

Translate this page into:
Pharmacodynamic material basis and mechanism of Tenghuang Jiangu Wan on osteoarthritis using UPLC-Q-TOF-MS integrated with target network pharmacology
⁎Corresponding authors at: Changchun University of Chinese Medicine, No.1035, Boshuo Road, Jingyue National High-tech Industrial Development Zone, Changchun, China. 178521212@qq.com (Hao Yue) yuehao@ccucm.edu.cn (Hao Yue)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Tenghuang Jiangu Wan (THJGW) is a commonly utilized treatment for osteoarthritis (OA), yet its pharmacodynamic material basis and molecular mechanism remain inadequately understood. This study systematically characterized the in vitro and in vivo chemical components of THJGW using ultra-performance liquid chromatography-quadrupole time-of-flight mass spectrometry (UPLC-Q-TOF-MS). The pharmacodynamic material basis of THJGW in the treatment of OA was explored through a target network pharmacology approach and molecular docking technology. Cellular experiments were subsequently employed to validate the molecular mechanisms. Consequently, a total of 134 components from THJGW were identified in vitro, comprising 45 flavonoids, 10 iridoid glycosides, 39 phenylethanoid glycosides, 9 phenylpropanoids, 11 organic acids, 4 phenolics, and 16 other compounds. Additionally, 38 prototype absorbed components in serum were characterized for the purpose of network construction. 11 key components and 10 core targets (VEGFA, STAT3, RELA/NF-κB p65, PIK3R1, PIK3CA, MMP9, MMP1, IL-6, HDAC1, and FGF2) were determined by the target network pharmacology. Through Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, it was found that these intersection targets are associated with various signaling pathways such as pathways in cancer, lipid and atherosclerosis, and prostate cancer. Notably, the lipid and atherosclerosis pathway was closely linked to OA. The molecular docking results demonstrated strong binding affinities of the three core targets: IL-6, STAT3, and RELA/NF-κB p65. These results suggest that these targets may play a significant role for THJGW in treating OA. In vitro experiments showed that THJGW had notable protective effects on LPS-induced RAW264.7 macrophages by reducing levels of TNF-α and IL-6, and inhibiting phosphorylation expression of STAT3 and NF-κB p65 proteins related to the lipid and atherosclerosis pathway. This study provides insight into the pharmacodynamic material basis and mechanism of action for THJGW in the treatment of OA, which provide scientific evidence for the scientific application and improvement of quality standards for THJGW.
Keywords
Tenghuang Jiangu Wan
UPLC-Q-TOF-MS
Osteoarthritis
Pharmacodynamic material basis
Target network pharmacology
Mechanism

1 Introduction
Osteoarthritis (OA) is a prevalent degenerative joint disease in clinical settings, characterized by cartilage degeneration, bone remodeling, osteophyte formation, joint inflammation, and loss of normal joint function. This leads to symptoms like joint pain, swelling, and limited movement, significantly impacting individuals' overall health and quality of life (Kraus et al., 2015). Currently, common drugs used to manage symptoms and restore joint function, such as non-steroidal anti-inflammatory drugs and glucocorticoids, not only harm the digestive tract, liver, and kidney function but also accelerate arthritis progression in the long term (Sharma, 2021). Current medical research reveals that traditional Chinese medicine (TCM) has shown great potential and advantages in the prevention and treatment of OA, owing to its abundant resources, good efficacy, and fewer side effects (Chen et al., 2023). Tenghuang Jiangu Wan (THJGW) is a TCM formula, derived from the extensive clinical experience of renowned Chinese medicine expert Professor Bailing Liu, which has shown good clinical efficacy in the treatment of OA. Its prescription has seven herbal medicines, including Rehmannia glutinosa (Gaertn.) Libosch ex Fisch. et Mey. (Radix Rehmanniae Praeparata), Pyrola calliantha H. Andres (Pyrola), Epimedium brevicornu Maxim. (Epimedium brevicornum), Drynaria fortunei (Kunze) J. Sm. (Rhizoma Drynariae), Cistanche deserticola Y. C. Ma. (Desertliving Cistanche), Spatholobus suberectus Dunn (Spatholobus), and Raphanus sativus L. (Radish Seed). Oral administration of the Tenghuang Jiangu (THJG) formula has been shown in studies to slow OA progression by inhibiting catabolic factor expression and subchondral bone sclerosis (Li et al., 2021). Furthermore, the THJG capsule has been shown to improve articular cartilage tissue injury in rats by decreasing the expression of inflammatory factors, resulting in fewer OA-related pain symptoms (Yan et al., 2020). However, more research is required to determine the pharmacodynamic material basis and mechanism of action for THJGW in treating OA.
