This research also used Mpro (PDB ID: 6LU7) to measure the effectiveness of prepared therapeutic agents.
To save valuable time, in-silico molecular modeling studies were used to confirm the ability of drug candidates. It is an effective model and has been used by researchers to help combat Covid-19. Nelfinavir, for example, was the recommended antiviral based on the homology models and 3D binding similarity to Mpro (Xu et al., 2020; Hatada et al., 2020). The docking affinity between the candidates and [PDB ID: 6LU7] (main protease for SARS-CoV-2) was investigated using AutoDock 4.2 from MGL v 1.5.6. (Morris et al., 1998; Morris et al., 2009; Ansari et al., 2020). Computational assessment of ADME, Drug-likeness and evaluation of toxicity (Daina et al., 2017; Pires et al., 2015; Jamal et al., 2020) were also carried out along with target prediction (Gfeller et al., 2014).
2 Materials and methods
2.1 Materials
Pure grade reagents were utilized, procured from Sigma Aldrich. SMP10 (Stuart) melting point apparatus was used for the determination of melting points. IR spectra were acquired on a Perkin Elmer instrument employing ATR accessory. NMR (1H and 13C) was performed via Bruker 400 MHz NMR spectrometers. “δ“, ppm (parts per million) was used to report proton chemical shifts while “J”, coupling constant was denoted by Hz (Hertz) and the terms singlet “s“, doublet “d”, triplet “t“, multiplet “m” and broad “br“ were used. TLC on silica gel Al foil was visualized under UV lamp.
2.2 General procedure for the synthesis of 2-amino-4-chloro-pyrimidine derivatives (1–7)
2 mmol of 2-amino-4-chloro-pyrimidine was weighed and transferred into microwave (CEM, Discovery, USA) reaction vial; 1 mL of anhydrous propanol was added to it and keeps stirring at room temperature. 2 mmol of different substituted amine was added to the reaction vial. After stirring 200 µL triethylamine was also introduced. Reaction was performed at 120–140 °C for 15–30 min and monitored by TLC. After cooling the obtained precipitate was dispersed in saturated sodium bicarbonate in water for the product to be extracted using ethyl acetate. Anhydrous sodium sulfate (Na2SO4) was used for drying of extract, following filtration, and being concentrated under reduced pressure to yield 1–7 derivatives (Scheme 1).
Synthesis of 2-amino-4-chloro-pyrimidine derivatives (1–7).
2.2.1 4-(4-methylpiperazin-1-yl)pyrimidin-2-amine (1)
Yield: (1.736 g, 54 %); mp:180 °C; IR (ATR, cm−1): 3318(—NH2), 3155(—CH aromatic),2868 (—CH aliphatic), 1651 (—C⚌N aromatic), 1582 (—C⚌C aromatic),1469 (—C⚌C aromatic); 1H NMR (400 MHz, CDCl3): δ 2.20 (s, 3H),2.29–2.30(m, 4H), 3.47–3.48 (m, 4H), 5.94 (s br, —NH2) 5.99 (AB quartet, 1H, J = 5.6 Hz)), 7.73 (AB quartet, 1H, J = 6.0 Hz); 13C NMR (100 MHz, CDCl3): δ 43.57, 46.24, 54.77, 93.53, 157.28, 162.80, 163.45; elemental analysis calc for C9H15N5, %C 55.89, %H 7.76, %N 36.2; found %C 55.08, %H 7.78, %N 34.7.
2.2.2 4-(4-methylpiperidin-1-yl)pyrimidin-2-amine (2)
Yield: (1.736 g, 54 %); mp: 150 °C; IR (ATR, cm−1): 3311(—NH2), 3125(—CH aromatic) 2946(—CH aliphatic), 2921(—CH aliphatic),1647(—C⚌N aromatic),1582(—C⚌C aromatic),1438 (—C⚌C aromatic); 1H NMR (400 MHz, CDCl3): δ 0.89 (d, J = 6.4 Hz, 3H),0.96–0.99 (m, 2H), 1.59–1.62 (m, 3H), 2.73–2.76(m, 2H), 4.24–4.27 (m, 2H), 5.89 (s br, —NH2) 5.98 (AB quartet, 1H, J = 6.0 Hz)), 7.70 (AB quartet, 1H, J = 6.0 Hz); 13C NMR (100 MHz, CDCl3): δ 22.23, 31.11, 33.86,43.94,93.46,157.11,162.47,163.52; elemental analysis calc for C10H16N4, %C 62.41, %H 8.32, %N 29.13; found %C 61.8, %H 8.43, %N 28.06.
2.2.3 4-(4-(pyrimidin-2-yl) piperazine-1-yl) pyrimidin-2-amine (3)
Yield: (1.736 g, 54 %); mp: 200 °C; IR (ATR, cm−1): 3434(—NH2), 2848(—CH aliphatic),1635(—C⚌N aromatic),1583 (—C⚌C aromatic); 1H NMR (400 MHz, CDCl3): δ 3.60–3.61 (m, 4H), 3.77–3.79 (m, 4H), 6.03 (s br, —NH2), 6.05 (AB quartet, 1H, J = 5.6 Hz), 6.64–6.66 (m, 1H), 7.83 (AB quartet, 1H, J = 5.6 Hz), 8.38 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 39.99, 43.38,93.59, 110.81, 157.32, 158.43, 161.63, 162.78, 163.43; elemental analysis calc for C12H15N7, %C 55.97, %H 5.83, %N 38.09; found %C 55.55, %H 5.93, %N 36.95.
2.2.4 4-(4-(4-fluorophenyl)piperazine-1-yl) pyrimidin-2-amine (4)
Yield: (1.736 g, 54 %); mp: 220 °C; IR (ATR, cm−1): 3405(—NH2), 3297(—CH aromatic), 3137(—CH aromatic), 2884(—CH aliphatic), 2844(—CH aliphatic), 1636(—C⚌N aromatic), 1546(—C⚌C aromatic), 1507(—C⚌C aromatic); 1H NMR (400 MHz, CDCl3): δ 3.09 (t, 4H, 4.8 Hz), 3.64 (t, 4H, 4.8 Hz), 6.02 (s, —NH2), 6.06 (AB quartet, 1H, J = 6.0 Hz), 6.97–7.00 (m, 2H), 7.03–7.08 (m, 2H), 7.76 (AB quartet, 1H, J = 6.0 Hz); 13C NMR (100 MHz, CDCl3): δ 43.57, 49.24, 96.58, 115.89, 118.06, 118.13, 157.42, 162.75, 163.48; elemental analysis calc for C14H16FN5, %C 61.47, %H 5.85, %N 25.61; found %C 61.58, %H 6.03, %N 25.75.
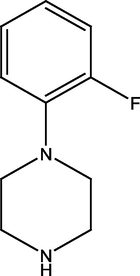
2.2.5 4-(4-(2-fluorophenyl)piperazine-1-yl) pyrimidin-2-amine (5)
Yield: (1.736 g, 54 %); mp: 165 °C; IR (ATR, cm−1): 3480(—NH2), 3264(—CH aromatic), 3112(—CH aromatic),1624(—C⚌N aromatic),1585(—C⚌C aromatic), 1546(—C⚌C aromatic); 1H NMR (400 MHz, CDCl3): δ 3.01–3.03 (m, 4H), 3.65–3.67 (m, 4H), 6.02 (s, —NH2), 6.05 (AB quartet, 1H, J = 6.0 Hz), 6.98–7.16 (m, 4H), 7.77 (AB quartet, 1H, J = 5.6 Hz); 13C NMR (100 MHz, CDCl3): δ 43.74, 50.45, 93.52, 116.34, 116.54, 119.98, 123.14, 125.34, 140.01, 157.45, 162.78, 163.48; elemental analysis calc for C14H16FN5, %C 61.49, %H 5.85, %N 25.61; found %C 58.66, %H 5.67, %N 24.61.
