

Translate this page into:
Organocatalyst as a synthetic gadget for pharmaceutically potent molecules
⁎Corresponding author at: Department of Chemistry, Government College University, Faisalabad 38000, Pakistan. almeerazia817@gmail.com (Nasir Rasool),
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
Organocatalysts have become a third main method of catalyzing chemical reactions aside from bio and metal catalysts. The potential for green chemistry and some striking benefits, including stability, low cost, easy availability, excellent enantioselectivity, and simple recovery, have made organocatalyzed reactions a fruitful approach. Recent developments in the field of organocatalysis include the enhancement of enantioselectivity, the development of novel catalyst classes such as NHCs, and dual catalysis, which combines organo- with metal or photocatalysis to enable novel reactivity. This review focuses on organocatalyzed chemical synthesis of pharmacophores such as benzoxazinones, pyrrolidines, triazoles, pyrazolinones, tricyclic coumarins, (+)-paroxetine, and (+)-femoxetine. The review has been outlined into six categories i.e. Lewis base catalysts, Lewis acid catalysts, Bronsted base catalysts, Bronsted acid catalysts, bifunctional catalysts, and organo-photocatalysts. Less reusability due to catalyst deactivation demands higher catalytic loading. Thus scalability in this field is still challenging. Moreover, achieving higher catalytic efficiency needs future efforts.
Keywords
Organocatalysts
Organo-photocatalyst
Bronsted acid/base catalysis
Lewis acid/base catalysis
Drug candidates

1 Introduction
Chemists can create new scaffolds with unique features and significant effects on human life by using the excellent toolkit that organic synthesis offers. Since Wöhler's urea synthesis, the discipline has fundamentally aided the medical sciences and seen enormous advancements in the synthesis of small to large compounds with natural origins (Garcia-Castro et al., 2016). Small organic molecules have the potential to significantly influence biology and medicine, serving as both pharmacophores and probes that help to elucidate the macromolecules controlling biological systems. Particularly, functionalized heterocyclic small molecules have been used in drug design applications in the pharmaceutical industry (Pitt et al., 2009). Both synthetic and naturally occurring biologically active products frequently contain heterocycles. Due to their ability to bind selectively to pharmacophores, their significance in drug development is continually growing. The majority of synthetic medicines in the market have a heterocyclic moiety (Baumann and Baxendale, 2013) which is synthesized by several traditional named reactions (Li, 2004). The majority of those traditional methods rely on the employment of high-boiling solvents, costly or hazardous catalysts, extended reaction durations, and thermal heating. To replace the antiquated classical methods that date back a long way, researchers from all around the world have devised alternative sustainable synthetic procedures (Taylor et al., 2016; Nishanth Rao et al., 2021).
According to the 1987 Brundtland Commission on Environment and Development of the UN (Brundtland, 1985), the term “sustainable development” refers to development that meets present needs without endangering the capacity of future generations to fulfill their necessities. Creating more renewable energy sources and lowering pollution are two of the most important components of sustainable development. The modern chemical industry faces a challenge in maintaining its socioeconomic benefits and applications while protecting the environment. Synthesis of biologically active moieties by using organocatalysis reduces the environmental impact of chemical processes (Clark and Rhodes, 2007; Shaikh, 2014).
Organocatalysts have broad applications across multiple fields due to their efficiency, selectivity, and eco-friendly properties. They play a crucial role in drug discovery and asymmetric synthesis in the pharmaceutical sector, facilitating the synthesis of enantiomerically pure molecules (Burke, 2023). Organocatalysts help in the sustainable synthesis of green insecticides and agrochemicals in agriculture. They are also important in material science for synthesizing biodegradable polymers and nanomaterials. In fine chemicals, these catalysts enhance the production of fragrances and flavors, while in renewable energy, improve biofuel production. Furthermore, organocatalysts are essential to green chemistry since they support solvent-free and sustainable processes (Aukland and List, 2021).
Metal-catalyzed reactions offer a greater range of substrates, but they also come with certain disadvantages, including the high expense of catalyst synthesis and the potential toxicity of the metals to the products (List, 2007; Davies, 2013). In contrast to metals, organic compounds are more affordable, stable, non-toxic, widely accessible, and ecologically benign. Furthermore, compared to metal-catalyzed reactions, organocatalyzed reactions are less susceptible to the presence of air or water. As a result, these reactions have improved operational simplicity and reproducibility. Both metal and biocatalysts can be better substituted by organocatalysts with greater effectiveness.
Unlike enzymes, that are extremely substrate-specific and cannot withstand even a slight alteration in the reactant's configuration, organocatalysts offer a wide range of substrates. Furthermore, an additional benefit that organic catalysts have over both metal and enzyme catalysts is the ease of amenability to solid support, which facilitates catalyst recovery and streamlines the reaction work-up process. Since enzymes are recognized because of high stereospecificity and strong catalytic activity, similar properties exhibited by tiny organic molecules have always been considered one of the reasons for their remarkable activity (Breslow, 1982). These organic compounds operate via interacting with substrates either through strong interactions like covalent bonding or weak interactions such as Van der Waals forces or H-bonding (Benaglia et al., 2003; Berkessel and Gröger, 2006; Barbas, 2008). The reaction rate is significantly accelerated as a result of these interactions (Akiyama et al., 2006; Taylor and Jacobsen, 2006). Even though organic molecules have long been employed as catalysts, it has only been in the last few years that the employment of these molecules in enantioselective synthesis has become a significant idea (Raj and Singh, 2009).
Recently Zafar et al. (Zafar et al., 2024) and Ahmad et al. (Ahmad et al., 2024)wrote reviews on the synthesis of organic compounds by using organocatalysts with focus on mechanistic studies. While this review majorly focuses on the pharamaceitical advantages of organocatalyzed reactions.
This review illustrates the synthesis of several bioactive molecules such as pyrrolidines, triazoles, pyrazolinones, tricyclic coumarins, (+)-paroxetine, and (+)-femoxetine by using different organocatalysts. It has been categorized into Lewis base catalysis, Lewis acid catalysis, Bronsted base catalysis, Bronsted acid catalysis, bifunctional catalysis, and organo-photocatalysis. These classes are further categorized based on the names of the organocatalysts that are used Table 1.
Catalyst type
Catalyst name
Catalyst code
Synthesized compounds
Bioactivity
Reference
Lewis base catalysts
Hayashi-Jørgensen catalyst
C1
14
Antibacterial
(Rodriguez et al., 2020)
C2
22-trans, 27-trans
Antidepressant
(Szcześniak et al., 2019)
C3
31a
Anticancer
(Zhang et al., 2018b )
C4
34a
Anticancer
(Li et al., 2019)
N-heterocyclic carbene (NHC)
C5
38
Topoisomerase I inhibitor
(Zimmerman et al., 2020)
C6
50a
Antiparasitic
(Coelho et al., 2019)
C7
60
Antirheumatic
(Murugesh et al., 2020)
Tert-leucinamide
C8
64
Precursor of vitamin B5
(Du et al., 2021)
Trimethylimidazolidin-4-one hydrochloride
C9
70c
Anticancer
(Pham et al., 2020)
Triphenylphosphine
C10
73
Antibacterial
(Gholami et al., 2019)
74b
Antioxidant
Lewis acid catalysts
Trimethylsilyl trifluoromethane
sulfonateC11
103
Anticancer
(Štadániová et al., 2020)
Hyamine
C12
101, 102
Antibacterial
(Arora et al., 2018)
Cinchonidine-derived quaternary ammonium
C13
111
Antiandrogenic
(Guerrini et al., 2014)
Boron trifluoride diethyl etherate
C14
115
Anti-inflammatory,
Treat sleeping sickness,
Postsynaptic-selective
alpha-1-antagonist(Mahecha-Mahecha et al., 2020)
Ethylaluminum dichloride
C15
120
Antiangiogenic and antioxidizing
(Wu et al., 2022)
Bronsted base
catalystsPyrrolidine
C16
138
Antirenal fibrosis
(Simek et al., 2022).
C17
151
Anticancer
(Zhang et al., 2019c)
C18
153, 155
Anticancer
(Xu et al., 2019b)
165
Anti-inflammatory
C19
167
Cathepsin K inhibitors
(Silva et al., 2020)
DBU
C20
172, 174
Antifungal
(Khan and Saigal, 2018)
177c
Antioxidant
(Gomes et al., 2020)
179
Anti-HCMV
(Herrmann et al., 2023)
Cinchona primary amine
C21
C22(±)-199
Antibacterial
(Ernouf et al., 2018)
Quinidine
C23
202
Anticancer
(He et al., 2022)
Cinchonine-derived squaramide
C24
211
HRV protease inhibitors
(Wu et al., 2019)
DABCO
C25
229
Ab 1-X inhibitor in PDAPP
(Winneroski et al., 2019)
232
HIV-1 NNRTI inhibitor
(Chen and He, 2020)
4-pyrrolidinopyridine
C26
239
Dithiolated O-antigen of E. coli serogroup 64
(Wan et al., 2023)
TBD
C27
245
Photosensitizers in cancer therapy
(de Oliveira et al., 2019)
Bronsted acid
catalystsTakemoto's thiourea
C28
255
Treat Chagas disease
(Guerrero-Corella et al., 2021)
Quinine-derived thiourea
C29
261a
Anticancer
(Wang et al., 2023)
C30
264
(Li et al., 2023)
Chiral phosphoric acid
C31
267
Antiviral and Anticancer
(Yan et al., 2022)
C32
272
Anticancer
(Wang et al., 2022)
α-angelica lactone
C33
277
Anti-platelet
(Thatikonda et al., 2020)
DES
C34
290
Anti-inflammatory and Analgesic
(Imran et al., 2020)
p-toluenesulfonic acid
C35
304,305
Potential enzyme inhibitors
(Repetto et al., 2019)
Triflouoroacetic acid
C36
310a
Anticancer
(Srinivasulu et al., 2018)
Pyridine-2-carboxylic acid
C37
321
AChE inhibitor
(Pervaiz et al., 2020)
Bifunctional organo-catalysts
Cinchona alkaloid catalyst
C38
324
Anticancer
(Liu et al., 2021)
Cinchona-based thiourea
C39
332
Antidepressant and Antiparasitic
(Park et al., 2019)
Cinchona alkaloids-derived squaramide
C40
340
Anti-proliferative
(Chang et al., 2022)
Quinine
C41
343
Anticancer
(Hammer et al., 2018)
Hydroquinine
C42
355
Anticancer
(Kovalevsky et al., 2023)
Quinine-derived squaramide
C43
358
Anticancer
(Zhao et al., 2023a )
Proline
C44
361
COX-2 enzyme inhibitor
(Akhtar and Lee, 2020)
Prolinol
C45
375
SPHK inhibitors
(Escudero-Casao et al., 2018)
C46
394
Antidepressant
(Sharma and Pandey, 2022)
β-amino alcohol
C47
407
Dopamine agonists
(Chavan et al., 2019a)
Chiral carbamate
C48
410
RNA polymerase inhibitor
(Gannedi et al., 2021)
Organo-photo catalysts
1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene
C49
414
Anticancer
(Sherwood et al., 2018)
CC-CMP
C50
424
Cholinesterase inhibitor,
Antitumor and
Treat glaucoma(Ou et al., 2019)
Lumiflavin
C51
431
Anti-convulsant, Thrombin inhibitor and Phosphodiesterase-4 inhibitor
(Chilamari et al., 2020)
Anthraquinone
C52
437
Treat cardiovascular disease, Analgesic and Antimalarial
(Zhao et al., 2023b)
Eosin Y
C53
444
Inhibitors of liver X receptor
(Vidyacharan et al., 2019)
Remarkably, a large number of the very recently created asymmetric organocatalysis syntheses meet crucial requirements for a commercial process. In general, developing scalable catalyst preparation processes, further reducing catalyst loading, and technically appealing downstream processing methods will be major challenges to the greater usage of organocatalysts. Finding extremely appealing and cost-effective synthetic pathways to the target molecules, such as avoiding the use of protective groups (especially those with challenging cleavage), is another problem (Gröger, 2008).
2 Lewis base catalysis
Lewis bases organocatalysts based on nitrogen, carbon, oxygen, phosphorus, and sulfur make up the majority of organocatalysts. These bases react via a variety of processes to change substrates into either electrophiles or activated nucleophiles. Iminium ions, enamines, acyl ammonium ions, 1-, 2-, or 3-ammonium enolates, etc. are examples of typical reactive intermediates (Seayad and List, 2005). Moreover, Lewis base organocatalysis includes the enolate reaction modes of ammonium (Gaunt and Johansson, 2007)and azolium (Douglas et al., 2012), which employ tert-amine or NHC catalysts, respectively (Douglas, 2012). Lewis base catalysis has several benefits, including mild reaction conditions, metal-free residue, and environmental friendliness (Meng et al., 2022). In 2018, Li and colleagues synthesized the annulated 4H-pyran and annulated 3, 4-dihydro-2H-pyran by a TEDA-promoted chemo-divergent (4 + 2) cycloaddition reaction of allenoates with 2-arylidene-1H-indene-1,3(2H)-diones (Xi et al., 2018). Afterward, the authors discovered that the same materials could be used to form a formal (3 + 2) cycloadduct under phosphine catalysis (Ma et al., 2019).
2.1 Hayashi-Jørgensen catalyst
Pyrrolidine derivatives show potent antimicrobial activities (Le Goffic, 1985; Raj et al., 2003). Le Goffic established that pyrrolidine moiety boosted lincosamide drugs' antibacterial action (Le Goffic, 1985; Odagiri et al., 2013). Pyrrolidine cores are synthesized by various synthetic methods, including reductive amination and dipolar cycloadditions of azomethine ylides (Bdiri et al., 2017). Nevertheless, organocatalyzed Michael additions are one of the strongest and most reliable techniques. Rodriguez et al. used organocatalytic Michael addition and reductive cyclization to synthesize a library of 3,4-disubstituted pyrrolidine derivatives. First, aldehyde 5 and nitroalkene 6 undergo Hayashi-Jørgensen C1 catalyzed Michael addition to give the Michael adduct 7, which then goes through reductive cyclization, reducing one carbonyl group in the isoindoline-1,3-dione scaffold to produce pyrrolidine 8. The Tertbutoxycarbonyl group protects the secondary amino group of pyrrolidine 8, resulting in Boc-protected pyrrolidines 9. Then 3-hydroxyisoindolin-1-one moiety of compound 9 is oxidized to isoindoline-1,3-dione moiety to give product 10 which undergoes reductive amination to afford corresponding benzyl derivatives 12. These derivatives undergo hydrazinolysis to give 13 (Scheme 1) (Rodriguez et al., 2020). Hydrochlorides 14 of these products 13 are produced to improve their solubility in aqueous solutions for biological testing. The key features of this methodology are that only one-column chromatography is required for this four reaction steps sequence, the catalyst is readily available, stable and typically shows constantly good catalytic activity, and the solvent Et2O preserves good enantiomeric and diastereomeric purities. The synthesized compounds were tested for methicillin-resistant strains of S. aureus and E. coli bacteria. Compound 14 was shown to be the most effective with MIC100 = 37 μmol/L and MIC50 = 17 μmol/L respectively for both bacteria.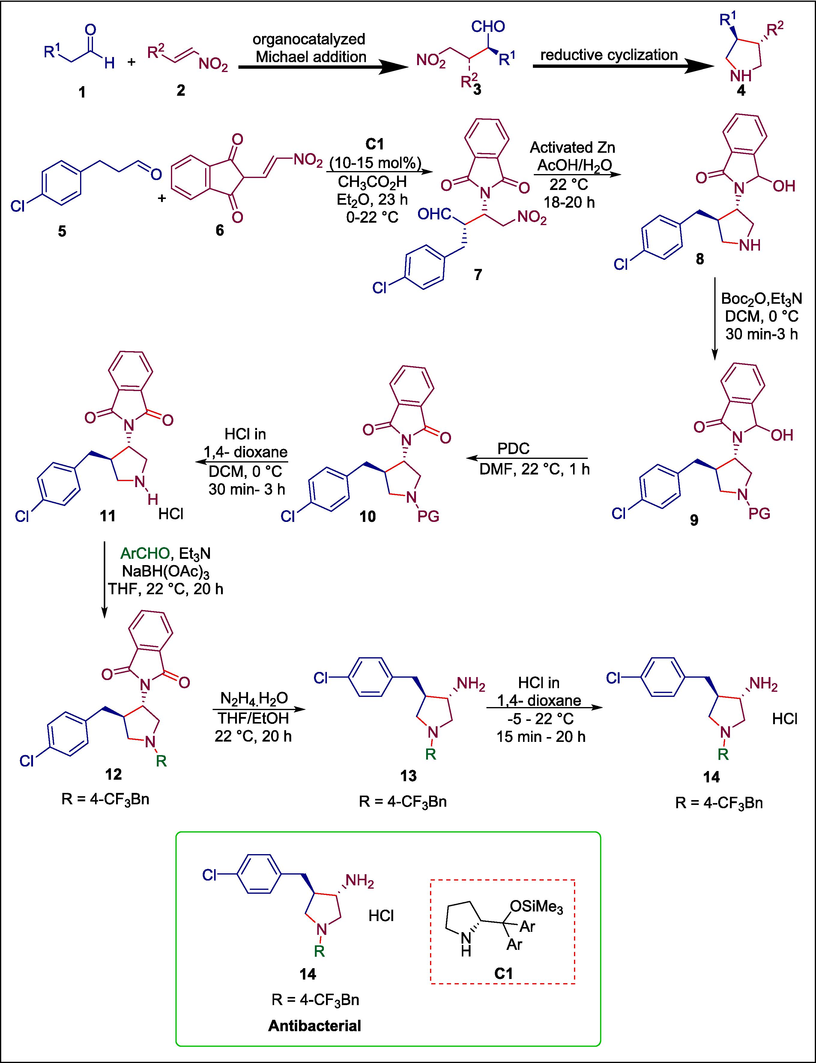
Hayashi-Jørgensen catalyzed the synthesis of pyrrolidine derivatives.
(+)-Paroxetine and (+)-femoxetine are used to treat depression. Their synthesis is frequently achieved using enzymatic asymmetric desymmetrization, asymmetric catalysis, chiral pool, chiral base, and chiral auxiliary (Greenhalgh and Simpkins, 2002; Gill et al., 2003). These techniques have shown to be successful, but their primary flaw is the lengthy and intricate synthetic sequencing. Szcześniak et al. devised a method for the asymmetric formation of these antidepressants by employing organocatalytic Michael addition of aldehydes to trans-nitroalkenes achieved in bath or continuous flow. The synthesis starts when aldehydes 15a/b and nitroolefins 16a/b undergo a Michael addition reaction in the presence of Hayashi-Jørgensen catalyst C2 in a continuous flow to give the γ-nitroaldehyde 19a-syn/anti which undergoes Wittig olefination to form the product 20-syn/anti. After that, olefin 20-syn/anti is hydrolyzed to produce aldehyde 21-syn/anti, which is then subjected to reductive cyclization to produce paroxetine 22-trans (Scheme 2) (Szcześniak et al., 2019).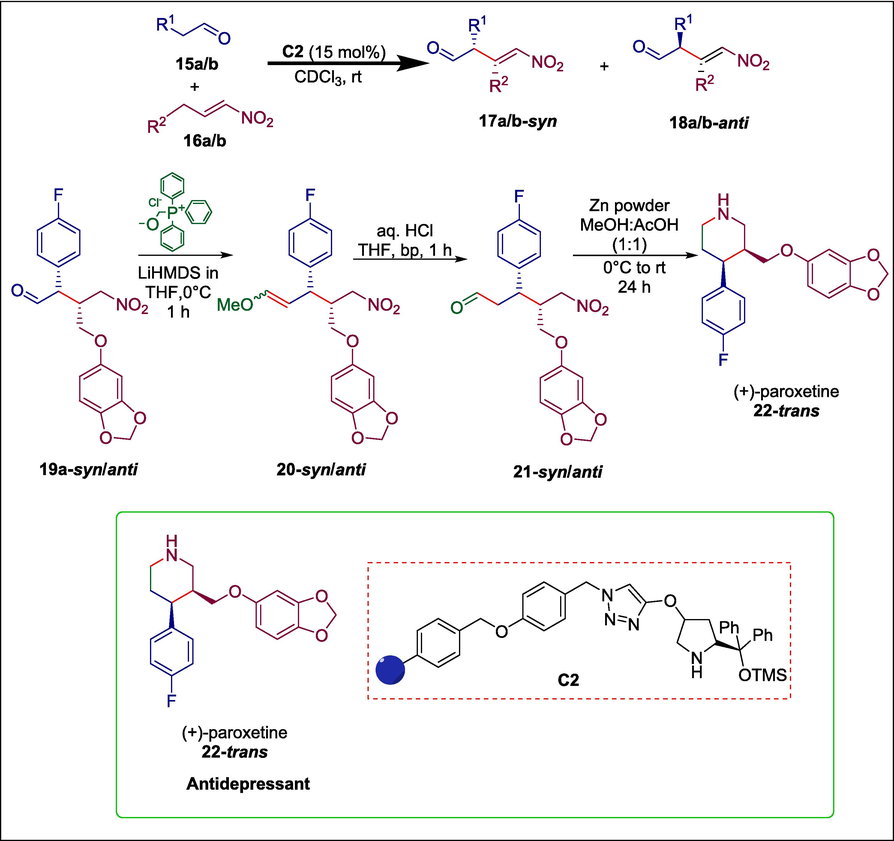
Hayashi-Jørgensen catalyzed synthesis of (+)-paroxetine.
(+)-Femoxetine’s 27-trans synthesis is carried out using the same chemical process. The crude mixture of γ-nitroaldehyde 23b-syn/anti is subjected to Wittig olefination, producing compound 24-syn/anti, which, upon acidic hydrolysis, provides δ-nitroaldehyde 25-syn/anti. Without purification, this reaction was then employed directly for the subsequent step. After that, δ-nitroaldehyde 25-syn/anti is treated with zinc powder to produce 26-trans, which is then treated with formaldehyde and NaCNBH3 to produce (+)-femoxetine 27-trans (Scheme 3). This method's simple procedure and use of an affordable, easily accessible substrate are among its main advantages.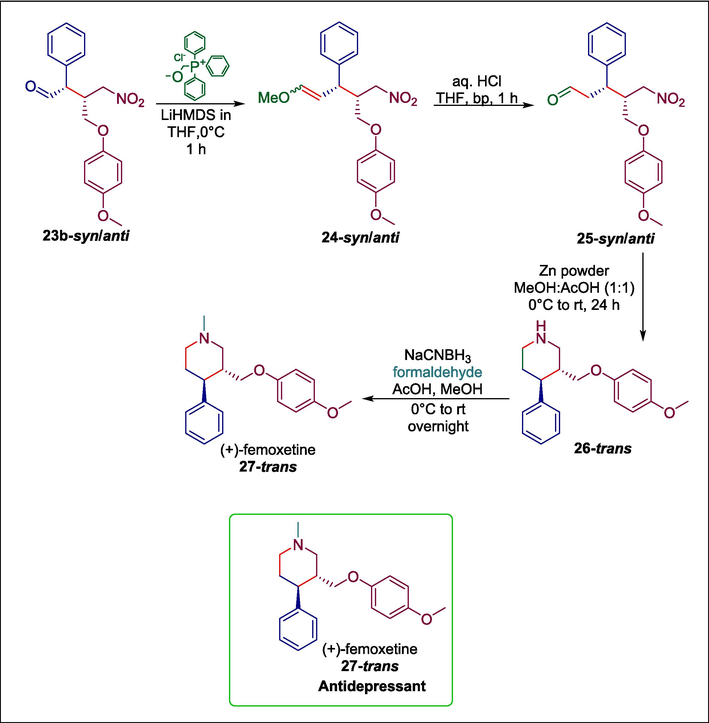
Hayashi-Jørgensen catalyzed synthesis of (+)-femoxetine.
An organometallic framework called lipophilic “sandwich” ferrocene is frequently used in the synthesis of anti-parasitic, antibacterial, and antitumor medicines (Hillard et al., 2010). Bioactive ferrocenes that have been synthesized through different techniques in the past few years can be either achiral or racemic or simple conjugates of ferrocenyl that already have chiral moiety (Van Staveren and Metzler-Nolte, 2004). The stereoselective construction of ferrocene compounds with potential applications in pharmacology has received relatively little attention. Zhang et al. used organocatalytic [2 + 2 + 2] annulation to create a series of chiral spirocyclic pyrazolone-ferrocene hybrids with numerous stereocenters in high yields and exceptional diastereo- and enantioselectivity. Aldehyde 28 and nitroalkene 29 undergo Hayashi-Jørgensen secondary amine C3 catalyzed Michael addition. Subsequently, spirocyclic pyrazolone-ferrocene hybrids 31 are produced by adding compound 30 and DBU (Scheme 4) (Zhang et al., 2018b). This method is atom-economic, safe for the environment, and works well with many functional groups. Compound 31a, found in this series, exhibited strong inhibition of RalA, with an IC50 of 1.20 ± 0.19 μM. It also caused reactive oxygen species to accumulate and prevented the growth of pancreatic cancer cells with IC50 = 1.6 μM.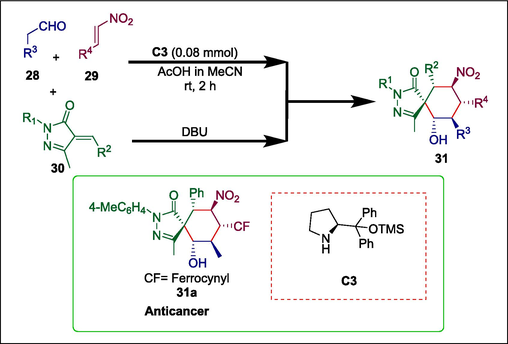
Hayashi-Jørgensen catalyzed the synthesis of pyrazolone-ferrocene hybrids.
Multifunctional spiropyrazolones fused with cyclohexane(ene) have appealing biological actions like antibacterial, anticancer, anti-inflammatory, and analgesic characteristics. Previously, asymmetric organocatalytic processes were used to create spiropyrazolone scaffolds from Pyrazolin-5-one substrates; however, in these methods, pyrazolinones function as adaptable electrophilic C2 synthons (Zheng et al., 2015b Li et al., 2016; Leng et al., 2018). Li et al. developed an asymmetric [4 + 2] annulation method using the α,β,γ,δ-unsaturated pyrazolone substrate 32, aldehyde 33 and a Hayashi-Jørgensen amine catalyst C4 to synthesize α,β,γ,δ-unsaturated pyrazolone 34. The compound 33 is activated in this reaction using a HOMO-raising approach (Li et al., 2012; Li et al., 2018), while pyrazolone substrate 32 functions as the electron-deficient C4 scaffold and branched diene for spiro-construction (Li et al., 2018) (Scheme 5) (Li et al., 2019). Theafforded chiral compounds exhibited strong cytotoxic activity. The compound 34a caused apoptotic cell death in HCT116 colorectal cancer cells and has IC50 = 0.37 μM.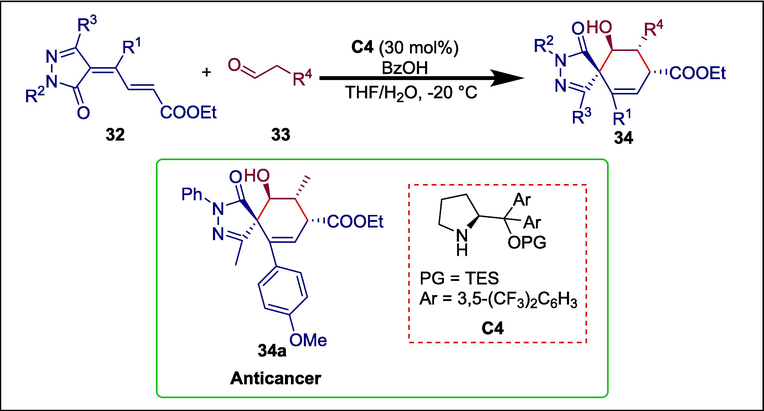
Hayashi-Jørgensen amine-catalyzed synthesis of spirocyclohexene pyrazolones.
2.2 N-heterocyclic carbene catalyst (NHC)
Ketonitrile, a vital framework of numerous N-containing bioactive compounds like piperidine, pyridine, pyridin-2(1H)-one, and 2-aminothiophene, is produced via N-heterocyclic carbene catalyzed ring-opening coupling between an aldehyde and a cyclic oxime derivative in the presence of the NHC catalyst (Streuff et al., 2012; Nguyen et al., 2020). The problem of employing hazardous transitional metals in C–C bond activation has led to the development of this NHC-catalyzed approach. Yang and Wan developed thiazolium C5 catalyzed reaction between oxime derivative 35 with benzaldehyde 36 to produce ketonitrile 37 which was then transformed into CWJ-a-5 38. Adding NHC C5 to benezenecarbaldehyde 36 in the presence of Cs2CO3 initiates the catalytic cycle. The obtained intermediate 39 goes through a single-electron transfer process with cyclic oxime ether 35 to give the intermediates 40 and 41. In iminyl radical 41, the hyperconjugation effect between the unpaired-electron-occupied orbit on the N atom and the σ orbital of the α-C–C bond leads to a quick radical-type ring-opening process, yielding the nucleophilic alkyl radical 42. Then the cross-coupling between 42 and 40 affords the compound 43, which experiences an intramolecular fragmentation to furnish the desired compound 37 and regenerates the C5 (Scheme 6) (Yang and Wan, 2021). This technique showed several good synthetic properties, including as high yields, readily available reactants, and environmental friendliness (Zimmerman et al., 2020). A formal synthesis of the strong topoisomerase I inhibitor CWJ-a-5 38 demonstrates the synthetic potential of this new technique.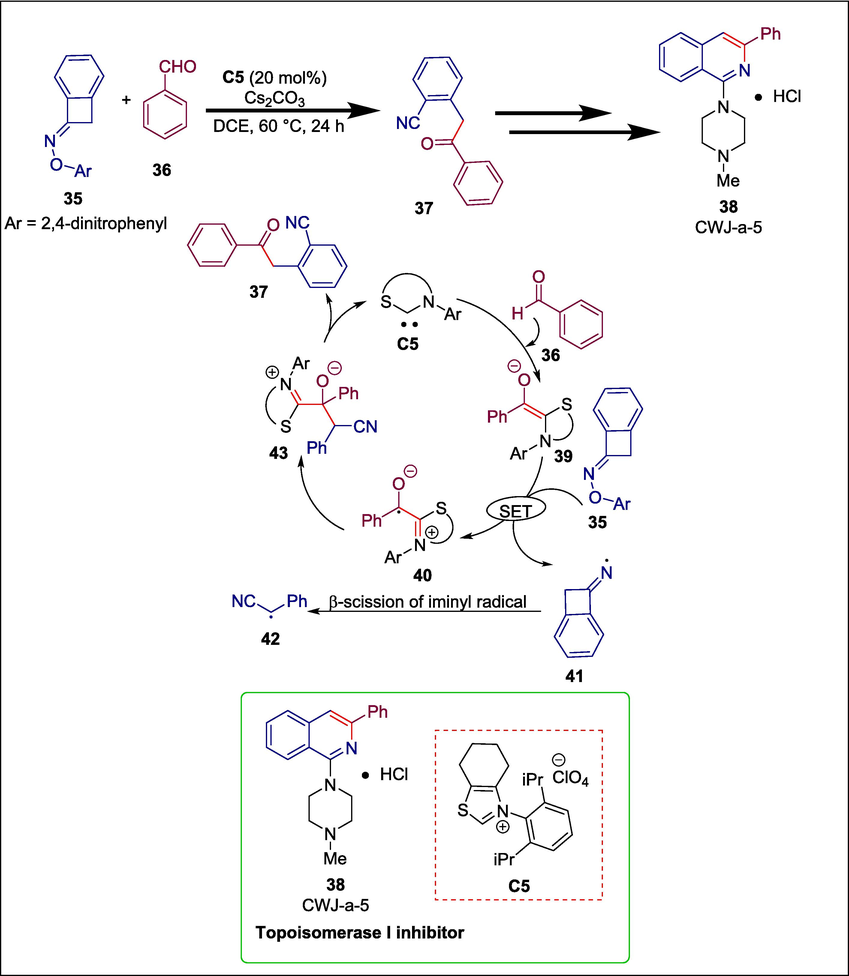
NHC catalyzed synthesis of CWJ-a-5.
Chagas disease, which affects around 8 million individuals worldwide, is brought on by infection with the protozoan parasite Trypanosoma cruzi. There are currently only two medications that are approved to cure Chagas disease: nifurtimox (NFX) and benznidazole (BZ). Both medications show poor chronic phase cure rates and frequently cause major side effects (de Fátima Oliveira et al., 2008). Coelho et al. synthesized eighteen tricyclic coumarins using NHC organocatalysis to fill this gap in novel drug candidates for Chagas disease. The first step in the synthesis of target compound 50 is to prepare α; β-unsaturated aldehydes 49 using the Wittig reaction, which produces substituted cinnamaldehydes in a high yield. Cross-aldol condensation between aromatic aldehydes 45 and substituted 2-hydroxyacetophenones 44 yields the required chalcones 46. The next step is a homoenolate annulation reaction between cinnamaldehydes 49 and chalcones 46, which is catalyzed by NHC C6 and results in good to outstanding yields of tricyclic coumarins 50 (Scheme 7) (Coelho et al., 2019). The cytotoxic efficacy of these synthesized compounds against Trypanosoma cruzi on L929 cells of mice was assessed. Ten of the eighteen compounds that were examined had IC50 values that were lower than those of the reference drug benznidazole. 50a with IC50 = 0.4 µM was the most potent compound.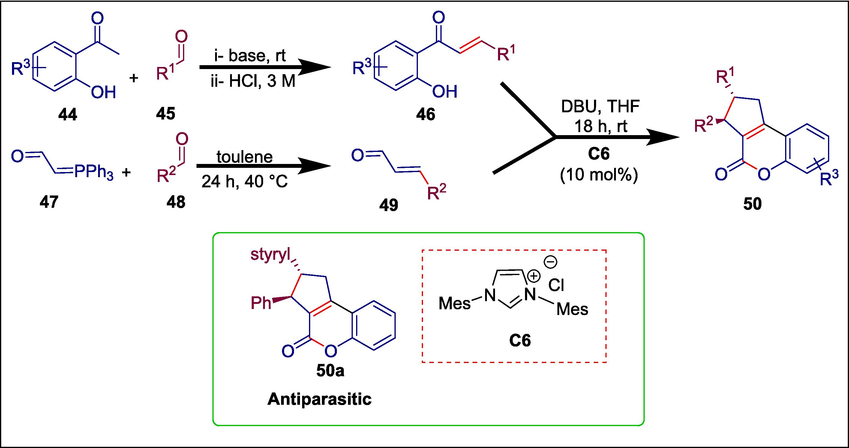
NHC catalyzed synthesis of tricyclic coumarins.
A synthetic drug called iguratimod (T-614) treats rheumatic diseases (Tanaka et al., 2015). It also lessens severe pancreatitis by blocking the NLRP3 inflammasome and the NF-kB pathway (Hou et al., 2019; Ishikawa and Ishikawa, 2019). Previously Takano et al. synthesized T-614 amine precursor utilizing 3-nitro-4-phenoxyphenol through NO2 reduction, mesylation, O-alkylation using 3-chloropropionic acid, intramolecular ring closure using polyphosphoric acid, bromination, nucleophilic azidation and dehydrogenative amination. The overall yield under these protocols was unsatisfactory. Murugesh et al. devised a proficient synthetic technique to synthesize iguratimod 60 with high yield through regioselective Vilsmeier-Haack formylation and intramolecular N-heterocyclic carbene-catalyzed aldehyde-nitrile cross-coupling reactions. The first step of this synthesis is the nucleophilic aromatic substitution (SNAr) of 1-chloro-4-methoxy-2-nitrobenzene 51 with C6H5OH 52 to yield 4-methoxy-2-nitro-1-phenoxybenzene 53 which then goes through NO2 reduction followed by mesylation to afford N-(5-methoxy-2-phenoxyphenyl) methanesulfonamide 54. Then 54 undergoes Vilsmeier-Haack formylation and demethylation to yield N-(4-formyl-5-hydroxy-2-phenoxyphenyl)methanesulfonamide 57 which undergoes classical O-alkylation to give compound 58 that is converted into amine precursor of iguratimod 59 via aldehyde-nitrile cross-coupling using triazolium pre-catalyst (Scheme 8) (Murugesh et al., 2020).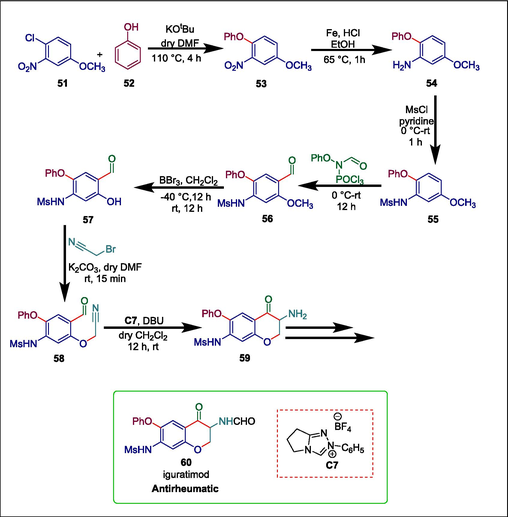
NHC catalyzed synthesis of iguratimod.