Ultra-performance liquid chromatography-quadrupole time-of-flight mass spectrometry (UPLC-Q-TOF-MS) is an analytical tool with high resolution, high sensitivity, and structural characterization capabilities that has been widely used to analyze chemical components analysis in TCM (Kang et al., 2012). Network pharmacology converts the “one-target, one-drug” model into a “multi-components, multi-targets” model and has emerged as a powerful tool for investigating complex diseases and revealing the complex relationships between proteins, diseases, and drugs (Cheng et al., 2018). Traditional network pharmacology usually combines with pharmacokinetic parameters to predict absorption in vivo and mechanisms of drugs. Lin et al. analyzed the pharmacokinetic features of 16 absorbed components in rat plasma after oral administration of Wendan decoction at three different dosages, and subsequently, a total of 10 core components and targets were screened out through network pharmacology (Lin et al., 2023). Liu et al. explored the mechanism of pharmacokinetic target components of San-Ye-Tang-Zhi-Qing formula in the treatment of type 2 diabetes mellitus on the basis of network pharmacology, which indicated five analytes were absorbed and eliminated quickly in vivo, and explained their pharmacological mechanisms (Liu et al., 2020). Another study demonstrated a network pharmacology-integrated pharmacokinetics strategy to explore the pharmacological mechanisms of the major nine ingredients of Xianglian pill in ulcerative colitis treatment (Liu et al., 2021). However, the traditional network pharmacology process is cumbersome and the results may not be accurate. Target network pharmacology uses absorbed components in blood as candidates and integrates network pharmacology methods to investigate the mechanism of action for Chinese medicine, potentially reducing the number of false positive target sites discovered by traditional network pharmacology (Xu et al., 2017). Molecular docking is primarily used to investigate the intermolecular binding manner and interaction relationship, allowing prediction for the docking pattern and binding affinity of drug ligands and protein receptors, which is one of the most important methods to ensuring the reliability of network pharmacology predictions (Wang et al., 2021).
In this study, firstly, the in vitro and in vivo chemical components of THJGW were systematically analyzed and characterized by using UPLC-Q-TOF-MS technology combined with UNIFI software to shed light on its in vitro and in vivo chemical profile. Secondly, using target network pharmacology and molecular docking, potential active ingredients, targets, and mechanisms of action in the treatment of OA with THJGW were identified. Finally, experimental validation was performed on the predicted key targets, revealing the pharmacodynamic material basis and mechanism of action of THJGW in the treatment of OA.
2 Materials and methods
2.1 Chemicals and reagents
THJGW was provided by Changchun People Pharmaceutical Group Co., Ltd. (Changchun, China). Reference standards for icariin, icariside II, naringin, acteoside, echinacoside, D-(-)-quinic acid, and geniposidic acid were obtained from Shanghai Yuanye Bio-Technology Co., Ltd. (Shanghai, China). The purity of the aforementioned standards exceeded 98 % (HPLC grade). Methanol (HPLC grade) was purchased from Tedia (Fairfield, OH, USA). Formic acid (HPLC grade) was purchased from Aladdin (Shanghai, China). Ultrapure water was prepared using a Milli-Q system (Millipore, USA). Sodium formate and leucine enkephalin were obtained from Waters (Milford, USA). Dulbecco’s minimum essential medium (DMEM) was purchased from Gibco (CA, USA). Fetal bovine serum (FBS) was supplied by Clark Biotechnology Institution (Richmond, VA, USA). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and lipopolysaccharides (LPS) were obtained from Solarbio (Beijing, China). Dimethyl sulfoxide (DMSO) was bought from Macklin Biochemical Co., Ltd. (Shanghai, China). Mouse TNF-α and IL-6 ELISA kits were obtained from Huangshi Yanke Biotechnology Co., Ltd. (Hubei, China). The Enhanced BCA Protein Assay Kit, RIPA Lysis Buffer, Phenylmethylsulfonyl Fluoride (PMSF), Primary Antibody Dilution Buffer, Secondary Antibody Dilution Buffer, Blocking Buffer, and Western Wash Buffer were obtained from Beyotime Biotechnology (Shanghai, China). The hypersensitivity enhanced chemiluminescence (ECL) substrate kit was purchased from Beijing Labgic Technology Co., Ltd. (Beijing, China). Rabbit polyclonal antibodies of STAT3 (10253–2-AP), NF-κB p65 (10745–1-AP), β-actin (20536–1-AP), and HRP-conjugated Affinipure Goat Anti-Rabbit IgG (H + L) (SA00001-2) were purchased from Proteintech Group (Wuhan, China). Rabbit anti-Phospho-STAT3 (P-STAT3) (AF3293) and Phospho-NF-κB p65 (P-p65) (AF2006) were obtained from Affinity Biosciences (Jiangsu, China). All other chemicals and organic solvents were of analytical grade or better.