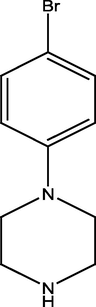
2.2.6 4-(4-(4-bromophenyl)piperazine-1-yl) pyrimidin-2-amine (6)
Yield: (1.736 g, 54 %); mp: 245 °C; IR (ATR, cm−1): 3464(—NH2), 3402(—NH2),3285(—CH aromatic), 2835(—CH aliphatic), 1546(—C⚌C aromatic), 1438(—C⚌C aromatic); 1H NMR (400 MHz, CDCl3): δ 3.16 (t.4H, J = 4.8 Hz), 3.63 (t.4H, J = 4.8 Hz), 6.03 (s, —NH2), 6.05 (AB quartet, 1H, J = 6.0 Hz), 6.92 (AB quartet, 2H, J = 8.8 Hz), 7.35 (AB quartet, 2H, J = 8.8 Hz)., 7.77 (AB quartet, 1H, J = 6.0 Hz); 13C NMR (100 MHz, CDCl3): δ 43.34, 48.14, 93.56, 110.75, 118.04, 131.99, 150.50, 157.42, 162.72, 163.47; elemental analysis calc for C14H16BrN5, %C 50.26, %H 4.78, %N 20.94; found %C 49.28, %H 4.71, %N 20.45.
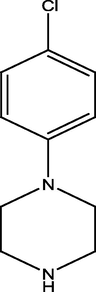
2.2.7 4-(4-(4-chlorophenyl)piperazine-1-yl) pyrimidin-2-amine (7)
Yield: (1.736 g, 54 %); mp: 270 °C; IR (ATR, cm−1): 3414(—NH2), 3291(—CH aromatic), 2981(—CH aliphatic),2817(—CH aliphatic),1633(—C⚌N aromatic),1586(—C⚌C aromatic),1544(—C⚌C aromatic); 1H NMR (400 MHz, CDCl3): δ 3.38–3.40 (m, 4H), 3.62–3.66 (m, 4H), 6.02 (s, —NH2), 6.04(AB quartet, 1H, J = 6.4 Hz), 6.82 (AB quartet, 2H, J = 6.4 Hz), 7.78 (AB quartet, 1H, J = 5.6 Hz), 8.16 (AB quartet, 1H, J = 5.2 Hz); 13C NMR (100 MHz, CDCl3): δ 43.95, 45.33, 93.51, 108.73, 150.30, 154.71, 156.0, 157.43, 164.30, 166.90; elemental analysis calc for C14H16ClN5, %C 57.98, %H 5.52, %N 24.16; found %C 57.95, %H 5.47, % N 24.12.
2.3 Anticancer activity
2.3.1 Cell culture and cell viability assay
Anticancer activity of synthesized derivatives (1–7) was studied against human colon colorectal-HCT116 and breast cell line-MCF7 (ATCC®) and were maintained at 37° C in DMEM medium accompanied with 1 percent penicillin–streptomycin, 1 percent l-glutamine, and 10 % fetal bovine serum in a 5 % CO2 humid chamber. After trypsinization with Trypsin-EDTA 0.25 %, cells were incubated for 5 min at 5 % CO2 humid, then neutralized with a 1:1 of DMEM culture medium and centrifuged at 1000 rpm for 5 min. After 3 to 6 passages, cells were plated in 96-well plates (104 cells) and maintained in DMEM for 24 h. The cells were individually exposed to various concentrations (50, 100, 200 and 300 μM) of synthesized derivatives (1–7) and incubated for 24 h.. The viability assay was checked thrice by adding MTT solution (10 µL) into each well including untreated cells as positive controls. The reaction was performed for four hours at 37 °C. Afterwards, dimethyl sulfoxide (100 µL) was added following absorbance measurement at 570 nm on SYNERGY Neo2 multi-mode reader (Biotek), and cell viability was computed as:
2.4 Docking studies
2.4.1 Preparation of derivatives for the docking
The chemical structure of synthesized derivatives (1–7) was drawn using ChemDraw and Remdesivir (REM) (Wishart, et al., 2017) was used as standards for the docking study. Corina molecular structure generator tool (Andronico, et al., 2011) was employed to produce 3-Dimensional (3D) structures following use of Discovery Studio visualizer 2020 (Jamal et al., 2021) to apply CHARMM force field.
2.4.2 Preparation of receptors
The configuration of crystal SARS-CoV-2 main protease N3 (inhibitor) complex [PDB ID: 6LU7] was downloaded from Protein Data Bank (Berman, et al., 2000; Ansari et al., 2020) and 3-D construction was achieved by X-ray diffraction method (observed resolution 2.16 Å, R-Value Free 0.235, R-Value Work 0.202 and R-Value Observed 0.204). After removal of N3 inhibitor, HETATOM and water molecules from the indigenous SARS-CoV-2 protease 3D structure, CHARMM force fields simulations were carried out on both 3D structures using Discovery Studio visualizer 2020 for energy minimization process.
2.4.3 AutoDock calculation
The computational prognostics for the tested derivatives and [PDB ID: 6LU7] (SARS-CoV-2 main protease) were performed using AutoDock 4.2 (MGL v 1.5.6). AutoDock (Lamarckian Genetic Algorithm) employs empirical free energy force field for the receptor interaction as score. The AutoDock was executed after realizing the default docking parameters on the active site, yet, for the coverage of maximum area inside the grid box which can accommodate the selected active site key residues His41, Cys145 and Glu166 identified through literature survey in the grid box (60x60x60 Å), the grid center point co-ordinates X (−15.253), Y (14.22) and Z (65.592) were set for PDB ID:6LU7 with the default grid points spacing (0.375 Å).
2.4.4 ADME, drug-likeness and toxicity
In Silico investigations for ADME (absorption, distribution, metabolism, and excretion), drug-likeness, and pharmacokinetics attributes of derivatives 1–7 were examined by means of SwissADME online tool from SIB, Switzerland (Daina et al., 2017). Further executed toxicity analysis was done by using pkCSM online server (Pires, et al., 2015) which can quickly assess and predict the toxicity properties of derivatives of interest.
3 Results
3.1 Anticancer activity
The anticancer activities of synthesized derivatives (1–7) were tested against human colon colorectal (HCT116) and breast cancer (MCF7) cell lines by employing MTT assay. EC50 of derivatives are presented in Table 1 and cell viability (%) is shown in the Figs. 1–2. Among the synthesized derivatives, derivative 6 having bromophenyl piperazine moiety at 4 position of the pyrimidine ring exhibited highest anticancer activity on both human colon colorectal (HCT116) with EC50 89.24 ± 1.36 µM and breast cancer (MCF7) cell lines with EC50 89.37 ± 1.17 µM. Second most active derivative was 1, with 4-methyl piperazine moiety and EC50 values were 209.17 ± 1.23 µM and 221.91 ± 1.37 µM against human colon colorectal (HCT116) and on breast cancer cell lines (MCF7) respectively. While EC50 of doxorubicin as a positive control was 2 µM and 0.98 µM against human colon colorectal (HCT116) and on breast cancer cell lines (MCF7) respectively.
S.No.