2.3 Tert-leucinamide catalyst
(R)- pantolactone is an essential component of medicines and is the synthetic precursor of vitamin B5, often known as calcium pantothenate (Müller et al., 2000). Previous strategies for catalytic asymmetric transformations include the aldol transformation between silyl ether of S-ethyl 2-methylpropanethioate and isobutyraldehyde, as well as the chiral ligand–metal complex-catalyzed asymmetric reduction of ketopantolactone, (Takahashi et al., 1986)a five-step process bearing on Sharpless dihydroxylation, (Upadhya et al., 1999)and an aldol transformation of ketopantolactone (Evans et al., 2002). Harmful transition metals, very low reaction temperatures (−78 °C), poor yields, and high costs of reactants are the main drawbacks of these techniques. Du et al. described tert-leucine-based 2-phenolic anilide C8 catalyzed asymmetric cross-aldol reactions between glyoxylate 62 and aldehydes 61 to produce (R)-pantolactone 64. The process begins when isobutyraldehyde 61 and phenolic tertiary-leucinamide catalyst C8 combine to generate intermediate imine 65, which reversibly transforms into intensely nucleophilic enamine 66. To efficiently activate the aldehyde scaffold and quicken the aldol reaction, the catalyst's amide and hydroxy groups create two H-bonds with ethyl glyoxylate 62. The bulky tertiary-butyl group of C8 acts as a steric hindrance, so enamine’s 66 attack to the 61 primarily occurs in the Re-face of the aldehyde skeletal of 62. This results in the R configurational aldol product 63, which is then reduced and immediately lactonized to produce (R)-pantolactone 64 in a high yield (Scheme 9) (Du et al., 2021). A simple recrystallization can easily enrich the resulting (R)-pantolactone's enantiopurity to 99 %. The high step economy, readily available reactants, cost-effectiveness, no use of hazardous chemicals, low reaction conditions, and ease of use of this process make it superior to conventional techniques.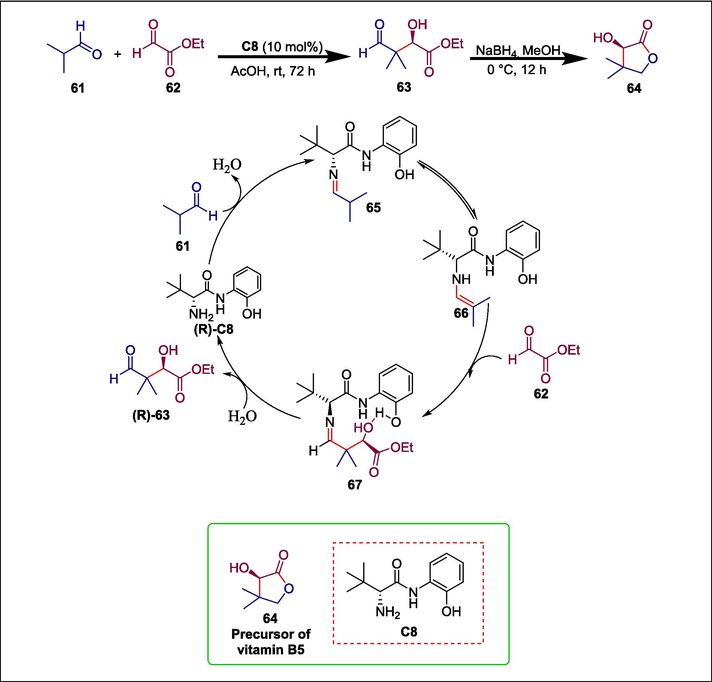
Tert-leucinamide catalyzed synthesis of (R)- pantolactone.
2.4 Trimethylimidazolidin-4-one hydrochloride catalyst
Several pharmaceutical molecules, including amlodipine, isradipine, dioscorine, and (+)-calanolide, contain 1,4-dihydropyridines and 4H-pyran derivatives as significant structural elements (Shehab and Ghoneim, 2016; Nazari et al., 2017). Research on hybrid compounds with combinations of these two heterocyclic functionalities is an intriguing way to make use of the bioactivities of both 1,4-dihydropyridines and 4H-pyran moieties. Pham et al. synthesized new 1,4-dihydropyridine and 4H-pyran hybrid compounds i.e. 4H-pyrano[2,3-b]pyridine derivatives 70a-f. The required products were produced through the activation of an organocatalyst C9 in the reaction between 1,4-dihydropyridines 68 and acrolein 69. The reaction begins when acrolein 69 reacts with trimethylimidazolidin-4-one hydrochloride catalyst C9 to form iminium ion 71 which goes through Michael addition to the 1,4-dihydropyridine 68 to afford cationic adduct 72. After the enamine's hydrolysis and potential hydrate production, cyclization produces the bicyclic compound 74, which, upon dehydration, yields the desired 4-hydroxypyrano[2,3-b]pyridine hybrid molecule 70 (Scheme 10) (Pham et al., 2020). When these synthetic products were examined for cytotoxicity against HepG2 and KB cancer cell lines, the results were extremely encouraging: every molecule had an IC50 of at least moderate, three compounds (70a, 70b, and 70c) had IC50 < 10 mM and one compound 70c had IC50 < 1 mM.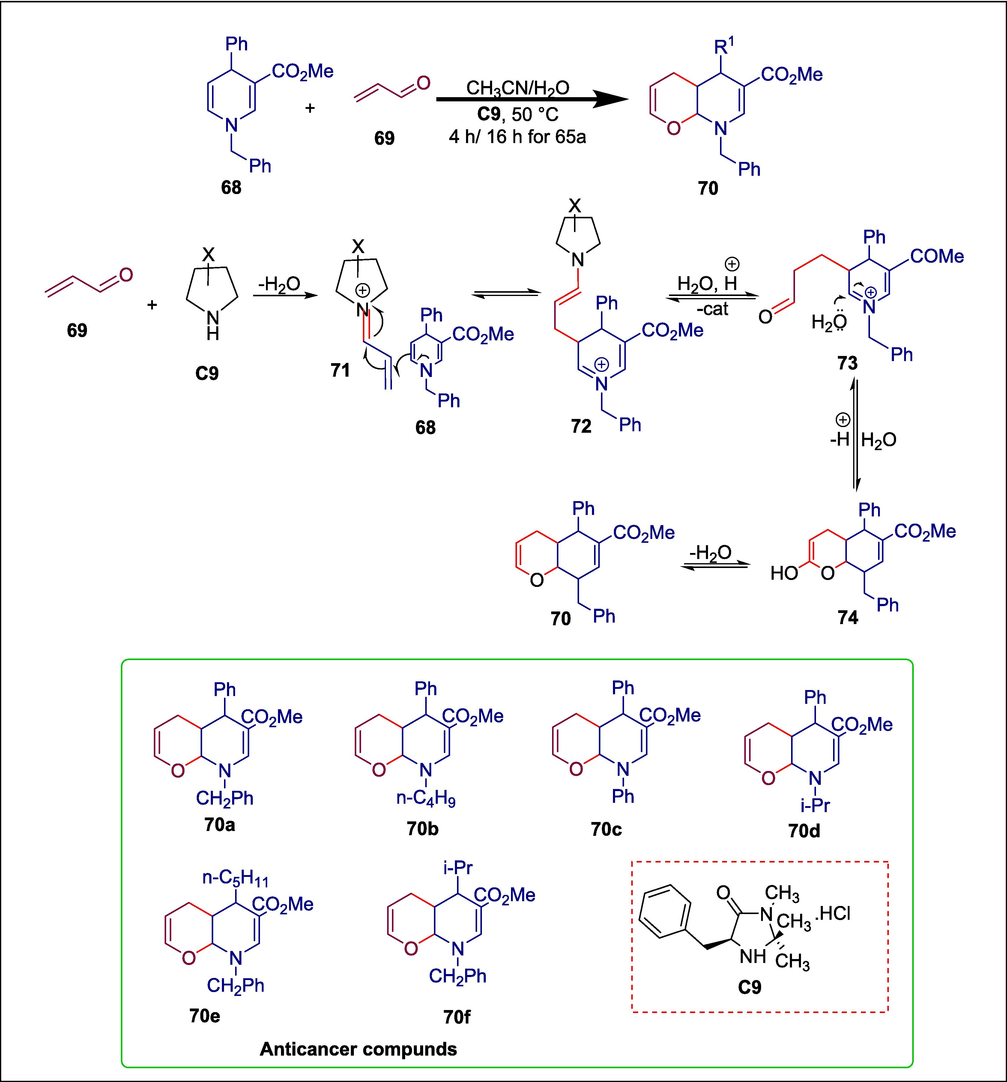
Trimethylimidazolidin-4-one hydrochloride catalyzed synthesis of 1,4-dihydropyridines and 4H-pyran derivatives.
2.5 Triphenylphosphine catalyst
One significant class of biologically active heterocycles is composed of oxazinones and oxathiinones (Miyauchi et al., 1996; Piergentili et al., 2007; Singh et al., 2011; Méndez‐Rojas et al., 2018). Oxazinones and their benzoid derivatives have a range of medicinal properties, including anticancer (Bolognese et al., 2002), antiviral (Abood et al., 1997; Jarvest et al., 1997), antithrombotic (Hsieh et al., 2005), antimycobacterial (Waisser et al., 2000), anti-inflammatory (Hsieh et al., 2004), antidiabetic, and hypolipidemic (Madhavan et al., 2006) actions. Phosphine-induced organocatalyzed reactions are a potent method for producing heterocycles of sulfur, nitrogen, and oxygen. Gholami et al. produced novel benzoxazinones 73 and benzoxathiinones 74 via PPh3 C10 catalyzed reaction of aminophenol 71a or 2-mercaptophenol 71b with alkyl X-phenylpropiolates 72 (Scheme 11). The process begins as Ph3P C10 is added to alkyl X-phenylpropiolate 72 to generate zwitterionic intermediate 75 which is protonated by 71a and 71b to yield vinylphosphonium cation 76 and the anionic intermediates 77 and 78, respectively. Path a involves a nucleophilic substitution reaction between 76 and 77, forming intermediate 79, which is then transformed into compound 80 through intramolecular Michael addition. By losing a proton, intermediate 80 produces compound 82, which is then changed to 84 by removing PPh3 and tautomerization. In path b, 78 undergoes Michael addition to 76 to form ylide 85, which then experiences 1,2-proton exchange and the loss of PPh3 to yield the required product 87 (Scheme 12) (Gholami et al., 2019). The antioxidant and antibacterial properties of the produced compounds were assessed. Compounds 73a; 73b and 74a possess greater antibacterial activity against S. aureus, B. subtilis, and E. coli bacteria, however, compound 74b has the most potent antioxidant activity (93.6 %).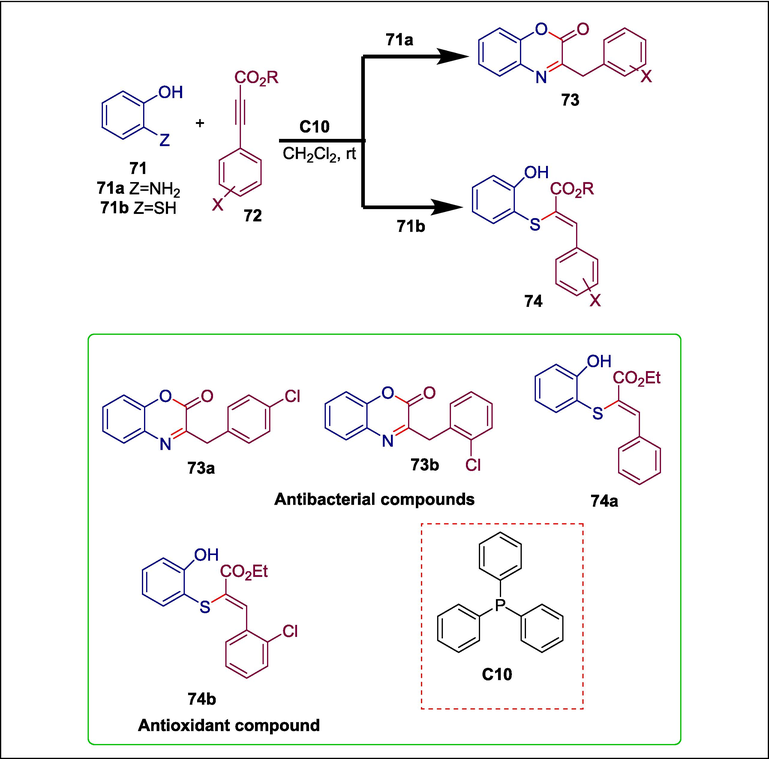
PPh3 catalyzed synthesis of benzoxazinones and benzoxathiinones.
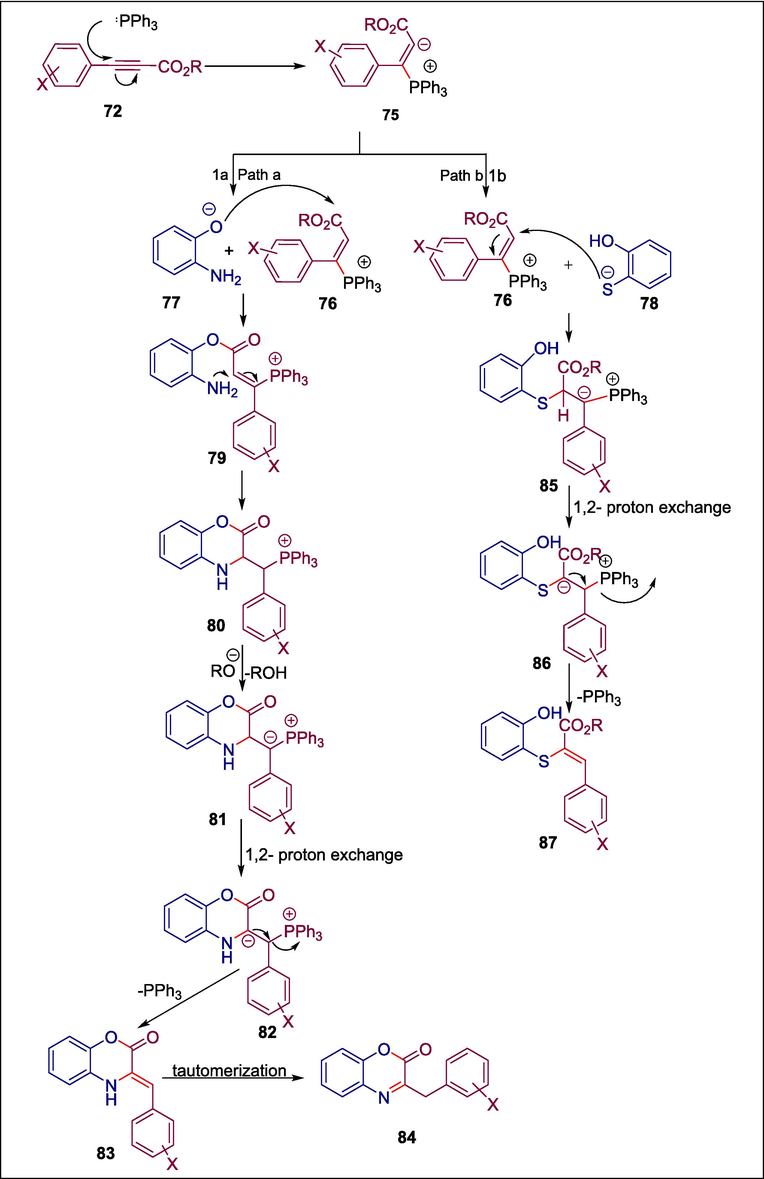
Mechanism of synthesis of benzoxazinones and benzoxathiinones.
Summary
Lewis base organocatalysts are an essential tool in modern synthesis because of their remarkable efficiency in stimulating organic reactions by donating electron pairs to electrophilic substrates. These catalysts, often containing nucleophilic centers such as nitrogen, phosphorus, or oxygen, operate by interacting with electrophiles to increase their reactivity or stabilize transition states. Lewis base catalysts enhance nucleophilicity of the substrate to start the catalytic cycle. The resulting complex undergoes a reaction and then releases the product and the catalyst for further turnover (Fig. 1). Lewis base catalysts’ metal-free nature, cost-effectiveness, and potential to minimize waste make them an excellent choice for sustainable and green chemistry. In this section, we have described different Lewis base catalysts such as Hayashi-Jørgensen, N-heterocyclic carbene, tert-leucinamide, trimethylimidazolidin-4-one hydrochloride, and tri-phenylphosphine catalysts for the synthesis of valuable pharmacophores showing various bioactivities i.e. antibacterial, antidepression, anticancer, antioxidizing, and anti topoisomerase I.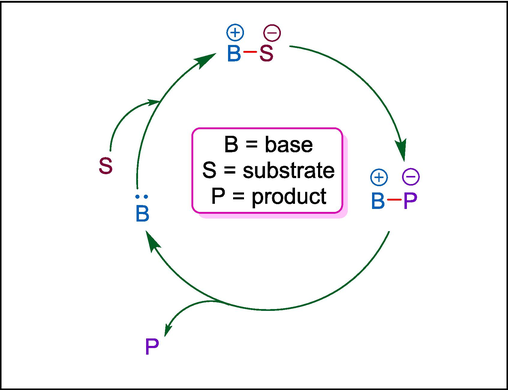
Mechanism of Lewis base catalysis.
3 Lewis acid catalysis
Lewis acids are of particular interest when discussing the synthesis of novel chiral organocatalysts since they often stimulate reactions by electrophilically activating organic functional groups (such as carbonyl compounds, imines, and epoxides) toward nucleophilic attack (Schenker et al., 2011). Lewis acid catalysis allows important chemical reactions such as Friedel-Crafts, Diels-Alder, and several aldol, Mannich, and Michael reactions. As a result, extensive research has been done on enantiopure Lewis acids, which has made significant asymmetric modifications of these reactions possible (Hollis et al., 1993). A major drawback of enantioselective Lewis acid catalysis, despite the abundance of sophisticated catalysts and techniques developed in this field, is the necessity for frequently high catalyst loadings. This is due to problems like insufficient Lewis acidity, which prevents some substrate classes from being accessible at all, product inhibition, hydrolytic instability, and competition with nonenantioselective background catalysis (Carreira and Singer, 1994; Hollis and Bosnich, 1995). List et al. synthesized in situ silylated disulfonimide Lewis acid organocatalysts with catalyst loadings as low as 10 parts per million (ppm) (10–15) for highly enantioselective Mukaiyama-type reactions involving silylated nucleophiles (Gatzenmeier et al., 2016). Recently, a new class of catalysts i.e. the C–H acids, which are composed of binaphthyl-allyl-tetrasulfones has been developed. These catalysts were found to be extraordinarily active Lewis acid catalysts for enantioselective Diels-Alder reactions of cinnamates with cyclopentadiene upon in-situ silylation.
3.1 Trimethylsilyl trifluoromethanesulfonate catalyst
Isoxazolidines have been identified as viable therapeutic candidates because of their intriguing pharmacological characteristics such as antiviral (Loh et al., 2010)and anticancer (Bonanomi et al., 2020).Štadániová et al. reported a novel synthetic method utilizing the organocatalyst trimethylsilyl trifluoromethanesulfonate (TMSOTf) C11 to produce 1,2,3-triazoles containing the 3-hydroxymethylated 4-hydroxyisoxazolidine moiety 95. The mechanism of this protocol involves elimination reactions of 5-acetoxyisoxazolidines 88 with a catalytic amount of Trimethylsilyl trifluoromethanesulfonate in the presence of BSTFA in anhydrous N-methyl-2-pyrrolidone (NMP) to afford 2,3-dihydroisoxazoles 89 which are treated with of K2OsO4·2H2O in the presence of aqueous N-methylmorpholine-N-oxide (NMO) in CH3COCH3/H2O to yield diastereomeric mixtures of isoxazolidine-4,5-diols 90, consisting of four inseparable isomers, which are then converted into the corresponding O-benzoylated isoxazolidines 91a,b via utilizing DMAP. After reacting with acetonitrile, the isoxolidines 91a,b form diastereomeric mixtures of azidoisoxazolidines 92a,b. The synthesized azidoisoxazolidine 92a is further treated with phenylacetylene to give soxazolidinyl triazole 93 which undergoes debenzoylation to yield isoxazolidin-4-ol 94 which is then desilylated to yield the required 4-hydroxyisoxazolidinyl triazoles 95 (Scheme 13) (Štadániová et al., 2020). The inhibitory action of this produced compound was assessed on the MOLM-13 cell line and found that compound 95 displayed a moderate increase in cell death and suppression of metabolic activity.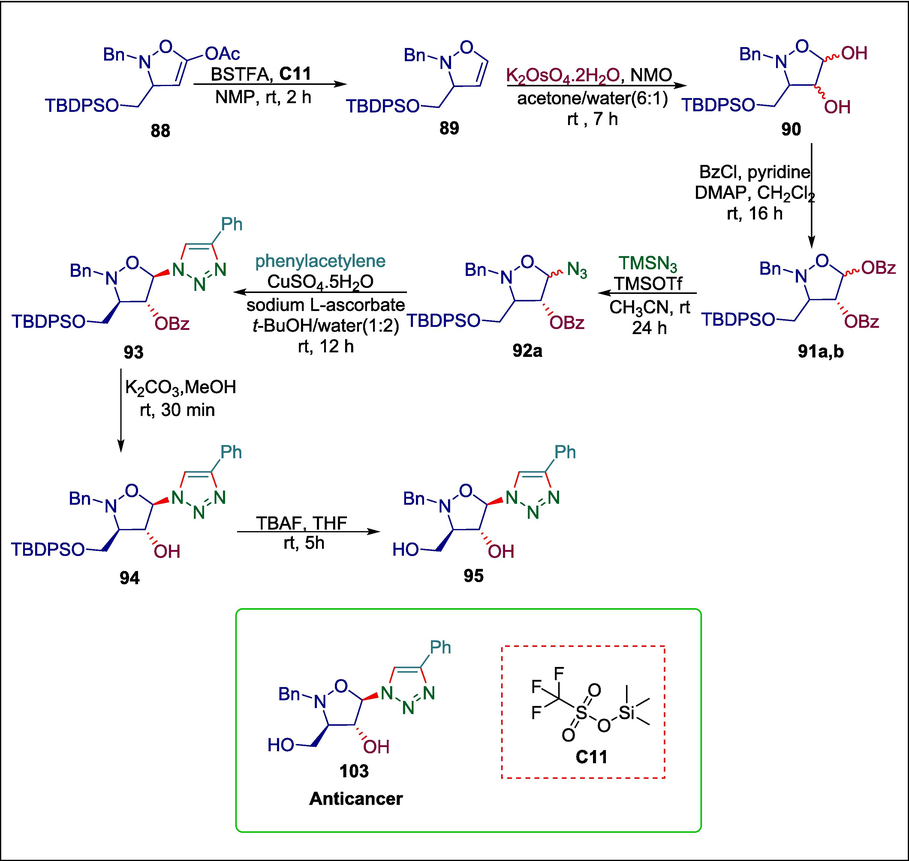
Trimethylsilyl trifluoromethanesulfonate catalyzed synthesis of 1,2,3-triazoles bearing the 3-hydroxymethylated 4-hydroxyisoxazolidine moiety.
3.2 Hyamine catalyst
Quinoline and its derivatives, primarily quinolones, have pharmacological properties (Wen et al., 2015; Gu et al., 2017). Many medicines, including ciprofloxacin, sparfloxacin, ofloxacin, norfloxacin, and gatifloxacin, are quinoline-based. Numerous attempts have been made to eliminate the negative effects of these drugs and antibiotics. The need to find novel antimicrobials arises from this situation. Arora et al. presented a sophisticated hyamine-catalyzed heteroannulation of the quinoline ring with dimedone to synthesize the corresponding azepinone analogues (Scheme 14) (Arora et al., 2018). The procedure entails reacting dimedone 96 with o-aminobenzoic acid 97 and isatoic anhydride 98 to produce acridine dione derivatives (105 and 106 respectively) which are then agitated with NH2OH.HCl in water and with a hyamine catalyst C12 to produce the corresponding oximes (99 or 100). When a freshly produced organocatalyst is added, these oximes (99 or 100) undergo Beckmann rearrangement, yielding the corresponding quinolino-annulated azepinones (101 or 102), respectively (Scheme 15) (Arora et al., 2018). This catalyst is prepared by heating cyanuric chloride (TCT) with dimethylformamide. It exhibits excellent catalytic activity, high efficiency, and ease of handling throughout the rearrangement process, which is one of its benefits. These synthetic azepinones were screened against two Gram-positive and two Gram-negative bacteria to assess their antibacterial activity. Compounds 101a and 102b demonstrated MICs of 15 μg/mL in the case of Staphylococcus aureus, indicating greater potency than the reference antibiotic streptomycin. Comparably, compound 101b's MIC of 10 μg/mL for Bacillus subtilis showed that it was more effective than the standard drugs tetracycline (30 μg/mL) and streptomycin (40 μg/mL). Significantly lower MIC values (05 and 01 μg/mL, respectively) were observed for compounds 101a and 102a compared to tetracycline (25 μg/mL) and reference streptomycin (55 μg/mL). Similarly, compounds 101a and 102c had remarkable minimum inhibitory concentrations (MIC) of 10 and 20 μg/mL, respectively, in contrast to reference drugs streptomycin (20 μg/mL) and tetracycline (55 μg/mL).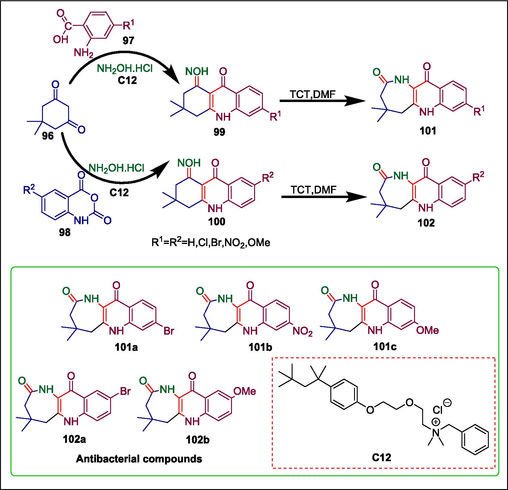
Hyamine-catalyzed synthesis of quinolino annulated azepinones.
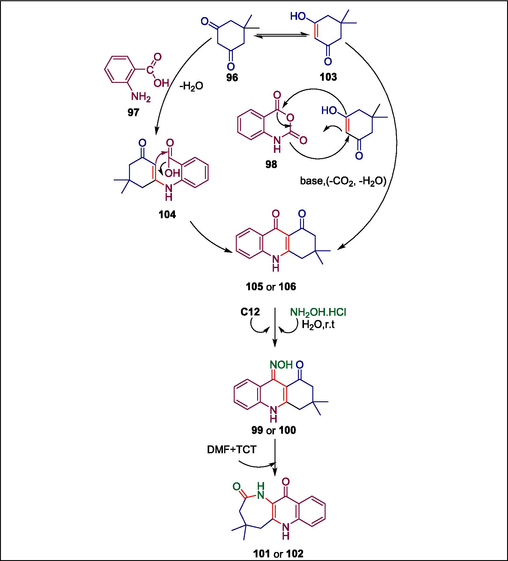
Mechanism of synthesis of quinolino-annulated azepinones.
3.3 Cinchonidine-derive quaternary ammonium catalyst
Development of enantiomerically pure compounds is a persistent need in the current chiral pharmaceutical market primarily due to the pure enantiomers' better therapeutic efficacy and safety profile over their racemates. One such example is bicalutamide (Fradet, 2004), a first-generation nonsteroidal androgen receptor (AR) antagonist that is mainly used to treat prostate cancer by selectively blocking the AR. The R isomer of bicalutamide has an almost exclusive antiandrogenic action, which can be explained by the (R)-enantiomer's 30-fold greater binding affinity to AR than the S isomer. Chen et al. used cinchonine/cinchonidine-derive quaternary ammonium salt C13 as a catalyst to develop a method for the asymmetric oxohydroxylation of readily available methyl/ethyl α-benzenesulfonylmethyl-β-aryl enoates and α-bromomethyl-β-aryl enoates with permanganate (Chen et al., 2024).This reaction demonstrated strong enantioselectivities and good tolerance for a broad range of substrates under mild conditions. In just three to five steps, the synthesized enantioenriched α-hydroxy-β-ketone esters (108a and 108b) were successfully used in the diversity-oriented synthesis of 2-arylmethyl-substituted bicalutamide analogue 111. After undergoing reductive deoxygenation (Lopez and Salazar, 2013), the asymmetric oxohydroxylation products 108a and 108b yield the α-hydroxy ethyl esters 109a and 109b, respectively. Then through ester hydrolysis and amide production, (R)-111, a reported (R)-bicalutamide analogue (Guerrini et al., 2014), was effectively synthesized (Scheme 16).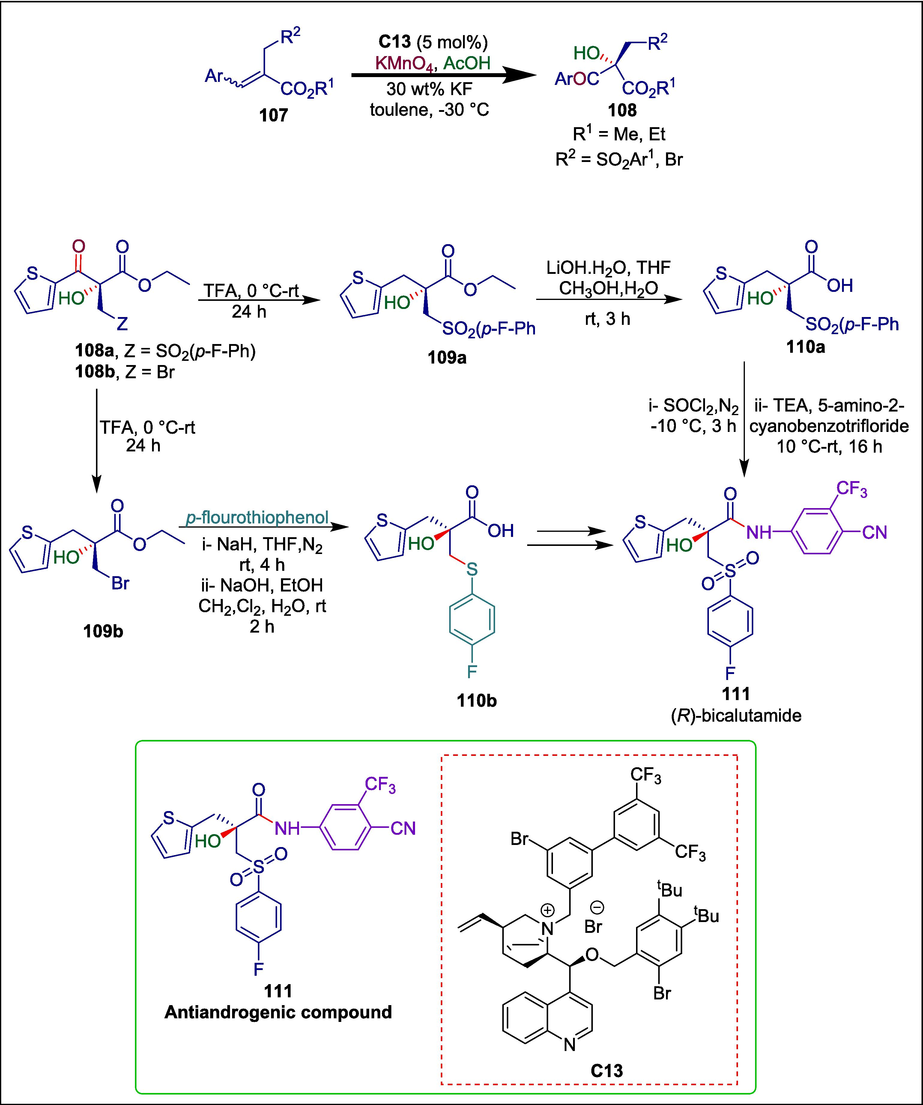
Cinchonidine-derive quaternary ammonium catalyzed synthesis of (R)-bicalutamide analogue.
3.4 Boron trifluoride diethyl etherate catalyst
Many natural compounds and active pharmaceutical products, such as naftopidil, naproxen, and suramin (Wang et al., 2012), are made up of fused naphthalenes, which are also actively used as ligands (Bai et al., 2019).So, it is preferable to develop simple synthetic methods for synthesizing functionalized naphthalenes. Mahecha-Mahecha et al. reported a simple methodology for the synthesis of functionalized cycloalkane-fused naphthalenes by using Lewis acid catalyst C14 and readily available substrates. Arylboronic acid pinacol esters 112 undergo Suzuki-Miyaura coupling reaction with cycloalkenyl triflates 113 to form aryl-substituted cyclohexene derivatives 114which are further cyclized by using boron trifluoride diethyl etherate catalyst C14 and yield desired fused naphthalenes 115 (Scheme 17) (Mahecha-Mahecha et al., 2020).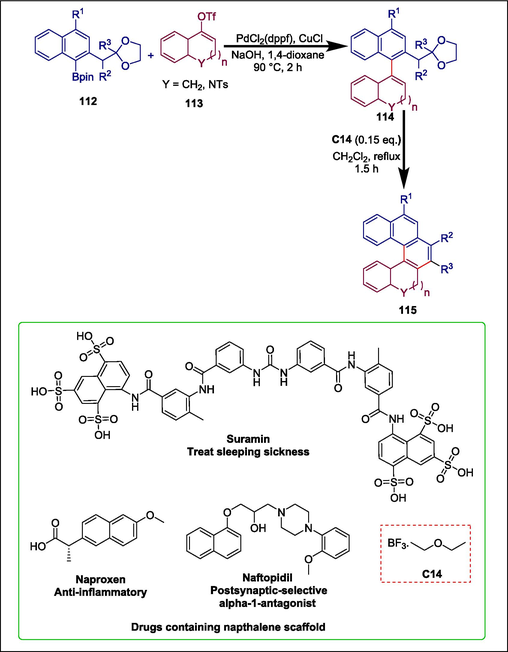
Boron trifluoride diethyl etherate catalyzed synthesis of fused naphthalenes.
3.5 Ethylaluminum dichloride catalyst
Fluorinated compounds are widely found in organic materials, agrochemicals, and pharmaceuticals1 because of their special biophysical characteristics such as lipophilicity (Smart, 2001), metabolic stability, bioavailability. Therefore, the synthetic community has been consistently interested in developing effective and practical methods to access fluorinated compounds. In this regard, gem-difluorinated cyclopropanes have become useful fluorine-containing building blocks for organic synthesis. Wu et al. reported a Lewis acid-catalyzed cross-coupling reaction between mono- or disubstituted gem-difluorinated cyclopropanes 116 and electron-rich arenes and allylsilanes 117 to synthesize fluoroallylic products 120 and fluorinated 1,5-dienes 121. First, an aluminum complex 122 is formed because of the strong affinity between fluorine of 116 and the Lewis acid EtAlCl2 C15, which then undergoes fluoride abstraction to produce the cyclopropyl cation intermediate 123. The ring tension is then released, causing C–C bond cleavage to generate a more stable fluoroallyl cation intermediate 124. Ultimately, the nucleophiles 125 attack the electrophilic fluoroallylic cation species 124 to form the C–C bond, which delivers the allylation product 126 and regenerates the Lewis acid catalyst by releasing a molecule of HF (with arene nucleophiles) or TMSF (with allylsilane nucleophiles) as a byproduct (Scheme 18) (Wu et al., 2022). The synthesis of a bioactive-related molecule 120a with a fluoroalkene moiety further illustrated the practicality of this method.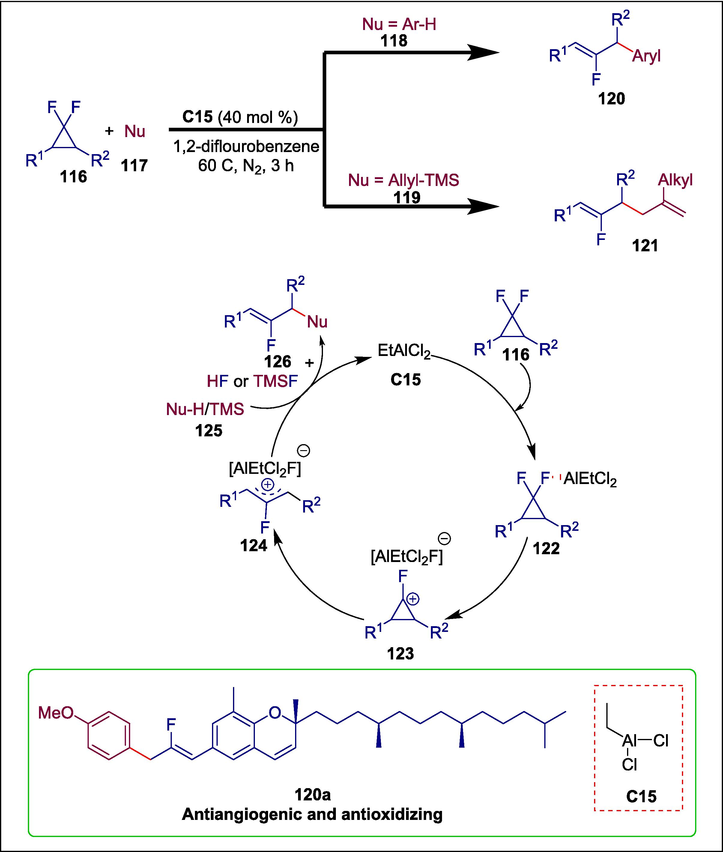
Ethylaluminum dichloride catalyzed synthesis of flouronated compounds.
Summary
Lewis acid organocatalysts are a class of catalysts that increase the electrophilicity of the reaction intermediates by accepting electron pairs from nucleophilic substrates. Usually composed of atoms like silicon, aluminum, or boron, these catalysts have electron-deficient centers that can coordinate to lone pairs of electrons on nucleophiles. This coordination lowers the energy barrier for the reaction by stabilizing negative charges or activating electrophiles for nucleophilic attack. A common mechanism includes the Lewis acid binding to carbonyl oxygen, increasing the electrophilic nature of the carbonyl carbon, rendering it more susceptible to nucleophilic attack (Fig. 2). These catalysts offer an effective and environmentally friendly substitute for conventional metal-based Lewis acids by enabling reactions to continue at milder conditions with improved selectivity. This section describes the usage of various Lewis acid catalysts e.g. trimethylsilyl trifluoromethanesulfonate and hyamine for synthesizing anticancer and antibacterial compounds.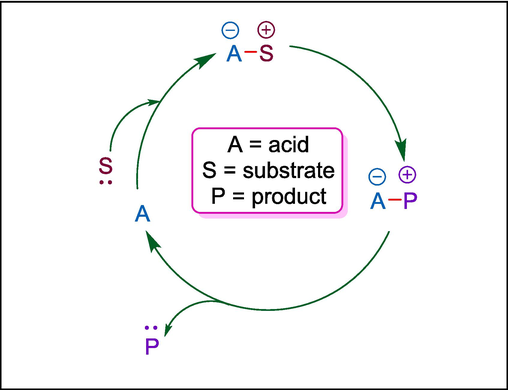
Mechanism of Lewis acid catalysis.