2.2 Extractions
1.5 g of THJGW powder was accurately weighed and placed in a 50 mL conical flask with stopper. Then, 50 mL of methanol was precisely measured and added to dissolve, then the flask was re-weighed to determine its mass. After ultrasound extracted (40 kHz, 100 W) for 1 h, any weight loss was compensated by adding methanol. The resulting mixture was shaken and filtered, then 1 mL of the filtrate was precisely transferred to a 50 mL volumetric flask with the volume set to the mark. Before analysis, the prepared solution was filtered through a 0.22 µm microporous membrane filter.
2.3 Animals handling
15 male Sprague-Dawley rats (180–220 g) were supplied by Liaoning Changsheng Biotechnology Co., Ltd. (Dalian, China). All rats were housed in a controlled environment (12 h light/dark cycle, temperature: 25 ± 1 ℃, relative humidity: 50 ± 5 %) with freely available food and water. After 7 days of adaptive feeding, the rats were randomly divided into five groups of three. One group was designated as the control group. Rats were given THJGW powder (3.0 g/kg/d) via gavage for 7 days, while the control group received the same amount of saline. All animal experiments were approved by the Bioethics Committee of Changchun University of Chinese Medicine and performed in accordance with the guidelines of the Institutional Animal Care (Approval No. 2023452).
2.4 Serum collection and preparation
Prior to the day of last administration, the rats were fasted for 12 h with only access to water. Abdominal aortic blood samples were collected at 0.25, 0.5, 1, and 2 h after the last dose. These samples were then clotted for 30 min at room temperature before being centrifuged at 3000 rpm for 10 min at 4 °C. The resulting serum samples were then divided into smaller aliquots and stored at −80 °C until further analysis.
At each time point, 900 μL serum sample was collected and mixed with a four-fold volume of methanol. The mixture was vortexed for 2 min and then centrifuged at 3000 rpm for 10 min at 4 °C. The supernatant was collected and dried with nitrogen. The residue was then dissolved in 100 μL of methanol. Finally, the solution was centrifuged at 13000 rpm for 10 min at 4 °C before injecting 2 μL of the supernatant for UPLC-Q-TOF-MS analysis.