R
EC50 (µM ± SD)
HCT116
MCF7
1
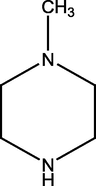
209.17 ± 1.23
221.91 ± 1.37
2
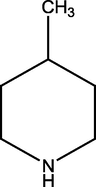
272.48 ± 1.59
271.20 ± 1.27
3
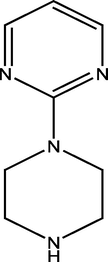
368.20 ± 2.02
335.23 ± 1.47
4
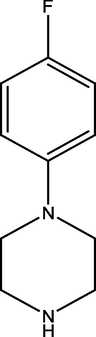
334.54 ± 1.06
191.35 ± 1.22
5
349.41 ± 1.14
252.07 ± 1.78
6
89.24 ± 1.36
89.37 ± 1.17
7
458.89 ± 1.41
275.79 ± 1.51
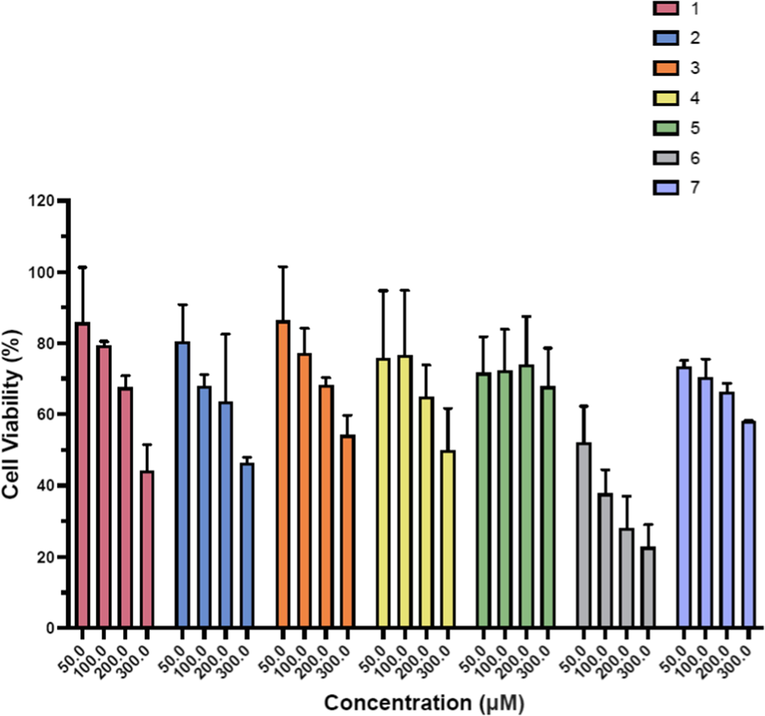
% Cell viability against colon colorectal (HCT116) using pyrimidine derivatives (1–7).
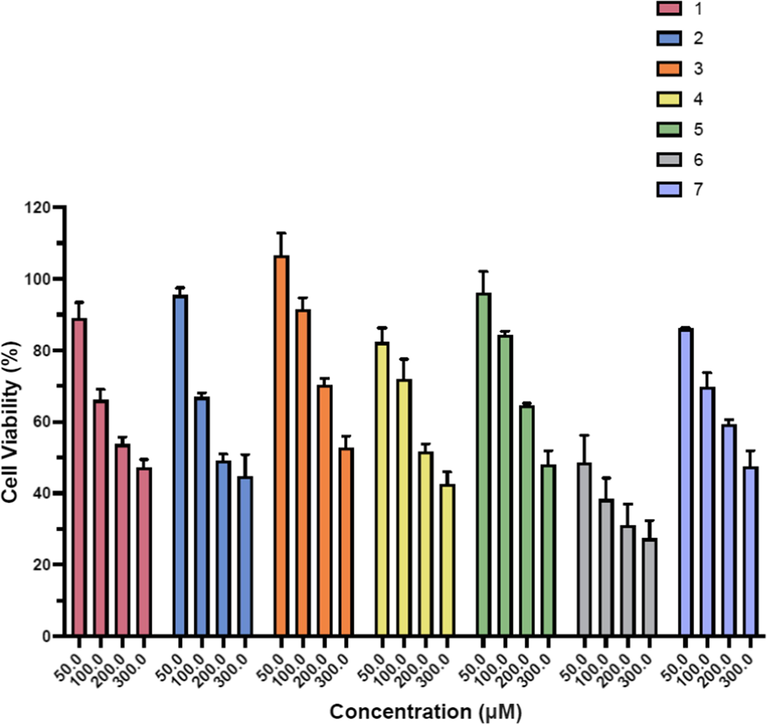
% Cell viability against breast cancer cell lines (MCF7) using pyrimidine derivatives (1–7).
3.2 Molecular docking
Because of the tremendous therapeutic and biomedical importance, a series of seven pyrimidine substituted derivatives were synthesized and characterized in order to determine their binding efficiency versus the main protease of SARS-CoV-2 using molecular docking and ADMET studies. The molecular interaction results of synthesized pyrimidine derivatives (1–7) with 3CLpro (PDB ID: 6LU7) analyzed by docking experimentation are shown in Table 2 and Figs. 3 and 4. The results revealed that most of the pyrimidine derivatives had superior or equivalent affinity for the 3CLpro, as determined by binding energy scores. Derivative 6 was the top candidate with a binding energy docking score of −8.12 kcal/mol along with inhibition constant of 1.11 µM. 6 interacted with the 3CLpro via formation of three hydrogen bonds (bond lengths 1.98–3.74 Å). Amino acids Phe140 and Asn142 were crucially involved in the hydrogen bonds formation. His41, Met49, Tyr54, Leu141, His163, His164, Met165, Glu166, His172, Asp187, Arg188 amino acid residues participated in hydrophobic interaction (Table 2) It was also observed that Met165 formed pi-sulfur bond, His42 formed Pi-Pi stacking while Met49 was involved in Pi-Alkyl interaction (Fig. 4f1). The derivative 6 was closely followed by 7 with binding energy score of −7.83 kcal/mol and inhibition constant 1.83 µM having two hydrogen bond formations after the involvement of Phe140 and Asn142 and bond length of 2.06–2.93 Å were noted. Leu141, His172, His163, Glu166, Gln189, Arg188, Asp187, Tyr54, His41 amino acid residues participated in hydrophobic interaction (Table 2). The amino acid residues like Cys145, Met49 and Pro52 were involved in Alkyl/pi-alkyl interaction while Met165 was form Pi-Sulfur bond (Fig. 4g1). The binding energy scores of derivative 3, 4 and 5 were −6.61, −7.03 and −7.06 and kcal/mol, respectively, with the formation of five, two and five hydrogen bonds together with hydrophobic interactions. It is interesting to note that derivative 3, 4, 5, 6 and 7 exhibited better binding affinity than the control standard Remdesivir drug, which exhibited binding energy of −6.41 kcal/mol, inhibition constant of 19.91 µM and His41, Asn142, Gly143, Cys145, Met165, Glu166, Leu167, Pro168, His172, Gln189, Thr190, Gln192 amino acid residues participated in hydrophobic interaction (Table 2).