4 Bronsted base catalysis
Chiral organic Bronsted bases have been identified as extremely effective and selective catalysts for enantioselective synthesis. They are frequently employed in asymmetric organic synthesis, and the key factor of the Bronsted base catalysis is the presence of acidic “hydrogen” in the substrates (Zhang et al., 2018a). Common examples of organic Brønsted base catalysis in asymmetric synthesis are hydrocyanation reactions e.g. the Strecker reaction and cyanohydrin synthesis (Seayad and List, 2005). An ongoing problem in the study of Bronsted base catalysis is broadening the range of pronucleophiles that can be used in enantioselective reactions. Typically, chiral tert-amines have been frequently used as chiral Brønsted base catalysts (Marcelli and Hiemstra, 2010). P1-phosphazenes, cyclopentenimines, and chiral guanidines are examples of chiral uncharged organobases that have higher basicity than tert-amines and have recently become effective chiral Brønsted base catalysts (Uraguchi et al., 2007; Leow and Tan, 2009; Bandar and Lambert, 2012; Nunez et al., 2013). However, the limited basicity of these traditional chiral organobases restricts pronucleophiles to extremely acidic molecules like β-dicarbonyl compounds and nitroalkanes, which limits the feasible chemical conversions that can be catalyzed by chiral Brønsted base catalysis. In 2020 Kondoh et al. overcame the intrinsic limitations of pronucleophiles by developing a new chiral Brønsted base catalysts which comprise a P2-phosphazene as an organosuperbase and a chiral guanidine as a hydrogen bond donor for substrate recognition. Its strong catalytic activity was shown in the less acidic α-phenylthioacetate enantioselective direct Mannich-type reaction (Kondoh et al., 2020).
4.1 Pyrrolidine catalyst
The meroterpenoid applanatumol B is generated by the therapeutic fungus Ganoderma applanatum. It has a fused dioxacyclopenta[cd]indene motif and exhibits antirenal fibrosis activity (Luo et al., 2016) Simek et al. created a unified organocatalyzed method for synthesizing meroterpenoid applanatumols B. The process begins with 2,5-dihydroxyacetophenone 127 being protected as dibenzyl ether which undergoes an aldol addition with 4-pentenal 128 and subsequent acidic dehydration to give dienone 129. Afterward, α-aminoxy ketone 131 is produced by a novel tandem reaction comprising nucleophilic allylation/alkoxide-accelerated oxy-Cope rearrangement and radical oxygenation. Then 131 is heated to a 5-exo-trig cyclization temperature, yielding an inseparable mixture of cyclopentanes 132a, b, and c and trace amounts of 6-endo cyclization product 132d. The alkoxyamine function in the inseparable crude mixture is removed by oxidative means, resulting in a mixture of aldehydes 133a, b, and c which is transformed by Pinnick oxidation, furnishing a separable mixture of carboxylic acids 134a, b, c in and cyclohexanone 133d. Then the esterification of carboxylic acid 133a and subsequent Johnson-Lemieux cleavage of the terminal olefin yield aldehyde 135 (Yu et al., 2004). The introduction of the remaining carbon atom is achieved by pyrrolidine C16 catalytic α-hydroxymethylation of 135 leading initially to a 1,3- dioxan-4-ol intermediate which is in situ broken down and reduced to the 1,3-diol unit. Following methyl ester hydrolysis, acid 136 is produced, and it is epimerized to produce tricyclic product 137. After that, applanatumol B 138 is produced through a small amount of epimerization brought on by the removal of the benzyl-protecting groups (Scheme 19) (Simek et al., 2022).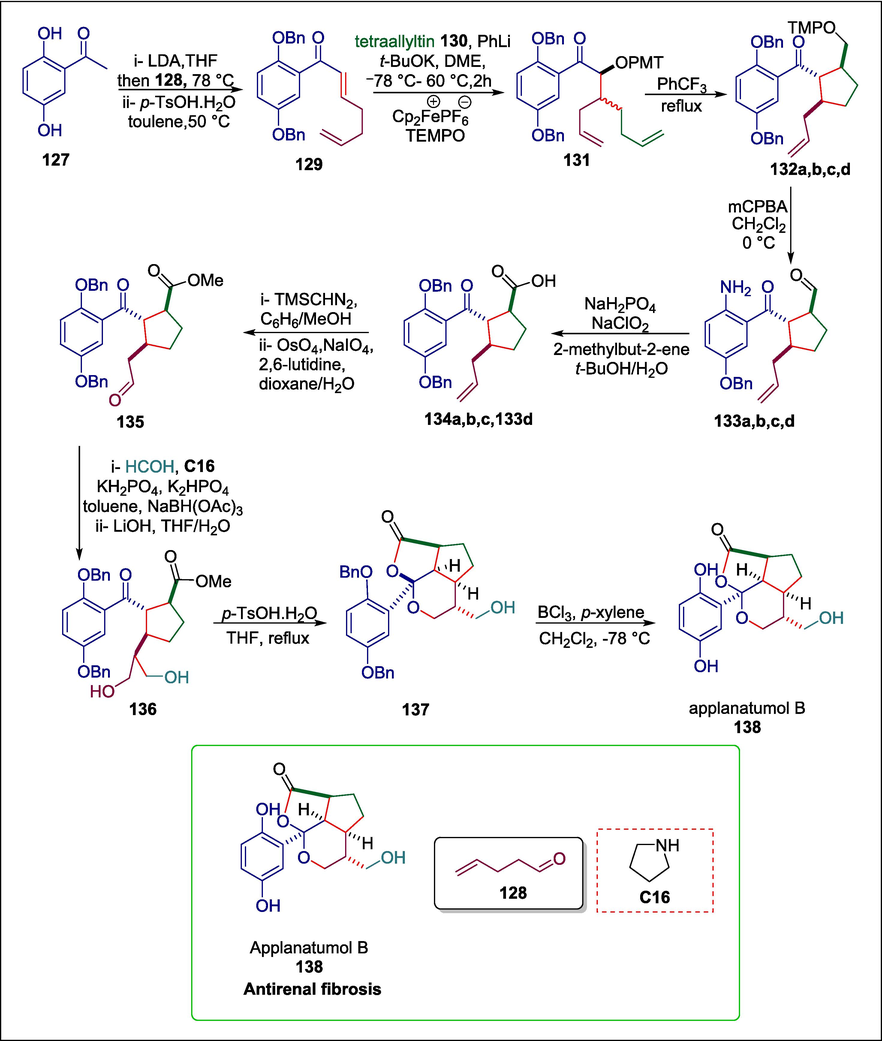
Pyrrolidine catalyzed synthesis of applanatumol B.
(−)-Pavidolide B is a natural marine bioactive compound. Tetracyclic diterpenoid (−)-pavidolide B 139 exhibits specific inhibition against the human promyelocytic leukemia cell line HL-60 (IC50 = 2.7 μg/mL), according to preliminary biological research conducted on tumor cells (Shen et al., 2012). Zhang et al. devised a succinct method for the asymmetric total synthesis of (−)-pavidolide B 139 (Zhang et al., 2017), which was made possible by an intramolecular [3 + 2] annulation reaction of a vinylcyclopropane (VCP) mediated by thiyl radical. 2,4-hexadienal 139 and dimethyl 2-bromomalonate 140 react first in the presence of an organocatalyst (R)-2-(diphenyl((trimethylsilyl)oxy)methyl)pyrrolidine C17 to produce an aldehyde 141. This aldehyde then goes through hydrolysis and Mitsunobu reaction sequences to form compound 145, which then goes through the [3 + 2] annulation reaction to produce the desired tricyclic skeletal 146. Following treatment of this precursor under the circumstances of the Krapcho decarboxylation reaction, aldehydes 147a and 147b are produced. Aldehyde 147b is converted to a homoallylic product via Dess-Martin periodinane (DMP) oxidation, which yields diene 149b which combines with Grubbs II catalyst to produce the tetracyclic skeleton 150b. Pavidolide B 151 is then obtained by treating 150b with RhCl3·3H2O (Scheme 20) (Zhang et al., 2019c). In this strategy, low-cost energy is used to stimulate chemical reactions.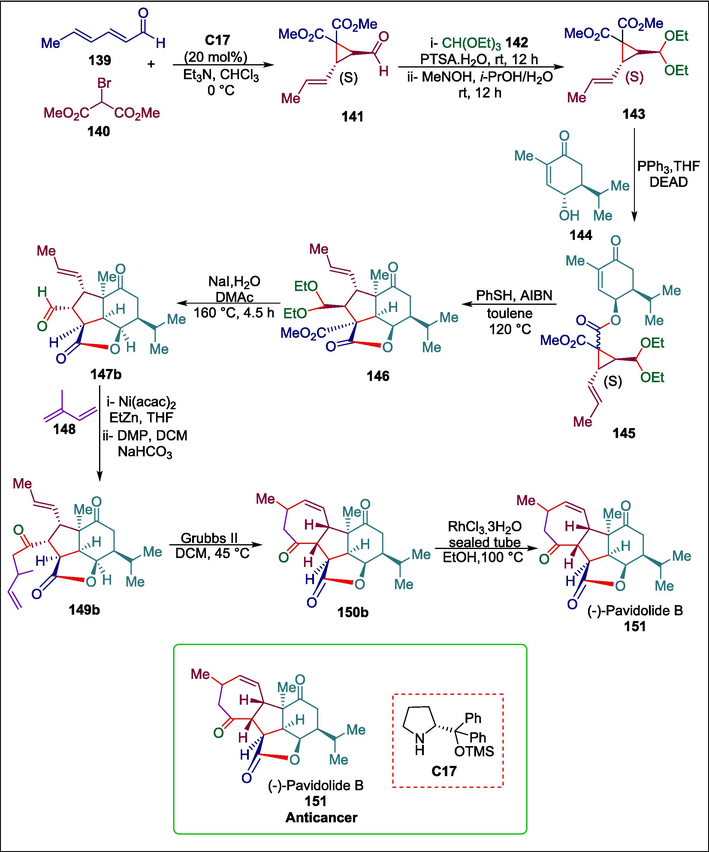
Pyrrolidine catalyzed synthesis of (−)-pavidolide B.
Bridged N-heterocycles like morphans and normorphans are significant skeleton of several bioactive compounds (Zhang et al., 2006; Betou et al., 2014). Dixon et al. recently synthesized enantioenriched morphans by co-catalyzing the desymmetrization of 4-propargylamino cyclohexanones with chiral amine and chiral silver complexes. Only transition metal catalysts were able to initiate these asymmetric carbocyclization reactions. To create a variety of desirable morphans 153 and normorphans 155 with a broad substrate range and outstanding enantioselectivity (up to 97 % ee), Xu et al. developed a metal-free organocatalytic enantioselective desymmetrizing cycloisomerization of arylsulfonyl-protected ynamide-cyclohexanones 152 and 154. The reaction begins when pyrrolidine catalyst C18 and the ynamide-tethered cyclohexanone 156 undergo an amine-ketone condensation reaction through intermediate 157 to give the enamine intermediate 158. The nucleophilic carbon site of 158′s enamine group can target the α or β position of the ynamide group, resulting in the formation of vinyl anion intermediates 159 or 161 (Marien et al., 2018). The α and β carbons of the Tosyl-containing ynamide are both positively charged because Ts is more electron-withdrawing than Ms. The nucleophilic attack favors the β site to create a sterically less hindered 6-membered ring intermediate 159, which eventually leads to morphan 160. Due to the negatively charged β carbon in PG = Ms, the positively charged α carbon site is favored by the nucleophilic addition, forming a sterically more hindered 5-membered ring intermediate, which is the precursor to normorphan 162 (Scheme 21) (Xu et al., 2019b). The greater electron-withdrawing capacity of Tosyl than Mesyl in the ynamide moiety is responsible for the observed protecting-group-dependent regiodivergence. This procedure provides the first amine-catalyzed ynamide reaction, devoid of both Brønsted acid and transition metals, and also the first metal-free asymmetric Conia-ene-type carbocyclization. Furthermore, the bioactivity of the recently synthesized normorphans and morphans as anticancer drugs was examined. Some morphans showed cytotoxic effects on esophageal cancer cells, such as SK-GT-4 and KYSE-450, and nearly half of these morphans demonstrated remarkable cytotoxicity in breast cancer cells, MDA-MB-231 and melanoma cells A375, while the normorphan moieties showed only weak antitumor activity against these five cell lines.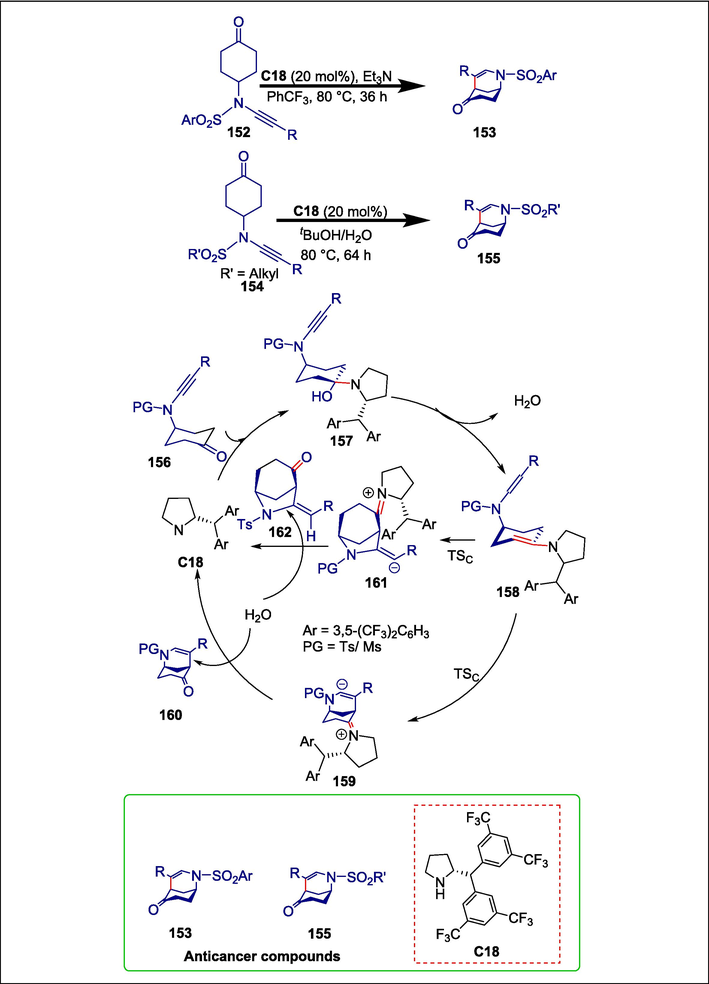
Pyrrolidine catalyzed synthesis of morphans and normorphans.
Starting from morphan 163, the anti-inflammatory drug 165 was also synthesized by reducing the alkenyl and carbonyl groups, oxidizing the CH2 group next to the nitrogen, and deprotecting the Tosyl group (Scheme 22) (Xu et al., 2019b ).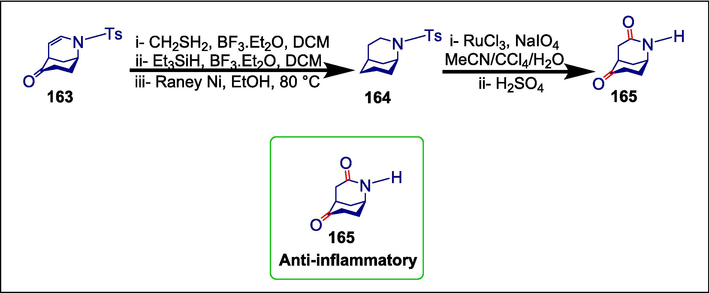
Pyrrolidine catalyzed synthesis of an anti-inflammatory drug.
Cathepsin K (CatK) is a key target in the development of treatments for bone diseases due to its strong collagenolytic activity (Brömme and Lecaille, 2009; Boonen et al., 2012). It has been reported that epoxypeptidomimetics are strong cathepsin inhibitors. Silva et al. described a green synthesis of novel peptidomimetics via a one-pot asymmetric epoxidation/Ugi multicomponent reaction. This process permits the use of ethanol/water mixtures as green solvents. These scientists synthesized epoxy-α-acyloxycarboxamides (LSPN690-694) through an asymmetric proline-catalyzed epoxidation ofα,β-unsaturated aldehydes followed by the Passerini multi-component reaction. The reaction begins when α,β-unsaturated aldehyde 166 undergoes proline-derived C19 catalyzed epoxidation to yield the crude epoxyaldehyde which is then subjected to the Ugi reaction conditions using benzylamine, benzoic acid, and n-butyl isonitrile to give the desired product epoxy-α-acyloxycarboxamides 167 (Scheme 23) (Silva et al., 2020). The synthesized moieties were assessed against CatK and found that the compounds LSPN690 167a and LSPN694 167b were active with IC50 values of 18.45 ± 1.34 µM and 6.44 ± 0.54 µM respectively.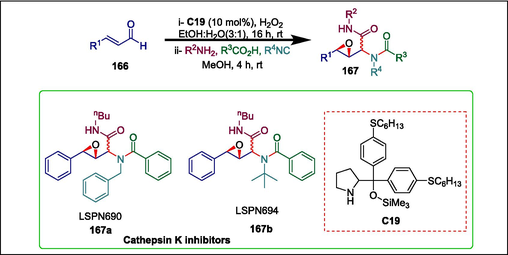
Pyrrolidine catalyzed synthesis of epoxy-acyloxycarboxamides.
4.2 1,8-diazabicyclo[5.4.0]undec-7-ene catalyst
Pyridine derivatives are used as antifungal agents (Enoch et al., 2006). Recently, Zhang et al. used the ester of substituted α-amino acid to manufacture tetrahydroimidazo (Dömling, 2002; Zhu and Bienaymé, 2006) pyridine derivatives. Long reaction times (Sun et al., 2010; Hadjebi et al., 2011; Pal et al., 2013a, 2013b, Zhang et al., 2017), the employment of stoichiometric amounts of catalyst (Sun et al., 2010; Zhang et al., 2017), and complicated product isolation processes are some of these methods' drawbacks (Kiruthika and Perumal, 2014; Pal et al., 2014; Zhang et al., 2017; Borah et al., 2023). Musawwer et al. created a simple one-pot synthesis of N-aryl-1,4-dihydropyridines (1,4- DHPs) by reacting easily obtainable starting ingredients such as malononitrile, aromatic aldehydes, dialkyl acetylenedicarboxylates, and arylamines with organocatalyst DBU C20. Initially, dimethyl acetylenedicarboxylate (DMAD) 168, arylamines 169, malononitrile 170, and aromatic aldehydes 171 were reacted under ideal circumstances, yielding products 172. Compared to electron-donating groups, aromatic aldehydes with electron-withdrawing groups produced better yields in a shorter amount of time. The same process for diethyl acetylenedicarboxylate (DEAD) 173 was next investigated using various aromatic amines 169, malononitrile 170, and aromatic aldehydes 171, resulting in the production of the desired compounds 174 (Scheme 24) (Khan and Saigal, 2018). The current methodology stands out for its good to exceptional yield, fast reaction time, lack of column chromatography required for product purification, and easy experimental setup. The antifungal activity of the products was examined against Aspergillus niger (MTCC-281), Aspergillus fumigates (MTCC-343), and Claviceps purpure. All the products demonstrated moderate to good activity but compounds 160a, 160b, 162a, 162b, 162c, and 162d showed exceptional activity in comparison with the standard drug Nystatin.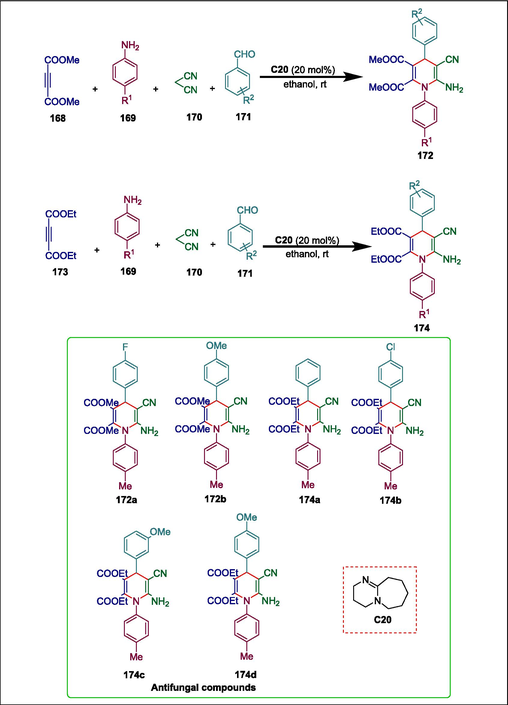
DBU catalyzed synthesis of N-aryl-1,4-dihydropyridines.
The drug zidovudine, also known as 3′-azido-3′-deoxythymidine (AZT), was first created as an antitumoral agent (Ostertag et al., 1974)and now it is also used to treat and prevent AIDS. Zidovudine derivatives exhibit a range of pharmacological characteristics, including antiviral, antioxidant, anticancer, and antibacterial effects (Zhang et al., 2012; de Souza et al., 2015; da Rosa et al., 2017). Gomes et al. produced a variety of 1,2,3-triazoyl-zidovudine derivatives 177 using organocatalyzed reactions. By reacting zidovudine 175 with various functionalized keto compounds 176, such as β-keto-amides, β-diketones, β-keto-esters, α-keto-nitriles, and β-keto-sulfones, in the presence of DBU C20, the hybrid compounds 177 were produced in moderate to good yields (Scheme 25) (Gomes et al., 2020). The afforded products were examined for their antioxidant activity. In mice with equal potency and efficacy, compounds 177a, 177b, 177c, and 177d reduced the production of reactive oxygen species (ROS) and lipid peroxidation in the prefrontal cortex and hippocampal regions. The derivative 177b demonstrated the best results in the cortex, with an IC50 value of 200.80 + 48.55 μM, whereas in the hippocampal region, 177c was the most potent with an IC50 value of 70.95 ± 32.01 μM.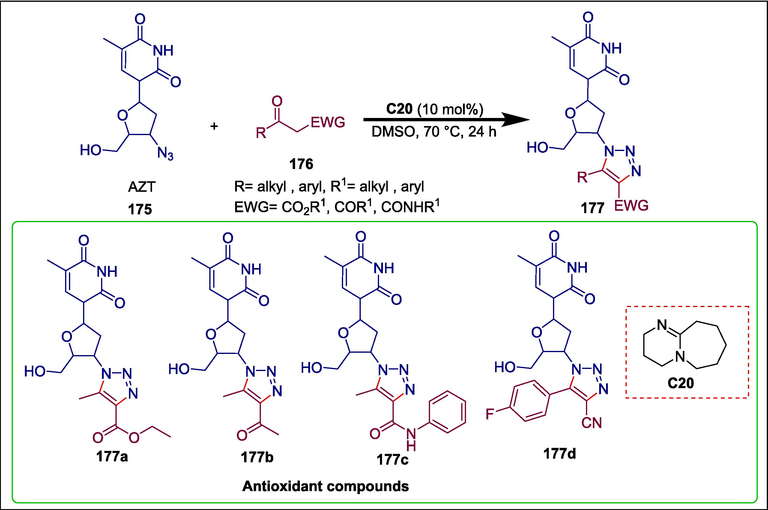
DBU catalyzed synthesis of zidovudine derivatives.
Fluorescent labeling of drugs is a widely used method for investigating their mechanisms of action. The solubility, polarity, and biological activity of the drug under investigation can all be changed by the application of a fluorescent label, which could lead to an inaccurate representation of the mechanism of action under investigation. Therefore, the creation of drugs with built-in fluorescence that can be monitored directly within cells is highly beneficial. Herrmann et al. reported a simple process for developing fluorescent hybrid medicines 178–185. This involved combining two easily accessible non-fluorescent pharmacophores with a non-cleavable linker by an organo-click reaction known as azide-carbonyl [3 + 2] cycloaddition, which was catalyzed by Ramachary-Bressy-Wang. The non-fluorescent benzimidazole azide compounds 188–192 fuse with non-fluorescent arylacetaldehyde 186 in the presence of a DBU catalyst C20 to furnish the required fluorescent hybrid drugs 178–182. Following the ester derivative 182′s deprotection to provide carboxylic acid 193, the carboxy group of 193 combines with the amine moiety of the readily accessible E3 Ligase ligand 194 to generate the new PROTAC (Proteolysis targeting chimera) 183. The non-fluorescent benzimidazole azide compounds 188 and 189 undergo DBU-catalyzed fusion to aldehyde 187 to form the required 2- deoxy-artemisinin-benzimidazole hybrid compounds 184 and 185 respectively (Scheme 26) (Herrmann et al., 2023). All freshly obtained fluorescent compounds displayed stronger anti-HCMV activity (EC50 down to 0.07 ± 0.00 μM) than the standard drug ganciclovir (EC50 = 2.60 ± 0.50 μM) and 179 was the most potent compound with EC50 = 0.07 ± 0.00 μM.Scheme 27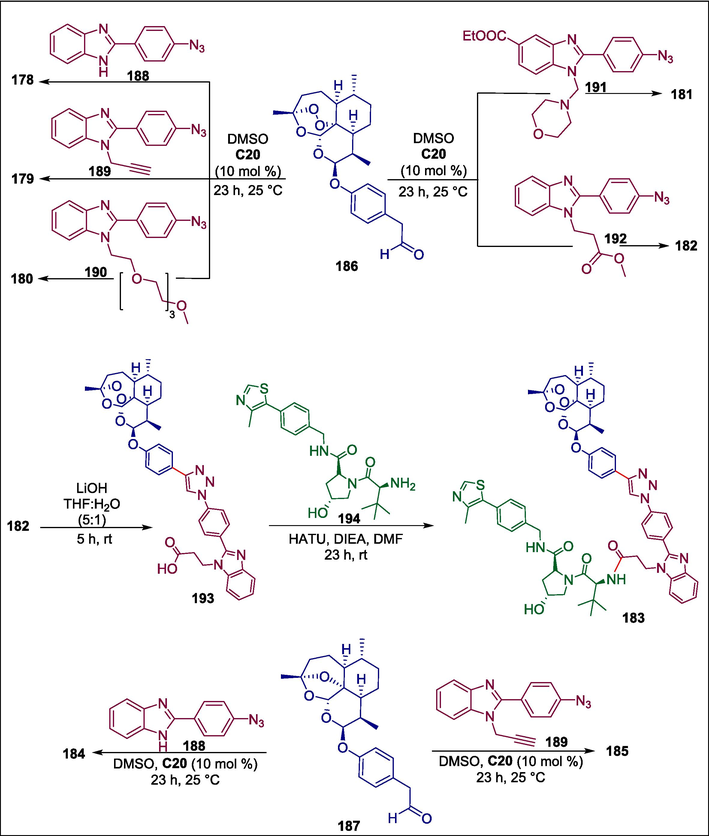
DBU catalyzed synthesis of fluorescent hybrid drugs.
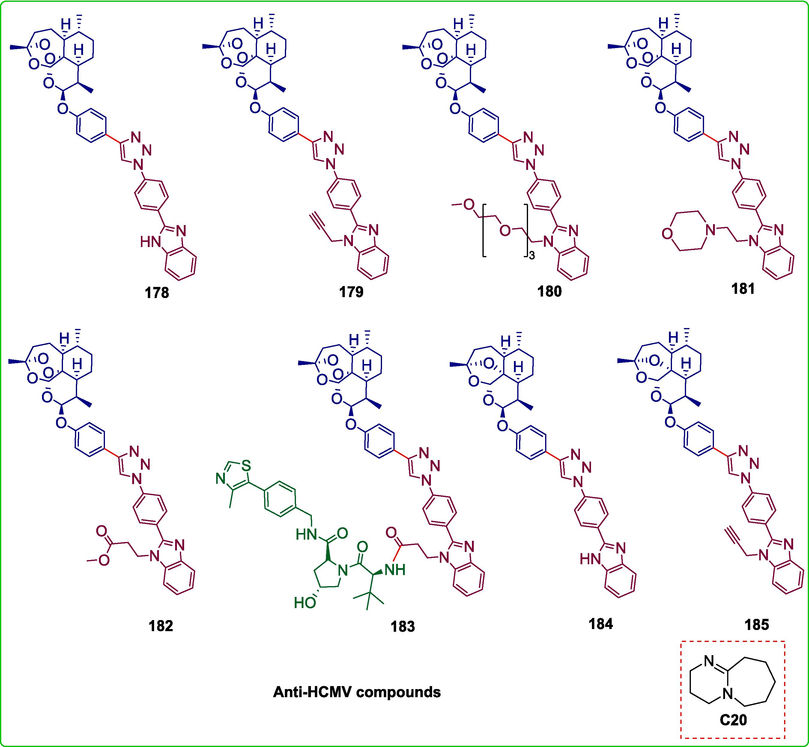
Structures of fluorescent hybrid drugs.
4.3 Cinchona alkaloids-derived catalyst
Epoxy ketone moieties are important pharmacophores e.g. cerulenin, a known FabB/F inhibitor (Price et al., 2001). Ernouf et al. synthesized α; β-unsaturated epoxy ketone via organocatalyzed asymmetric epoxidation (Lifchits et al., 2013). The terminal alkene 195 and methyl vinyl ketone (MVK) 196 undergo cross-metathesis to yield the α,β-unsaturated ketone 197 which undergoes organocatalyzed asymmetric epoxidation in the presence of cinchona primary amine catalysts C21 and C22 to produce optically enhanced epoxides (±)-198. Following the aldolization of (±)-198 and acetaldehyde, a diastereoisomeric mixture of alcohols is produced. Methanesulfonyl chloride is then added to the mixture to yield the enone (±)-199 (Scheme 28) (Ernouf et al., 2018). This α; β-unsaturated epoxy ketone (±)-199 was investigated for antibacterial activity and found active against hospital-acquired methicillin-resistant S. aureus (HA-MRSA) and community-acquired methicillin-resistant S. aureus (CA-MRSA) albeit with IC50 values of 43 µg/mL and 32 µg/mL.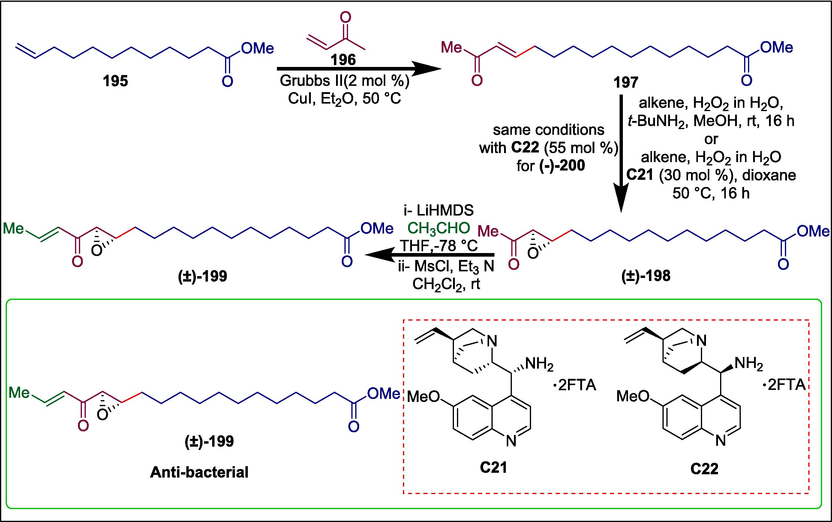
Cinchona primary amine catalyzed synthesis of α,β-unsaturated epoxy ketones.
In pharmaceutical chemistry, α-carbolines are intriguing structural motifs (Debnath et al., 2021). They are extensively present in several natural products and exhibit a variety of biological properties, for example cytotoxic (Moquin-Pattey and Guyot, 1989; Lin et al., 2016), antibacterial, and antifungal properties (Saundane et al., 2013). It has also been demonstrated that C3-modified spiro-oxindoles are very effective antitumor agents (Fan et al., 2017; Boddy and Bull, 2021). He et al. provided the first deprotected organocatalytic asymmetric [3 + 3] annulations of isatin-derived Morita-Baylis-Hillman (MBH) carbonates 200 and indolin-2-imines 201 to create a range of multifunctionalized α-carboline-spiro-oxindole hybrids 202 in good to excellent yields with high stereoselectivities. The chiral organocatalyst (quinidine) C23 is added to the MBH carbonate of isatin 200 to initiate the reaction, resulting in the electrophilic onium salt 203. Subsequently, the tertiary-butoxide anionic base generated in situ takes a CH2 proton from 201 to give the carbanion intermediate 204, which attacks the γ-position of the nitrogen ylide 206 through an SN2′ pathway, leading to the allylic adduct 207 once the C23 is eliminated. Next, in the presence of a base, intermediate 207 is changed into intermediate 208, which then goes through an intramolecular aza-Michael reaction to produce the required spiro-oxindole α-carboline 202 (Scheme 29) (He et al., 2022). Certain afforded compounds suppressed the proliferation of colorectal cancer cells. It was discovered that the most effective compound, 202, activated autophagy, induced the creation of autophagosomes, and activated the overall autophagy flux in HCT116 cells.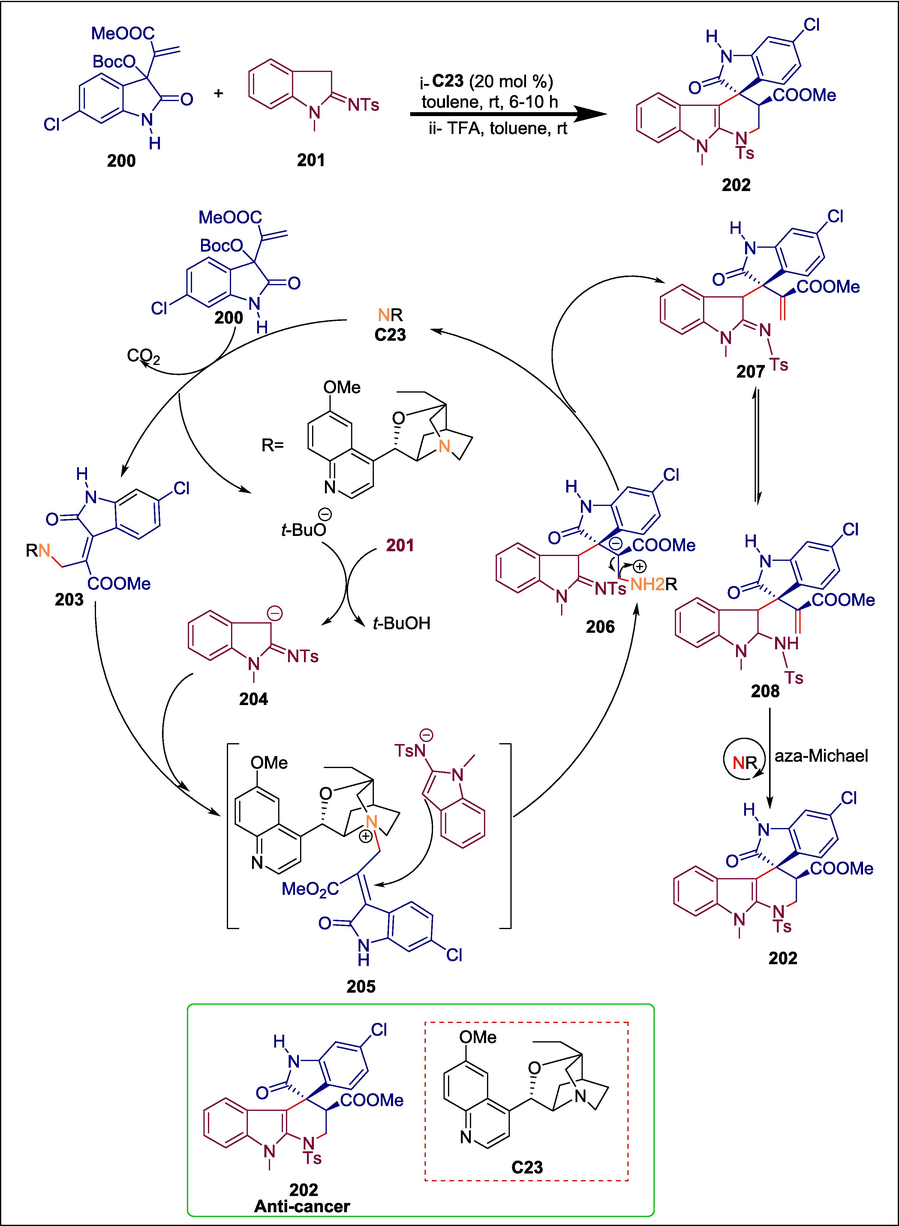
Quinidine catalyzed synthesis of α −carboline-spiro-oxindole hybrids.
N-substituted 2-pyridones and their derivatives are commonly present in pharmaceutical compounds and physiologically active natural products, such as human rhinovirus protease inhibitors, CG400549 (anti-Staphlococcus), and glucokinase inhibitors (Schiebel et al., 2014; Ramirez et al., 2017; Jia et al., 2017; Liu et al., 2017). Several metal-catalyzed reactions have been used in the past to synthesize N-substituted 2-pyridones; however, organocatalyzed Michael addition has not been documented (Zhang et al., 2015; Huang et al., 2017; Xu et al., 2019a). Wu et al. first time described the synthesis of N-substituted 2-pyridones 211 by enantioselective, organocatalytic aza-Michael additions of halogenated 2-hydroxypyridines (pyridin-2(1H)-ones) 209 to α,β-unsaturated-1,4-diketones or 1,4-ketoesters 210. The reaction begins when 212 and 213 undergo cinchonine-derived squaramide C24 catalyzed Michael addition to give Michael adduct 214 which is reduced via the Baeyer-Villiger reaction to phenyl ester 215. This ester is then hydrolyzed and reduced selectively to alcohol 216 (Kenner and Seely, 1972; Frein et al., 2009), whose hydroxyl group is then brominated and reduced to get 217 (Hutchins et al., 1977) whose bromo-substituent is transformed to the NH2 and then isoxazole-3-carboxamido-group (Messaoudi et al., 2010) to prepare desired compound 218 (Scheme 30) (Wu et al., 2019). The reaction conditions are moderate and simple to carry out. High enantioselectivities and good yields were obtained from the Michael adducts. The synthesis of important intermediate 218 (Dragovich et al., 2003; Martinez et al., 2004), for the production of human rhinovirus (HRV) protease inhibitors demonstrated the synthetic application of this approach.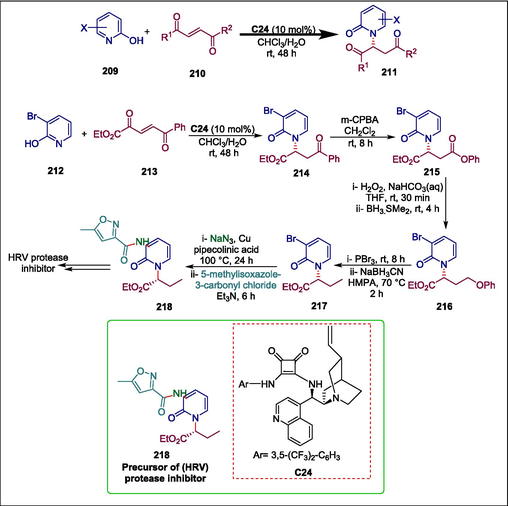
Cinchonine-derived squaramide-catalyzed synthesis of HRV protease inhibitor.