2.5 UPLC-MS analysis
A Waters Acquity UPLC system equipped with a Q-TOF SYNAPT G2-Si HDMS mass spectrometer (Waters, USA) was utilized to analyze the in vitro and in vivo (absorbed in blood) chemical components of THJGW. A Supelco Ascentis® Express C18 Column (2.7 μm, 3.0 × 50 mm) was used at 40 °C with 0.1 % (v/v) formic acid in water (A) and 0.1 % (v/v) formic acid in methanol (B) as mobile phase. The gradient elution conditions for positive ion mode were as follows: 0–2 min, 10–32 % B; 2–3 min, 32–65 % B; 3–5 min, 65–75 % B; 5–7 min, 75–85 % B; 7–12 min, 85–90 % B; 12–15 min, 90–95 % B; 15–17 min, 95 % B; 17–22 min, 95–10 % B; 22–25 min, 10 % B. And for negative ion mode, the gradient elution conditions were as follows: 0–2 min, 10–25 % B; 2–4 min, 25–32 % B; 4–8 min, 32–43 % B; 8–11 min, 43–65 % B; 11–13 min, 65–85 % B; 13–18 min, 85–90 % B; 18–21 min, 90–95 % B; 21–23 min, 95 % B; 23–28 min, 95–10 % B; 28–31 min, 10 % B. The flow rate was 0.4 mL/min and the injection volume was 2 μL for all the cases above. Specific and accurate masses of all samples in both positive and negative ion modes were determined using mass spectrometry with an electrospray ion source (ESI). The mass range was 50–2000 Da and a scan speed of 1.5 s was used in MSE resolution mode. The temperature of ion source was 120 °C in positive ion mode and 110 °C in negative ion mode. The desolvation temperature was 400 °C. The flow rates for desolvation gas and cone gas were 800 and 50 L/h, respectively. The capillary voltage was set to 3.0 kV in the positive ion mode and 2.5 kV in the negative ion mode. The sampling cone voltage was 40 V. The low collision energy was 6 V, while the high collision energy was set to 20–30 V and 30–45 V for MSE mode. Sodium formate was used in calibrating the mass spectrometer, and leucine enkephalin (m/z 556.2771 in positive ion mode, m/z 554.2615 in negative ion mode) was used as the lock mass.
2.6 Target network pharmacology
2.6.1 Screening for active component targets of THJGW
The prototype components absorbed in blood of THJGW were used as candidates. Their 2D structures were searched and downloaded from the PubChem compound database, and then component targets were predicted following the method of Zhao et al. (Zhao et al., 2023).
2.6.2 Acquisition of OA targets
Using “osteoarthritis” as the keyword, OA-related targets were retrieved following the method of Zhao et al. (Zhao et al., 2023). Then, components-related targets were intersected with OA-related targets and visualized by the OmicShare tools, a free online platform for data analysis (https://www.omicshare.com/tools) to identify potential targets for THJGW in the treatment of OA.
2.6.3 Protein–protein interaction network
Above intersection targets following the method of Zhao et al. were uploaded into the STRING database and Cytoscape 3.7.2 to visualize a protein–protein interaction (PPI) network (Zhao et al., 2023). Furthermore, the CytoHubba plug-in with the maximal clique centrality (MCC) algorithm was utilized to predict 10 probable hub genes of THJGW against OA.
2.6.4 Network construction
A complex “herbs-components-targets-disease” network was constructed in Cytoscape 3.7.2 following the method of Zhao et al. (Zhao et al., 2023). The network was later topologically analyzed by the CytoNCA plug-in to screen out the key components based on values of degree centrality (DC) and between centrality (BC).
2.6.5 GO enrichment analysis and KEGG enrichment analysis
Intersection targets were following the method of Zhao et al. submitted into the Metascape database for GO enrichment and KEGG pathway analysis to explore the core mechanisms and pathways of THJGW anti-OA (Zhao et al., 2023). GO enrichment analysis mainly includes biological processes (BP), cellular components (CC), and molecular functions (MF). Subsequently, the top 10 GO terms and top 20 KEGG pathways were screened, and the results were visualized on a bioinformatics network platform (https://www.bioinformatics.com) for GO analysis bar charts and KEGG pathway enrichment analysis bubble charts.
2.7 Molecular docking
Molecular docking was performed on key components predicted by degree centrality (DC) and between centrality (BC) values, as well as core targets obtained using the MCC algorithm. The 3D structures of compounds were downloaded in SDF format from the PubChem database and converted into MOL2 format using Open Babel software. The 3D crystal structures of targets VEGFA (PDB ID: 1MKK), STAT3 (PDB ID: 6NJS), RELA/NF-κB p65 (PDB ID: 3QXY), PIK3R1 (PDB ID: 1PBW), PIK3CA (PDB ID: 6GVF), MMP9 (PDB ID: 1ITV), MMP1 (PDB ID: 1CGE), IL-6 (PDB ID: 1ALU), HDAC1 (PDB ID: 4BKX), and FGF2 (PDB ID: 1BAS) were retrieved from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB) database (https://www.rcsb.org/). All protein and ligand structures were pre-processed using AutoDockTools 1.5.6 and saved as a PDBQT format file. The docking simulation was performed with AutoDock Vina 1.1.2 to estimate the potential binding modes between active compounds and core targets. The 2D and 3D structures of the ligands and targets with a high binding score were visualized by Discovery Studio software, and the heatmap for binding affinity of molecular docking was plotted on a bioinformatics network platform (http://www.bioinformatics.com).