Compounds
Estimated Free Energy of Binding
(kcal/mol)Estimated Inhibition Constant (Ki)
Hydrogen bonds formation details
Hydrogen bonds length
(Angstrom)Amino acid residues involved in Van der waals interaction
Standard (REM)
−6.41
19.91 uM
A:HIS163:HE2 -:UNK1:N23
2.33
His41,Asn142,Gly143,Cys145,,Met165,Glu166,Leu167,Pro168,His172,Gln189,Thr190,Gln192
:UNK1:H66 - A:PHE140:O
2.20
A:PRO168:CA -:UNK1:O9
3.22
A:HIS172:CD2 -:UNK1:N23
3.28
:UNK1:C24 - A:ASN142:OD1
3.07
1
−4.74
335.62 uM
A:GLU166:HN -:UNK1:N5
2.05
WAALS = Ser144,Leu141,Met165,Gln189,His172
:UNK1:H26 - A:GLU166:OE2
2.11
:UNK1:H27 - A:PHE140:O
2.11
:UNK1:C7 - A:GLU166:O
3.04
2
−5.65
71.95 uM
A:GLU166:HN -:UNK1:N7
2.10
TYR54, ASP187, ARG188
:UNK1:H15 - A:GLU166:O
2.05
:UNK1:C14 - A:GLN189:OE1
3.08
A:GLU166:HN -:UNK1
2.87
3
−6.61
14.24 uM
A:GLY143:HN -:UNK1:N11
1.89
THR26,THR25,HIS41,HIS163,MET165,HIS172,GLU166
:UNK1:H20 - A:PHE140:O
2.18
:UNK1:C9 - A:LEU141:O
3.04
:UNK1:C9 - A:SER144:OG
3.59
:UNK1:C13 - A:ASN142:OD1
3.31
4
−7.03
7.02 uM
A:TYR54:HH -:UNK1:F1
2.66
PHE140,LEU141,HIS164,PRO52,ARG188,GLN189,GLU166,HIS163
:UNK1:H33 - A:ASN142:OD1
1.94
5
−7.06
6.67 uM
A:GLY143:HN -:UNK1:N8
1.85
HIS41,THR25,THR26, MET165, HIS163, HIS172, GLU166
:UNK1:H33 - A:PHE140:O
2.26
:UNK1:C10 - A:ASN142:OD1
3.18
:UNK1:C12 - A:LEU141:O
3.16
:UNK1:C12 - A:SER144:OG
3.69
6
−8.12
1.11 uM
:UNK1:H33 - A:PHE140:O
3.03
His41,Met49,Tyr54,Leu141,,His163,His164,Met165,Glu166,His172,Asp187,Arg188
:UNK1:H34 - A:ASN142:OD1
1.98
:UNK1:C13 - A:GLN189:OE1
3.74
7
−7.83
1.83 uM
:UNK1:H33 - A:PHE140:O
2.93
LEU141,HIS172,HIS163,GLU166,GLN189,ARG188,ASP187,TYR54,HIS41
:UNK1:H34 - A:ASN142:OD1
2.06
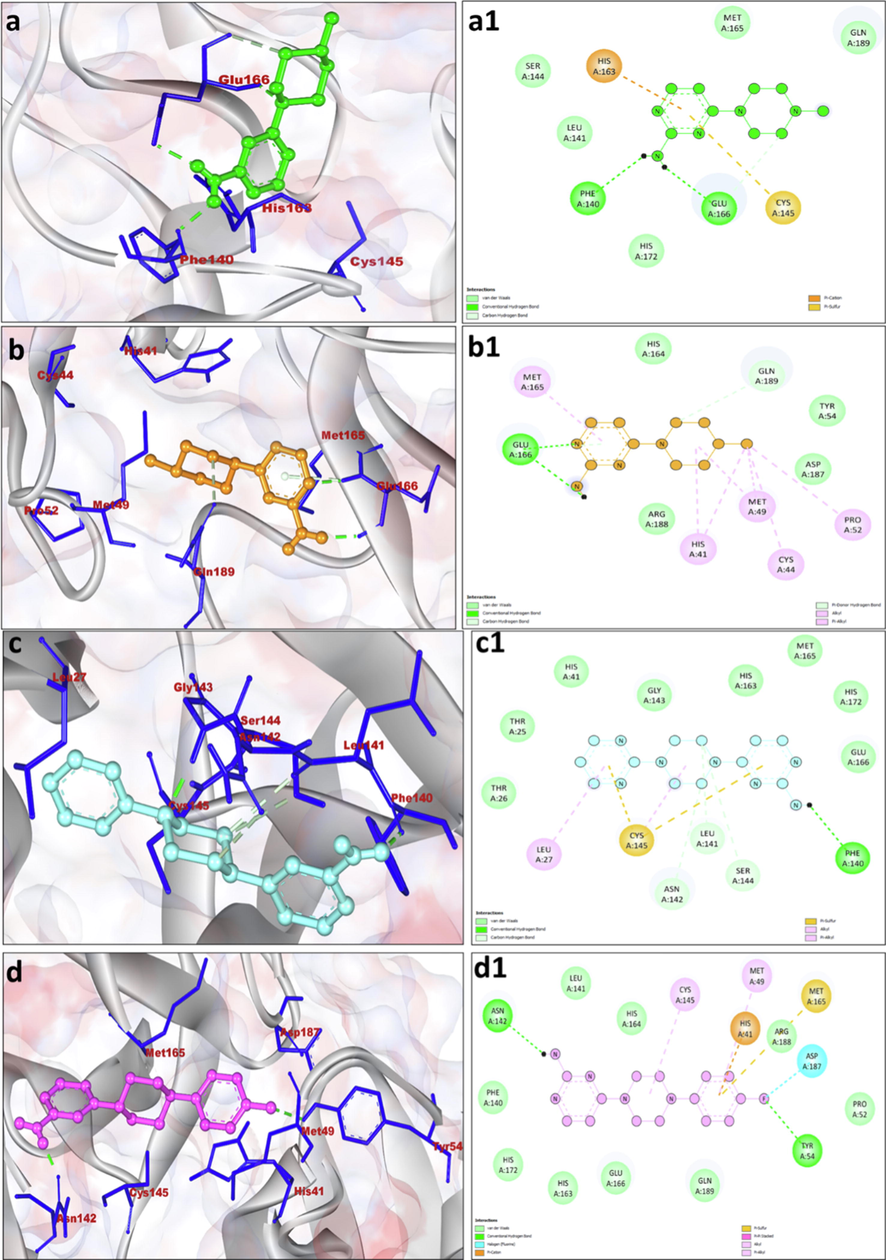
a, b, c and d displaying the 3D interaction of SARS-CoV2 main protease (PDB ID: 6LU7) with compound 1, 2, 3 and 4. Compounds were shown by ball stick pattern in the center and main protease were shown in light grey color. Close interacting residues are shown by stick pattern with dark blue color. a1, b1, c1 and d1 displaying 2D representation of hydrophobic interaction with amino acid residues. Hydrogen bonds are shown by green color dotted lines. 3D and 2D graphics were produced by Discovery Studio Visualizer 2020.