4.4 1,4-diazabicyclo[2.2. 2]octane catalyst
Alzheimer's is a neurodegenerative disease. An extracellular plaque of amyloid-beta (Aβ) forming in the brain is one of the disease's clinical features. Since BACE1 is essential for the synthesis of Aβ, it has been actively pursued in the search for novel treatments for Alzheimer's disease (Koh et al., 1990; Haass and Selkoe, 2007; Masters and Selkoe, 2012; Vassar et al., 2014; Grujičić and Nikolić, 2021). Winneroski et al. previously reported the fragment-based discovery of LY2811376, the first BACE1 inhibitor known to have a significant decrease in human CSF Aβ (May et al., 2011; May et al., 2015). Currently, this group has reported the synthesis of a variety of BACE1 inhibitors through organocatalyzed reactions by using trans-cyclopropyl scaffolds as structural constraints. The reaction starts when 1,3-dichloroacetone 219 is de-symmetrized to phosphorane 220, and the leftover chloride is subsequently replaced with the protected allylamine anion to yield β-ketophosphorane 221, which undergoes Wittig olefination to give α-amino enone 222. Next, enone 222 is cyclopropanated by an organocatalyst DABCO C25, yielding the racemic trans-cyclopropyl intermediate 223, which is then subjected to intramolecular [3 + 2] nitrone cycloaddition using Ti(OEt)4 and 4-methoxybenzyl hydroxylamine, resulting in a racemic mixture favoring diastereomer 224. This diastereomer then undergoes reductive ring opening to yield 225, which is then processed through two steps to give protected aminothiazine 226 that undergoes subsequent SNAr reaction with 2-chloropyrimine to form 227. The CH3MgBr double-addition to the ester group of 227 results in tert-alcohol 228 (Scheme 31) (Winneroski et al., 2019). This is then followed by the removal of the protecting group and an acid-catalyzed etherification using (1R,2R-2-methylcyclopropyl] methanol to yield ether 229. Compound 229 showed a 40 % decrease in cortical Ab 1-X in PDAPP mice having a dose of 100 mg/kg.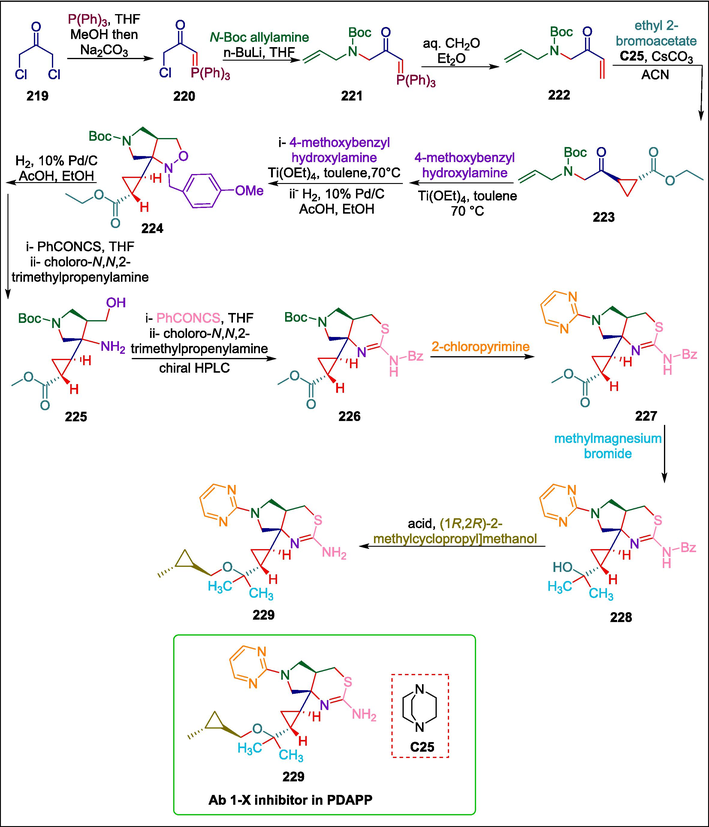
DABCO catalyzed synthesis of BACE1 inhibitors.
Cyclopropane frameworks are significant structural building blocks found in various natural products and bioactive moieties (Sandanayaka et al., 2003; Brackmann and de Meijere, 2007; Tripathi et al., 2017). Previously cyclopropane building blocks were synthesized through metal carbenoids to electron-rich or electron-neutral alkenes and Michael-intiated cyclization of alkyl halides (Lee et al., 2012; Rueping et al., 2012) or ylides (Kakei et al., 2007) with electron-deficient alkenes. Chen and He presented the first method to synthesize spirocyclopropyl oxindoles via triethylenediamine −catalyzed Michael/alkylation cascade reaction (Xie et al., 2007) of N-unprotected 3-bromooxindoles (Zheng et al., 2015a) with α,β-unsaturated acyl phosphonates involving α-substituted ammonium ylide intermediates. 3-bromooxindole 230 reacts with β,β-disubstituted acyl phosphonates 231 (Esteban et al., 2018) in the presence of DABCO catalyst C25 to form spirocyclopropyl oxindole 232 (Scheme 32) (Chen and He, 2020). One of these synthesized compounds, compound 232a is a diastereomer of HIV-1 NNRTI inhibitor 233.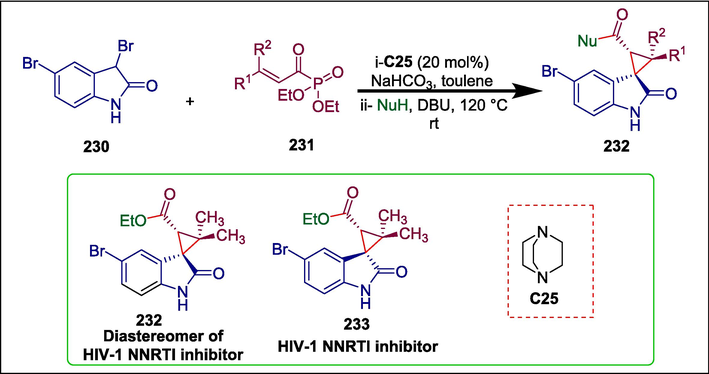
DABCO catalyzed synthesis of spirocyclopropyl oxindoles.
4.5 4-pyrrolidinopyridine catalyst
The World Health Organization has recognized E. coli as one of the “critical priority” antibiotic resistance bacteria (Tacconelli, 2017). The development of anti-E. coli vaccinations is getting a lot of attention due to their resistance to antibiotics. The O-antigen is a significant part of the lipopolysaccharide, and a desirable antigen for vaccine development (Micoli et al., 2019; Li and Li, 2020; Seeberger, 2021). A novel one-pot organo-catalysis relay glycosylation method was created by Wan et al. to synthesize the dithiolated O-antigen of E. coli serogroup 64. 3,6-di-O-acetyl-2-nitroglucal 234, trisaccharide glycosyl thiol 235, and mannosyl acceptor 236 are the three essential components needed for the synthesis. In the presence of the organocatalyst 4-pyrrolidinopyridine (PPY) C26, trisaccharide thiol 235 (Tacconelli, 2017) reacts with 234 to produce the tetrasaccharide intermediate. This intermediate is then attached with the 236 to yield the required pentasaccharide 237, which is then converted into pentasaccharide 238 by removing the bulky TIPS groups and converting the NO2 group into the corresponding acetamide. Following that, compound 238′s methyl ester and all benzoyl groups are eliminated to yield compound 239, which is the dithiolated O-antigen of E. coli serogroup 64 (Scheme 33) (Wan et al., 2023). This procedure is compatible with a broad range of substrates, mild reaction conditions, moderate to outstanding yields, and high site- and stereoselectivity.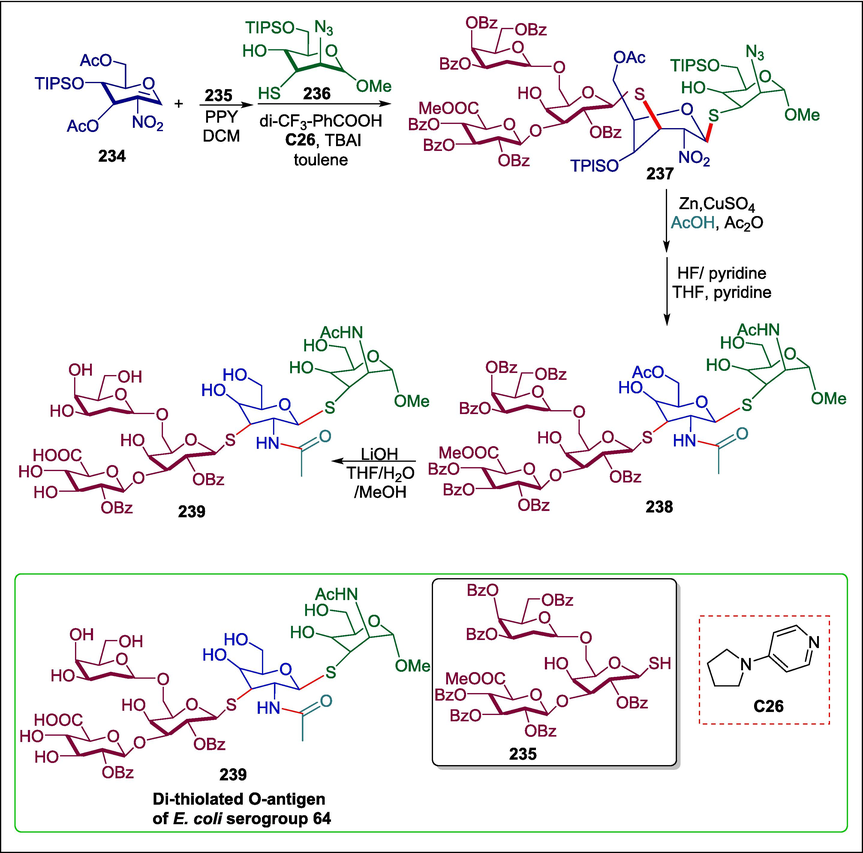
4-pyrrolidinopyridine catalyzed synthesis of di-thiolated O-antigen of E. coli serogroup 64.
4.6 Triazabicyclodecene catalyst
Near-infrared dyes can act as photosensitizers in cancer photodynamic therapy and help monitor nanocarriers (Pais-Silva et al., 2017). It is necessary to develop modern synthetic techniques to ensure that the drug delivery system's loading and dye photophysical characteristics stay stable for reliable monitoring in biological systems. Oliveira et al. disclosed a simple chemical conjugation of the carbocyanine heptamethine near-infrared dye IR780 to polylactide to create a polymeric near-infrared probe (IR-PLA) which allows stable fluorescent tagging of biodegradable polyester nanocarriers. D,L-lactide 240 undergoes Triazabicyclodecene (TBD) C27 organocatalyzed ring-opening polymerization with a cyclooctyne initiator 241 to afford “clickable” polylactide 242. Then IR780 243 is derivatized and conjugated to polylactide 242 through a copper-free one-pot azide-alkyne cycloaddition reaction to furnish IR-PLA 245 (Scheme 34) (de Oliveira et al., 2019).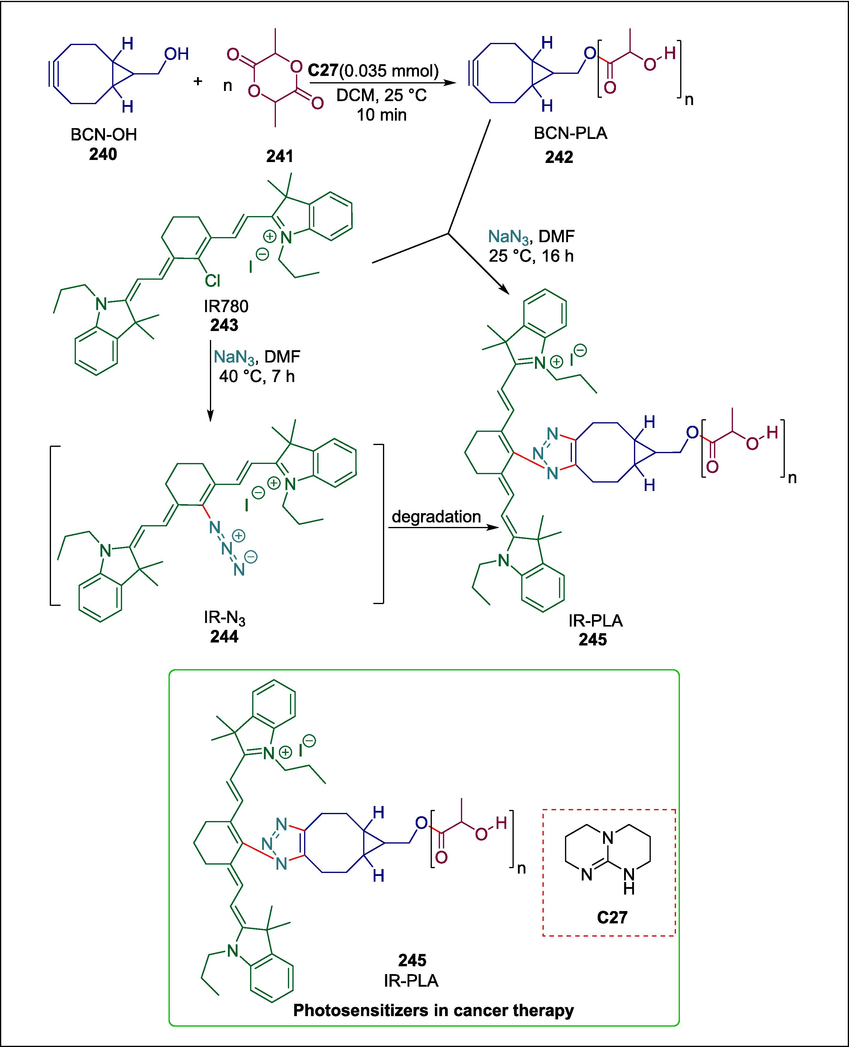
TBD catalyzed synthesis of IR-PLA.
Summary
A type of catalyst known as Bronsted base organocatalysts works by picking up protons (H+) from substrates and converting them into nucleophilic entities that can take part in further chemical transformations. Usually, these organocatalysts have simple functional groups that serve as proton acceptors, like amidines or amines. One common method includes the deprotonation of acidic protons, such as those in α-carbonyl compounds, leading to the production of reactive enolate intermediates. After that, these enolates can undergo nucleophilic attack on electrophiles, promoting reactions like Michael additions or aldol condensations. The regeneration of the Bronsted base usually marks the end of the catalytic cycle and permits repetition of the procedure (Fig. 3). These catalysts operate under mild conditions, offer high selectivity, and avoid the use of metal-based systems, making them attractive for environmentally benign and sustainable synthetic applications. This section covers the synthesis of remarkable therapeutic agents like applanatumol B, cathepsin inhibitors, human rhinovirus (HRV) protease inhibitors, BACE1 inhibitors, HIV-1 NNRTI inhibitors, dithiolated O-antigen of E. coli, cytotoxic, anti-inflammatory, antifungal, anti-HCMV, antioxidizing, and antibacterial compounds by using different organocatalysts such as pyrrolidine, 1,8-diazabicyclo[5.4.0]undec-7-ene, 1,4-diazabicyclo[2.2. 2]octane, 4-pyrrolidinopyridine, triazabicyclodecene, and cinchona alkaloid-derived catalysts.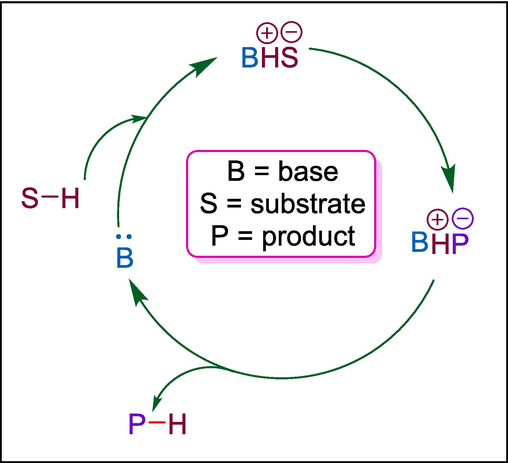
Mechanism of Bronsted base cataltalysis.
5 Bronsted acid catalysis
Brønsted acid catalysts increase the reactivity of electrophilic species by lowering their electron density. The exceptional versatility of enantioselective Brønsted acid catalysis stems from the incredibly tiny and labile nature of the catalytic particle, which is an acidic hydrogen atom (Merad et al., 2018). They have mostly been used as catalysts for C-O bond formation and breakage, including hydrolysis and the synthesis of esters and acetals. In the twentieth century, the catalytic activity of Brønsted acids in carbon–carbon bond formation was underestimated. However, Brønsted acids have become effective catalysts for the synthesis of C–C bonds at the start of the twenty-first century. Brønsted acids catalyze the formation of iminium salt, oxonium salt, carbocation, and vinylic carbocation by activating carbonyl, imine, alkene, alkyne, and OH groups, all of which promote the nucleophilic addition (Akiyama and Mori, 2015). In 2020, Lu et al. found that azoalkenes could function as carbon–carbon-nitrogen 1,3-dipole surrogates rather than four-atom synthons in asymmetric [3 + 2] cycloaddition reactions with 3-vinylindoles in the presence of chiral phosphoric acid. This resulted in the production of enantioenriched 2,3-dihydropyrroles in high yields and good stereoselectivities (Mei et al., 2020).
5.1 Thiourea catalyst
Asymmetric organocatalytic aza-Michael addition of a ketimine with an ortho hydroxyl group (Esteban et al., 2018) to nitroalkenes under batch and flow conditions gives 1,2-diamines. Diamines are significant components for the production of bioactive precursors. It has not yet been reported how these diamine derivatives were synthesized organocatalytically under flow circumstances. Corella et al. carried out a Takemoto’s thiourea C28 catalyzed reaction between trans-2,4-dichloro-β-nitrostyrene 249 and a ketimine 246 in batch and flow to obtain compound 250. Following an hour of residence, 250 undergoes an addition reaction before being exposed to HCl for acidic hydrolysis, which yields the corresponding amine hydrochloride 251, which is directly protected as a Boc carbamate 253. After that, its nitro group is reduced to produce amine 254, which is the intermediate for the synthesis of VNI 255 that has been described in the literature. This procedure, which uses acetone as a green solvent, also recovers the catalyst C28 in 85 % and the precursor to a ketimine i.e. 2-hydroxybenzophenone 246, in 90 % (Scheme 35) (Guerrero‐Corella et al., 2021). The synthesis of VNI, the drug-like equivalent of posaconazole for the treatment of Chagas disease, illustrates the efficacy of this process.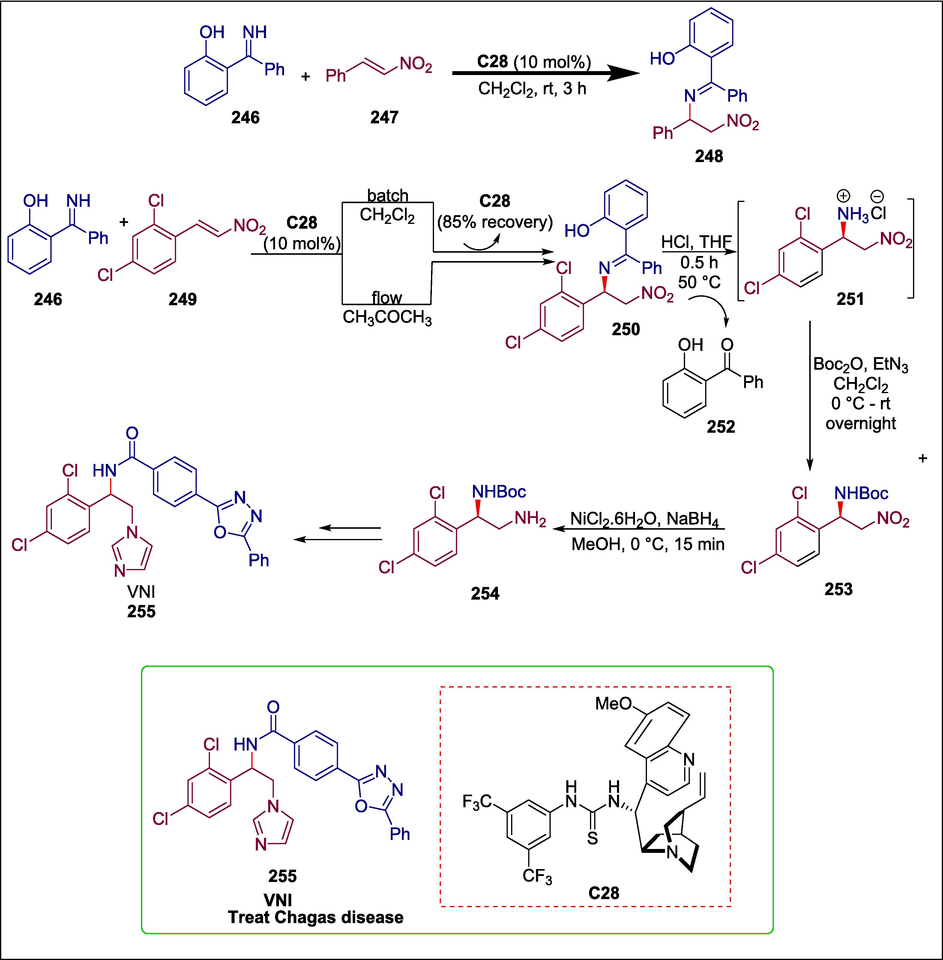
Takemoto's thiourea catalyzed synthesis of VNI.
Oxazole compounds exhibit various bioactivities e.g. antifungal (Tomi et al., 2015; Zhang et al., 2017), antibacterial (Škedelj et al., 2013; Patil et al., 2016), anti-inflammatory (Otrubova et al., 2014; Perrone et al., 2015), antiviral (Kim et al., 2013; Zhong et al., 2013), antituberculosis (Meissner et al., 2013; Abhale et al., 2017), anti-cancer (Lu and Wu, 2013; Maini et al., 2015), antiparasiticda Rosa et al. (da Rosa et al., 2017; Taha et al., 2017), and anti-diabetic (Yoon et al., 2014; Kalwat et al., 2016). They are also prone to attach to different enzymes and receptors of the biological systems. Wang et al. used an organocatalyst to produce derivatives of oxazol-5-one that contain chiral trifluoromethyl and isoxazole scaffolds through molecular hybridization. Tert-leucine 256 reacts with trifluoroacetic anhydride 257 to form N-trifluoroalkyl acetylated tert-leucine 258, which then cyclizes to yield intermediate 259. Next, 259 and 4-nitro-5-styrylisoxazole 260 undergo quinine-derived thiourea C29 catalyzed Michael addition to yield the desired compounds 261 (Scheme 36) (Wang et al., 2023). The antiproliferative properties of these produced compounds were tested against A549, HepG2, MCF-7, HeLa, and 5637 cancer cell lines. Compound 261a demonstrated significant anti-cancer activity (IC50 = 1.8 ± 0.1 μM) against HepG2 cells and suppressed the growth of cells.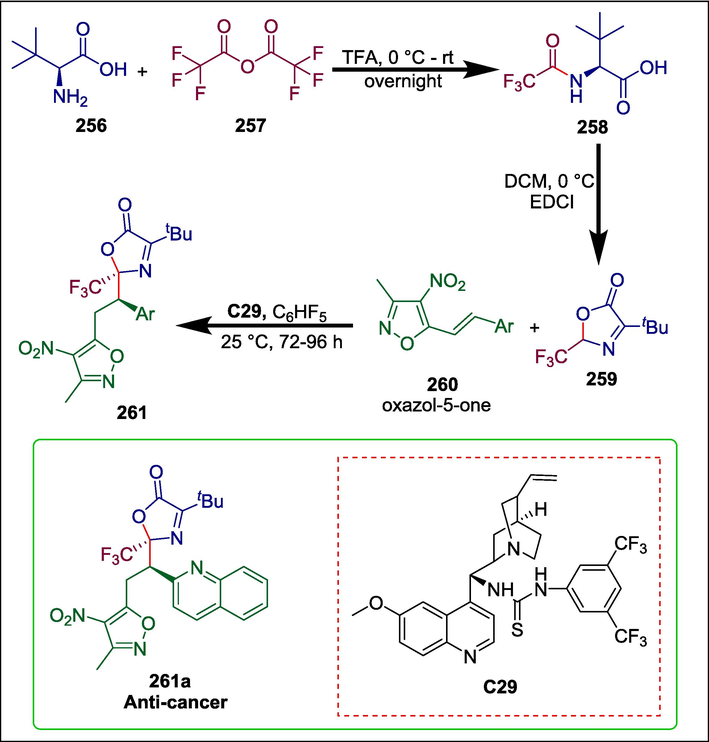
Quinine-derived thiourea catalyzed synthesis of oxazol-5-ones.
Spirooxindoles and pyran are vital heterocyclic compounds found in many natural products and have displayed a wide spectrum of bioactivities (Nazhand et al., 2020; Zhang et al., 2021a). Prior research by Jin and colleagues revealed the effective NHC-catalyzed [2 + 4] cycloaddition protocol that produced spirooxindole-pyrano[2,3-c]pyrazole compounds with antibiotic properties (Yang et al., 2022). Li et al. presented a new and advanced organocatalyzed protocol for the synthesis of spirooxindole containing a tricyclic pyran-annulated benzopyran skeleton. A thiourea-catalyzed C30 domino reaction between isatylidene malononitrile 262 and 1-(2-hydroxyphenyl)-butane-1,3-dione 263 results in multicyclic spirooxindole products 264 with an O,O-acetal-fused tricyclic motif or tetrahydroxanthone (Scheme 37) (Li et al., 2023). The obtained compounds show encouraging anticancer activity with low IC50 values. The most active compounds are 264a and 264b, which have IC50 values of 16.47 μM and 17.92 μM, respectively.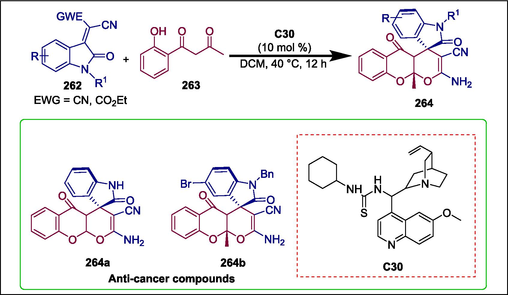
Quinine-derived thiourea catalyzed spiro oxindole containing a tricyclic pyran-annulated benzopyrans.
5.2 Chiral phosphoric acid catalyst
Triarylmethanes show remarkable biological activities, such as the ability to inhibit histidine protein kinases and have antiviral, antifungal, antioxidant, and anticancer effects (Ameen and Snape, 2013; Mondal et al., 2018). A key problem in asymmetric catalysis is the successful discriminating between two (typically) sterically identical aryl groups, which is necessary for the production of such large molecules (Besset et al., 2013). Yan et al. tackled this problem by implementing an efficient organocatalytic technique that produced effective enantioselection between aryl and heteroaryl groups. Racemic tertiary alcohol 265 undergoes asymmetric reduction with benzothiazoline 266 as the hydride source to form indole-containing triarylmethane 267. The reaction starts with phosphoric acid-catalyzed C31 dehydration to form cation 268, paired with a counter anion. This pair of ions may be in pseudo-resonance, or equilibrium, with the activated indole imine methide form 269. The hydride source then moves near the benzylic carbon to yield the required molecule 267 (Scheme 38) (Yan et al., 2022). The reaction occurs in mild circumstances with great efficiency and enantiocontrol, which are key aspects of this method. The antiviral and cytotoxic properties of the obtained products were examined. It was found that compound 267a had a significant antiviral action with an IC50 value of 2.27 µM while compound 267b exhibited high cytotoxicity, with 50 % cytotoxic concentration (CC50) values ranging from 5.6 to 18.2 µM.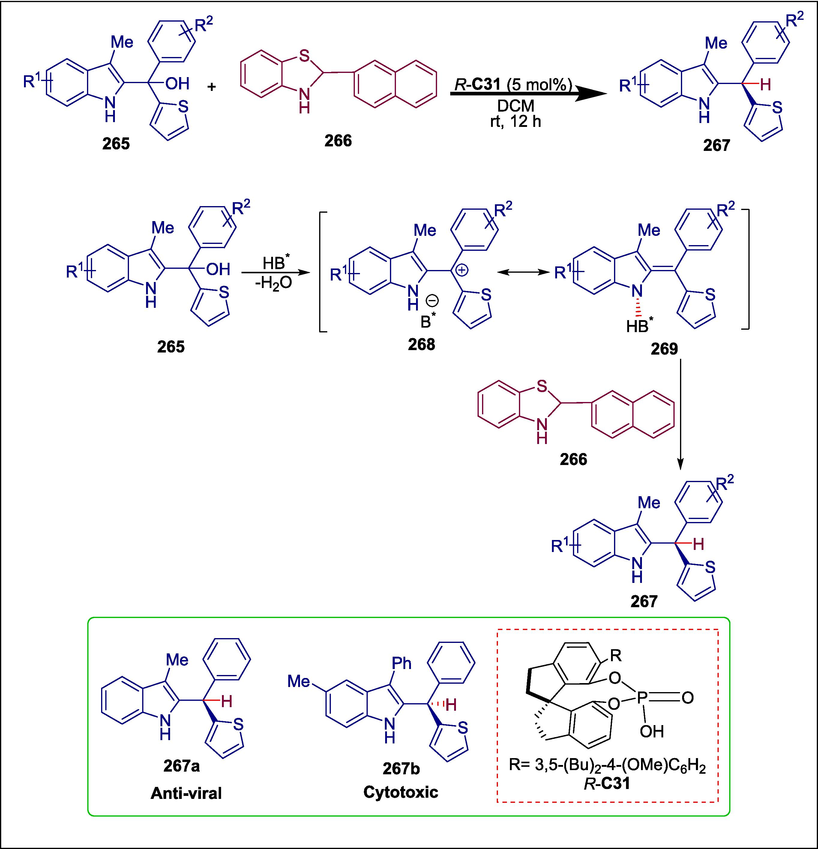
Chiral phosphoric acid-catalyzed synthesis of triarylmethane.
Axially chiral allene scaffolds are commonly found in natural products (Hoffmann-Röder and Krause, 2004; Pu et al., 2009; Cai et al., 2011; Rivera-Fuentes and Diederich, 2012). In the past, Li's group used thiazolones as appropriate nucleophiles in CPA-catalyzed 1,8-addition processes with alkynyl para-quinone methides (p-QMs) or alkynyl aza-p-QMs to create axially chiral tetrasubstituted allenes (Zhang et al., 2019a, b). For the formation of axially chiral allene scaffolds, finding suitable nucleophiles under Brønsted acid catalysis is a crucial problem (Kwon et al., 2018). In the presence, Wang et al. developed a chiral phosphoric acid (CPA) R-C32 catalyzed asymmetric construction of hexahydropyrrolo[2,3-b]indole-containing tetrasubstituted allene scaffolds bearing both axial chirality and central through a cascade 1,8-addition/dearomatization-cyclization reaction of para-aminophenyl propargylic alcohols 270 with tryptamines nucleophiles 271. This reaction resulted in various such tetrasubstituted allenes 272 bearing multiple chiral elements in good yields with high diastereo- and enantioselectivities. The reaction starts as para-aminophenyl propargylic alcohol 270 is dehydrated leading to the synthesis of alkynyl aza-para-QM intermediate 274. Then, by creating two hydrogen bonds, (R)–C32 activates tryptamine 271 and intermediate 274. This promotes an enantioselective 1,8-addition/CADA(catalytic asymmetric dearomatization) reaction between the two, resulting in the production of tetrasubstituted allene intermediate 275, which then goes through a stereoselective intramolecular cyclization process to yield the desired tetrasubstituted allene 272 (Scheme 39) (Wang et al., 2022). A cytotoxicity analysis of these produced products suggested that they can partially suppress the proliferation of pancreatic cancer cells and 272a is the most potent compound with an IC50 value of 86.6(μM).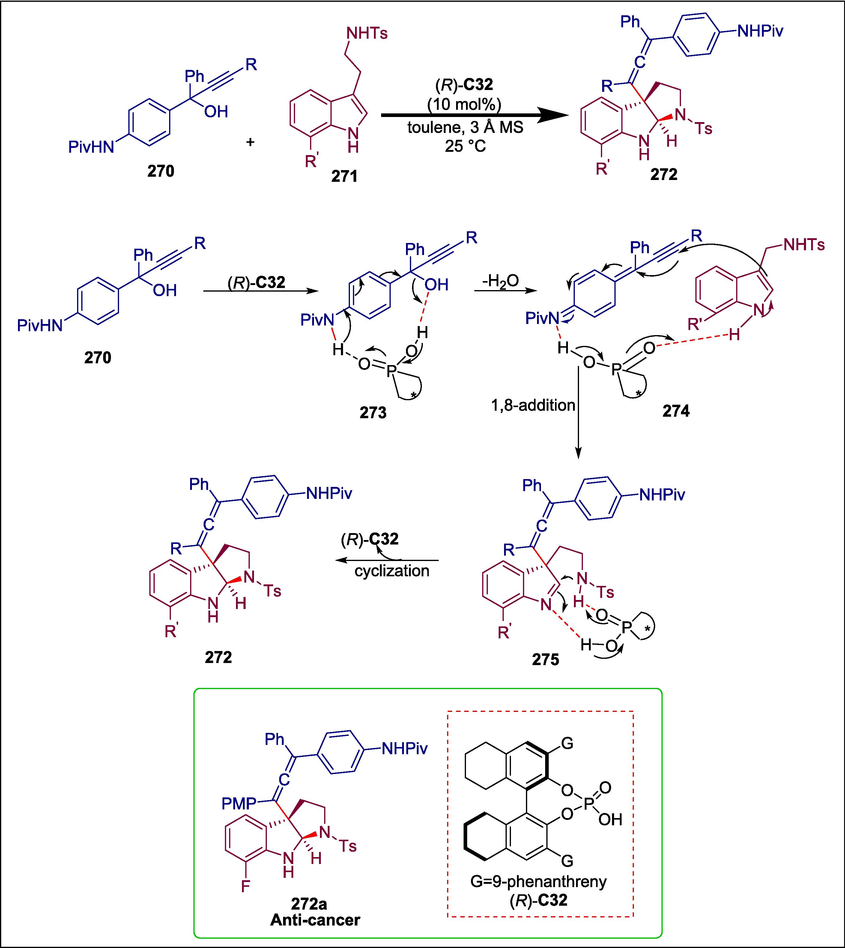
Chiral phosphoric acid-catalyzed synthesis of chiral allenes.
5.3 α-angelica lactone catalyst
Phthalide-derived scaffolds exist in many pharmaceutical and natural product compounds e.g. 3-butylphthalide, mycophenolate, and fumarate (Napolitano, 1997; Karmakar et al., 2014; Saikia and Gogoi, 2018). Recently, arbutin has been used as a pre-oxidant in the presence of Cu-salt and oxygen gas to create phthalide dervatives by an oxidation process mediated by visible light. The arbutin used in this method is toxic (Finney et al., 2018). Thatikonda et al. reported the synthesis of bioactive racemic 3-butylphthalide 277 in 35 % yield by benzylic oxidation of 1-butyl phthalan 276 using α-angelica lactone C33 as an organocatalyst. The process begins with the acidic proton of C33 being deprotonated by 4-(Dimethylamino)pyridine, resulting in the dienolate species 278. This species then takes triplet O2 to yield the peroxide species (Peixoto et al., 2013), whose homolytic cleavage produces the radical species 279 and 280. Following the H-atom abstraction of 276 by either the alkoxy radical 279 or the peroxy radical 280, radical species 283 is produced. This species combines with oxygen to produce hydroperoxide intermediate 284, which collapses to product 277 upon additional H-removal by the base. On the other hand, a single electron transfer from substrate 276 might produce a radical cation that can then undergo H-atom transfer to produce an oxocarbenium ion. This ion then recombines with peroxy radical 280 to produce intermediate 284, which dehydrates to produce 3-butylphthalide 277 (Scheme 40) (Thatikonda et al., 2020). This approach has the advantage that its reaction conditions work with a variety of cyclic ethers. A formal synthesis of a commercial anti-platelet drug demonstrates the synthetic potential of this new technology.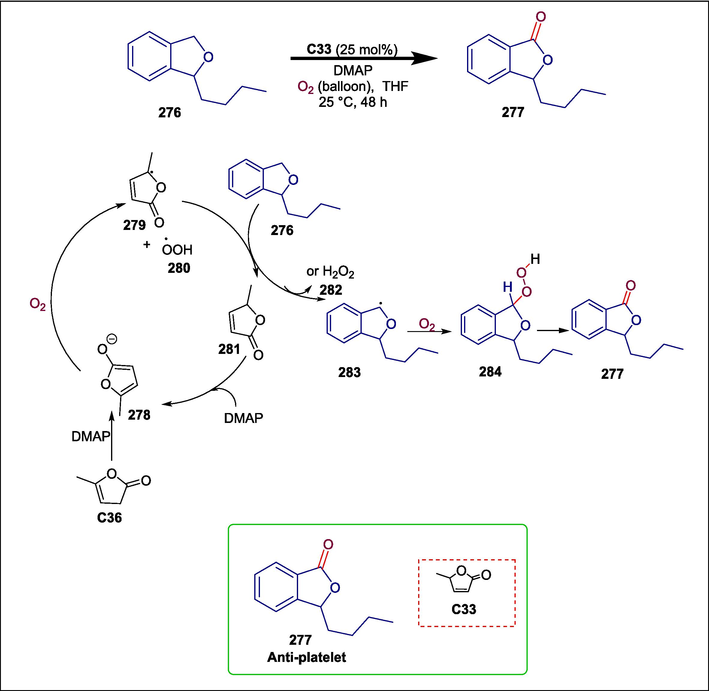
α-angelica lactone catalyzed synthesis of 3-butylphthalide.