2.8 Cell culture and viability assay
The RAW264.7 murine macrophage cell line was obtained from iCell Bioscience Inc. (Shanghai, China) and cultured with DMEM containing 10 % FBS at 37 °C in a 5 % CO2 humidified incubator.
Cells (1 × 104 cells/well) were seeded into 96-well plates and incubated for 24 h. Then, cells were treated with increasing concentrations of LPS (0.25, 0.5, 1, 2, 5, 10 µg/mL) and THJGW (6.25, 12.5, 25, 50, 100, 200 µg/mL) alone for 24 h, followed by 1 µg/mL of LPS was used to be co-treated with THJGW for 24 h. Cells untreated with LPS and THJGW were considered the control group. 50 μL of MTT solution (2 mg/mL) was added to each well, followed by cultivation for 3 h. The formaldehyde crystals formed were dissolved in 200 μL of DMSO after removing MTT, and the absorbance of the solutions was measured at 490 nm with a microplate reader.
2.9 ELISA of TNF-α and IL-6
RAW264.7 cells (2 × 105 cells/well) were inoculated on 6-well plates and cultured for 24 h. After pre-treatment by THJGW for 1 h, cells were stimulated with 1 μg/mL LPS for 24 h. Subsequently, the cell supernatant was collected, and the concentrations of TNF-α and IL-6 in the culture supernatant were determined using the appropriate ELISA kits according to the manufacturer’s instructions.
2.10 Western blot
RAW264.7 cells were seeded in 6-well plates at a density of 2 × 105 cells/well and treated with a combination of 1 μg/mL LPS and THJGW at varying concentrations for 24 h. Cells were harvested and fully lysed with the RIPA lysis buffer containing 1 % PMSF at 0 °C, followed by collecting supernatants. The protein concentration of lysates was measured using the BCA protein assay kit. Equal amounts of protein samples were separated using 12.5 % sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked in the blocking buffer for 1.5 h at room temperature before being incubated with the primary antibody at 4 °C overnight in a shaking incubator. The primary antibodies used were diluted as follows: STAT3 (1:2000), NF-κB p65 (1:1000), P-STAT3 (1:1000), P-p65 (1:1000), and β-actin (1:1000). The membranes were washed three times with the wash buffer before incubating with the HRP-conjugated secondary antibody (1:4000) for 50 min at room temperature. After three washes, the blots were visualized using the hypersensitivity ECL substrate kit. Then, the images were captured on a Tanon 5200 chemiluminescence imaging system (Shanghai, China) and analyzed with Image J software.