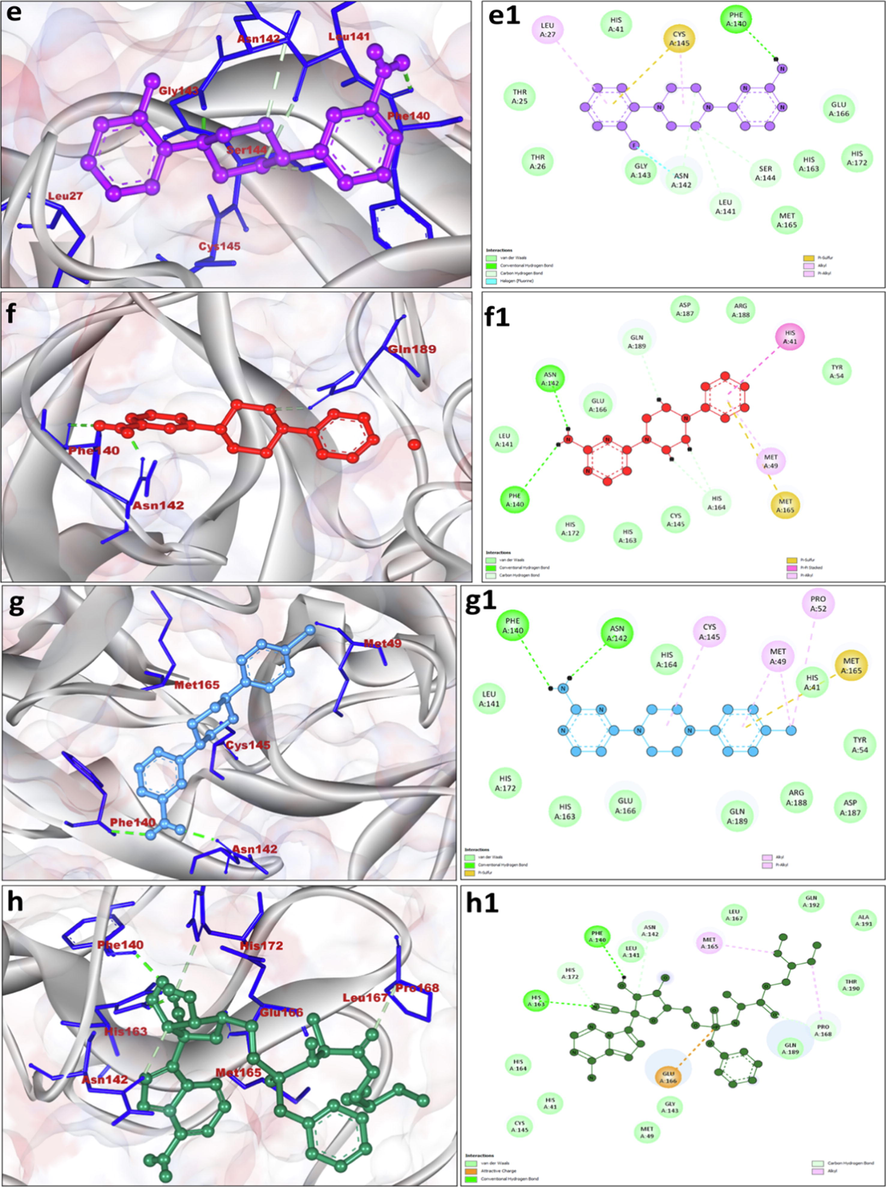
e, f, g and h displaying the 3D interaction of SARS-CoV2 main protease (PDB ID: 6LU7) with compound 5, 6, 7 and standard Remdesivir. Compounds were shown by ball stick pattern in the center and main protease were shown in light grey color. Close interacting residues are shown by stick pattern with dark blue color. e1, f1, g1 and h1 displaying 2D representation of hydrophobic interaction with amino acid residues. Hydrogen bonds are shown by green color dotted lines. The 3D and 2D graphics were produced by Discovery Studio Visualizer 2020.
3.3 ADME, drug-likeliness and toxicity results
ADME prediction data obtained from Swiss ADME are represented in Table 3. All the derivatives shown high GI absorption. It was found that except derivatives 1 and 3, all derivatives shown BBB permeability. Most of the derivatives shown negative log Kp values. Drug-likeness analysis revealed that all derivatives have zero violation for the required parameters of Lipinski’s rule of five (Lipinski et al., 2001) (Table 4). All the derivatives shown molecular weight in the ranged of 192.36–334.21 g/mol. No violation of rotatable bonds was observed, and the values were found between 1 and 2 bonds in comparison with standard cutoff value < 9 bonds. Observed TPSA values were between found in the range of 55.04–84.06 Å2. The n-octanol–water partition coefficient, log Po/w, is the standard used for Lipophilicity (Pliska et al., 1996). Consensus Log P was ranged between 0.14 and 1.89 for all the derivatives (Table 4). All derivatives showed bioavailability score of 0.55 and had a synthetic accessibility score of 2.21 to 2.44, which was within the typical standard range (Table 4).
Compounds
GI absorption
BBB permeant
Pgp substrate
CYP1A2 inhibitor
CYP2C19 inhibitor
CYP2C9 inhibitor
CYP2D6 inhibitor
CYP3A4 inhibitor
log Kp (cm/s)
1
High
N
N
N
N
N
N
N
−7.39
2
High
Y
N
N
N
N
N
N
−6.34
3
High
N
Y
Y
N
N
N
N
−7.5
4
High
Y
Y
Y
N
N
N
N
−6.55
5
High
Y
Y
Y
N
N
N
N
−6.55
6
High
Y
Y
Y
N
N
Y
N
−6.5
7
High
Y
Y
Y
N
N
Y
N
−6.27
Compounds
MW (g/mol)
Rotatable bonds
H-bond acceptors
H-bond donors
TPSA
(Å2)Consensus Log P
Lipinski violations
Ghose violations
Veber violations
Egan violations
Muegge violations
Bioavailability Score
Synthetic Accessibility
1
193.25
1
3
1
58.28
0.14
0
1
0
0
1
0.55
2.21
2
192.26
1
2
1
55.04
1.21
0
0
0
0
1
0.55
2.41
3
257.29
2
4
1
84.06
0.27
0
1
0
0
0
0.55
2.44
4
273.31
2
3
1
58.28
1.67
0
0
0
0
0
0.55
2.31
5
273.31
2
3
1
58.28
1.67
0
0
0
0
0
0.55
2.38
6
334.21
2
2
1
58.28
1.99
0
0
0
0
0
0.55
2.42
7
289.76
2
2
1
58.28
1.89
0
0
0
0
0
0.55
2.33
The BOILED-Egg model is used for the simultaneous assessment of two simple physicochemical characteristics i.e., brain access and gastrointestinal absorption of drugs/chemicals and presented in Fig. 5. Further the toxicity analysis performed using pkCSM server (Pires, et al., 2015) demonstrated no AMES toxicity and the Max. tolerated dose (Human) values was −0.03 and −0.018, respectively for derivate 6 and 7. The range of values was −0.018–0.354 for derivatives 3,4,5,6 and 7. Minnow toxicity analysis values for all derivatives were found ranged between 1.666 and 3.241 (Table 5).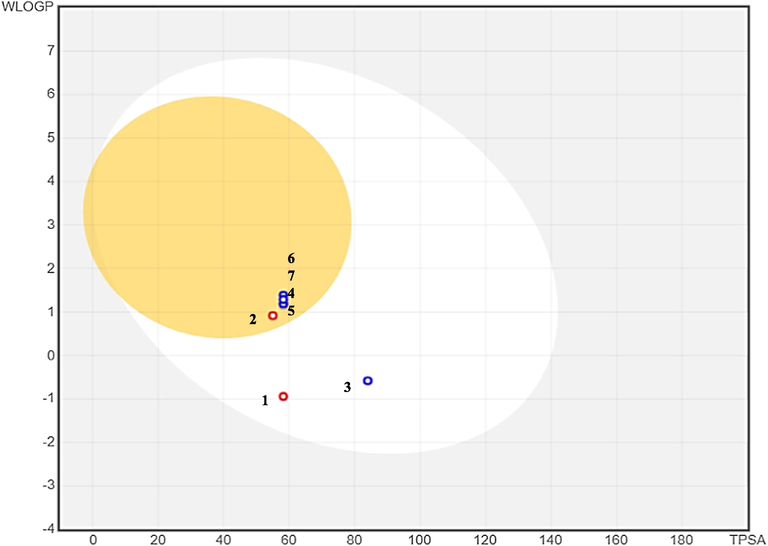
showing BOILED-Egg graph of compounds 1–7 Where yellow part showing the points situated in BOILED-Egg’s yolk are molecules predicted to passively permeate through the blood–brain barrier (BBB), White part showing the points situated in BOILED-Egg’s white are molecules predicted to passively absorbed by the gastrointestinal tract, Blue dots showing molecules predicted to be effluated from the central nervous system by the P-glycoprotein. Red dots showing molecules predicted not to be effluated from the central nervous system by the P-glycoprotein.