5.4 Deep eutectic solvent catalyst
The indole ring and its derivatives are now considered unique pharmacophores (Sujatha et al., 2009; Nirmal et al., 2010; Chavan et al., 2011). This ring is also an essential component of indomethacin, an NSAID currently on the market. Harsh chemicals and solvents were used to synthesize indole-type moieties, which resulted in serious health problems and environmental damage (Gomha and Khalil, 2012; Farghaly et al., 2015). Deep eutectic solvent (DES), an alternative to such solvents, is the best option for certain chemical reactions (Singh et al., 2013; Bakht et al., 2016). Imran et al. used ultrasound and DES (choline chloride: urea, 1:2) in a multistep reaction involving isatin 285 and thiosemicarbazide 286 to create derivatives of indolin-2-ones 290. The urea part of DES C34 catalyzes the reaction by forming H-bonds. Thus, the acetyl moiety of 3-bromoacetylcoumarin 288 is stabilized by urea in a DES through hydrogen bonding. This is then attacked by the amide functional group of hydrazine thioamide 287 to give the vital intermediate 289 in 95 % yield through the cyclization and dehydration process. This intermediate 289 is refluxed from 1 to 3 h in the presence of substituted aromatic amines and formaldehyde and 30 mL of ethylene glycol to synthesize indolin-2-ones derivatives 290 (Scheme 41) (Imran et al., 2020). The high yield, time and energy savings, use of DES, and a green solvent are the advantages of this protocol. The afforded compounds were evaluated for their lipid peroxidation, anti-inflammatory, analgesic, and ulcerogenic properties. In comparison to the reference medicine indomethacin, compounds 290a-d showed good anti-inflammatory activity, whereas 290e-g demonstrated extremely good analgesic action with % analgesic activity values of 69.36 ± 0.5845 %, 66.27 ± 1.0072 % and 69.14 ± 0.6892 % respectively(Scheme 42). The active compounds also exhibited lipid peroxidation activities and their ulcerogenicity was far less than the indomethacin.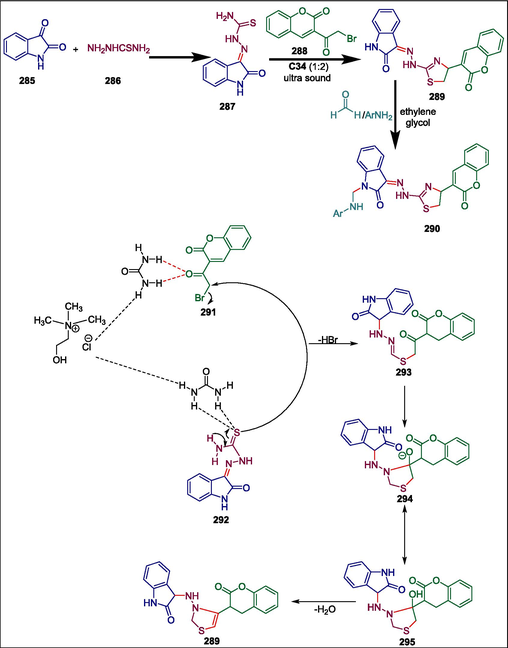
DES catalyzed synthesis of indolin-2-ones.
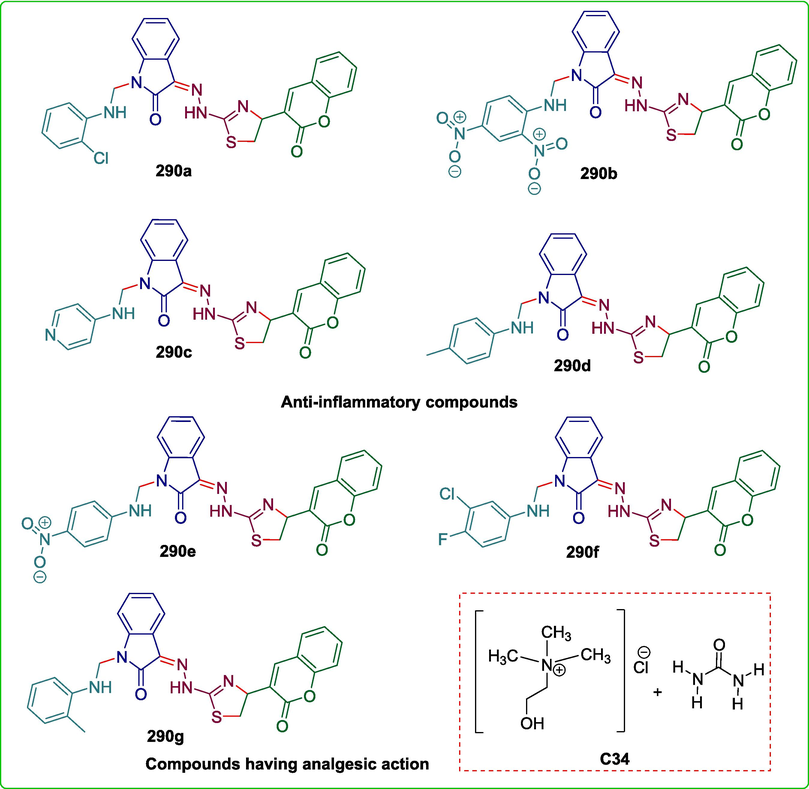
Structures of bioactive indolin-2-ones.
5.5 P-toluenesulfonic acid catalyst
Since 1,2,3-triazoles are stable in the presence of oxygen, light, and moisture, they are significant building blocks for synthesis (Bourne et al., 2004). Triazole scaffolds are found in different kinds of pharmaceuticals (Dalvie et al., 2002; Kharb et al., 2011). Because typical alkenes react slowly or not at all with azides, the synthesis of 1,2,3-triazolines from azides and alkenes is still restricted to highly activated alkenes (Scheiner, 1968; Kadaba and Edelstein, 1990). Repetto et al. synthesized 1,2,3-triazoles from the reaction of D-glucono-1,5-lactone 296 catalyzed by p-toluenesulfonic acid C35. The mechanism involves intramolecular ring closure through a 1,3-dipolar azide-alkene cycloaddition to form a 1,2,3- triazoline, followed by removal of pTsOH, leading to aromatization (Scheme 43) (Repetto et al., 2019). The practical application of this procedure is that triazole compounds are expected to exhibit action as enzyme inhibitors. Additionally, 2-hexenoate derivatives that were partially protected were created as helpful synthetic intermediates. Compounds 304 and 305 are potential enzyme inhibitors and partially unsaturated analogues of polyhydroxindolizidine alkaloids such as castanospermine and swainsonine [43,44].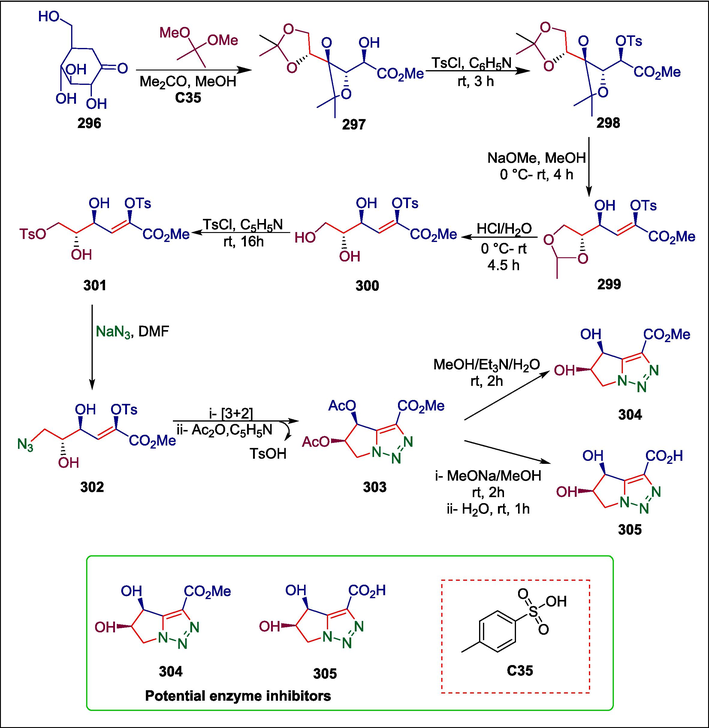
. p-toluenesulfonic acid-catalyzed synthesis of 1,2,3-triazoles.
5.6 Trifluoroacetic acid catalyst
The octahydroindolo[2,3-a]quinolizine ring system is the fundamental structure that underpins almost two thousand different natural chemical families. A sophisticated formation of these building blocks using the Pictet-Spengler reaction was reported by You (Wang et al., 2017)and Zhai (Luo et al., 2004). However, the utilization of lengthy and multistep syntheses to prepare the required scaffolds is a serious drawback of current techniques. Srinivasulu et al. demonstrated a one-step build/couple/pair technique to synthesize various octahydroindolo[2,3-a]quinolizine derivatives with more than three contiguous chiral centers and a wide diversity of molecular geometries through desymmetrization of the oxidative-dearomatization products of phenols. After the phenol precursor 306 is subjected to Dess-Martin oxidation and hyperiodinate-catalyzed oxidative-dearomatization, cyclohexadione pluripotent building block 307 is produced. This building block then reacts with tryptamine 308 in the presence of organocatalyst TFA C36 to give intermediate 309, which is then subjected to aza-Michael addition, resulting in a mixture of the diastereoisomers 310 (Scheme 44) (Srinivasulu et al., 2018). The distinct chemotype compound 310a that efficiently reduced glycolytic generation of ATP and membrane potential in Hepa1-6 was found most potent compound during the cellular screening of the synthesized compounds library. This compound represents prospective lead therapeutic candidates for the treatment of cancer.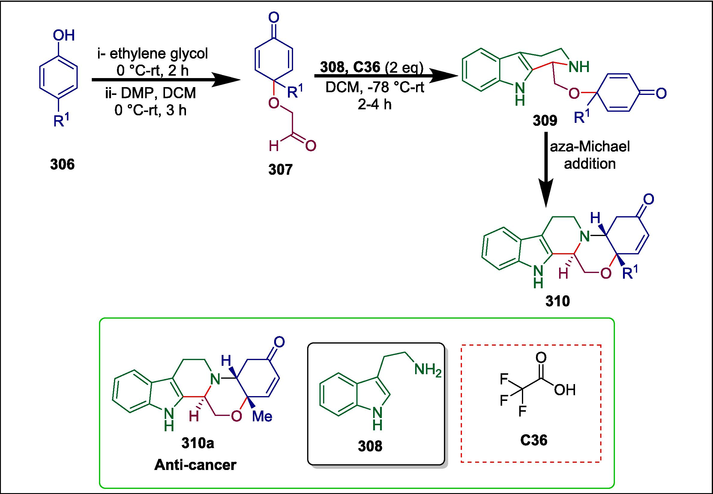
Triflouoroacetic acid-catalyzed synthesis of spirocyclic pyrazolone-ferrocene hybrids.
5.7 Pyridine-2carboxylic acid catalyst
Substituted imidazoles are employed as powerful BRAF kinase inhibitors (Niculescu-Duvaz et al., 2013), orally active 5-lipoxygenase (5-LO) inhibitors (Mano et al., 2003), and farnesyltransferase inhibitors (Lin et al., 2003). The typical method for synthesizing 2,4,5-trisubstituted imidazoles involves cyclo-condensing an aldehyde and ammonium acetate with an α-ketomonoxime, α-hydroxyketone, or 1,2-diketone. The use of hazardous solvents, a convoluted setup process, challenges with purification, low yield, and costly reagents are some of the disadvantages of this approach. Using pyridine-2-carboxylic acid C37 as an organocatalyst, Pervaiz et al. devised a one-pot, solvent-free formation of 2,4,5-trisubstituted imidazole derivatives 314 through a condensation reaction of substituted aromatic aldehydes 312, benzil 311, and ammonium acetate 313. The reaction begins when C37′s proton attacks the carbonyl group of aldehydes 316 and 311. Next, the protonated carbonyl group of aldehydes is exposed to the nitrogen of NH3, which binds as a nucleophile to generate the diamine intermediate 318. This diamine intermediate and protonated benzil 315 condense to give cyclic intermediate state 320 which undergoes [1, 5]-H shift to give the trisubstituted imidazoles 321 (Scheme 45) (Pervaiz et al., 2020). This technique stands out for its quick reaction time, ecologically benign synthesis, simple setup, and pure products. To investigate their potential to treat Alzheimer's disease, these afforded compounds were screened against the activity of the acetylcholinesterase (AChE) enzyme. Compounds 321a, 321b, 321c, 321d, 321e, 321f, 321 g, and 321 h were all active, but 321 h, with an ICs50 value of 102.56 ± 0.14 μM, found to be a strong AChE inhibitor.Scheme 46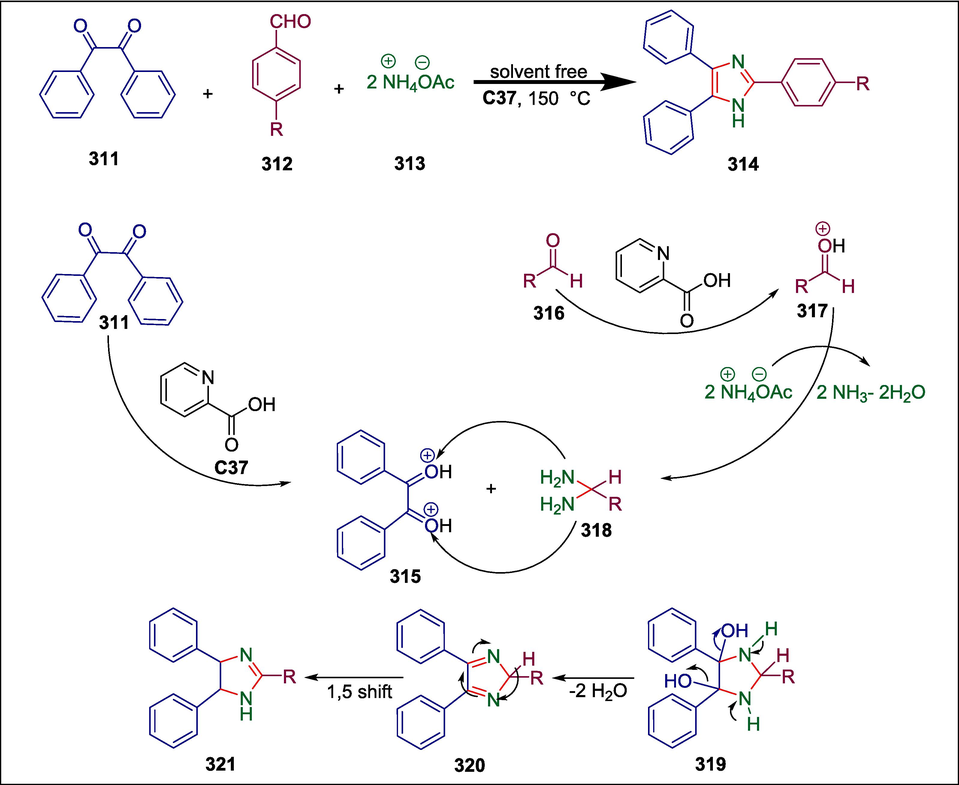
Pyridine-2-carboxylic acid-catalyzed synthesis of 2,4,5-trisubstituted imidazoles.
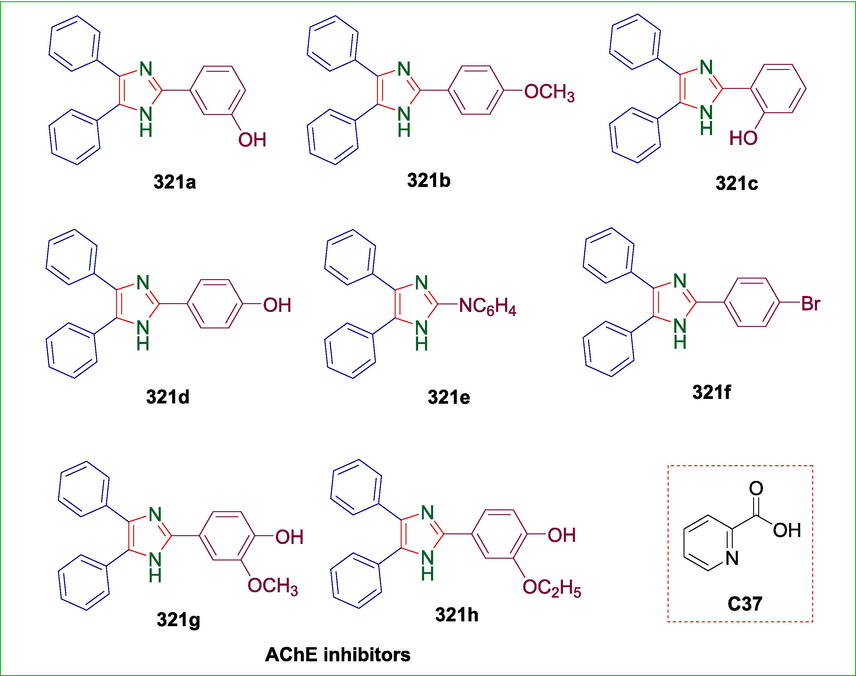
Structures of bioactive 2,4,5-trisubstituted imidazole.
Summary
A class of catalysts known as bronsted acid organocatalysts promotes reactions by donating protons (H+) to the substrates, which increases their electrophilicity and makes them more reactive toward nucleophilic attack. Typically, these organocatalysts have proton-donating acidic functional groups like phosphoric, sulfonic, or carboxylic acids. The mechanism of action generally involves the protonation of a key functional group, such as a carbonyl oxygen or an imine nitrogen, to increase the electrophilic character of the carbonyl carbon or the iminium ion. This protonation accelerates the reaction and decreases the energy barrier for nucleophilic assault (Fig. 4). The application of chiral phosphoric acids in asymmetric catalysis, where the acid not only activates the substrate but also causes stereoselectivity in the product, is a typical example. Bronsted acid organocatalysts are useful in environmentally friendly and sustainable synthesis processes because of their benefits, which include low reaction conditions, great selectivity, and metal-free catalytic cycles. In this section, the synthesis of many bioactive compounds i.e. VNI, the drug-like equivalent of Posaconazole, AChE inhibitors, analgesic, anticancer, antiviral, anti-platelet, and anti-inflammatory compounds via using various organocatalysts such as thiourea, chiral phosphoric acid, α-angelica lactone, deep eutectic solvent, p-toluenesulfonic acid, trifluoroacetic acid, and pyridine-2-carboxylic acid catalyst has been described.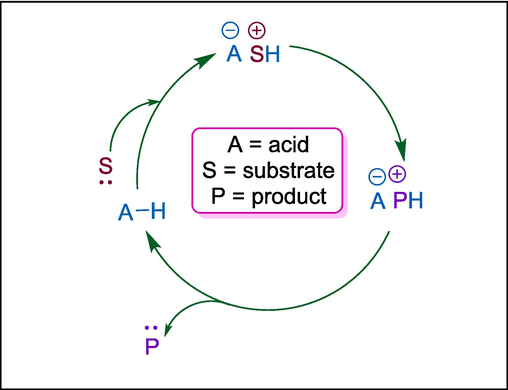
Mechanism of Bronsted acid catalysis.
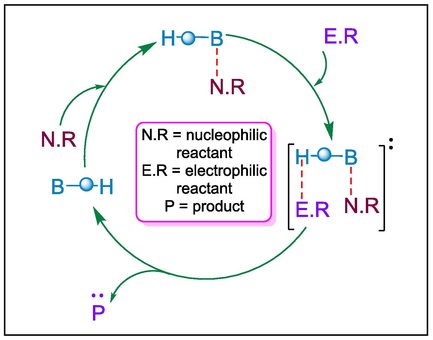
Mechanism of bifunctional organocatalysis.
6 Bifunctional organocatalysis
Bifunctional organocatalysts have been applied to numerous asymmetric synthesis processes (Siau and Wang, 2011; Fang and Wang, 2015). In these systems, two different functional groups act synergistically to bring an electrophile and a nucleophile together in a transition state that allows transformations that were previously difficult to carry out (Jiménez et al., 2017). In this type of catalysis, hydrogen-bonding interaction is commonly used. As an illustration, the squaramide, amide, (thio)urea, hydroxyl group, and guanidinium ion are frequently employed as donor functional groups for hydrogen bonding. Through their acidic protons, these groups develop H-bonds with a Lewis-basic moiety in the substrate. In 2021, Zhang et al. developed an effective halogen-free and green pathway for forming cyclic carbonates from CO2 and epoxides by using bifunctional organocatalysts bearing diamine and carboxylic acid groups (Zhang et al., 2021b).
6.1 Cinchona alkaloids-derived catalyst
Spirooxindoles are very important skeletal in many pharmacophores. The synthesis of these moieties has been the focus of significant research in recent times in an attempt to expand the boundaries of current drug and probe development (Hong and Wang, 2013; Cheng et al., 2014; Xie et al., 2018). Liu et al. synthesized many spirooxindoles derivatives via organocatalytic [3 + 2] annulations of the readily available 3-hydroxyoxindoles and pyrrolidone-derived cyclic ketolactams. 3-hydroxyoxindole 322 reacts with pyrrolidone-derived cyclic enone 323 in the presence of chiral bifunctional cinchona alkaloid catalyst C38 to yield a variety of tetrahydrofuranyl spirooxindole 324 (Scheme 47) (Liu et al., 2021). The one-step assembly of four continuous stereocenters, significant atom economy, and gentle (oxidant-free) reaction conditions are some of the intrinsic benefits of this synthetic technique. The anti-proliferative activity of these synthetic compounds was tested. The N-unprotected compound 324 remarkably suppressed the growth of MCF-7 breast cancer cells and the gastric cancer cells HGC27 and BGC823.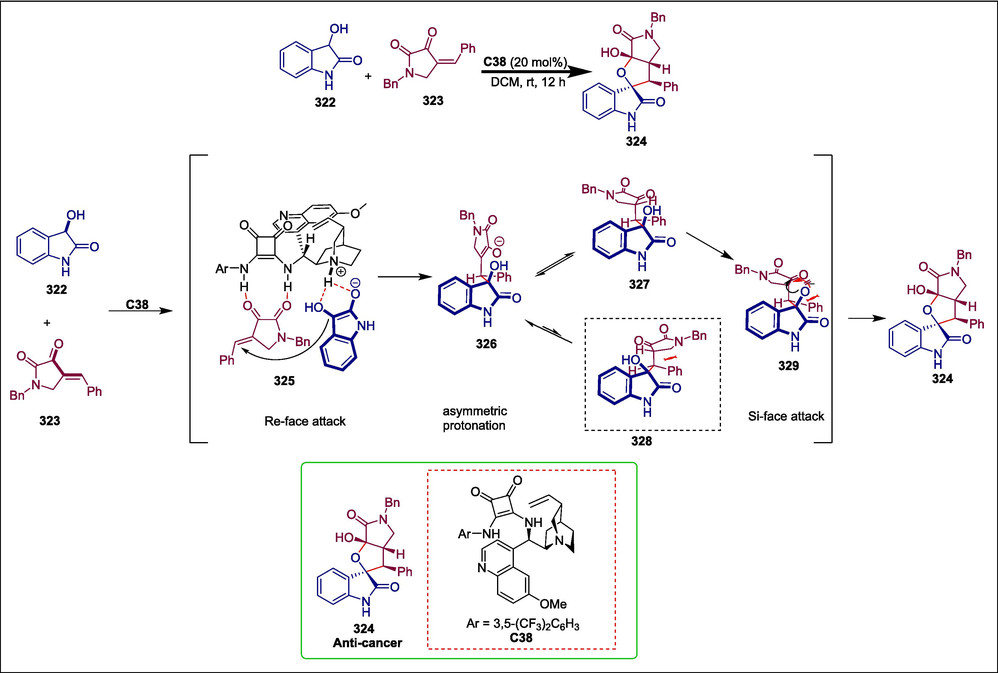
Cinchona alkaloid catalyzed synthesis of spirooxindoles.
Chiral secondary and tertiary β-hydroxy ester frameworks can be found in dicrotaline, atorvastatin, and epothilone B, among other pharmaceutical and natural medicines (Kusumi et al., 2013; Mahrwald, 2013; Dias et al., 2016; Cheng et al., 2018). In the biosynthesis of polyketides and fatty acids, nature freely employs the enzymatic decarboxylative activation of malonic acid half-thioesters (MAHTs) to produce simple ester enolates or their counterparts (Staunton and Weissman, 2001; Austin et al., 2004; Blaquiere et al., 2009; Bernardi et al., 2012). Drawing inspiration from nature, Park et al. effectively used MAHTs as ester enolate equivalents in the organocatalytic decarboxylative aldol reactions of MAHTs 331 with 2,2,2-trifluoroacetoketone 330 to produce enantio-enriched β-hydroxy thioesters 332. Cinchona-based thiourea (CD-TU) C39, which is an extremely effective polyketide synthase-mimic catalyst, is utilized in this reaction. In the presence of methanol, β-hydroxy thioesters 332 react with amines to form β-hydroxy amides, 333, 334, and 335. These amides are essential scaffolds for the development of CF3 analogues of antidepressant drugs, including (R)-tomoxetine, (R)-fluoxetine, and (R)-duloxetine. β-hydroxy thioester 332 can also be easily converted into the trifluoromethylated-analogue 336 of the antidepressant drug fenpentadiol (Elks, 2014; Park et al., 2019) by employing the Grignard process. Furthermore, β-hydroxy thioesters 332 also provide access to the ketones 337 and 338, important intermediates in the production of antiparasitic isoxazoline medicines like fluralaner and afoxolaner, respectively, through Liebe skind-Srogl coupling (Scheme 48) (Park et al., 2019). The easy conversion of the aldol adducts' thioester moiety into various chiral trifluoromethylated tert-aldol therapeutic agents highlighted the practical application of the current biomimetic aldol approach.
CD-TU catalyzed synthesis of trifluoromethylated tertiary aldol pharmacophores.
1,2-Azoles are important building blocks in ligand/catalyst development and are often present in different substantial natural products and pharmaceuticals (Adams, 1960; Vicentini et al., 2007; Lutter et al., 2020; Durand-Reville et al., 2021). Since it is challenging to manage the development of a heterocyclic ring and the atroposelective installation of a stereogenic axis at the same time, the atroposelective technique for 1,2-azoles, such as isoxazole, isothiazole, and its S-oxides, has not been examined (Lamers et al., 2016; Zhang et al., 2017; Yu et al., 2018). Chang et al. described an organocatalytic novel ring formation method for the synthesis of atropoisomeric naphthyl pyrazoles by using modified vinylidene ortho-quinone methide intermediates. This approach offers a simple way to create a variety of atropisomeric heterobiaryls that could not be obtained with previous methods. (Z)-N'-(1-cyclopropyl-3-(2-hydroxy-7-methoxynaphthalen-1-yl)prop-2-yn-1-ylidene)-P,P-diphenylphosphinic hydrazide 339 reacts with NBS in the presence cinchona alkaloids-derived squaramide C40 as an organocatalyst to give naphthyl-pyrazole (N, N-1,2-azole) 340 in moderate to high yields and with high enantioselectivities (Scheme 49) (Chang et al., 2022). In A375 cells, the produced pyrazole 340 with an IC50 value of 2.57 μM caused apoptosis and had strong antiproliferative action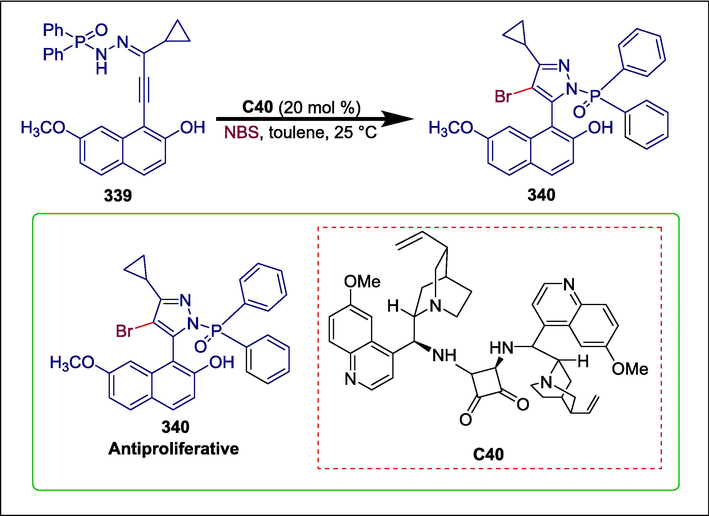
Cinchona alkaloids-derived squaramide catalyzed synthesis of naphthyl-pyrazole.
Seven-membered cyclic trienes, or tropolones, are a group of aromatic compounds with various bioactivities, including antifungal, antibacterial, and anticancer properties (Pauson, 1955; Morita et al., 2003; Zhao, 2007; Nakano et al., 2015). Hammer et al. documented the first organocatalytic approach involving HOMO-activation of tropolones through bifunctional Brønsted-base catalysis. A range of transformations, including photoisomerizations into complex chiral norcaranes, illustrate the synthetic value of the resulting compounds. Tropolone 341 and N-propargylmaleimide 342 undergo cycloaddition using the bifunctional Brønsted-base quinine catalyst C41 to give bridged bicyclic cycloadduct 343 (Scheme 50) (Hammer et al., 2018). Then this synthesized cycloadduct 343 was tested on MCF-7 cancer cells and showed cytotoxic activity.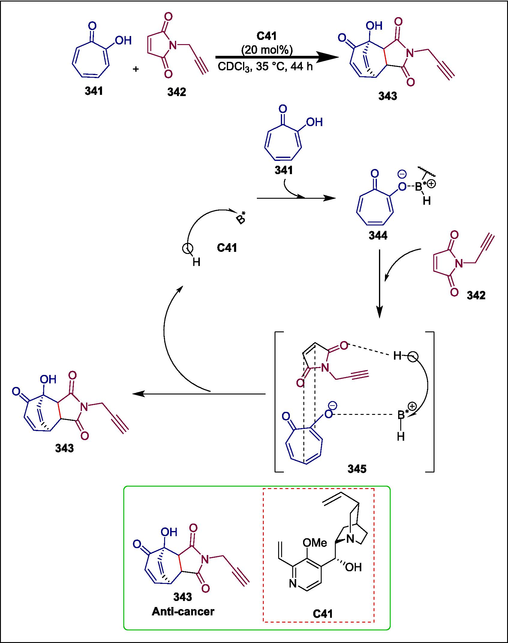
Quinine catalysed synthesis of tropolones.
(S)-forphenicinol is the active component of the immunomodulator (Ishizuka et al., 1982)and anticancer (Ishozika et al., 1982)medicine forfenimex. Total synthesis of (S)-forphenicinol was previously achieved by condensing 3-methoxybenzaldehyde with an (S,S)-2,2-dimethyl-5-amino-1,3-dioxane-based chiral auxiliary (Weinges et al., 1987). This approach has some drawbacks, such as low yield (less than 1 %), difficult chiral auxiliary synthesis, and the inability of its recycling due to oxidative damage during deprotection. A more practical and effective approach for the organocatalytic asymmetric formation of (+)-(S)-forphenicinol was disclosed by Kovalevsky et al. The readily available 2-hydroxy dimethylterephthalate 346 is reduced to diol 347, which then converts into cyclohexanone-based ketal 348. Following this, the distant methoxycarbonyl group is reduced to produce hydroxymethylarene 349, which is then further oxidized to produce aldehyde 350 that is transformed into N-protected imine 352. This Schiff base 352 is subjected to hydroquinine-catalyzed C42 asymmetric Mannich-type addition to allomaltol 353 produced from kojic acid. This results in an adduct 354, which then goes through successive oxidative fragmentation to yield the required enantiomerically pure (S)-forphenicinol hydrochloride 355 (Scheme 51) (Kovalevsky et al., 2023). Compared to yields obtained by already-known approaches, the yield obtained by this methodology is much higher (17 %).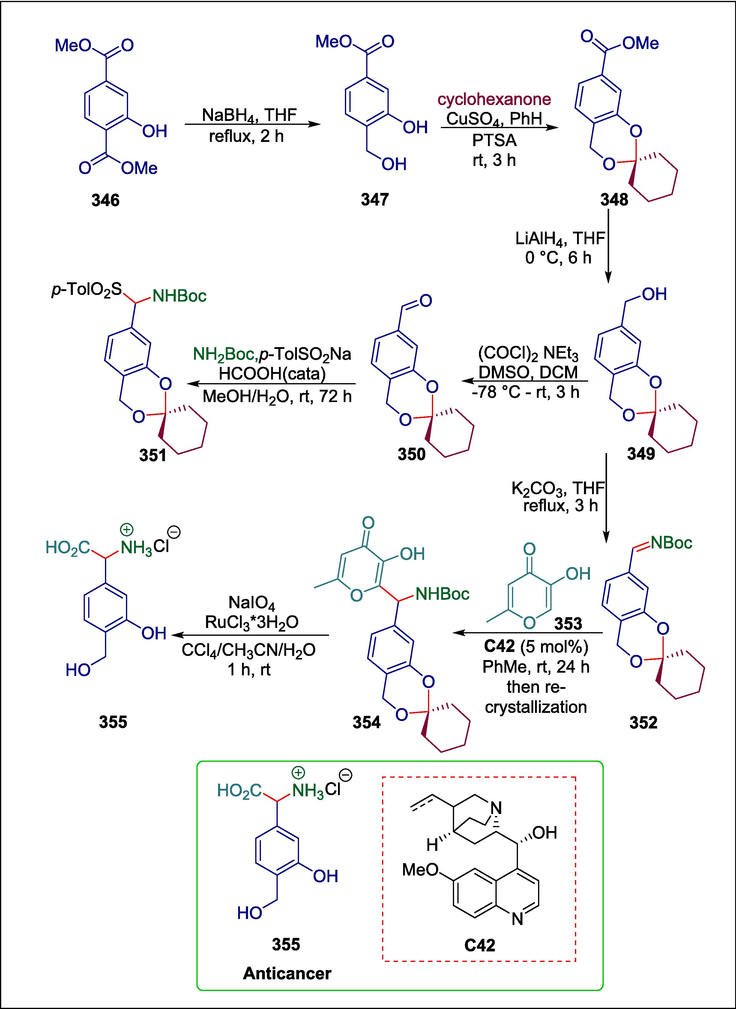
Hydroquinine-catalyzed synthesis of (S)-forphenicinol hydrochloride.
Organofluorine scaffolds commonly exist in numerous biologically active natural products and drugs (O'Hagan, 2008; Berger et al., 2011; Hiyama, 2013; Gillis et al., 2015). However, due to the inherent steric congestion, synthesizing vicinally bis(trifluoromethyl)-substituted compounds is a significant problem in current chemical synthesis (Gao et al., 2014; Molander and Ryu, 2014; Yang et al., 2015). To produce the vicinally bis(trifluoromethyl)-substituted spiro pyrrolidine-benzothiophenones 358, Zhao et al. proposed an organocatalytic C43 asymmetric [3 + 2] cycloaddition of β-trifluoromethyl enones 356 with 3-(N-2,2,2-trifluoroethyl) benzothiophene ketimines 357 (Scheme 52) (Zhao et al., 2023a). When the cytotoxicity of these produced compounds was tested against the A549 and K562 cancer cells, it was discovered that compounds 358a and 358b showed cytotoxicity, with IC50 values of 24.91 μM and 45.02 μM respectively.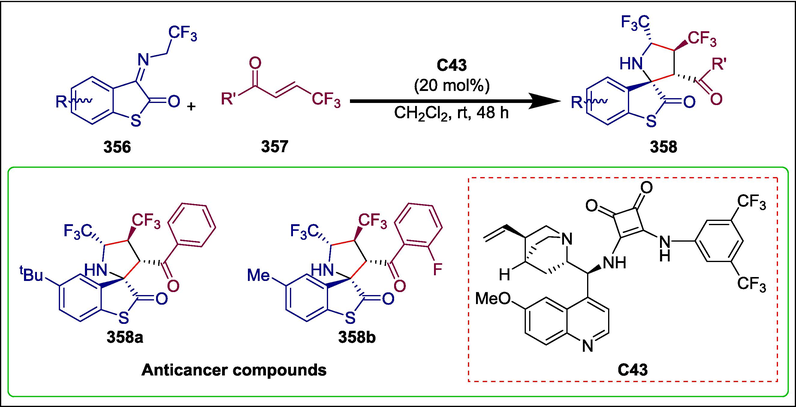
Quinine-derived squaramide catalyzed synthesis of spiro pyrrolidine-benzothiophenones.