3 Results
3.1 Identification of main constituents of THJGW
The high-resolution mass spectrometry data of THJGW were rapidly acquired using the UPLC-Q-TOF-MSE method equipped with MassLynx V4.1 software, and the base peak ion (BPI) chromatograms of THJGW in positive and negative ion modes are shown in Fig. 1. The prototype library of UNIFI platform has been enriched with literature, ChemSpider, and SciFinder Scholar to interpret each liquid peak. A total of 134 components including flavonoids, iridoid glycosides, phenylethanoid glycosides, phenylpropanoids, organic acids, phenolics, and other compounds in THJGW were inferred and identified via the UNIFI platform, comparison with standards, references from the literature, and the cleavage laws of mass spectrometry. Detailed mass spectrometry informations on these compounds are shown in Table 1. The chemical structures of the main compounds were summarized in Fig. 2 based on accurate molecular mass, mass spectrometry data, and relevant literature. a: Radix Rehmanniae Praeparata; b: Pyrola; c: Rhizoma Drynariae; d: Desertliving Cistanche; e: Epimedium brevicornum; f: Spatholobus; g: Radish Seed.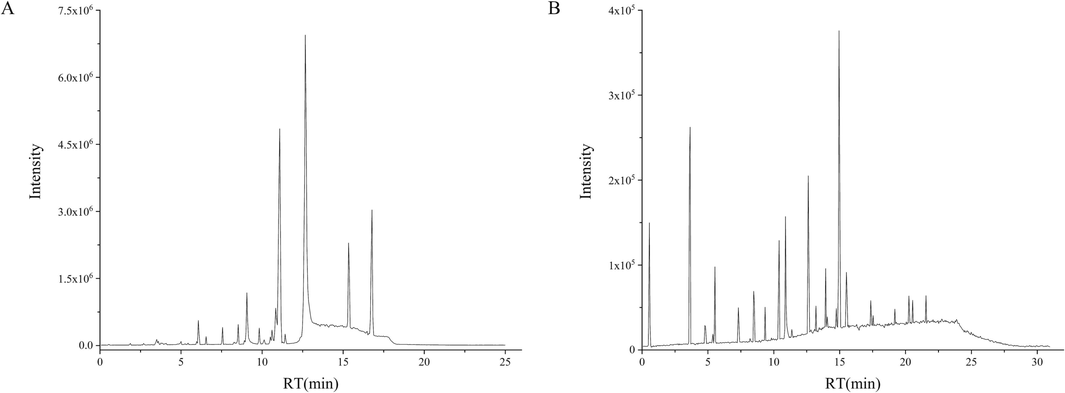
Base peak ion chromatograms of THJGW in positive ion mode (A) and negative ion mode (B).
NO.
Identified
Selected ion
RT
(min)
Formula
Measured mass
(m/z)
Mass error
(ppm)
Major product ions
Type
Attribution
P1
Melibiose
[M−H]-
0.56
C12H22O11
341.1084
−1.5
221.0663,179.0559
Other compounds
a
P2
Glucose
[M−H]-
0.56
C6H12O6
179.0551
−5.7
161.0448,89.0242
Other compounds
a,d
P3
Mannitol
[M−H]-
0.56
C6H14O6
181.0710
−3.9
149.0450,119.0345
Other compounds
d
P4
Quinic acid
[M−H]-
0.56
C7H12O6
191.0558
−1.7
173.0448,128.0434,85.0287,71.0133,59.0141
Organic acids
e
P5
5-GGMF
[M + HCOO]-
0.57
C18H26O13
495.1345
−2.2
161.0451,125.0242
Other compounds
a
P6
Citric acid
[M−H]-
0.58
C6H8O7
191.0194
−1.5
173.0033,87.0087
Organic acids
a,d,e
P7
Glucoraphenin
[M−H]-
0.60
C12H21NO10S3
434.0243
−2.8
383.1189,179.0559
Other compounds
g
P8
Kankanoside B
[M + HCOO]-
0.63
C15H24O10
409.1353
0.3
183.0625,165.0786,101.0240,71.0140
Iridoid glycosides
d
P9
Koaburaside
[M−H]-
1.05
C14H20O9
331.1038
1.0
179.0560
Phenolics
f
P10
Rehmannioside D
[M + HCOO]-
1.06
C27H42O20
731.2251
−0.1
541.1209,179.0560
Iridoid glycosides
a
P11
Acetylcatalpol
[M + HCOO]-
1.07
C18H26O10
447.1513
1.1
179.0560
Iridoid glycosides
a
P12
Geniposidic acid
[M−H]-
1.44
C16H22O10
373.1133
−2.0
211.0568,193.0480,149.0628
Iridoid glycosides
a,d
P13
Androsin
[M + HCOO]-
1.44
C15H20O8
373.1133
−2.0
149.0608
Phenolics
b
P14
Protocatechuic acid
[M−H]-
1.44
C7H6O4
153.0194
0.3
149.0608,101.0318
Organic acids
a,b,c,e,f
P15
Kankanose
[M−H]-
1.64
C27H38O18
649.1983
−0.4