Compounds
AMES toxicity
Max. tolerated dose (Human)
hERG I inhibitor
hERG II inhibitor
Oral Rat Acute Toxicity (LD50)
Oral Rat Chronic Toxicity (LOAEL)
Skin sensitization
Minnow toxicity
1
No
0.55
No
No
2.196
0.877
No
3.241
2
No
0.881
No
No
2.516
2.333
No
1.666
3
No
0.354
No
No
2.521
0.802
No
2.364
4
Yes
0.018
No
No
2.564
1.381
No
2.255
5
Yes
0.019
No
No
2.553
1.39
No
2.005
6
No
−0.03
No
No
2.645
1.302
No
1.776
7
No
−0.018
No
No
2.641
1.313
No
1.922
4 Discussion
4.1 Chemistry
The microwave synthesis of pyrimidine derivatives (1–7) was done in a single step. 2-amino-4-chloro-pyrimidine and different substituted amine was transferred into microwave reaction vial and reacted at 120–140 °C for 15–30 min in the presence of anhydrous propanol triethylamine (monitored by TLC). It further involved the products dispersion in aqueous saturated sodium bicarbonate solution and products extraction with ethyl acetate. After drying over anhydrous sodium sulfate (Na2SO4), filtered products were subjected to reduced pressure for concentrated yield of 1–7 derivatives (Scheme 1).
4.2 Anticancer activity
The replacement of 4-methyl piperazine by 4-methyl piperidine (derivative 2) resulted in decrease of anticancer activity and EC50 values were 272.48 ± 0.02 µM, 271.20 ± 0.06 µM for both cancer cell lines (HCT116 and MCF7) respectively. Substitution on pyrimidine ring with 4-Pyrimidin-2-yl-piperazine (Derivative 3), 4-flurophenyl piperazine (Derivatives 4), 2-flurophenyl piperazine (Derivative 5), and 4-chlorophenyl piperazine (Derivative 7) didn’t improve anticancer activity against HCT116 and derivative 7 exhibited least activity against human colon colorectal (HCT116). However, against MCF7, derivatives 4, 5, and 7 demonstrated improved activity with EC50 191.35 ± 1.22, 252.07 ± 1.78 and 275.79 ± 1.51 µM respectively. % cell viability as shown in Figs. 1–2 indicates the concentration dependent cell viability in all derivatives, at higher concentration (300 µM) % cell viability was observed lower (23–58 %) while at low concentration (50 µM) % cell viability was ranged 52–86 % against human colon colorectal (HCT116). While against breast cancer cell line (MCF7), % cell availability was 84–100 % at lower concentration, when concentration was increased to 300 µM, % cell viability was ranged 27–48 %.
4.3 Molecular docking
Pyrimidine derivatives have received a great deal of attention in the treatment of a broad spectrum of pharmacological conditions, including HIV, cardiovascular disease, diabetes, fungal, bacterial, viral, and cancer (Patil, 2018; Rane et al., 2021). It has been reported that pyrimidine derivatives are used as herbicides, antioxidants, bronchodilators, antipyretics, anti-leishmanials agents, as well as analgesics and anti-inflammatory medications (Rane et al., 2021). Because pyrimidine base is found in both DNA and RNA, therefore, it can be explored in the treatment of viruses that have both DNA and RNA as genetic materials (Sharma et al., 2014). The first step in drug development is to design the targeted therapeutic derivatives utilizing cheminformatics techniques. Over the last decade, simulated drug discovery approaches have surfaced as important tools in the drug development process, and they have been used to uncover protein inhibitors as well as analyze protein-drug and protein–protein interactions (Keretsu et al., 2020). The 3C-Like proteinase (3CLpro) of the SARS-CoV-2 has been characterized as a promising therapeutic target for COVID-19 treatment due to it plays an essential role in virus replication and post-translational modifications of replicase polyproteins.
The results of binding efficiency of 1–7 versus the main protease of SARS-CoV-2 revealed that most of the pyrimidine derivatives had superior or equivalent affinity for the 3CLpro, as determined by binding energy scores. Derivative 6 was the top candidate and interacted with the 3CLpro via formation of three hydrogen bonds (bond lengths 1.98–3.74 Å). Amino acids Phe140 and Asn142 were crucially involved in the hydrogen bonds formation. His41, Met49, Tyr54, Leu141, His163, His164, Met165, Glu166, His172, Asp187, Arg188 amino acid residues participated in hydrophobic interaction. It was also observed that Met165 formed pi-sulfur bond, His42 formed Pi-Pi stacking while Met49 was involved in Pi-Alkyl interaction (Fig. 4f1). The derivative 6 was closely followed by 7 having two hydrogen bond formations after the involvement of Phe140 and Asn142 and bond length of 2.06–2.93 Å were noted. Leu141, His172, His163, Glu166, Gln189, Arg188, Asp187, Tyr54, His41 amino acid residues participated in hydrophobic interaction. The amino acid residues like Cys145, Met49 and Pro52 were involved in Alkyl/pi-alkyl interaction while Met165 was form Pi-Sulfur bond (Fig. 4g1). It is interesting to note that derivative 3, 4, 5, 6 and 7 exhibited better binding affinity than the control standard Remdesivir drug, which exhibited binding energy of −6.41 kcal/mol, inhibition constant of 19.91 µM and His41, Asn142, Gly143, Cys145, Met165, Glu166, Leu167, Pro168, His172, Gln189, Thr190, Gln192 amino acid residues participated in hydrophobic interaction (Table 2).
GLU-166 has been shown to be required for maintaining the right shape of the S1 pocket and the active conformation of the enzyme. In general, N-terminal residues in the protein formed a H-bond with GLU-166 of the promotor, forming the S1 pocket of the main protease, which serves as a catalytic pocket (Zhang et al., 2020). In the present study, it has been found that derivatives 1, 2 formed hydrogen bond with Glu166, while derivatives 3, 4, 5, 6, 7 formed van der waals interactions (Fig. 3a1-d1 and Fig. 4e1-g1). It has been also reported that the main protease formation is quite analogous to that of serine proteases. However, instead of a triad of catalytic residues, main protease has a catalytic dyad (involving His41 and Cys145) (Griffin, 2020). In this present work, it was found that His41 was involved in Alkyl/pi-alkyl interaction with derivatives 2, Pi cation with derivatives 4, Pi-Pi stacking with derivatives 6 and Van der waals interaction with derivatives 3, 5 and 7 (Fig. 3a1-d1 and Fig. 4e1-g1). Derivatives 1, 3 and 5 formed Pi-Sulfur bond with Cys145 during interaction. Cys145 also involved in van der waals interaction with derivatives 6 while it formed alkyl/pi alkyl bonds with derivatives 4 and 7 (Fig. 3a1-d1 and Fig. 4e1-g1). In the current study, the key amino acids residues of SARS-CoV2 main protease i.e., His41, Cys145, and Glu166 were found to be actively involved in the interaction with our designed derivative.
4.4 ADME, drug-likeliness and toxicity results
The Lipinski rule is commonly used to assess a derivatives potential as a pharmacological candidate. This rule filters simulation assessments and aids in screening potential therapeutic agents during the early drug designing stages, thus reducing time, exercise and costs in clinical drug development (Gombar et al., 2003; Hughes et al., 2011). The rule assesses some of the fundamental molecular features that a derivative contains, such as absorption, distribution, metabolism, and excretion, for a specific spectrum of derivatives and if a derivative contains at least two of the characteristics, it is considered to be safe (Bojarska, et al., 2020; Lipinski, 2004). ADME prediction data obtained from Swiss ADME demonstrated that all derivatives had high GI absorption. It was found that except derivatives 1 and 3, all derivatives shown BBB permeability and most of the derivatives are less skin permeable.