6.2 Proline catalyst
Phthalimides are biologically active moieties and are included in certain commercial medicines that are used to treat multiple myeloma (Pomalyst), psoriasis (Otezla), and rheumatoid arthritis (Thalomid) (Gutiérrez-Rodríguez, 1984; Del Rosso and Kircik, 2016; Rios-Tamayo et al., 2017). Recent methods for synthesizing phthalimides have included benzannulation processes catalyzed by transition metals (Sheykhan et al., 2017; Yang et al., 2018). The use of transition metals is dangerous due to their toxicity. A simple, atom-efficient, one-pot, environmentally safe method for synthesizing phthalimide derivatives was presented by Akhtar and Lee. They carried out an L-proline catalyzed C47 reaction between two dienophiles of α,β-unsaturated aldehydes 360, and maleimides 359 to synthesize phthalimide derivatives 361 An effective benzannulation is involved in the reaction, which is carried out by a conventional [4 + 2] cycloaddition of azadiene intermediates produced in situ from N-substituted maleimides and enals. At first, α,β-unsaturated aldehyde 360 is transformed into iminium ion 362 in the presence of C44 and phenylmethanoic acid which is further transformed into azadiene intermediate 364 through 363. After that, the Diels-Alder adduct 365 is obtained by [4 + 2] cycloaddition of 364 with 359. This adduct then proceeds via L-proline elimination of desired phthalimide 361 (Scheme 53) (Akhtar and Lee, 2020). This approach has the benefit of yielding a wide range of functionalized phthalimides in moderate to exceptional yields, as it is compatible with α,β-unsaturated aldehydes, and N-substituted maleimides. The synthesis of biologically active compound 361, which exhibited strong activity (IC50 = 0.4 μM) as a COX-2 enzyme inhibitor, validates the importance of this effective procedure. (Alaa et al., 2011).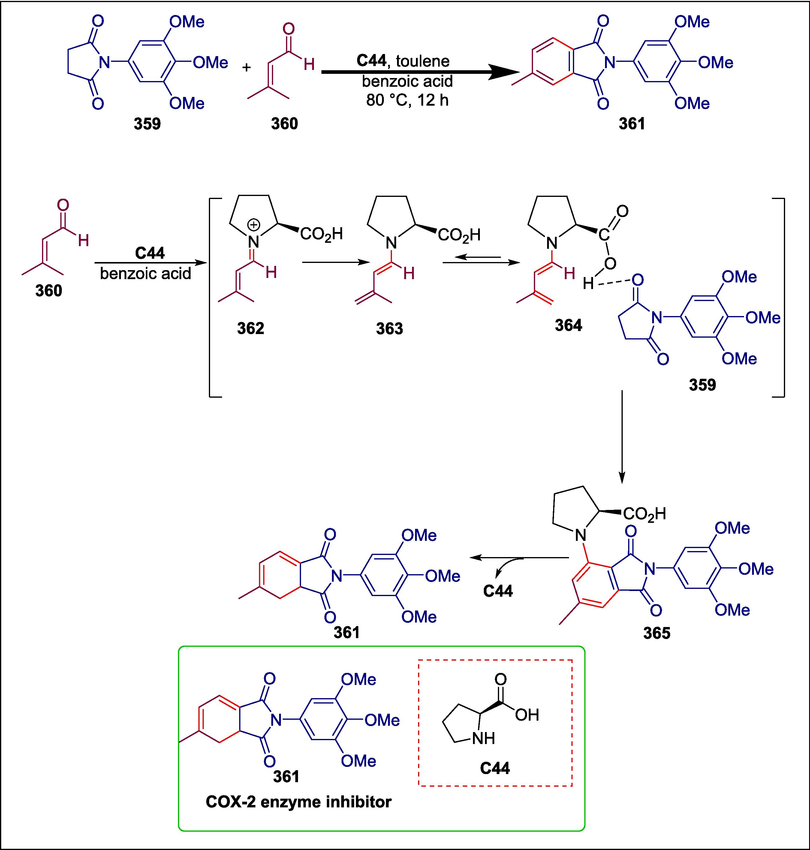
Proline catalyzed synthesis of phthalimides.
6.3 Prolinol catalyst
Sphingosine kinase (SPHK) inhibitors are promising therapeutic agents because SPHK1 is typically up-regulated in different cancer types and its genetic ablation makes cancer cells more susceptible to chemotherapeutic agents (Shida et al., 2008; Takabe et al., 2008; Gault and Obeid, 2011; Santos and Lynch, 2015). Casao et al. devised an effective method h for generating enantioenriched alkynylaziridines by using a prolinol-catalyzed aziridination reaction of α; β-unsaturated aldehydes followed by homologation reaction. This method produced a series of sixteen sphingosine analogues with a common triazole scaffold. The process begins with monoprotected diol 366 oxidizing to α,β-unsaturated aldehyde 367, which then goes through an aziridination reaction catalyzed by Tetramethylsilane −protected diphenylprolinol (R)-C45 (Jensen et al., 2012) to yield β-formyl aziridine 368. Then 368 undergoes Corey-Fuchs conditions to afford dibromoalkene 369 which gives ethynylaziridine 371 through metal-halogen exchange and ulterior α-elimination. After aziridine 371′s ring opens, 372 is produced, which goes through basic hydrolysis to produce the N-protected amino alcohol 373. 1-Azidododecane 378 is produced when 1-bromododecane 376 reacts with sodium azide 377. The corresponding azido aldehyde 383 undergoes a Wittig reaction with corresponding trifluorinated phosphorus ylide 382 to give azide 384 (Scheme 55). Next, the azides 378 and 384 undergo a cycloaddition reaction with the alkyne 373 to produce triazoles 374. These triazoles are then converted to the required N-Boc derivatives 375 (Scheme 54) (Escudero-Casao et al., 2018). The in vitro activity of the synthesized compounds as SPH inhibitors was examined and all compounds except two show activity. But two of them (375a and 375b) demonstrated an IC50 value near to that of standard inhibitor N,N-dimethylsphingosine (DMS) whose IC50 values for SPHK1 for SPHK2 are 27.3 µM and 3.8 µM respectively. Compound 375a exhibited an IC50 = 7.9 µM for SPHK2 while compound 375b was the most potent in this series in terms of activity with IC50 values of 60 µM and 18.1 µM for SPHK1 for SPHK2 respectively.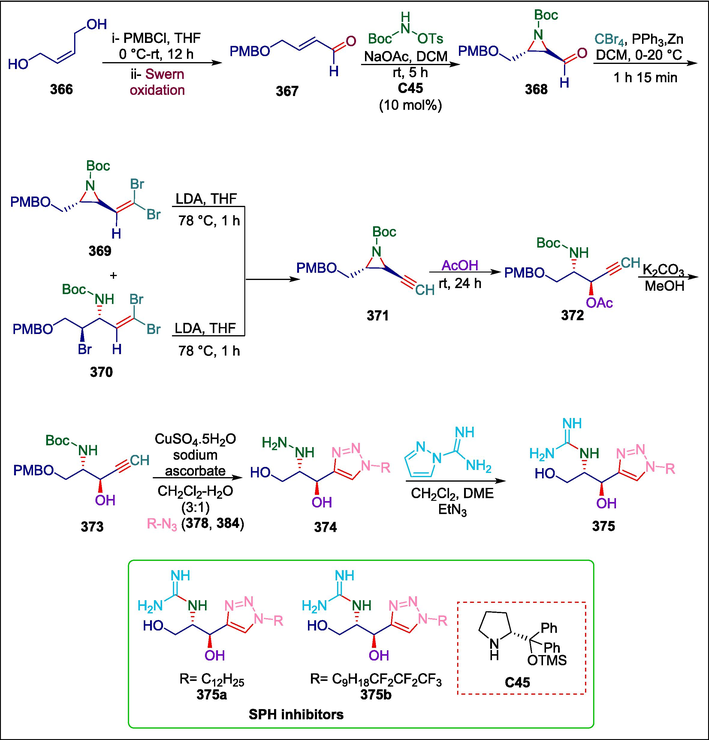
Prolinol catalyzed synthesis of sphingosine analogues.
Synthesis of azides.
Reboxetine is an antidepressant drug. It is also useful in the treatment of narcolepsy, panic disorder, ADHD (attention deficit hyperactivity disorder) (Melloni et al., 1984; Hajós et al., 2004), and cocaine dependence diseases (Ghanizadeh, 2015; Sharma and Pandey, 2022). It was previously synthesized using chiral starting materials, protection, deprotection, and multistep techniques. By using trans-cinnamaldehyde, Sharma and Pandey devised an effective, straightforward, and succinct prolinol-catalyzed protecting-group free synthetic approach for the total synthesis of (S,S)-reboxetine 394 and (S,R)-reboxetine 391. The asymmetric synthesis of 394 and 391 is started with trans-cinnamaldehyde 385, which upon treatment with H2O2 and tris-buffered saline-protected (R-bis[3,5- bis(trifluoromethyl)phenyl]prolinol C46 organocatalyst followed by subsequent reduction affords 2,3-epoxy-1-ol 386. The OH group of the epoxide compound 386 changes into O-mesylate, which experiences an epoxide opening at the C-3 position. After undergoing a K2CO3 treatment in MeOH and configuration inversion, compound 387 (Behrens and Sharpless, 1985) is produced which is then treated with 2-ethoxyphenol to produce the required (S,S)-epoxy ether 392 that undergoes the regioselective ring-opening with 2-aminoethyl hydrogen sulfate and 1,8-Diazabicyclo[5.4.0]undec-7-ene as the base to afford the zwitterion 393. Finally, compound 393′s cyclization under basic circumstances yields (S,S)-reboxetine 394. Under Mitsunobu esterification conditions, intermediate 381 may also offer a parallel route to the synthesis of 391 through configurational inversion at the C-3 position (Mitsunobu, 1981; Lipshutz et al., 2006), followed by basic hydrolysis (Scheme 56) (Sharma and Pandey, 2022).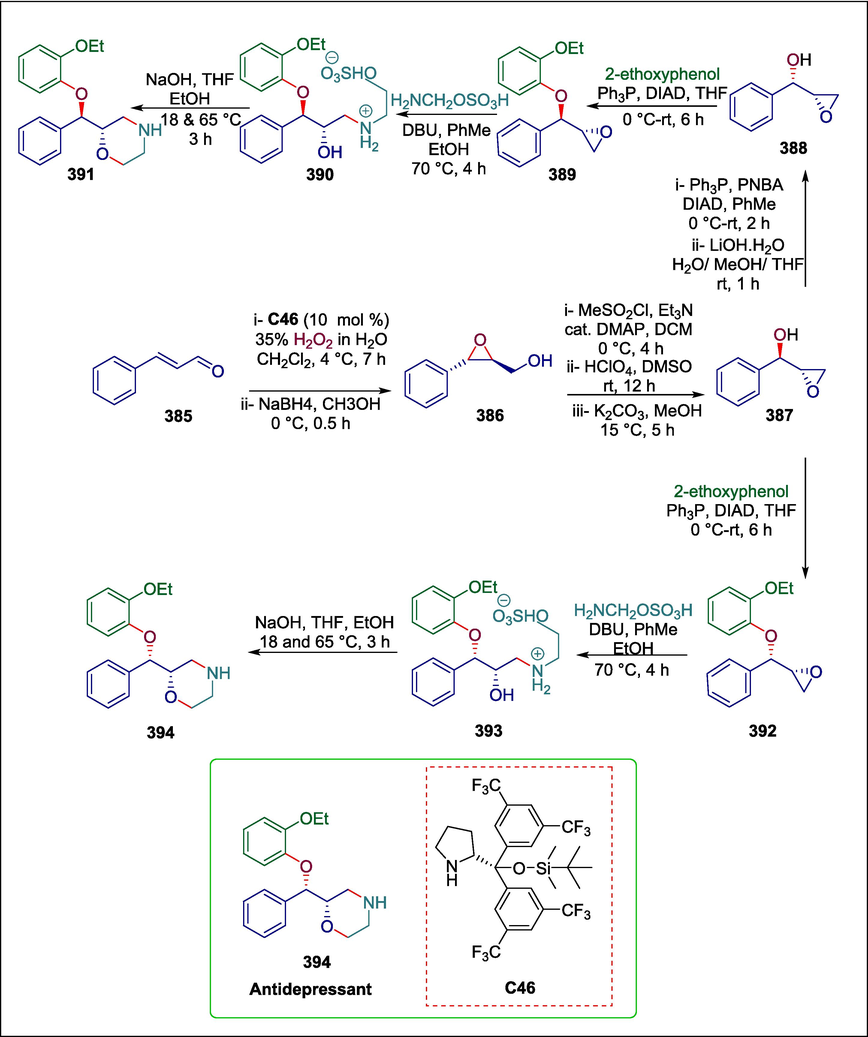
Prolinol catalyzed synthesis of reboxetine.
6.4 β-amino alcohol catalyst
Quinagolide 407 is a drug used to treat high prolactin levels. It combines the structural characteristics of ergolines CQ 32–084 (Satoh et al., 2007), pergolide (Barbe et al., 2011), and apomorphine (Kim and Batey, 2018), three common dopamine agonists. To date, enantioselective total synthesis of (−)-quinagolide 407 is not reported. Chavan et al. presented a completely different methodology for the total synthesis of (−)-quinagolide 407 (Chavan et al., 2018; Chavan et al., 2019b) from pyridine 395 by using racemic β-amino alcohol C47 (Suttibut et al., 2011)as an organocatalyst. The main steps used in the synthesis are organocatalyzed Diels-Alder reaction for fixing all three stereocenters on the piperidine ring, Birch deoxygenation, Lewis acid (TiCl4) catalyzed intramolecular Friedel-Crafts cyclization of dicarboxylic acid 401, and one-pot diastereoselective ketone reduction-intramolecular cyclization to synthesize tetracyclic oxazolidinone 403 that gives the desired trans-geometry (Scheme 57) (Chavan et al., 2019a). It has been found that under Birch reduction conditions, a C-N bond in an electron-deficient isoquinuclidine scaffold cleaves reductively. This is the first synthetic attempt to obtain the (−)-quinagolide 407 core framework.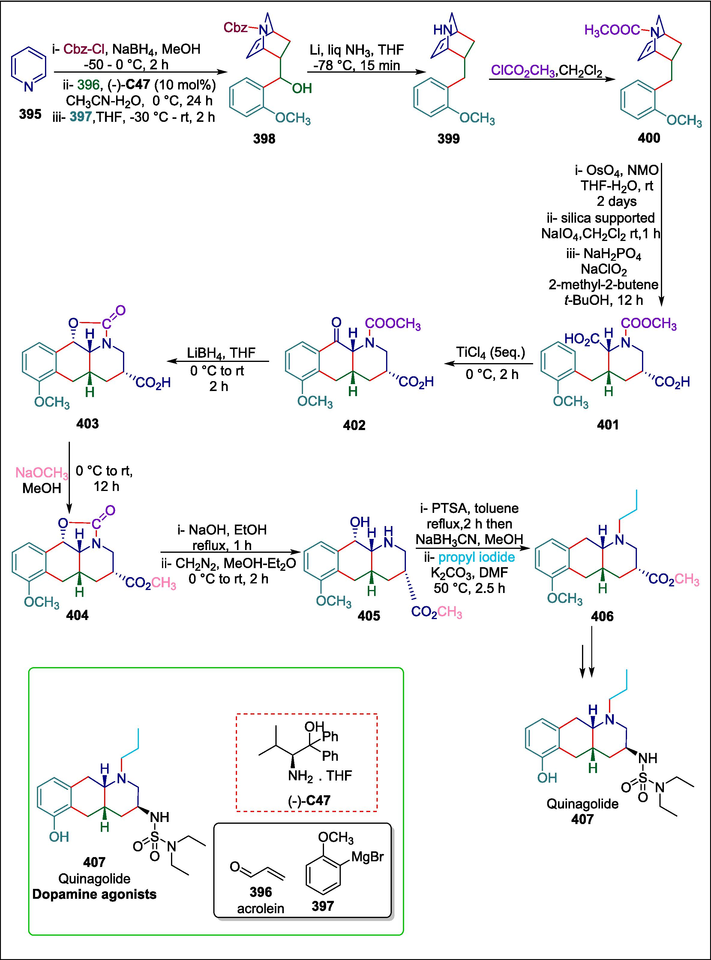
β −amino alcohol catalyzed synthesis of Quinagolide.
6.5 Chiral carbamate catalyst
Remdesivir, an RNA-dependent RNA polymerase inhibitor, has been employed in the treatment of COVID-19 (Cho et al., 2012; De Wit et al., 2020; Wang et al., 2020). Remdesivir is difficult to synthesize, and the net cost is very high. In particular, to achieve the required diastereoisomer (39 %) for subsequent coupling with the D-ribose-derived 5-alcohol, the stereoselective assembly of the P-chirogenic center needs recrystallization of a 1:1 isomeric p-nitrophenylphosphoramidate mixture multiple times. Gannedi et al. designed one-pot synthesis of remdesivir 384 by using chiral carbamate C48 as organocatalyst for stereoselective (S)-P-phosphoramidation employing a 1:1 diastereomeric mixture of phosphoramidoyl chloridates as the coupling reagent to prevent a waste of the other diastereomer. In the presence of catalyst C48, phosphoramidoyl chloridates 408 and D-ribose-derived 5-alcohol 409 are coupled in a 1:1 mixture which is subsequently subjected to acidic hydrolysis in p-TSA to remove isopropylidene group, to yield a mixture of 410 and its (R)-diastereomer, which is further purified by recrystallization to obtain remdesivir 410 (Scheme 58) (Gannedi et al., 2021).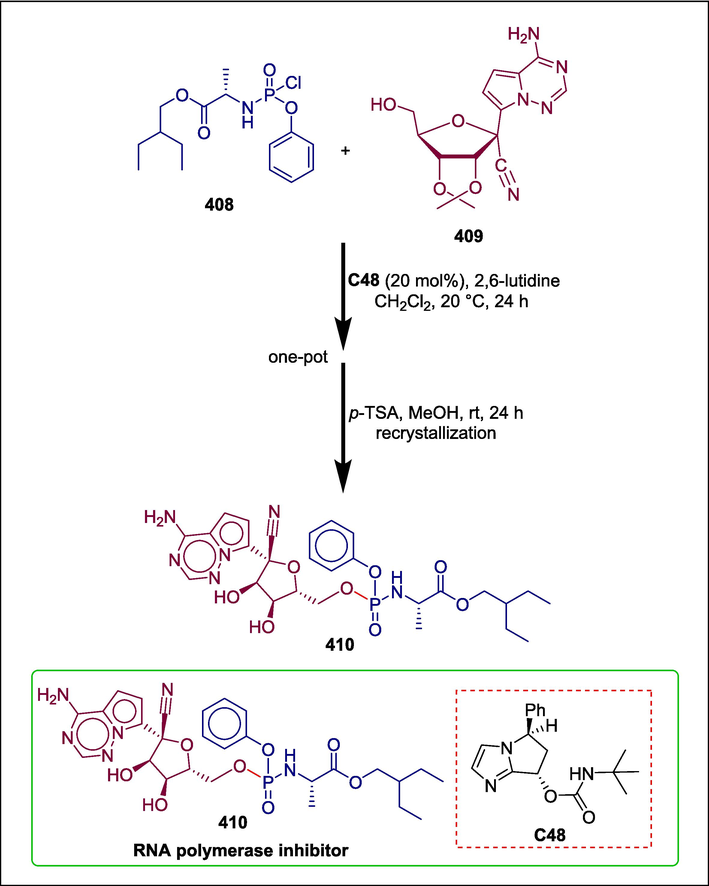
Chiral carbamate catalyzed synthesis of remdesivir.
Summary
Bifunctional organocatalysts are a special kind of catalysts that can simultaneously activate the electrophile and nucleophile in a reaction because they have both an electrophilic/acidic site and a nucleophilic/basic site. The catalyst's ability to cooperate with both reactants through dual activation increases the rate and selectivity of the reaction. A typical bifunctional organocatalyst has a basic group (imidazole or amine) paired with a hydrogen-bond donor (like a thiourea or squaramide group). In their mechanism of action, the acidic site, often a hydrogen bond donor, interacts with the electrophile such as a carbonyl group by stabilizing its partial negative charge. At the same time, the basic site deprotonates the nucleophile and generates a more reactive nucleophilic species. Simultaneously activating both reaction partners lowers the activation energy and facilitates reactions (Fig. 5). By avoiding harmful metals, operating in mild environments, and promoting extremely selective, effective reactions with little waste, bifunctional organocatalysts serve as green catalysts. Their capacity to be recycled and utilized in non-toxic solvents contributes even more to their environmental sustainability. This section covers the usage of a variety of bifunctional organocatalysts i.e. proline, prolinol, β-amino alcohol, chiral carbamate, and cinchona alkaloids-derived catalysts for the synthesis of many bioactive compounds such as analogous of (R)-tomoxetine, (R)-fluoxetine, and (R)-duloxetine, analogous of antidepressant drug fenpentadiol, fluralaner, afoxolaner, (S)-forphenicinol, reboxetine, remdesivir, quinagolide, COX-2 enzyme inhibitors, SPHK inhibitors, and anticancer compounds.
7 Organo-photo catalysis
Photocatalysis under UV activation facilitates the formation of a wide range of unconventional bonds in organic synthesis (Snyder et al., 1981; Dugave and Demange, 2003; Koike and Akita, 2016). Visible light photocatalysis, which offers mild conditions and high functional group tolerance, has recently become a popular approach for activating small molecules. In this case, the light-absorbing photocatalyst is primarily responsible for starting the chemical reaction (Zeitler, 2009; Meyer et al., 2016; Ochola and Wolf, 2016; Shaw et al., 2016; Hu et al., 2017; Zhan and Li, 2017). The use of visible light is a cheap, energy-efficient, and environmentally benign way to stimulate reactions (Brooks et al., 2013; Hager and MacMillan, 2014; Tasker and Jamison, 2015). It is also intriguing to consider using single-electron transfer (SET) pathways, which are made possible by visible light absorption, to speed up molecular activation at low temperatures. In contrast to traditional thermal conditions, the use of single electron transfer in conjunction with a catalyst enables the use of mild reaction conditions to generate required products in excellent yield for a wider substrate scope (Zuo and MacMillan, 2014). Even though the use of SET in photocatalysis has advanced significantly, more research is still needed to understand the photophysical characteristics of these systems and how they relate to catalytic efficiency. This will help to reveal crucial aspects of photocatalyst design (Ochola and Wolf, 2016). In 2020, Chu et al. synthesized heterogeneous organo-photocatalysts single-step coupling reaction of eosin Y (EY) with commercial cotton threads. Then the immobilized EY is used for photoinduced organic conversions and reversible deactivation radical polymerization over several reaction cycles with well-retained catalytic efficiency (Chu et al., 2020).
7.1 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene photocatalyst
A popular methodology for C–H functionalization of heteroarenes is the Minisci reaction (Minisci et al., 1989; Minisci et al., 1990; Duncton, 2011). Radical precursors and photoredox chemistry have been used to generate several latest variants of the Minisci reaction. Recently Okada (Okada et al., 1988; Okada et al., 1991) and Overman (Schnermann and Overman, 2012; Müller et al., 2015; Pratsch et al., 2015; Jamison and Overman, 2016) produced alkyl radicals by subjecting N-(acyloxy)phthalimides (NAPs) to light in the presence of a photosensitizer (Okada et al., 1988). An external oxidant is needed for the reductive production of radicals in this approach. To circumvent the need for an external oxidant and the usage of costly metal-based catalysts such as iridium-based complexes, Sherwood et al. accomplished the Minisci reaction using an organic photocatalyst C49. In their one-pot Minisci approach, they started with a carboxylic acid 411 that acts as a radical precursor and then undergoes NAP synthesis in situ. After that, they add a heteroarene 412 and photocatalyst C49. This technique avoided the requirement to isolate a pre-functionalized alkyl partner and allowed for the quick production of analogues employing a group of chemicals which are widely accessible in a high degree of structural variety. The process begins when NAP 415 uses single-electron transfer (SET) to oxidatively quench the excited photocatalyst. This leads to reductive fragmentation, which produces the alkyl radical 417 along with CO2 and phthalimide 416. By attacking a protonated heteroarene 418, radical intermediate 417 produces an adduct 419, which is then deprotonated to produce α-amino radical 420, which then undergoes single-electron transfer with the photocatalyst's oxidized form to produce the desired protonated product 421, which ends the catalytic cycle (Scheme 59) (Sherwood et al., 2018). The benefits of this approach are low conditions, a high level of functional group tolerance, and a low requirement of the carboxylic acid reactant. The synthesis of analogues of camptothecin 414, an anticancer drug, in 17 % yield using late-stage functionalization of the produced molecules 413 illustrates the practical application of this methodology.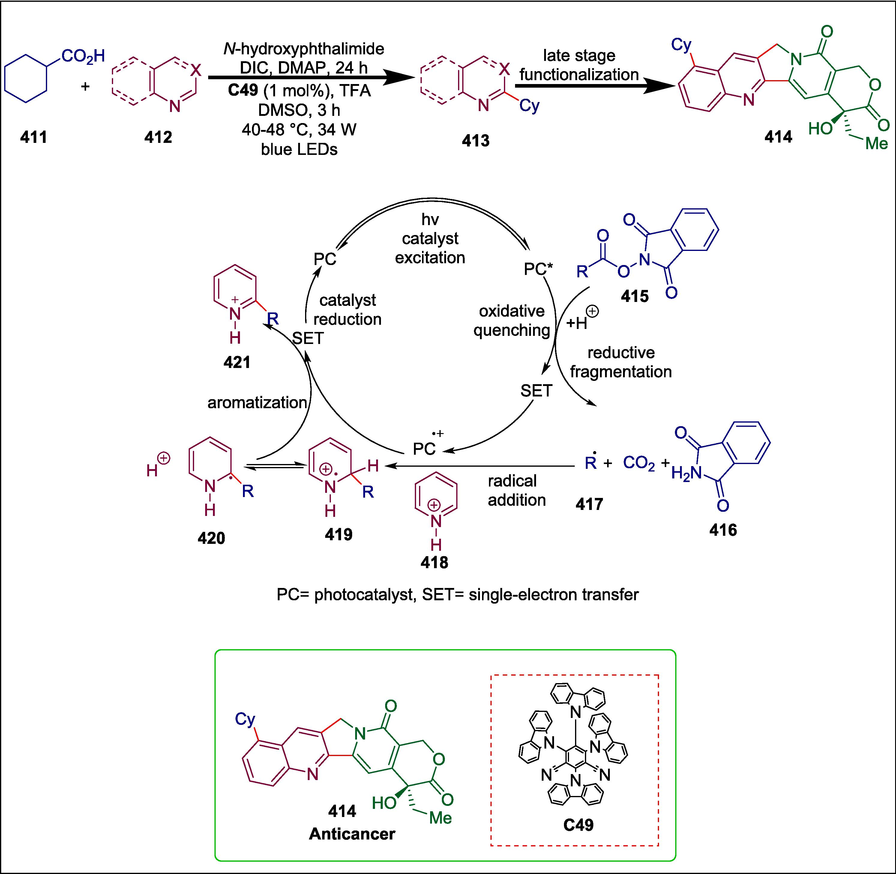
Organo-photocatalyzed synthesis of cyclohexyl quinolines.
7.2 Carbazolic-cyano conjugated microporous polymer (CC-CMP) photocatalyst
Natural products often contain heterocycle-fused indolines and spirocyclic oxindoles, which have a variety of pharmacological characteristics, including anticancer, antimigraine, and contraceptive effects. Because of this, these moieties are being valued for use in drug discovery and as building blocks in the synthesis of other alkaloids and drugs. But the majority of current synthetic approaches depend on developing vital fused indoline and spirocyclic oxindole skeletons using indole precursors.These methods are not widely applied because they require costly noble-metal catalysts and tedious procedures. Ou et al. developed a quick way to produce 3-formyloxindoles 424 by using a heterogeneous organophotocatalyst C50 and double C–H functionalization of N-arylacrylamides 423 and 1,3-dioxolane 422 in a metal-free, light-mediated process. The photoexcited catalyst undergoes SET to TBHP and gives a tert-butyloxy radical. After that, the tert-butyloxy radical's regioselectivity removes a proton from the carbon 2 of 1,3-dioxolane 422 to form radical specie 425 (Fan et al., 2018). This radical specie then combines with acrylamide 423 to form radical specie 426, which undergoes intramolecular cyclization to produce radical intermediate 427. By oxidizing radical intermediate 427 with PC•+, the dioxolane acetal intermediate 428 is formed, and aromaticity is subsequently restored. Acidic hydrolysis ultimately affords the requisite product 424 (Scheme 60) (Ou et al., 2019). This approach demonstrates the great potential of using CMPs as stable, reusable, and metal-free light-triggered photocatalysts that provide a sustainable means of producing therapeutic compounds and bioactive heterocycles. Compared to multistep processes, the resulting 3-formyloxindoles are more suited for the rapid synthesis of certain pharmaceutically necessary products, such as (±)-desoxyeseroline, (±)-esermethole, and (±)-N-methylerulescine.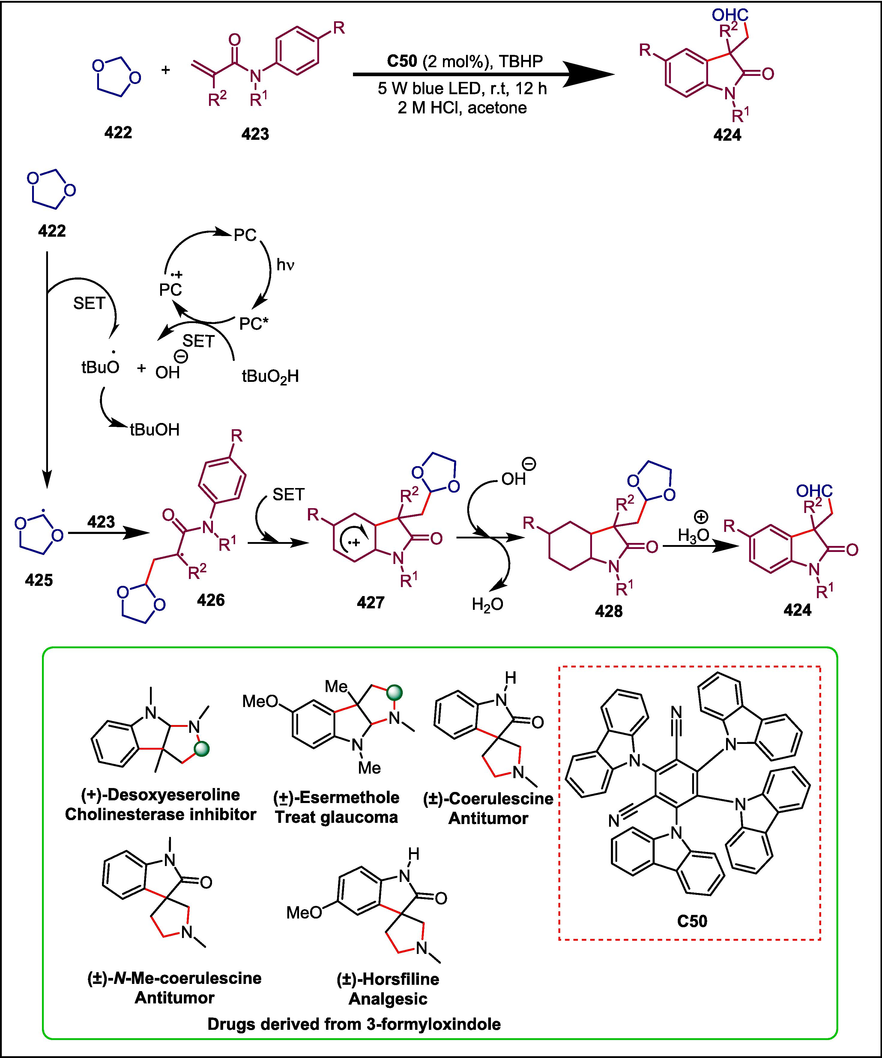
CC-CMP catalyzed synthesis of 3-formyloxindoles.
7.3 Lumiflavin photocatalyst
Carbon-centered radicals are essential components of advanced synthetic chemistry. Visible light organophotoredox catalysis has emerged as a potentially effective method for obtaining C-centered radicals from a wide range of latent functional groups, such as boronic acids. Chilamari et al. described an aqueous approach that used a organophotocatalyst lumiflavin C51 to convert aliphatic, aromatic, and heteroaromatic boronic acids 429 to C-centered radicals 433. Through open-shell conjugate addition to various Michael acceptors, these radicals were utilized to produce a broad pool of alkylated products 431, including three molecules with pharmacological significance. Boronic acid 429 coordinates with water to give aqua-boryl complex 432. The excited lumiflavin abstracts proton and an electron from 432 then 432 underwent fragmentation to afford heteroaryl radical 433. With the addition of openshell conjugates to diethyl ethylidene malonate (DEEM) 430, the former is rapidly captured. This results in the α-malonyl radical 434, which is then reduced to give the desired alkylated product 431, and the organophotocatalyst is regenerated (Scheme 61) (Chilamari et al., 2020).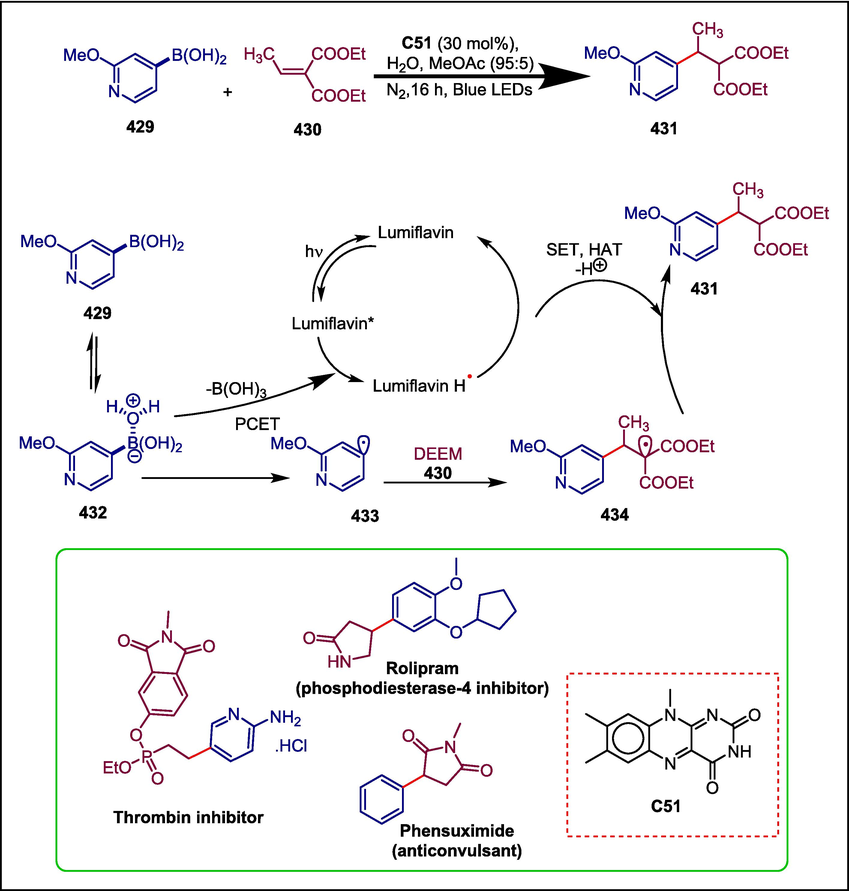
Lumiflavin catalysed synthesis of alkylated products.
7.4 Anthraquinone (AQ) photocatalyst
Quinoline has been considered a remarkable motif because of its promising biological properties, which have been demonstrated by approved pharmaceuticals including pitavastatin, cinchophen, camptothecin. Zhao et al. described the organophotocatalyzed synthesis of quinolines 437 through cyclizing secondary alcohol 436 into 2-aminobenzyl alcohols 435 at room temperature. By using DMSO and readily available anthraquinone as photocatalyst C52 this technique removesthe need of high thermal conditions and toxic metal catalysts. A hydrogen atom is extracted from 435 or 436 by the excited photocatalyst AQ via hyrdrogen atom transfer (HAT), producing 438 or 440 radicals, followed by another HAT to yield 2-aminobenzaldehyde 439 or acetophenone 441. Then protonated AQ is oxidized to AQ by the DMSO. Ultimately, condensation occurs between 439 and 441 to create the desired product 437 (Scheme 62) (Zhao et al., 2023b).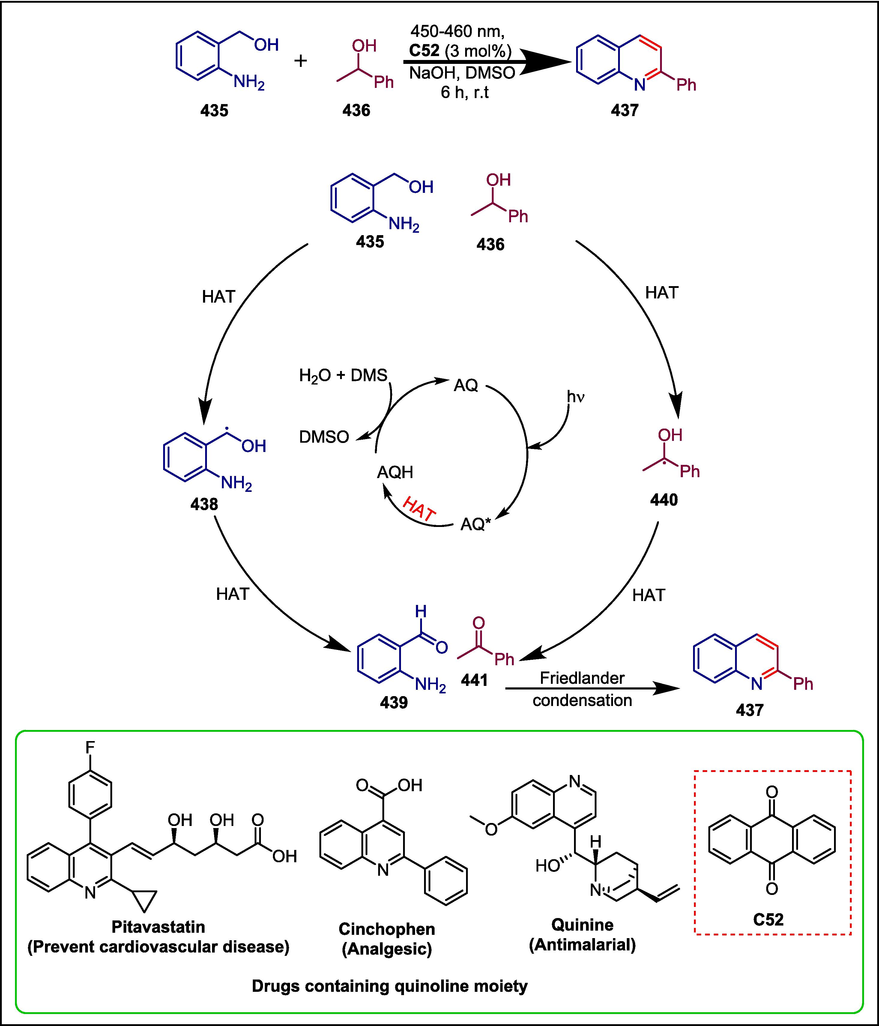
Anthraquinone catalyzed synthesis of quinolines.