Drug-likeness analysis revealed that all derivatives have zero violation for the required parameters of Lipinski’s rule of five (Lipinski, et al., 2001) (Table 4). No violation of rotatable bonds was observed, and the values were found between 1 and 2 bonds in comparison with standard cutoff value < 9 bonds. Observed TPSA values were optimum as the recommended values should be between 20 and 130 Å2 (Ertl, et al., 2000). The n-octanol–water partition coefficient, log Po/w, is the standard used for Lipophilicity (Pliska, et al., 1996). Consensus Log P should not be higher than 6 (Mannhold, et al., 2009), while, in the present work, it was not higher than 1.89 for all the derivatives (Table 4).
The determination of a bioavailability score is required in order to calculate the likelihood that a candidate will have at least 10 % oral bioavailability in rats or significant Caco-2 permeability, and it should not be<0.25 (Martin, 2005) and our derivatives showed bioavailability score of 0.55. The predicted synthetic accessibility score should be ranged from 1 (very easy) to 10 (very difficult) (Ertl and Schuffenhauer, 2009; Ansari, et al., 2020) and all the derivatives had scores of 2.21 to 2.44, which was within the typical standard range (Table 4).
The BOILED-Egg model is used for the simultaneous assessment of two simple physicochemical characteristics i.e., brain access and gastrointestinal absorption of drugs/chemicals and these two pharmacokinetic behaviors are crucial at various stages of the drug discovery and development processes (Daina and Zoete, 2016) In the present study, it was observed that derivatives 2, 4, 5, 6 and 7 were expected to pass across the blood–brain barrier (BBB), whereas derivatives 1 and 3 were predicted to be absorbed passively by the gastrointestinal tract (Fig. 5). Further investigation found that the P-glycoprotein was unable to effluate the derivatives 1 and 2 from the central neurological system, however the derivatives 3, 4, 5, 6 and 7 were able to be effluated from the central neurological system (Fig. 5).
Further the toxicity analysis performed using pkCSM server (Pires, et al., 2015) demonstrated that all derivatives have shown no skin sensitization properties. The best identified derivatives were 6 and 7 which shows no AMES toxicity and the Max. tolerated dose (Human) values was −0.03 and −0.018, respectively for derivate 6 and 7 which is within the standard range i.e., the value smaller than/equal to 0.477 log(mg/kg/day) is considered low, whereas more than 0.477 log(mg/kg/day) is considered to be high. In this present analysis the values found for derivatives 3,4,5,6 and 7 were within the standard range. Minnow toxicity analysis values for all derivatives were found satisfactory with the values ranged between 1.666 and 3.241 because LC50 values below 0.5 mM (Log LC50 < -0.3) will be considered as high acute toxicity (Table 5).
5 Conclusion
Pyrimidine derivatives (1–7) were synthesized successfully via microwave method, incorporating multifunctional amino derivatives. Synthesized derivatives (1–7) were tested against human colon colorectal (HCT116) and breast cancer cell line (MCF7). Derivative 6 exhibited highest anticancer activity on human colon colorectal (HCT116) with EC50 89.24 ± 1.36 µM and on breast cancer cell line (MCF7) with EC50 89.37 ± 1.17 µM.
Molecular docking between derivatives (1–7) and SARS-CoV-2 main protease revealed that derivative 6 was the best candidate with a binding energy score of −8.12 kcal/mol and inhibition constant of 1.11 µM. All derivatives displayed high GI absorption and except derivatives 1 and 3, all other derivatives displayed blood brain barrier permeability. Skin permeability of all derivatives was less and bioavailability score of all derivatives were 0.55. The toxicity analysis revealed that all derivatives have shown no skin sensitization properties. The best identified derivatives were 6 and 7 which shows no AMES toxicity and the Max. tolerated dose (Human) values was −0.03 and −0.018, respectively.
Acknowledgement
The authors would like to express gratitude and profoundly thank the Deanship of Scientific Research at Imam Abdulrahman Bin Faisal University for proving the funds for the Project Number 2019-126-DSR.
Ethics approval and consent to participate
Not Applicable.
Consent for publication
Not Applicable.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files, and also available from the corresponding author on reasonable request.
Funding
Project Number 2019-126-DSR to FQ. Funder; Deanship of Scientific Research at Imam Abdulrahman Bin Faisal University.
Authors' contributions
FQ and MN acquired the funds via university projects fund, designed and formulated the research plan and conducted the synthesis and characterization. SH and SAA helped with studies and data for biological studies. MAA and QMSJ performed computational analysis. NU and MT performed and analyzed NMR studies. OA is research student and helped FQ and MN with analysis in lab and formatting references section. MKB and BH edited the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Not Applicable.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- A review on the medicinal importance of pyridine derivatives. J. Drug Des. Med. Chem.. 2015;1:1-11.
- [CrossRef] [Google Scholar]
- Data-driven high-throughput prediction of the 3-D structure of small molecules: review and progress. J. Chem. Inf. Model.. 2011;51(4):760-776.
- [CrossRef] [Google Scholar]
- TAT-peptide conjugated repurposing drug against SARS-CoV-2 main protease (3CLpro): Potential therapeutic intervention to combat COVID-19. Arab. J. Chem.. 2020;13(11):8069-8079.
- [CrossRef] [Google Scholar]
- A supramolecular approach to structure-based design with a focus on synthons hierarchy in ornithine-derived ligands: review, synthesis, experimental and in silico studies. Molecules. 2020;25(5):1135.
- [CrossRef] [Google Scholar]
- Synthesis, characterization and computational study on potential inhibitory action of novel azo imidazole derivatives against COVID-19 main protease (Mpro: 6LU7) J. Mol. Struct.. 2021;1225:129230
- [CrossRef] [Google Scholar]
- Design, synthesis and anticancer studies of novel aminobenzazolyl pyrimidines as tyrosine kinase inhibitors. Bioorg. Chem.. 2018;77:84-100.
- [CrossRef] [Google Scholar]
- A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem. 2016;11(11):1117-1121.
- [CrossRef] [Google Scholar]
- SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep.. 2017;7:42717.
- [CrossRef] [Google Scholar]
- Design and synthesis of new anticancer pyrimidines with multiple-kinase inhibitory effect. Bioorg. Med. Chem.. 2010;18(11):3860-3874.
- [CrossRef] [Google Scholar]
- Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem.. 2000;43(20):3714-3717.
- [CrossRef] [Google Scholar]
- Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform.. 2009;1(1):8.
- [CrossRef] [Google Scholar]
- Coronaviruses: an overview of their replication and pathogenesis; section 2 genomic organization. In: Maier H.J., Bickerton E., Britton P., eds. Coronaviruses: Methods and Protocols. New York: Springer; 2015. p. :1-23.
- [Google Scholar]
- SwissTargetPrediction: a web server for target prediction of bioactive small molecules. Nucleic Acids Res. 42(Web Server. 2014;issue),:W32-W38.
- [CrossRef] [Google Scholar]
- Synthesis of bis-indolylmethane sulfonohydrazides derivatives as potent α-glucosidase inhibitors. Bioorg. Chem.. 2018;80:112-120.
- [CrossRef] [Google Scholar]
- Role of ADME characteristics in drug discovery and their in silico evaluation: in silico screening of chemicals for their metabolic stability. Curr. Top. Med. Chem.. 2003;3(11):1205-1225.
- [CrossRef] [Google Scholar]
- SARS-CoV and SARS-CoV-2 main protease residue interaction networks change when bound to inhibitor N3. J. Struct. Biol.. 2020;211(3):107575.
- [CrossRef] [Google Scholar]
- Fragment molecular orbital based interaction analyses on COVID-19 main protease− inhibitor N3 complex (PDB ID: 6LU7) J. Chem. Inf. Model.. 2020;60(7):3593-3602.
- [CrossRef] [Google Scholar]
- Principles of early drug discovery. Br. J. Pharmacol.. 2011;162(6):1239-1249.