7.5 Eosin Y photocatalyst
Indoles provide a wide range of bioactivity (Cerecetto et al., 2005)to the pharmaceutical sector, including antibacterial, anti-inflammatory, HIV protease inhibition, and anticancer medicines. Commercially significant pharmaceutical drugs like MK-4827 and pazopanib have indazole as a fundamental scaffold. The majority of cases of arylation by arylhalides and Negishi cross-coupling required expensive ligands, Cu/Pd as catalysts, and the use of base and oxidant as additives with long reaction durations. Therefore, there is less research available on the direct arylation of 2H-indazoles at the C-3 position. Vidyacharan et al. synthesized liver X-receptor inhibitor drugs 444 without converting DIPEA into its radical cation, and they proposed a simple, sustainable method for direct C-3 arylation of 2H-indazoles 442 with aryldiazonium salts 443 under milder conditions using organo-photoredox catalysis with green light. By synthesizing the aryl radical 445 and recovering the photocatalyst C53, the SET from reduced photocatalysts completes the catalytic cycle. After reacting with heteroarene 442 the resultant radical moiety 445 yields radical intermediate 446, which is then converted to carbocation intermediate 447 by donating electrons to the N,N-Diisopropylethylamine (DIPEA). The system is aromaticized during deprotonation, producing the desired product 444 (Scheme 63) (Vidyacharan et al., 2019). High yields were obtained with the phenyl moiety containing electron-donating groups, but very low yields were obtained with bromosubstitution.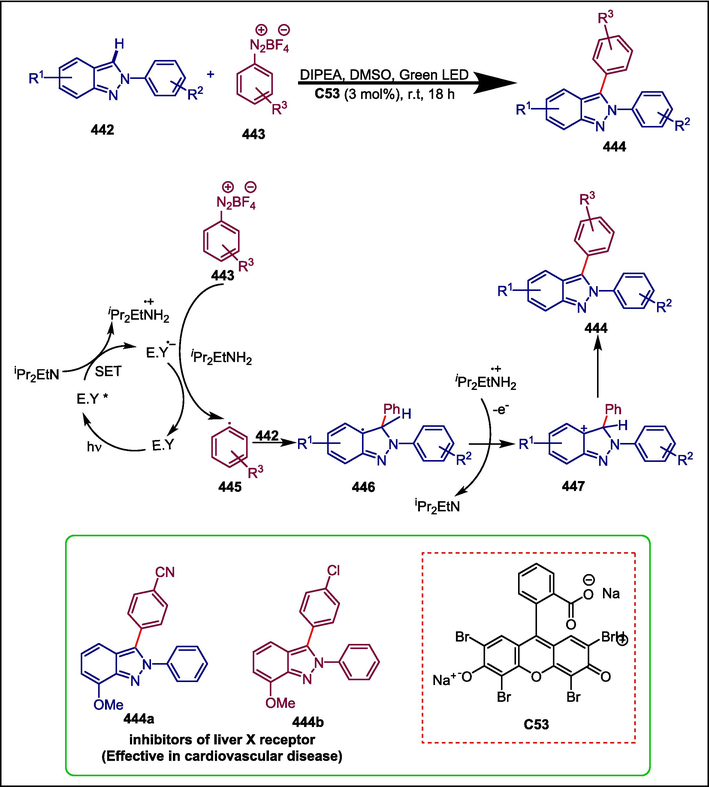
Eosin Y catalyzed synthesis of liver X-receptor inhibitor drugs.
Summary
A type of catalyst known as photo-organocatalysts uses visible light as a sustainable and environmentally friendly energy source to drive organic reactions. These catalysts, often organic molecules with conjugated structures, absorb light and achieve an excited state, enabling them to transmit electrons or energy to reactants. The mechanism of action generally involves generating reactive intermediates, such as radicals or excited-state species, which can then participate in bond-forming or bond-breaking processes (Fig. 6). For example, a photo-organocatalyst can absorb light and enter an excited triplet or singlet state, where it either donates an electron to an electrophile or accepts an electron from a nucleophile, initiating redox reactions like photocatalytic reductions, oxidations, or cycloadditions. These catalysts are important for sustainable and green chemistry because they are often very selective, adaptable to mild reaction conditions, and provide a metal-free and energy-efficient substitute for conventional photoredox systems. In this chapter, we described different photo-organocatalysts such as benzophenone and 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene photocatalyst for the synthesis of significant pharmaceutically potent molecules like anticancer, antioxidant and antibacterial compounds.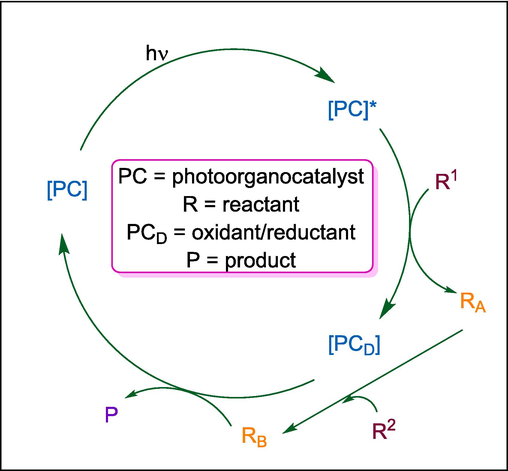
Mechanism of photo-organocatalysis.
8 Integrate cutting edge research
Advanced research in organocatalysis is pushing the boundaries of synthetic chemistry, with significant focus on enantioselective catalysis, sustainable practices, and novel reaction mechanisms. One of the most significant fields is the development of asymmetric organocatalysts that can perform highly selective, enantio-controlled reactions, important for producing complex drug moities. Scientists are currently investigating the concept of dual catalysis, which involves integrating metal or photoredox catalysis (Yao et al., 2022)with organocatalysis to provide novel and effective reaction pathways in mild conditions. Deeper understanding of reaction pathways is being gained through computational research and mechanistic studies, which makes it possible to rationally design catalysts with higher efficiency. Sustainable developments include the synthesis of enzyme-like biomimetic catalysts that encourage extremely selective reactions in aqueous or environmentally benign conditions. Organocatalysis is also transforming the field of polymer research by enabling metal-free, organocatalytic polymerization for biodegradable materials (Zhou et al., 2022). Organocatalysts are also improving efficiency in cascade and multicomponent reactions (Grondal et al., 2010) by facilitating challenging molecular syntheses in fewer steps with less waste. These developments are making organocatalysis a more effective and sustainable tool in modern synthetic chemistry Fig. 7.
Flow chart outlining the main points of this review.
9 Conclusion
Organocatalysis has emerged as the third main technique for catalyzing a diverse range of chemical processes apart from enzymes and metal catalysis. It enables eco-friendly, metal-free synthesis of chiral drugs, fine chemicals, and biodegradable polymers, while also facilitating complex natural product construction. Due to its sustainability and recyclability, it is a vital tool in green chemistry. According to a green chemistry viewpoint, the formation of a highly stereoselective organocatalytic approach in an aqueous medium has become the most appealing topic of organic chemistry. The previous few years have seen phenomenal growth in this sector, and more reactions are anticipated in the future. This review describes the potential and versatility of organocatalyzed reactions in the field of synthetic organic chemistry. It explains the use of different cost-effective, and environment-friendly organocatalysts for the synthesis of many remarkable bioactive molecules. Organocatalysts are green catalysts due to their ability to facilitate reactions without the need for toxic metals, harsh conditions, or excessive waste. For example, proline, a naturally occurring amino acid, is utilized in aldol reactions and frequently works in ambient and aqueous environments. Its nontoxic and biodegradable nature makes it highly sustainable. N-Heterocyclic carbenes (NHCs) can be used with greener solvents because they don't require metal catalysts and can function in milder environments. Similarly, without the need of transition metals, chiral phosphoric acids facilitate enantioselective processes. Furthermore, thiourea catalysts, which are employed in hydrogen-bonding catalysis, facilitate reactions in green solvents and mild conditions, supporting environment friendly synthetic methods. Even with its benefits, organocatalysis cannot replace the importance of metal and biocatalysis. Certain significant reactions like cross-coupling and metathesis, which are catalyzed by transition metals, are still improbable to occur through organocatalysis. Because of their narrow range of substrates, lower reactivity, and sensitivity to environmental factors, organocatalysts can result in longer reaction times and lower yields. In addition, achieving high enantioselectivity can be challenging, and their mechanisms are often not well understood.Thus, further research into organocatalysis is necessary to create an extremely efficient catalyst that can perform several reactions with improved practical applications.
CRediT authorship contribution statement
Labiqa Aman: Writing – review & editing, Writing – original draft. Shehla Khalid: Writing – review & editing, Visualization, Conceptualization. Nasir Rasool: Supervision, Funding acquisition. Almeera Zia: Visualization, Software. Muhammad Imran: Visualization, Project administration. Marius Irimie: Writing – review & editing. Codrut Ioan Ciurea: Visualization, Project administration.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Synthesis and antimycobacterial screening of new thiazolyl-oxazole derivatives. Eur. J. Med.. 2017;132:333-340.
- [CrossRef] [Google Scholar]
- Inhibition of human cytomegalovirus protease by benzoxazinones and evidence of antiviral activity in cell culture. Bioorg. Med. Chem. Lett.. 1997;7:2105-2108.
- [CrossRef] [Google Scholar]
- Sulphasomizole (5-p-aminobenzenesulphonamido-3-methyliso. thiazole): a new antibacterial sulphonamide. Nature, London. 1960;186:221-222.
- [CrossRef] [Google Scholar]
- Organocatalysis: a revolutionary approach in organic synthesis. African Multidisciplinary J. Sci. Art. Intell.. 2024;1:181-194.
- [CrossRef] [Google Scholar]
- Organocatalyzed synthesis of highly functionalized phthalimides via diels-alder reaction employing two dienophiles. J. Org. Chem.. 2020;85:15129-15138.
- [CrossRef] [Google Scholar]
- Recent progress in chiral Brønsted acid catalysis. Adv. Synth. Catal.. 2006;348:999-1010.
- [CrossRef] [Google Scholar]
- Stronger Brønsted acids: recent progress. Chem. Rev.. 2015;115:9277-9306.
- [CrossRef] [Google Scholar]
- Synthesis, anti-inflammatory activity and COX-1/COX-2 inhibition of novel substituted cyclic imides. Part 1: Molecular docking study. Eur. J. Med.. 2011;46:1648-1655.
- [CrossRef] [Google Scholar]
- Chiral 1, 1-diaryl compounds as important pharmacophores. MedChemComm.. 2013;4:893-907.
- [CrossRef] [Google Scholar]
- Organocatalyzed synthesis and antibacterial activity of novel quinolino annulated analogues of azepinones. J. Heterocycl. Chem.. 2018;55:2178-2187.
- [CrossRef] [Google Scholar]
- Organocatalysis emerging as a technology. Pure Appl. Chem.. 2021;93:1371-1381.
- [CrossRef] [Google Scholar]
- Crystal structure of a bacterial type III polyketide synthase and enzymatic control of reactive polyketide intermediates. J. Biol. Chem.. 2004;279:45162-45174.
- [CrossRef] [Google Scholar]
- Highly atroposelective synthesis of nonbiaryl naphthalene-1, 2-diamine NC atropisomers through direct enantioselective CH amination. Nat. Commun.. 2019;10:3063.
- [CrossRef] [Google Scholar]
- Physicochemical characterization of benzalkonium chloride and urea based deep eutectic solvent (DES): a novel catalyst for the efficient synthesis of isoxazolines under ultrasonic irradiation. J. Mol. Liq.. 2016;224:1249-1255.
- [CrossRef] [Google Scholar]
- Enantioselective Brønsted base catalysis with chiral cyclopropenimines. J. Am. Chem. Soc.. 2012;134:5552-5555.
- [CrossRef] [Google Scholar]
- Organocatalysis lost: modern chemistry, ancient chemistry, and an unseen biosynthetic apparatus. Angew. Chem. Int. Ed.. 2008;47:42-47.
- [CrossRef] [Google Scholar]
- Asymmetric total synthesis of (+)-luciduline: toward a general approach to related Lycopodium alkaloids. J. Org. Chem.. 2011;76:5354-5362.
- [CrossRef] [Google Scholar]
- An overview of the synthetic routes to the best selling drugs containing 6-membered heterocycles. Beilstein J. Org. Chem.. 2013;9:2265-2319.
- [CrossRef] [Google Scholar]
- Recent advances in the enantioselective 1, 3-dipolar cycloaddition of azomethine ylides and dipolarophiles. Tetrahedron Asymmetry. 2017;28:876-899.
- [CrossRef] [Google Scholar]
- Selective transformations of 2, 3-epoxy alcohols and related derivatives. Strategies for nucleophilic attack at carbon-3 or carbon-2. J. Org. Chem.. 1985;50:5696-5704.
- [CrossRef] [Google Scholar]
- Organic fluorine compounds: a great opportunity for enhanced materials properties. Chem. Soc. Rev.. 2011;40:3496-3508.
- [CrossRef] [Google Scholar]
- Asymmetric Organocatalysis: From Biomimetic Concepts to Applications in Asymmetric Synthesis. John Wiley & Sons; 2006.
- Bioinspired organocatalytic asymmetric reactions. Org. Biomol. Chem.. 2012;10:2911-2922.
- [CrossRef] [Google Scholar]
- Remotely controlled iridium-catalyzed asymmetric hydrogenation of terminal 1, 1-diaryl alkenes. Angew. Chem. Int. Ed.. 2013;34:8795-8797.
- [CrossRef] [Google Scholar]
- Carbamoyl radical-mediated synthesis and semipinacol rearrangement of β-lactam diols. Chem. Eur. J.. 2014;20:6505-6517.
- [CrossRef] [Google Scholar]
- Decarboxylative ketone aldol reactions: development and mechanistic evaluation under metal-free conditions. J. Org. Chem.. 2009;74:6190-6198.
- [CrossRef] [Google Scholar]
- Stereoselective synthesis and applications of spirocyclic oxindoles. Org. Chem. Front.. 2021;8:1026-1084.
- [CrossRef] [Google Scholar]
- Antitumor agents. 2. synthesis, structure− activity relationships, and biological evaluation of substituted 5 H-pyridophenoxazin-5-ones with potent antiproliferative activity. J. Med. Chem.. 2002;45:5217-5223.
- [CrossRef] [Google Scholar]
- Bonanomi, G., Estrada, A.A., Feng, J.A., et al., 2020. Isoxazolidine derived inhibitors of receptor interacting protein kinase 1 (ripk1), Google Patents.
- Inhibition of cathepsin K for treatment of osteoporosis. Curr. Osteoporos. Rep.. 2012;10:73-79.
- [CrossRef] [Google Scholar]
- Visible-light-induced organophotocatalytic and singlet oxygen-initiated domino construction of 1, 4-dihydropyridines, C-3 functionalized spiro [indoline-3, 4′-pyridines] and C-11 functionalized spiro [indeno-[1, 2-b] quinoxaline-11, 4′-pyridines] Org. Biomol. Chem.. 2023;21:1518-1530.
- [CrossRef] [Google Scholar]
- Freeze-frame inhibitor captures acetylcholinesterase in a unique conformation. Proc. Natl. Acad.. 2004;101:1449-1454.
- [CrossRef] [Google Scholar]
- Natural occurrence, syntheses, and applications of cyclopropyl-group-containing α-amino acids. 1. 1-Aminocyclopropanecarboxylic acid and other 2, 3-methanoamino acids. Chem. Rev.. 2007;107:4493-4537.
- [CrossRef] [Google Scholar]
- Cathepsin K inhibitors for osteoporosis and potential off-target effects. Expert Opin. Investig.. 2009;18:585-600.
- [CrossRef] [Google Scholar]
- Photon-driven reduction of Zn2+ to Zn metal. Inorg. Chem.. 2013;52:5794-5800.
- [CrossRef] [Google Scholar]
- World commission on environment and development. Environ. Law J.. 1985;14:26-30.
- [CrossRef] [Google Scholar]
- Asymmetric organocatalysis in drug discovery and development for active pharmaceutical ingredients. Expert Opin. Drug Discov.. 2023;18:37-46.
- [CrossRef] [Google Scholar]
- Chiral allene-containing phosphines in asymmetric catalysis. J. Am. Chem. Soc.. 2011;133:18066-18069.
- [CrossRef] [Google Scholar]
- Metal versus silyl triflate catalysis in the Mukaiyama aldol addition reaction. Tetrahedron Lett.. 1994;35:4323-4326.
- [CrossRef] [Google Scholar]
- Pharmacological properties of indazole derivatives: recent developments. Mini Rev. Med. Chem.. 2005;5:869-878.
- [CrossRef] [Google Scholar]
- Organocatalytic atroposelective construction of axially chiral N, N-and N, S-1, 2-azoles through novel ring formation approach. Nat. Commun.. 2022;13:1933.
- [CrossRef] [Google Scholar]
- Synthesis of 3-azidopiperidine skeleton employing ceric ammonium nitrate (CAN)-mediated regioselective azidoalkoxylation of enol ether: total synthesis of D2 Receptor agonist (±)-quinagolide. Org. Lett.. 2018;20:7011-7014.
- [CrossRef] [Google Scholar]
- Enantioselective formal total synthesis of (−)-quinagolide. Org. Lett.. 2019;21:9089-9093.
- [CrossRef] [Google Scholar]
- Total synthesis of (±)-quinagolide: a potent D2 receptor agonist for the treatment of hyperprolactinemia. ACS Omega. 2019;4:8231-8238.
- [CrossRef] [Google Scholar]
- Synthesis, characterization and evaluation of analgesic and anti-inflammatory activities of some novel indoles. Trop. J. Pharm. Res.. 2011;10:463-473.
- [CrossRef] [Google Scholar]
- DABCO-catalyzed michael/alkylation cascade reactions involving α-substituted ammonium ylides for the construction of spirocyclopropyl oxindoles: access to the powerful chemical leads against HIV-1. J. Org. Chem.. 2020;85:5203-5219.
- [CrossRef] [Google Scholar]
- Toward bicalutamide analogues with high structural diversity using catalytic asymmetric oxohydroxylation. J. Org. Chem.. 2024;89:3907-3911.
- [CrossRef] [Google Scholar]
- Synthesis and antitumor activity of epothilone B. Eur. J. Med.. 2018;157:925-934.
- [CrossRef] [Google Scholar]
- Organocatalytic asymmetric assembly reactions: synthesis of spirooxindoles via organocascade strategies. ACS Catal.. 2014;4:743-762.
- [CrossRef] [Google Scholar]
- General access to C-centered radicals: combining a bioinspired photocatalyst with boronic acids in aqueous media. ACS Catal.. 2020;10:12727-12737.
- [CrossRef] [Google Scholar]
- Synthesis and antiviral activity of a series of 1′-substituted 4-aza-7, 9-dideazaadenosine C-nucleosides. Bioorg. Med. Chem. Lett.. 2012;22:2705-2707.
- [CrossRef] [Google Scholar]
- Scalable and recyclable heterogeneous organo-photocatalysts on cotton threads for organic and polymer synthesis. ChemPhotoChem.. 2020;4:5201-5208.
- [CrossRef] [Google Scholar]
- Clean Synthesis Using Porous Inorganic Solid Catalysts and Supported Reagents. Royal Society of Chemistry; 2007.
- Design, synthesis, molecular modelling, and in vitro evaluation of tricyclic coumarins against Trypanosoma cruzi. Chem. Biol. Drug Des.. 2019;93:337-350.
- [CrossRef] [Google Scholar]
- Design and synthesis of a new series of 3, 5-disubstituted isoxazoles active against Trypanosoma cruzi and Leishmania amazonensis. Eur. J. Med.. 2017;128:25-35.
- [CrossRef] [Google Scholar]
- Synthesis, antioxidant and antitumoral activities of 5′-arylchalcogeno-3-aminothymidine (ACAT) derivatives. MedChemComm.. 2017;8:408-414.
- [CrossRef] [Google Scholar]
- Biotransformation reactions of five-membered aromatic heterocyclic rings. Chem. Res. Toxicol.. 2002;15:269-299.
- [CrossRef] [Google Scholar]
- Organotransition Metal Chemistry: Applications to Organic Synthesis: Applications to Organic Synthesis. Elsevier; 2013.
- Tratamento etiológico da doença de Chagas no Brasil. Revista De Patologia Tropical/journal of Tropical Pathology. 2008;37:209-228.
- [CrossRef] [Google Scholar]
- IR780-polymer conjugates for stable near-infrared labeling of biodegradable polyester-based nanocarriers. Eur. Polym. J.. 2019;120:109255
- [CrossRef] [Google Scholar]
- New organochalcogen multitarget drug: synthesis and antioxidant and antitumoral activities of chalcogenozidovudine derivatives. J. Med. Chem.. 2015;58:3329-3339.
- [CrossRef] [Google Scholar]
- Prophylactic and therapeutic remdesivir (GS-5734) treatment in the rhesus macaque model of MERS-CoV infection. Proc. Natl. Acad.. 2020;117:6771-6776.
- [CrossRef] [Google Scholar]
- Divergent reactivity of δ-and β′-acetoxy allenoates with 2-sulfonamidoindoles via phosphine catalysis: entry to dihydro-α-carboline, α-carboline, and spiro-cyclopentene motifs. J. Org. Chem.. 2021;86:11583-11598.
- [CrossRef] [Google Scholar]
- Oral apremilast for the treatment of plaque psoriasis. J. Clin. Aesthet. Dermatol.. 2016;9:43.
- [Google Scholar]
- The total synthesis of calcium atorvastatin. Org. Biomol. Chem.. 2016;14:2291-2296.
- [CrossRef] [Google Scholar]
- Recent advances in isocyanide-based multicomponent chemistry. Curr. Opin. Chem. Biol.. 2002;6:306-313.
- [CrossRef] [Google Scholar]
- NHCs in Organocatalysis: Azolium Enolate Generation and Synthetic Applications. University of St Andrews; 2012.
- NHCs in asymmetric organocatalysis: recent advances in azolium enolate generation and reactivity. Synthesis. 2012;44:2295-2309.
- [CrossRef] [Google Scholar]
- Hua Y, Luu HT, Sakata SK, Brown EL, Maldonado FC, Tuntland T. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 8. Pharmacological optimization of orally bioavailable 2-pyridone-containing peptidomimetics. J. Med. Chem.. 2003;46:4572-4585.
- [CrossRef] [Google Scholar]
- Organocatalyzed highly enantioselective aldol reaction of aldehydes for synthesis of (R)-pantolactone. Asian J. Org. Chem.. 2021;10:1167-1172.
- [CrossRef] [Google Scholar]
- Cis− trans isomerization of organic molecules and biomolecules: implications and applications. Chem. Rev.. 2003;103:2475-2532.
- [CrossRef] [Google Scholar]
- Minisci reactions: versatile CH-functionalizations for medicinal chemists. MedChemComm.. 2011;2:1135-1161.
- [CrossRef] [Google Scholar]
- Rational design of a new antibiotic class for drug-resistant infections. Nature. 2021;597:698-702.
- [CrossRef] [Google Scholar]
- The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer; 2014.
- Invasive fungal infections: a review of epidemiology and management options. J. Med. Microbiol.. 2006;55:809-818.
- [CrossRef] [Google Scholar]
- Epoxy isonitriles, a unique class of antibiotics: synthesis of their metabolites and biological investigations. Chembiochem. 2018;19:2448-2452.
- [CrossRef] [Google Scholar]
- Fluorinated triazole-containing sphingosine analogues. Syntheses and in vitro evaluation as SPHK inhibitors. Org. Biomol. Chem.. 2018;16:7230-7235.
- [CrossRef] [Google Scholar]
- Intramolecular hydrogen bond activation: thiourea-organocatalyzed enantioselective 1, 3-dipolar cycloaddition of salicylaldehyde-derived azomethine ylides with nitroalkenes. ACS Catal.. 2018;8:1884-1890.
- [CrossRef] [Google Scholar]
- A general method for the enantioselective synthesis of pantolactone derivatives. Org. Lett.. 2002;4:3379-3382.
- [CrossRef] [Google Scholar]
- Eosin Y as a direct hydrogen-atom transfer photocatalyst for the functionalization of C− H bonds. Angew. Chem.. 2018;130:8650-8654.
- [CrossRef] [Google Scholar]
- Tertiary amine-catalyzed difluoromethylthiolation of morita–baylis–hillman carbonates of isatins with zard's trifluoromethylthiolation reagent. Adv. Synth. Catal.. 2017;359:49-57.
- [CrossRef] [Google Scholar]
- Recent advances in asymmetric organocatalysis mediated by bifunctional amine–thioureas bearing multiple hydrogen-bonding donors. Chem. Commun.. 2015;51:1185-1197.
- [CrossRef] [Google Scholar]
- New and efficient approach for synthesis of novel bioactive [1, 3, 4] thiadiazoles incorporated with 1, 3-thiazole moiety. Eur. J. Med.. 2015;97:320-333.
- [CrossRef] [Google Scholar]
- Visible light mediated oxidation of benzylic sp 3 C-H bonds using catalytic 1, 4-hydroquinone, or its biorenewable glucoside, arbutin, as a pre-oxidant. Green Chem.. 2018;20:2242-2249.
- [CrossRef] [Google Scholar]
- Bicalutamide (Casodex®) in the treatment of prostate cancer. Expert Rev. Anticancer Ther.. 2004;4:37-48.
- [CrossRef] [Google Scholar]
- New sources of chemical diversity inspired by biosynthesis: rational design of a potent epothilone analogue. Org. Lett.. 2009;11:3186-3189.
- [CrossRef] [Google Scholar]
- Practical remdesivir synthesis through one-pot organocatalyzed asymmetric (S)-P-Phosphoramidation. J. Org. Chem.. 2021;86:4977-4985.
- [CrossRef] [Google Scholar]
- AgF-mediated fluorinative homocoupling of gem-difluoroalkenes. Org. Lett.. 2014;16:102-105.
- [CrossRef] [Google Scholar]
- Scaffold diversity synthesis and its application in probe and drug discovery. Angew. Chem. Int. Ed.. 2016;55:7586-7605.
- [CrossRef] [Google Scholar]
- Asymmetric Lewis acid organocatalysis of the Diels-Alder reaction by a silylated C-H acid. Science. 2016;351:949-952.
- [CrossRef] [Google Scholar]
- Still benched on its way to the bedside: sphingosine kinase 1 as an emerging target in cancer chemotherapy. Crit. Rev. Biochem. Mol. Biol.. 2011;46:342-351.
- [CrossRef] [Google Scholar]
- Recent developments in the use of catalytic asymmetric ammonium enolates in chemical synthesis. Chem. Rev.. 2007;107:5596-5605.
- [CrossRef] [Google Scholar]
- A systematic review of reboxetine for treating patients with attention deficit hyperactivity disorder. Nord. J. Psychiatry.. 2015;69:241-248.
- [CrossRef] [Google Scholar]
- Synthesis, characterization, and evaluation of antibacterial and antioxidant activities of novel benzoxazinones and benzoxathiinones. J. Heterocycl. Chem.. 2019;56:1505-1513.
- [CrossRef] [Google Scholar]
- Application of the chiral base desymmetrisation of imides to the synthesis of the alkaloid jamtine and the antidepressant paroxetine. Tetrahedron. 2003;59:9213-9230.
- [CrossRef] [Google Scholar]
- Applications of fluorine in medicinal chemistry. J. Med. Chem.. 2015;58:8315-8359.
- [CrossRef] [Google Scholar]
- Organocatalysis in the synthesis of 1, 2, 3-triazoyl-zidovudine derivatives: synthesis and preliminary antioxidant activity. ChemistrySelect. 2020;5:12255-12260.
- [CrossRef] [Google Scholar]
- A convenient ultrasound-promoted synthesis of some new thiazole derivatives bearing a coumarin nucleus and their cytotoxic activity. Molecules. 2012;17:9335-9347.
- [CrossRef] [Google Scholar]
- A novel chiral base mediated glutarimide desymmetrisation: application to the asymmetric synthesis of (-)-paroxetine. Synlett. 2002;2002:2074-2076.
- [CrossRef] [Google Scholar]
- Asymmetric organocatalysis on a technical scale: current status and future challenges. Organocatalysis 2008:227-258.
- [CrossRef] [Google Scholar]
- Organocatalytic cascade reactions as a new tool in total synthesis. Nat. Chem.. 2010;2:167-178.
- [CrossRef] [Google Scholar]
- Alzheimers's disease: epidemiological characteristics and its prevention. Zdravstvena Zaštita.. 2021;50
- [CrossRef] [Google Scholar]
- Design, synthesis and in vitro anticancer activity of novel quinoline and oxadiazole derivatives of ursolic acid. Bioorg. Med. Chem. Lett.. 2017;27:4128-4132.
- [CrossRef] [Google Scholar]
- Enantioselective organocatalyzed aza-michael addition reaction of 2-hydroxybenzophenone imines to nitroolefins under batch and flow conditions. Adv. Synth. Catal.. 2021;363:3845-3851.
- [CrossRef] [Google Scholar]
- A new avenue toward androgen receptor pan-antagonists: C2 sterically hindered substitution of hydroxy-propanamides. J. Med. Chem.. 2014;57:7263-7279.
- [CrossRef] [Google Scholar]
- Thalidomide a promising new treatment for rheumatoid arthritis. Arthrit. Rheumat.: Off. J. American College Rheumatol.. 1984;27:1118-1121.
- [CrossRef] [Google Scholar]
- Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat. Rev. Mol. Cell Biol.. 2007;8:101-112.
- [CrossRef] [Google Scholar]
- Novel four-component approach for the synthesis of polyfunctionalized 1, 4-dihydropyridines in aqueous media. Helv. Chim. Acta.. 2011;94:382-388.
- [CrossRef] [Google Scholar]
- Activation of C-H bonds via the merger of photoredox and organocatalysis: a coupling of benzylic ethers with Schiff bases. J. Am. Chem. Soc.. 2014;136:16986-16989.
- [CrossRef] [Google Scholar]
- The selective norepinephrine reuptake inhibitor antidepressant reboxetine: pharmacological and clinical profile. CNS Drug Rev.. 2004;10:23-44.
- [CrossRef] [Google Scholar]
- Catalytic asymmetric [4+ 2]-cycloadditions using tropolones: developments, scope, transformations, and bioactivity. Angew. Chem. Int. Ed.. 2018;57:13216-13220.
- [CrossRef] [Google Scholar]
- Organocatalytic asymmetric synthesis of multifunctionalized α-carboline-spirooxindole hybrids that suppressed proliferation in colorectal cancer cells. Org. Chem. Front.. 2022;9:1048-1055.
- [CrossRef] [Google Scholar]
- Autofluorescent artemisinin-benzimidazole hybrids via organo-click reaction: study of antiviral properties and mode of action in living cells. Chem. Eur. J.. 2023;29:e202301194.
- [Google Scholar]
- Ferrocene functionalized endocrine modulators as anticancer agents. Med. Org. Chem. 2010:81-117.
- [CrossRef] [Google Scholar]
- Organofluorine Compounds: Chemistry and Applications. Springer Science & Business Media; 2013.
- Synthesis and properties of allenic natural products and pharmaceuticals. Angew. Chem. Int. Ed.. 2004;43:1196-1216.
- [CrossRef] [Google Scholar]
- Homogeneous catalysis. Mechanisms of the catalytic Mukaiyama aldol and Sakurai allylation reactions. J. Am. Chem. Soc.. 1995;117:4570-4581.
- [CrossRef] [Google Scholar]
- Homogeneous catalysis. transition metal based lewis acid catalysts. Tetrahedron. 1993;49:5415-5430.
- [CrossRef] [Google Scholar]
- Recent advances in asymmetric organocatalytic construction of 3, 3′-spirocyclic oxindoles. Adv. Synth. Catal.. 2013;355:1023-1052.
- [CrossRef] [Google Scholar]
- Iguratimod (T-614) attenuates severe acute pancreatitis by inhibiting the NLRP3 inflammasome and NF-κB pathway. Biomed. Pharmacother.. 2019;119:109455
- [CrossRef] [Google Scholar]
- 2-Substituted benzoxazinone analogues as anti-human coronavirus (anti-HCoV) and ICAM-1 expression inhibition agents. Bioorg. Med. Chem. Lett.. 2004;14:4751-4754.
- [CrossRef] [Google Scholar]
- The evaluation of 2, 8-disubstituted benzoxazinone derivatives as anti-inflammatory and anti-platelet aggregation agents. Bioorg. Med. Chem. Lett.. 2005;15:2786-2789.
- [CrossRef] [Google Scholar]
- Controllable remote C–H bond functionalization by visible-light photocatalysis. Angew. Chem. Int. Ed.. 2017;56:1960-1962.
- [CrossRef] [Google Scholar]
- Gold-catalyzed highly regio-and enantioselective vinylcarbene insertion into O-H bonds of 2-pyridones. Chem. Commun.. 2017;53:3197-3200.
- [CrossRef] [Google Scholar]
- Selective reductive displacement of alkyl halides and sulfonate esters with cyanoborohydride reagents in hexamethylphosphoramide. J. Org. Chem.. 1977;42:82-91.
- [CrossRef] [Google Scholar]
- An improved synthesis of key intermediate to the formation of selected indolin-2-ones derivatives incorporating ultrasound and deep eutectic solvent (DES) blend of techniques, for some biological activities and molecular docking studies. Molecules. 2020;25:1118.
- [CrossRef] [Google Scholar]
- Iguratimod, a synthetic disease modifying anti-rheumatic drug inhibiting the activation of NF-κB and production of RANKL: its efficacy, radiographic changes, safety and predictors over two years’ treatment for Japanese rheumatoid arthritis patients. Mod. Rheumatol.. 2019;29:418-429.
- [CrossRef] [Google Scholar]
- Studies on effects of forphenicinol on immune responses. J. Antibiot.. 1982;35:1042-1048.
- [CrossRef] [Google Scholar]
- Antitumor effect of forphenicinol, a low molecular weight immunomodifier, on murine transplantable tumors and microbial infections. J. Antibiot.. 1982;35:1049-1054.
- [CrossRef] [Google Scholar]
- Fragment coupling with tertiary radicals generated by visible-light photocatalysis. Acc. Chem. Res.. 2016;49:1578-1586.
- [CrossRef] [Google Scholar]
- Potent selective thienoxazinone inhibitors of herpes proteases. Bioorg. Med. Chem. Lett.. 1997;7:1733-1738.
- [CrossRef] [Google Scholar]
- The diarylprolinol silyl ether system: a general organocatalyst. Acc. Chem. Res.. 2012;45:248-264.
- [CrossRef] [Google Scholar]
- Design, synthesis and primary biological evaluation of the novel 2-pyridone derivatives as potent non-nucleoside HBV inhibitors. Eur. J. Med.. 2017;136:144-153.
- [CrossRef] [Google Scholar]
- Design and application of a bifunctional organocatalyst guided by electron density topological analyses. Catal. Sci. Technol.. 2017;7:4470-4477.
- [CrossRef] [Google Scholar]
- Triazolines. 19. Nickel peroxide oxidation of. DELTA. 2–1, 2, 3-triazolines. A versatile general synthetic route to 1H–1, 2, 3-triazoles. J. Org. Chem.. 1990;55:5891-5894.
- [CrossRef] [Google Scholar]
- Catalytic asymmetric cyclopropanation of enones with dimethyloxosulfonium methylide promoted by a La-Li3−(biphenyldiolate) 3+ NaI complex. J. Am. Chem. Soc.. 2007;129:13410-13411.
- [CrossRef] [Google Scholar]
- Isoxazole alters metabolites and gene expression, decreasing proliferation and promoting a neuroendocrine phenotype in β-cells. ACS Chem. Biol.. 2016;11:1128-1136.
- [CrossRef] [Google Scholar]
- Phthalides and phthalans: synthetic methodologies and their applications in the total synthesis. Chem. Rev.. 2014;114:6213-6284.
- [CrossRef] [Google Scholar]
- Phenyl esters for C-terminal protection in peptide synthesis. J. Am. Chem. Soc.. 1972;94:3259-3260.
- [CrossRef] [Google Scholar]
- Organocatalyzed synthesis and antifungal activity of fully substituted 1, 4-dihydropyridines. ChemistrySelect. 2018;3:6830-6835.
- [CrossRef] [Google Scholar]
- Pharmacological significance of triazole scaffold. J. Enzyme Inhib. Med. Chem.. 2011;26:1-21.
- [CrossRef] [Google Scholar]
- Enantioselective isoquinuclidine synthesis via sequential Diels–Alder/visible-light photoredox C-C bond cleavage: a formal synthesis of the indole alkaloid catharanthine. Org. Chem. Front.. 2018;5:2934-2939.
- [CrossRef] [Google Scholar]
- Discovery of a new HIV-1 inhibitor scaffold and synthesis of potential prodrugs of indazoles. Bioorg. Med. Chem. Lett.. 2013;23:2888-2892.
- [CrossRef] [Google Scholar]
- One-pot four-component approach for the construction of dihydropyridines and dihydropyridinones using amines and activated alkynes. RSC Adv.. 2014;4:3758-3767.
- [CrossRef] [Google Scholar]
- β-Amyloid protein increases the vulnerability of cultured cortical neurons to excitotoxic damage. Brain Res.. 1990;533:315-320.
- [CrossRef] [Google Scholar]
- Fine design of photoredox systems for catalytic fluoromethylation of carbon–carbon multiple bonds. Acc. Chem. Res.. 2016;49:1937-1945.
- [CrossRef] [Google Scholar]
- Development of chiral organosuperbase catalysts consisting of two different organobase functionalities. Angew. Chem. Int. Ed.. 2020;59:7472-7477.
- [CrossRef] [Google Scholar]
- Total synthesis of (S)-forphenicinol via asymmetric organocatalysis. New J. Chem.. 2023;47:20814-20817.
- [CrossRef] [Google Scholar]
- Total synthesis of vineomycin B2. J. Am. Chem. Soc.. 2013;135:15909-15912.
- [CrossRef] [Google Scholar]
- Divergent control of point and axial stereogenicity: catalytic enantioselective C–N bond-forming cross-coupling and catalyst-controlled atroposelective cyclodehydration. Angew. Chem.. 2018;130:6359-6363.
- [CrossRef] [Google Scholar]
- Benzo [c] isothiazole 2-oxides: three-dimensional heterocycles with cross-coupling and functionalization potential. Adv. Synth. Catal.. 2016;358:3649-3653.
- [CrossRef] [Google Scholar]
- Structure activity relationships in lincosamide and streptogramin antibiotics. J. Antimicrob. Chemother.. 1985;16:13-21.
- [CrossRef] [Google Scholar]
- Enantioselective synthesis of nitrocyclopropanes via conjugate addition of bromomalonate to nitroalkenes catalyzed by Ni (II) complexes. Tetrahedron Lett.. 2012;53:3437-3439.
- [CrossRef] [Google Scholar]
- Asymmetric construction of spiropyrazolone skeletons via amine-catalyzed [3+ 3] annulation. Adv. Synth. Catal.. 2018;360:229-234.
- [CrossRef] [Google Scholar]
- Chiral guanidine catalyzed enantioselective reactions. Chem. Asian J.. 2009;4:488-507.
- [CrossRef] [Google Scholar]
- Name Reactions in Heterocyclic Chemistry. John Wiley & Sons; 2004.
- Stereoselective assembly of multifunctional spirocyclohexene pyrazolones that induce autophagy-dependent apoptosis in colorectal cancer cells. J. Org. Chem.. 2019;84:9138-9150.
- [CrossRef] [Google Scholar]
- Diastereo-and enantioselective construction of cyclohexanone-fused spirospyrazolones containing four consecutive stereocenters through asymmetric sequential reactions. Org. Chem. Front.. 2016;3:1087-1090.
- [CrossRef] [Google Scholar]
- Chemical strategies to boost cancer vaccines. Chem. Rev.. 2020;120:11420-11478.
- [CrossRef] [Google Scholar]
- Asymmetric synthesis of multicyclic spirooxindole derivatives bearing stereogenic quaternary carbon atoms. Org. Lett.. 2023;25:2642-2646.
- [CrossRef] [Google Scholar]
- Aminocatalytic asymmetric Diels-Alder reactions via HOMO activation. Acc. Chem. Res.. 2012;45:1491-1500.
- [CrossRef] [Google Scholar]
- The cinchona primary amine-catalyzed asymmetric epoxidation and hydroperoxidation of α, β-unsaturated carbonyl compounds with hydrogen peroxide. J. Am. Chem. Soc.. 2013;135:6677-6693.
- [CrossRef] [Google Scholar]
- Synthesis and structure-activity relationship studies of novel 3, 9-substituted α-carboline derivatives with high cytotoxic activity against colorectal cancer cells. Eur. J. Med.. 2016;110:98-114.
- [CrossRef] [Google Scholar]
- Synthesis and biological evaluation of 4-[3-biphenyl-2-yl-1-hydroxy-1-(3-methyl-3H-imidazol-4-yl)-prop-2-ynyl]-1-yl-benzonitrile as novel farnesyltransferase inhibitor. Bioorg. Med. Chem. Lett.. 2003;13:1293-1296.
- [CrossRef] [Google Scholar]
- Simplification of the Mitsunobu reaction. Di-p-chlorobenzyl azodicarboxylate: a new azodicarboxylate. Org. Lett.. 2006;8:5069-5072.
- [CrossRef] [Google Scholar]
- Introduction: Organocatalysis. ACS Publications; 2007. p. :5413-5415.
- A new indole alkaloid with anti-inflammatory activity from Nauclea officinalis. Nat. Prod. Res.. 2017;31:2107-2112.
- [CrossRef] [Google Scholar]
- Organocatalytic enantioselective synthesis of tetrahydro-furanyl spirooxindoles via [3 + 2] annulations of 3-hydroxyoxindoles and cyclic ketolactams. Adv. Synth. Catal.. 2021;363:2177-2182.
- [CrossRef] [Google Scholar]
- Inhibition of HIV-1 replication by isoxazolidine and isoxazole sulfonamides. Chem. Biol. Drug Des.. 2010;75:461-474.
- [CrossRef] [Google Scholar]
- Trifluoroacetic acid: uses and recent applications in organic synthesis. J. Fluorine Chem.. 2013;156:73-100.
- [CrossRef] [Google Scholar]
- On the structure of aspongopusin recently isolated from Aspongopus chinensis. Fitoterapia. 2013;84:318-320.
- [CrossRef] [Google Scholar]
- Applanatumols A and B, meroterpenoids with unprecedented skeletons from Ganoderma applanatum. RSC Adv.. 2016;6:45963-45967.
- [CrossRef] [Google Scholar]
- A fast assembly of pentacyclic benz [f] indolo [2, 3-a] quinolizidine core by tandem intermolecular formal Aza-[3+ 3] cycloaddition/pictet− spengler cyclization: a formal synthesis of (±)-tangutorine. J. Org. Chem.. 2004;69:4548-4550.
- [CrossRef] [Google Scholar]
- Regioselective functionalization of aryl azoles as powerful tool for the synthesis of pharmaceutically relevant targets. Nat. Commun.. 2020;11:4443.
- [CrossRef] [Google Scholar]
- Phosphine-catalyzed [3+ 2] annulations with γ-methyl allenoates. Chin. J. Chem.. 2019;39:2196.
- [CrossRef] [Google Scholar]
- Dual PPAR-α and-γ activators derived from novel benzoxazinone containing thiazolidinediones having antidiabetic and hypolipidemic potential. Biorg. Med. Chem.. 2006;14:584-591.
- [CrossRef] [Google Scholar]
- Sequential suzuki-miyaura coupling/lewis acid-catalyzed cyclization: an entry to functionalized cycloalkane-fused naphthalenes. Org. Lett.. 2020;22:6267-6271.
- [CrossRef] [Google Scholar]
- Modern Methods in Stereoselective Aldol Reactions. John Wiley & Sons; 2013.
- Ribosome-mediated incorporation of dipeptides and dipeptide analogues into proteins in vitro. J. Am. Chem. Soc.. 2015;137:11206-11209.
- [CrossRef] [Google Scholar]
- Novel imidazole compounds as a new series of potent, orally active inhibitors of 5-lipoxygenase. Biorg. Med. Chem.. 2003;11:3879-3887.
- [CrossRef] [Google Scholar]
- Cinchona alkaloids in asymmetric organocatalysis. Synthesis 2010:1229-1279.
- [CrossRef] [Google Scholar]
- Metal-free cyclization of ortho-nitroaryl ynamides and ynamines towards spiropseudoindoxyls. Angew. Chem.. 2018;130:5762-5766.
- [CrossRef] [Google Scholar]
- An efficient enzymatic preparation of rhinovirus protease inhibitor intermediates. Tetrahedron. 2004;60:759-764.
- [CrossRef] [Google Scholar]
- Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb. Perspect. Med.. 2012;2:a006262
- [CrossRef] [Google Scholar]
- Robust central reduction of amyloid-β in humans with an orally available, non-peptidic β-secretase inhibitor. J. Neurosci.. 2011;31:16507-16516.
- [CrossRef] [Google Scholar]
- The potent BACE1 inhibitor LY2886721 elicits robust central Aβ pharmacodynamic responses in mice, dogs, and humans. J. Neurosci.. 2015;35:1199-1210.
- [CrossRef] [Google Scholar]
- Catalytic asymmetric formal [3+ 2] cycloaddition of azoalkenes with 3-vinylindoles: synthesis of 2, 3-dihydropyrroles. Iscience. 2020;23
- [CrossRef] [Google Scholar]
- Structure–activity relationships of 2-aminothiazoles effective against Mycobacterium tuberculosis. Biorg. Med. Chem.. 2013;21:6385-6397.
- [CrossRef] [Google Scholar]
- Potential antidepressant agents. α-Aryloxy-benzyl derivatives of ethanolamine and morpholine. Eur. J. Med.. 1984;19:235-242.
- [CrossRef] [Google Scholar]
- Synthesis and biological evaluation of potential acetylcholinesterase inhibitors based on a benzoxazine core. Arch. Pharm.. 2018;351:1800024.
- [CrossRef] [Google Scholar]
- Recent advances in lewis base-catalysed chemo-, diastereo-and enantiodivergent reactions of electron-deficient olefins and alkynes. Chem. Rec.. 2022;22:e202100276.
- [Google Scholar]
- Enantioselective Brønsted acid catalysis as a tool for the synthesis of natural products and pharmaceuticals. Chem. Eur. J.. 2018;24:3925-3943.
- [CrossRef] [Google Scholar]
- An expeditious copper-catalyzed access to 3-aminoquinolinones, 3-aminocoumarins and anilines using sodium azide. Adv. Synth. Catal.. 2010;352:1677-1687.
- [CrossRef] [Google Scholar]
- Eosin Y (EY) photoredox-catalyzed sulfonylation of alkenes: scope and mechanism. Chem. Eur. J.. 2016;22:8694-8699.
- [CrossRef] [Google Scholar]
- Glycoconjugate vaccines: current approaches towards faster vaccine design. Expert Rev. Vaccines.. 2019;18:881-895.
- [CrossRef] [Google Scholar]
- Recent developments of free-radical substitutions of heteroaromatic bases. Heterocycles. 1989;28:489-519.
- [CrossRef] [Google Scholar]
- Substitutions by nucleophilic free radicals: a new general reaction of heteroaromatic bases. J. Heterocycl. Chem.. 1990;27:79-96.
- [CrossRef] [Google Scholar]
- The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products. Synthesis. 1981;1981:1-28.
- [CrossRef] [Google Scholar]
- Synthesis and antifungal activity of new azole derivatives containing an oxathiane ring. Bioorg. Med. Chem. Lett.. 1996;6:2377-2380.
- [CrossRef] [Google Scholar]
- Diastereoselective synthesis of vicinally bis (trifluoromethylated) alkylboron compounds through successive insertions of 2, 2, 2-trifluorodiazoethane. Angew. Chem.. 2014;126:14405-14409.
- [CrossRef] [Google Scholar]
- Overview on the recent strategies for the enantioselective synthesis of 1, 1-diarylalkanes, triarylmethanes and related molecules containing the diarylmethine stereocenter. ChemCatChem. 2018;10:1941-1967.
- [CrossRef] [Google Scholar]
- Grossularine-1 and grossularine-2, cytotoxic α-carbolines from the tunicate: dendrodoa grossularia. Tetrahedron. 1989;45:3445-3450.
- [CrossRef] [Google Scholar]
- Biological activity of tropolone. Biol. Pharm. Bull.. 2003;26:1487-1490.
- [CrossRef] [Google Scholar]
- Vitamins, 9. Vitamin B5. Ullmann's encyclopedia of industrial. Chemistry 2000:1-16.
- [CrossRef] [Google Scholar]
- Constructing quaternary stereogenic centers using tertiary organocuprates and tertiary radicals. Total synthesis of trans-clerodane natural products. J. Am. Chem. Soc.. 2015;137:660-663.
- [CrossRef] [Google Scholar]
- A convenient synthesis of iguratimod-amine precursor via NHC-catalyzed aldehyde-nitrile cross coupling reaction. ChemistrySelect. 2020;5:13916-13918.
- [CrossRef] [Google Scholar]
- Discovery and characterization of natural tropolones as inhibitors of the antibacterial target CapF from Staphylococcus aureus. Sci. Rep.. 2015;5:15337.
- [CrossRef] [Google Scholar]
- The synthesis of isocoumarins over the last decade. A review. Org. Prep. Proced. Int.. 1997;29:631-664.
- [CrossRef] [Google Scholar]
- Synthesis and evaluation of the antimicrobial activity of spiro-4h-pyran derivatives on some Gram positive and Gram negative bacteria. Iranian J. Pharm. Res.: IJPR. 2017;16:943.
- [CrossRef] [Google Scholar]
- Human health-related properties of chromones: an overview. Nat. Prod. Res.. 2020;34:137-152.
- [CrossRef] [Google Scholar]
- Access to 2-amino-3-arylthiophenes by base-catalyzed redox condensation reaction between arylacetonitriles, chalcones, and elemental sulfur. Adv. Synth. Catal.. 2020;362:160-165.
- [CrossRef] [Google Scholar]
- Potent BRAF kinase inhibitors based on 2, 4, 5-trisubstituted imidazole with naphthyl and benzothiophene 4-substituents. Biorg. Med. Chem.. 2013;21:1284-1304.
- [CrossRef] [Google Scholar]
- Synthesis pharmacological evaluation of novel Schiff base analogues of 3-(4-amino) phenylimino) 5-fluoroindolin-2-one. J. Young Pharm.. 2010;2:162-168.
- [CrossRef] [Google Scholar]
- Green synthesis of biologically active heterocycles of medicinal importance: a review. Environ. Chem. Lett.. 2021;19:3315-3358.
- [CrossRef] [Google Scholar]
- Bifunctional iminophosphorane organocatalysts for enantioselective synthesis: application to the ketimine nitro-Mannich reaction. J. Am. Chem. Soc.. 2013;135:16348-16351.
- [CrossRef] [Google Scholar]
- The effect of photocatalyst excited state lifetime on the rate of photoredox catalysis. Org. Biomol. Chem.. 2016;14:9088-9092.
- [CrossRef] [Google Scholar]
- Design, synthesis, and biological evaluations of novel 7-[7-amino-7-methyl-5-azaspiro [2.4] heptan-5-yl]-8-methoxyquinolines with potent antibacterial activity against respiratory pathogens. J. Med. Chem.. 2013;56:1974-1983.
- [CrossRef] [Google Scholar]
- Understanding organofluorine chemistry. An introduction to the C-F bond. Chem. Soc. Rev.. 2008;37:308-319.
- [CrossRef] [Google Scholar]
- A new and practical method of decarboxylation: photosensitized decarboxylation of N-acyloxyphthalimides via electron-transfer mechanism. J. Am. Chem. Soc.. 1988;110:8736-8738.
- [CrossRef] [Google Scholar]
- Photosensitized decarboxylative Michael addition through N-(acyloxy) phthalimides via an electron-transfer mechanism. J. Am. Chem. Soc.. 1991;113:9401-9402.
- [CrossRef] [Google Scholar]
- Induction of endogenous virus and of thymidine kinase by bromodeoxyuridine in cell cultures transformed by Friend virus. Proc. Natl. Acad.. 1974;71:4980-4985.
- [CrossRef] [Google Scholar]
- Design, synthesis, and characterization of α-ketoheterocycles that additionally target the cytosolic port Cys269 of fatty acid amide hydrolase. J. Med. Chem.. 2014;57:1079-1089.
- [CrossRef] [Google Scholar]
- Photocatalytic cascade radical cyclization approach to bioactive indoline-alkaloids over donor–acceptor type conjugated microporous polymer. ACS Catal.. 2019;9:5178-5183.
- [CrossRef] [Google Scholar]
- IR780-loaded TPGS-TOS micelles for breast cancer photodynamic therapy. Eur. J. Pharm. Biopharm.. 2017;113:108-117.
- [CrossRef] [Google Scholar]
- One-pot multicomponent reactions for the efficient synthesis of highly functionalized dihydropyridines. Synth. Commun.. 2013;43:986-992.
- [CrossRef] [Google Scholar]
- PEG-mediated one-pot multicomponent reactions for the efficient synthesis of functionalized dihydropyridines and their functional group dependent DNA cleavage activity. Bioorg. Chem.. 2013;48:8-15.
- [CrossRef] [Google Scholar]
- KF/Al2O3 mediated multicomponent reactions for the efficient synthesis of highly substituted dihydropyridines. J. Heterocycl. Chem.. 2014;51:E156-E162.
- [CrossRef] [Google Scholar]
- Direct access to β-trifluoromethyl-β-hydroxy thioesters by biomimetic organocatalytic enantioselective aldol reaction. Org. Lett.. 2019;21:4567-4570.
- [CrossRef] [Google Scholar]
- 1, 2, 3-Triazole-based inhibitors of Porphyromonas gingivalis adherence to oral streptococci and biofilm formation. Biorg. Med. Chem.. 2016;24:5410-5417.
- [CrossRef] [Google Scholar]
- Formal enantioselective synthesis of aplykurodinone-1. Angew. Chem. Int. Ed.. 2013;52
- [CrossRef] [Google Scholar]
- General role of the amino and methylsulfamoyl groups in selective cyclooxygenase (COX)-1 inhibition by 1, 4-diaryl-1, 2, 3-triazoles and validation of a predictive pharmacometric PLS model. Eur. J. Med.. 2015;94:252-264.
- [CrossRef] [Google Scholar]
- Organocatalyzed solvent free and efficient synthesis of 2, 4, 5-trisubstituted imidazoles as potential acetylcholinesterase inhibitors for Alzheimer's disease. Chem. Biodivers.. 2020;17:e1900493.
- [Google Scholar]
- Organocatalyzed synthesis and biological activity evaluation of hybrid compounds 4 H-pyrano [2, 3-b] pyridine derivatives. Synth. Commun.. 2020;50:1845-1853.
- [CrossRef] [Google Scholar]
- Dioxane and oxathiane nuclei: suitable substructures for muscarinic agonists. Biorg. Med. Chem.. 2007;15:886-896.
- [CrossRef] [Google Scholar]
- Constructing quaternary carbons from N-(acyloxy) phthalimide precursors of tertiary radicals using visible-light photocatalysis. J. Org. Chem.. 2015;80:6025-6036.
- [CrossRef] [Google Scholar]
- Inhibition of β-ketoacyl-acyl carrier protein synthases by thiolactomycin and cerulenin: structure and mechanism. J. Biol. Chem.. 2001;276:6551-6559.
- [CrossRef] [Google Scholar]
- Allenes in asymmetric catalysis: asymmetric ring opening of meso-epoxides catalyzed by allene-containing phosphine oxides. J. Am. Chem. Soc.. 2009;131:10364-10365.
- [CrossRef] [Google Scholar]
- Synthesis, antimicrobial and antifungal activity of a new class of spiro pyrrolidines. Biorg. Med. Chem.. 2003;11:407-419.
- [CrossRef] [Google Scholar]
- Rearranged terpenoids from the marine sponge Darwinella cf. oxeata and its predator, the nudibranch Felimida grahami. J. Nat. Prod.. 2017;80:720-725.
- [CrossRef] [Google Scholar]
- Intramolecular azide-alkene cycloaddition-elimination reaction in an aldohex-2-enonic acid derivative. Carbohydr. Res.. 2019;483:107751
- [CrossRef] [Google Scholar]
- Pomalidomide in the treatment of multiple myeloma: design, development and place in therapy. Drug Des. Devel. Ther. 2017:2399-2408.
- [CrossRef] [Google Scholar]
- Allenes in molecular materials. Angew. Chem. Int. Ed.. 2012;51:2818-2828.
- [CrossRef] [Google Scholar]
- Synthesis of chiral 3, 4-disubstituted pyrrolidines with antibacterial properties. Eur. J. Org. Chem.. 2020;2020:2565-2575.
- [CrossRef] [Google Scholar]
- Asymmetric oxidative Lewis base catalysis—unifying iminium and enamine organocatalysis with oxidations. Chem. Commun.. 2012;48:2201-2203.
- [CrossRef] [Google Scholar]
- Isocoumarins: general aspects and recent advances in their synthesis. Adv. Synth. Catal.. 2018;360:2063-2075.
- [CrossRef] [Google Scholar]
- Spirocyclopropyl β-lactams as mechanism-based inhibitors of serine β-lactamases. Synthesis by rhodium-catalyzed cyclopropanation of 6-diazopenicillanate sulfone. J. Med. Chem.. 2003;46:2569-2571.
- [CrossRef] [Google Scholar]
- A practical synthesis of (-)-oseltamivir. Angew. Chem. – Int. Ed. In English. 2007;46:5734.
- [CrossRef] [Google Scholar]
- Synthesis of some new indolo [2, 3-c] isoquinolinyl pyrazoles,-1, 3, 4-oxadiazoles and their biological activities. Med. Chem. Res.. 2013;22:3787-3793.
- [CrossRef] [Google Scholar]
- The addition of aryl azides to unstrained olefins. Tetrahedron. 1968;24:349-356.
- [CrossRef] [Google Scholar]
- Developments in chiral binaphthyl-derived Brønsted/Lewis acids and hydrogen-bond-donor organocatalysis. Eur. J. Org. Chem.. 2011;2011:2209-2222.
- [CrossRef] [Google Scholar]
- Rational design of broad spectrum antibacterial activity based on a clinically relevant enoyl-acyl carrier protein (ACP) reductase inhibitor. J. Biol. Chem.. 2014;289:15987-16005.
- [CrossRef] [Google Scholar]
- A concise synthesis of (−)-aplyviolene facilitated by a strategic tertiary radical conjugate addition. Angew. Chem.. 2012;124:9714-9718.
- [CrossRef] [Google Scholar]
- Discovery of semi-and fully-synthetic carbohydrate vaccines against bacterial infections using a medicinal chemistry approach: focus review. Chem. Rev.. 2021;121:3598-3626.
- [CrossRef] [Google Scholar]
- Organocatalysis: key trends in green synthetic chemistry, challenges, scope towards heterogenization, and importance from research and industrial point of view. J. Catal.. 2014;2014
- [CrossRef] [Google Scholar]
- Organocatalyzed protecting-group-free total synthesis of (S, S)-and (S, R)-reboxetine, an antidepressant drug. J. Org. Chem.. 2022;87:10430-10434.
- [CrossRef] [Google Scholar]
- Photoredox catalysis in organic chemistry. J. Org. Chem.. 2016;81:6898-6926.
- [CrossRef] [Google Scholar]
- Synthesis and biological activities of some fused pyran derivatives. Arab. J. Chem.. 2016;9:S966-S970.
- [CrossRef] [Google Scholar]
- Pavidolides A-E, new cembranoids from the soft coral Sinularia pavida. Tetrahedron Lett.. 2012;53:5759-5762.
- [CrossRef] [Google Scholar]
- Organocatalyzed, visible-light photoredox-mediated, one-pot Minisci reaction using carboxylic acids via N-(acyloxy) phthalimides. J. Org. Chem.. 2018;83:3000-3012.
- [CrossRef] [Google Scholar]
- C-H activation under the guise of Diels-Alder reaction: annulation toward the synthesis of benzo [e] isoindole-1, 3-diones. Org. Lett.. 2017;19:1270-1273.
- [CrossRef] [Google Scholar]
- Targeting SphK1 as a new strategy against cancer. Curr. Drug Targets.. 2008;9:662-673.
- [CrossRef] [Google Scholar]
- Asymmetric organocatalytic reactions by bifunctional amine-thioureas. Catal. Sci. Technol.. 2011;1:1298-1310.
- [CrossRef] [Google Scholar]
- Green asymmetric synthesis of epoxypeptidomimetics and evaluation as human cathepsin K inhibitors. Biorg. Med. Chem.. 2020;28:115597
- [CrossRef] [Google Scholar]
- Unified total synthesis of diverse meroterpenoids from Ganoderma applanatum. Org. Lett.. 2022;24:4552-4556.
- [CrossRef] [Google Scholar]
- Ultrasound and deep eutectic solvent (DES): a novel blend of techniques for rapid and energy efficient synthesis of oxazoles. Ultrason. Sonochem.. 2013;20:287-293.
- [CrossRef] [Google Scholar]
- LiBr catalyzed solvent-free ring expansion of epoxides to 1, 4-oxathian-2-ones with α-mercaptocarboxylic acids. Tetrahedron Lett.. 2011;52:3614-3617.
- [CrossRef] [Google Scholar]
- Discovery of the first inhibitors of bacterial enzyme D-aspartate ligase from Enterococcus faecium (Aslfm) Eur. J. Med.. 2013;67:208-220.
- [CrossRef] [Google Scholar]
- Fluorine substituent effects (on bioactivity) J. Fluorine Chem.. 2001;109:3-11.
- [CrossRef] [Google Scholar]
- Photochemistry of alkenes. 7. E. dblarw. Z isomerization of alkenes sensitized with benzene and derivatives. J. Org. Chem.. 1981;46:3609-3611.
- [CrossRef] [Google Scholar]
- Multidirectional desymmetrization of pluripotent building block en route to diastereoselective synthesis of complex nature-inspired scaffolds. Nat. Commun.. 2018;9:4989.
- [CrossRef] [Google Scholar]
- Synthesis of 1, 2, 3-triazoles bearing a 4-hydroxyisoxazolidine moiety from 4, 5-unsubstituted 2, 3-dihydroisoxazoles. Eur. J. Org. Chem.. 2020;2020:4775-4786.
- [CrossRef] [Google Scholar]
- Polyketide biosynthesis: a millennium review. Nat. Prod. Rep.. 2001;18:380-416.
- [CrossRef] [Google Scholar]
- Enantioselective titanium (III)-catalyzed reductive cyclization of ketonitriles. Angew. Chem. Int. Ed. Engl.. 2012;51:8661-8664.
- [CrossRef] [Google Scholar]
- Sujatha, K., Perumal, P.T., Muralidharan, D., et al., 2009. Synthesis, analgesic and anti-inflammatory activities of bis (indolyl) methanes. doi: 10.1002/chin.200924107.
- Synthesis of polysubstituted dihydropyridines by four-component reactions of aromatic aldehydes, malononitrile, arylamines, and acetylenedicarboxylate. Org. Lett.. 2010;12:3678-3681.
- [CrossRef] [Google Scholar]
- A highly enantioselective Diels-Alder reaction of 1, 2-dihydropyridine using a simple β-amino alcohol organocatalyst for a practical synthetic methodology of oseltamivir intermediate. Tetrahedron Lett.. 2011;52:4745-4748.
- [CrossRef] [Google Scholar]
- Total asymmetric synthesis of (+)-paroxetine and (+)-femoxetine. Eur. J. Org. Chem.. 2019;2019:6973-6982.
- [CrossRef] [Google Scholar]
- Tacconelli, E., 2017. Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development.
- Synthesis and molecular modelling studies of phenyl linked oxadiazole-phenylhydrazone hybrids as potent antileishmanial agents. Eur. J. Med.. 2017;126:1021-1033.
- [CrossRef] [Google Scholar]
- “Inside-out” signaling of sphingosine-1-phosphate: therapeutic targets. Pharm. Rev.. 2008;60:181-195.
- [CrossRef] [Google Scholar]
- Preparation of new chiral pyrrolidinebisphosphines as highly effective ligands for catalytic asymmetric synthesis of R-(−)-pantolactone. Tetrahedron Lett.. 1986;27:4477-4480.
- [CrossRef] [Google Scholar]
- Iguratimod for the treatment of rheumatoid arthritis in Japan. Expert Rev. Clin. Immunol.. 2015;11:565-573.
- [CrossRef] [Google Scholar]
- Highly regioselective indoline synthesis under nickel/photoredox dual catalysis. J. Am. Chem. Soc.. 2015;137:9531-9534.
- [CrossRef] [Google Scholar]
- Asymmetric catalysis by chiral hydrogen-bond donors. Angew. Chem. Int. Ed.. 2006;45:1520-1543.
- [CrossRef] [Google Scholar]
- Modern advances in heterocyclic chemistry in drug discovery. Org. Biomol. Chem.. 2016;14:6611-6637.
- [CrossRef] [Google Scholar]
- α-Angelica lactone catalyzed oxidation of benzylic sp 3 C-H bonds of isochromans and phthalans. Org. Biomol. Chem.. 2020;18:4046-4050.
- [CrossRef] [Google Scholar]
- Synthesis, characterization and comparative study the microbial activity of some heterocyclic compounds containing oxazole and benzothiazole moieties. J. Saudi Chem. Soc.. 2015;19:392-398.
- [CrossRef] [Google Scholar]
- ClbS is a cyclopropane hydrolase that confers colibactin resistance. J. Am. Chem. Soc.. 2017;139:17719-17722.
- [CrossRef] [Google Scholar]
- A new and short enantioselective synthesis of (R)-pantolactone. Tetrahedron Asymmetry. 1999;10:2899-2904.
- [CrossRef] [Google Scholar]
- Chiral tetraaminophosphonium salt-mediated asymmetric direct Henry reaction. J. Am. Chem. Soc.. 2007;129:12392-12393.
- [CrossRef] [Google Scholar]
- Bioorganometallic chemistry of ferrocene. Chem. Rev.. 2004;104:5931-5986.
- [CrossRef] [Google Scholar]
- Function, therapeutic potential and cell biology of BACE proteases: current status and future prospects. J. Neurochem.. 2014;130:4-28.
- [CrossRef] [Google Scholar]
- Synthetic pyrazole derivatives as growth inhibitors of some phytopathogenic fungi. J. Agric. Food. Chem.. 2007;55:10331-10338.
- [CrossRef] [Google Scholar]
- Continuous-flow visible light organophotocatalysis for direct arylation of 2H-Indazoles: fast access to drug molecules. ChemSusChem. 2019;12:2581-2586.
- [CrossRef] [Google Scholar]
- New groups of antimycobacterial agents: 6-chloro-3-phenyl-4-thioxo-2H-1, 3-benzoxazine-2 (3H)-ones and 6-chloro-3-phenyl-2H-1, 3-benzoxazine-2, 4 (3H)-dithiones. Eur. J. Med.. 2000;35:733-741.
- [CrossRef] [Google Scholar]
- Regio-and stereoselective organocatalyzed relay glycosylations to synthesize 2-amino-2-deoxy-1, 3-dithioglycosides. Org. Lett.. 2023;25:3611-3617.
- [CrossRef] [Google Scholar]
- Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res.. 2020;30:269-271.
- [CrossRef] [Google Scholar]
- Synthesis of novel oxazol-5-one derivatives containing chiral trifluoromethyl and isoxazole moieties as potent antitumor agents and the mechanism investigation. Bioorg. Chem.. 2023;135:106505
- [CrossRef] [Google Scholar]
- Construction of chiral tetrahydro-β-carbolines: asymmetric pictet-spengler reaction of indolyl dihydropyridines. Angew. Chem.. 2017;129:7548-7551.
- [CrossRef] [Google Scholar]
- Nonhematotoxic naphthalene diimide modified by polyamine: synthesis and biological evaluation. J. Med. Chem.. 2012;55:3502-3512.
- [CrossRef] [Google Scholar]
- Organocatalytic asymmetric synthesis of bioactive hexahydropyrrolo [2, 3-b] indole-containing tetrasubstituted allenes bearing multiple chiral elements. Tetrahedron Chem.. 2022;1:100007
- [CrossRef] [Google Scholar]
- Nichtproteinogene Aminosäuren, IV. EPC-synthese von l-(+)-forphenicin. Liebigs Ann. Chem.. 1987;1987:833-838.
- [CrossRef] [Google Scholar]
- Synthesis and evaluation of the anti-inflammatory activity of quinoline derivatives. Med. Chem. Res.. 2015;24:2591-2603.
- [CrossRef] [Google Scholar]
- Winneroski, L., Erickson, J., Green, S., et al., 2019. Preparation and biological evaluation of BACE1 inhibitors: leveraging trans-cyclopropyl moieties as ligand efficient conformational constraints, 2020, 28, 115194. DOI: https://doi.org/10.1016/j.bmc.115-119. Doi: 10.1016/j.bmc.2019.115194.
- Organocatalyzed enantioselective michael addition of 2-hydroxypyridines and α, β-unsaturated 1, 4-dicarbonyl compounds. Adv. Synth. Catal.. 2019;361:4966-4982.
- [CrossRef] [Google Scholar]
- Lewis acid-catalyzed ring-opening cross-coupling reaction of gem-difluorinated cyclopropanes enabled by C-F bond activation. Org. Lett.. 2022;24:8429-8434.
- [CrossRef] [Google Scholar]
- Brønsted acid tuned, lewis base promoted [4+ 2] annulation reactions of allenoates with electron-deficient olefins. Eur. J. Org. Chem.. 2018;2018:4917-4925.
- [CrossRef] [Google Scholar]
- Organocatalytic asymmetric synthesis of six-membered carbocycle-based spiro compounds. Adv. Synth. Catal.. 2018;360:194-228.
- [CrossRef] [Google Scholar]
- Organocatalytic enantioselective cascade michael-alkylation reactions: synthesis of chiral cyclopropanes and investigation of unexpected organocatalyzed stereoselective ring opening of cyclopropanes. J. Am. Chem. Soc.. 2007;129:10886-10894.
- [CrossRef] [Google Scholar]
- Access to N-substituted 2-pyridones by catalytic intermolecular dearomatization and 1, 4-acyl transfer. Angew. Chem. Int. Ed.. 2019;58:1980-1984.
- [CrossRef] [Google Scholar]
- Organocatalytic enantioselective conia-ene-type carbocyclization of ynamide cyclohexanones: regiodivergent synthesis of morphans and normorphans. Angew. Chem. Int. Ed.. 2019;58:16252-16259.
- [CrossRef] [Google Scholar]
- Organocatalytic discrimination of non-directing aryl and heteroaryl groups: enantioselective synthesis of bioactive indole-containing triarylmethanes. Chem. Sci.. 2022;13:5767-5773.
- [CrossRef] [Google Scholar]
- Asymmetric synthesis of structurally sophisticated spirocyclic pyrano [2, 3-c] pyrazole derivatives bearing a chiral quaternary carbon center. Org. Lett.. 2022;24:5474-5479.
- [CrossRef] [Google Scholar]
- C-C bond acylation of oxime ethers via NHC catalysis. Org. Lett.. 2021;23:1049-1053.
- [CrossRef] [Google Scholar]
- Copper-mediated radical 1, 2-bis (trifluoromethylation) of alkenes with sodium trifluoromethanesulfinate. Org. Lett.. 2015;17:1906-1909.
- [CrossRef] [Google Scholar]
- Construction of benzene rings by copper-catalyzed cycloaddition reactions of oximes and maleimides: an access to fused phthalimides. Org. Lett.. 2018;20:1216-1219.
- [CrossRef] [Google Scholar]
- Asymmetric photocatalysis enabled by chiral organocatalysts. ChemCatChem. 2022;14:e202101292.
- [Google Scholar]
- Discovery of pyridyl sulfonamide 11-beta-hydroxysteroid dehydrogenase type 1 (11β-HSD1) inhibitors for the treatment of metabolic disorders. Bioorg. Med. Chem. Lett.. 2014;24:5045-5049.
- [CrossRef] [Google Scholar]
- Three-dimensional heterocycles by iron-catalyzed ring-closing sulfoxide imidation. Angew. Chem. Int. Ed.. 2018;57:12053-12056.
- [CrossRef] [Google Scholar]
- Improved procedure for the oxidative cleavage of olefins by OsO4–NaIO4. Org. Lett.. 2004;6:3217-3219.
- [CrossRef] [Google Scholar]
- Advances in organocatalyzed synthesis of organic compounds. RSC Adv.. 2024;14:20365-20389.
- [CrossRef] [Google Scholar]
- Photoredox catalysis with visible light. Angew. Chem. Int. Ed.. 2009;48:9785-9789.
- [CrossRef] [Google Scholar]
- Visible-Light photocatalytic E to Z isomerization of activated olefins and its application for the syntheses of coumarins. Catalysts. 2017;7:337.
- [CrossRef] [Google Scholar]
- Halogen-free fixation of carbon dioxide into cyclic carbonates via bifunctional organocatalysts. Green Chem.. 2021;23:1147-1153.
- [CrossRef] [Google Scholar]
- Recent advances of chromone-based reactants in the catalytic asymmetric domino annulation reaction. Org. Chem. Front.. 2021;8:3968-3989.
- [CrossRef] [Google Scholar]
- Organocatalytic remote stereocontrolled 1, 8-additions of thiazolones to propargylic aza-p-quinone methides. Org. Lett.. 2019;21:7415-7419.
- [CrossRef] [Google Scholar]
- Remote stereocontrolled construction of vicinal axially chiral tetrasubstituted allenes and heteroatom-functionalized quaternary carbon stereocenters. Org. Lett.. 2019;21:503-507.
- [CrossRef] [Google Scholar]
- Asymmetric total synthesis of (−)-pavidolide B via a thiyl-radical-mediated [3+ 2] annulation reaction. J. Org. Chem.. 2019;84:15958-15971.
- [CrossRef] [Google Scholar]
- Practical asymmetric organocatalysis. Green Tech. Org. Synth. Med. Chem. 2018:185-217.
- [CrossRef] [Google Scholar]
- Application of organocatalysis in bioorganometallic chemistry: asymmetric synthesis of multifunctionalized spirocyclic pyrazolone–ferrocene hybrids as novel RalA inhibitors. Org. Chem. Front.. 2018;5:2229-2233.
- [CrossRef] [Google Scholar]
- Daphniyunnines A–E, alkaloids from daphniphyllum yunnanense. J. Nat. Prod.. 2006;69:553-557.
- [CrossRef] [Google Scholar]
- Highly regio-and enantioselective synthesis of N-substituted 2-pyridones: iridium-catalyzed intermolecular asymmetric allylic amination. Angew. Chem.. 2015;127:1893-1896.
- [CrossRef] [Google Scholar]
- Synthesis and evaluation of a dipeptide–drug conjugate library as substrates for PEPT1. ACS Comb. Sci.. 2012;14:108-114.
- [CrossRef] [Google Scholar]
- Plant troponoids: chemistry, biological activity, and biosynthesis. Curr. Med. Chem.. 2007;14:2597-2621.
- [CrossRef] [Google Scholar]
- Photochemical oxidation of alcohols to ketones or aldehydes using DMSO as an oxidant without activated agent. Tetrahedron. 2023;131:133208
- [CrossRef] [Google Scholar]
- Catalytic asymmetric synthesis of vicinally bis (trifluoromethyl)-substituted molecules via normal [3+ 2] cycloaddition of N-2, 2, 2-trifluoroethyl benzothiophene ketimines and β-trifluoromethyl enones. Org. Lett.. 2023;25:8027-8032.
- [CrossRef] [Google Scholar]
- Enantioselective [4+ 1] annulation reactions of α-substituted ammonium ylides to construct spirocyclic oxindoles. J. Am. Chem. Soc.. 2015;137:9390-9399.
- [CrossRef] [Google Scholar]
- Asymmetric synthesis of spiro [chroman-3, 3′-pyrazol] scaffolds with an all-carbon quaternary stereocenter via a oxa-Michael–Michael cascade strategy with bifunctional amine-thiourea organocatalysts. RSC Adv.. 2015;5:91108-91113.
- [CrossRef] [Google Scholar]
- Synthesis and antiviral activity of a novel class of (5-oxazolyl) phenyl amines. Eur. J. Med.. 2013;69:32-43.
- [CrossRef] [Google Scholar]
- Recent advances in asymmetric organocatalysis based on helical polymers. Polym. Chem.. 2022;13:3967-3974.
- [CrossRef] [Google Scholar]
- Multicomponent Reactions. John Wiley & Sons; 2006.
- Decarboxylative arylation of α-amino acids via photoredox catalysis: a one-step conversion of biomass to drug pharmacophore. J. Am. Chem. Soc.. 2014;136:5257-5260.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2024.106027.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary Data 1
Supplementary Data 1