- [CrossRef] [Google Scholar]
- Synthesis, characterization and electrochemical properties of some biologically important indole-based-sulfonamide derivatives. BMC Chem.. 2020;14(1):38.
- [CrossRef] [Google Scholar]
- A computational study of natural compounds from bacopa monnieri in the treatment of alzheimer's disease. Curr. Pharm. Des.. 2020;26(7):790-800.
- [CrossRef] [Google Scholar]
- Therapeutic development by repurposing drugs targeting SARS-CoV-2 spike protein interactions by simulation studies. Saudi J. Biol. Sci.. 2021;28(8):4560-4568.
- [CrossRef] [Google Scholar]
- Rational approach toward COVID-19 main protease inhibitors via molecular docking, molecular dynamics simulation and free energy calculation. Sci. Rep.. 2020;10(1):17716.
- [CrossRef] [Google Scholar]
- Pharmacological potential and synthetic approaches of imidazo[4,5-b]pyridine and imidazo[4,5-c]pyridine derivatives. Molecules. 2017;22(3):399.
- [CrossRef] [Google Scholar]
- Lead-and drug-like compounds: the rule-of-five revolution. Drug Discov. Today Technol.. 2004;1(4):337-341.
- [CrossRef] [Google Scholar]
- Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev.. 2001;46(1–3):3-26.
- [CrossRef] [Google Scholar]
- Design. synthesis and bioevaluation of N-trisubstituted pyrimidine derivatives as potent aurora A kinase inhibitors. Eur. J. Med. Chem.. 2014;78:65-71.
- [CrossRef] [Google Scholar]
- Design and synthesis of novel 1,2,3-triazole-pyrimidine hybrids as potential anticancer agents. Eur. J. Med. Chem.. 2014;86:368-380.
- [CrossRef] [Google Scholar]
- Calculation of molecular lipophilicity: State-of-the-art and comparison of log P methods on more than 96,000 compounds. J. Pharm. Sci.. 2009;98(3):861-893.
- [CrossRef] [Google Scholar]
- SARS-CoV-2: structure, biology, and structure-based therapeutics development. Front. Cell. Infect. Microbiol.. 2020;25(10):587269.
- [CrossRef] [Google Scholar]
- Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem.. 1998;19(14):1639-1662.
- [CrossRef] [Google Scholar]
- AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem.. 2009;30(16):2785-2791.
- [CrossRef] [Google Scholar]
- Part-1: Design, synthesis and biological evaluation of novel bromopyrimidine analogs as tyrosine kinase inhibitors. Eur. J. Med. Chem.. 2016;119:70-82.
- [CrossRef] [Google Scholar]
- Synthesis of new derivatives of benzofuran as potential anticancer agents. Molecules. 2019;24(8):1529.
- [CrossRef] [Google Scholar]
- Synthesis and spectroscopic properties of pyridones—experimental and theoretical insight. J. Mol. Liq.. 2014;193:51-59.
- [CrossRef] [Google Scholar]
- Synthesis, spectral studies and biological evaluation of 2-aminonicotinic acid metal complexes. Spectrochim. Acta A Mol. Biomol.. 2016;161:39-43.
- [CrossRef] [Google Scholar]
- Preparation of indium-cadmium sulfide nanoparticles with diverse morphologies: Photocatalytic and cytotoxicity study. J Mol. Str.. 2022;1253:132288.
- [CrossRef] [Google Scholar]
- Structural elucidation, molecular docking, α-amylase and α-glucosidase inhibition studies of 5-amino-nicotinic acid derivatives. BMC Chem.. 2020;14:43.
- [CrossRef] [Google Scholar]
- Biological and medicinal significance of pyrimidines: A Review. Int. J. Pharm. Sci. Res.. 2018;9(1):44-52.
- [CrossRef] [Google Scholar]
- pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem.. 2015;58(9):4066-4072.
- [CrossRef] [Google Scholar]
- Lipophilicity: The Empirical Tool and the Fundamental Objective. An Introduction. In: Pliska V., Testa B., van de Waterbeemd H., eds. Lipophilicity in drug action and toxicology. Wiley-VCH Verlag GmbH; 1996. p. :1-6.
- [CrossRef] [Google Scholar]
- Synthesis and characterization of cadmium-bismuth microspheres for the catalytic and photocatalytic degradation of organic pollutants, with antibacterial, antioxidant and cytotoxicity assay. J. Photochem. Photobiol. B.. 2020;202:111723.
- [CrossRef] [Google Scholar]
- Isatin based thiosemicarbazide derivatives as potential inhibitor of α-glucosidase, synthesis and their molecular docking study. J. Mol. Struct.. 2020;1222:128922.
- [CrossRef] [Google Scholar]
- Targeting virus–host interaction by novel pyrimidine derivative: an in silico approach towards discovery of potential drug against COVID-19. J. Biomol. Struct. Dyn.. 2021;39(15):5768-5778.
- [CrossRef] [Google Scholar]
- New hybrid hydrazinyl thiazole substituted chromones: as potential α-amylase inhibitors and radical (DPPH & ABTS) scavengers. Sci. Rep.. 2017;7(1):16980.
- [CrossRef] [Google Scholar]
- Significance and biological importance of pyrimidine in the microbial world. Int. J. Med. Chem.. 2014;2014:202784
- [CrossRef] [Google Scholar]
- Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease tmprss2. J. Virol.. 2013;87(23):12552-12561.
- [CrossRef] [Google Scholar]
- Pyridine derivatives possessing hypoglycemic and analgesic activity. Pharm. Chem. J.. 1989;23:739-743.
- [CrossRef] [Google Scholar]
- Computational study on peptidomimetic inhibitors against SARS-CoV-2 main protease. J. Mol. Liq.. 2021;322:114999
- [CrossRef] [Google Scholar]
- Michael cyclization of polarized systems: synthesis and in vitro anti-diabetic evaluation of some novel pyrimidine, pyridine, pyrazole and pyrazolo[3,4-b]pyridine derivatives. Croat. Chem. Acta.. 2017;90(3):461-469.
- [CrossRef] [Google Scholar]
- Design and synthesis of anticancer 1-hydroxynaphthalene-2-carboxanilides with a p53 independent mechanism of action. Sci. Rep.. 2019;9:6387.
- [CrossRef] [Google Scholar]
- Synthesis and in vitro study of benzofuran hydrazone derivatives as novel alpha-amylase inhibitor. Bioorg. Chem.. 2017;75:78-85.
- [CrossRef] [Google Scholar]
- Synthesis, α-amylase inhibitory potential and molecular docking study of indole derivatives. Bioorg. Chem.. 2018;80:36-42.
- [CrossRef] [Google Scholar]
- Synthesis, in vitro urease inhibitory potential and molecular docking study of benzofuran-based-thiazoldinone analogues. Sci. Rep.. 2020;10(1):10673.
- [CrossRef] [Google Scholar]
- Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov.. 2006;5:835-844.
- [CrossRef] [Google Scholar]
- DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res.. 2017;46(D1):D1074-D1082.
- [CrossRef] [Google Scholar]
- Worldometer, W., 2020. Coronavirus Update: Cases And Deaths From COVID-19 Virus Pandemic–Worldometer. info (2022). [online] Available at:[Accessed 27 July 2022].
- Nelfinavir was predicted to be a potential inhibitor of 2019-nCov main protease by an integrative approach combining homology modelling, molecular docking and binding free energy calculation. BioRxiv.. 2020;2020
- [CrossRef] [Google Scholar]
- Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368(6489):409-412.
- [CrossRef] [Google Scholar]
- Coronaviruses-drug discovery and therapeutic options. Nat. Rev. Drug. Discov.. 2016;15:327-347.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2022.104366.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary Data 1
Supplementary Data 1