485.1235,179.0347,135.0449
Other compounds
d
P16
6-O-Caffeoylglucopyranose
[M−H]-
1.69
C15H18O9
341.0871
−2.2
179.0347,135.0449
Phenylpropanoids
a
P17
Caffeic acid
[M−H]-
1.71
C9H8O4
179.0347
−1.4
135.0449,113.0242
Organic acids
a,c,e,g
P18
Decaffeoyl acteoside
[M−H]-
1.73
C20H30O12
461.1660
−0.9
179.0342,135.0449
Phenylethanoid glycosides
a,d
P19
3-O-sinapicoylquinic acid
[M + H]+
1.74
C18H22O10
399.1268
−4.4
369.1153,251.0916
Organic acids
g
P20
Cistanoside F or its isomer
[M−H]-
1.83
C21H28O13
487.1452
−1.0
294.1338,179.0356
Phenylethanoid glycosides
a,d
P21
Salidroside
[M + HCOO]-
1.87
C14H20O7
345.1178
−3.7
294.1338,264.0889
Phenylethanoid glycosides
d
P22
6-Deoxycatalpol
[M + HCOO]-
1.91
C15H22O9
391.1251
1.3
264.0889
Iridoid glycosides
d
P23
8-Epiloganic acid
[M−H]-
1.94
C16H24O10
375.1289
−2.0
294.1338,264.0889
Iridoid glycosides
a,d
P24
Mussaenosidic acid
[M−H]-
1.94
C16H24O10
375.1289
−2.0
294.1338,264.0889
Iridoid glycosides
d
P25
6-O-Feruloylajugol
[M + HCOO]-
1.97
C25H32O12
569.1868
−1.4
407.1330,294.1338
Phenylpropanoids
a
P26
Kankanoside E
[M + H]+
[M−H]-
2.32
6.55C16H28O8
349.1837
347.1707−5.7
−1.2179.0340
329.1520,167.0670Other compounds
d
P27
Sibirioside B
[M + HCOO]-
2.40
C22H30O13
547.1673
0.8
179.0346
Phenylpropanoids
a
P28
Cistanoside F or its isomer
[M−H]-
2.51
C21H28O13
487.1458
0.3
251.0576,179.0346
Phenylethanoid glycosides
a,d
P29
5,7-Dihydroxychromone-7-O-rutinoside
[M−H]-
2.67
C21H26O13
485.1294
−1.3
177.0191
Phenylpropanoids
c
P30
Gluroside
[M + HCOO]-
2.68
C15H24O8
377.1451
−0.6
177.0191
Iridoid glycosides
d
P31
Cistantubuloside C1/C2
[M−H]-
2.75
C35H46O21
801.2470
1.4
623.2148,179.0347
Phenylethanoid glycosides
d
P32
Forsythoside B
[M + HCOO]-
2.75
C34H44O19
801.2470
1.4
179.0347
Phenylethanoid glycosides
a
P33
Darendoside B
[M−H]-
2.81
C21H32O12
475.1824
0.6
461.1674,375.1435
Phenylethanoid glycosides
a
P34
7,8,3′,4′-Tetrahydroxyflavanone
[M + H]+
3.03
C15H12O6
289.0709
0.9
204.9809,163.0389
Flavonoids
f
P35
Neoeriocitrin
[M + H]+
[M−H]-
3.04
4.36C27H32O15
597.1817
595.16650.5
−0.7525.2881,289.0723,163.0389
459.1137,235.0245,151.0030Flavonoids
c
P36
Naringenin-7-O-glucoside
[M + H]+
3.18
C21H22O10
435.1285
−0.3
273.0763,153.0181
Flavonoids
c
P37
Liquiritin
[M + H]+
3.19
C21H22O9
419.1346
2.3
332.0912,228.1596
Flavonoids
c,e
P38
Naringenin
[M + H]+
3.19
C15H12O5
273.0759
0.6
153.0181,147.0442,119.0492
Flavonoids
f
P39
Naringin
[M + H]+
[M−H]-
3.19
5.36C27H32O14
581.1882
579.17123.0
−1.2435.3332,273.0763,153.0181
271.0607,151.0033Flavonoids
c
P40
Campneoside Ⅱ or its isomer
[M−H]-
3.27
C29H36O16
639.1932
0.3
621.1796,593.1485,161.0240
Phenylethanoid glycosides
a,d
P41
Diphylloside A
[M + H]+
3.45
C38H48O20
825.2789
−2.8
663.2299,517.1704,355.1180,299.0554
Flavonoids
e
P42
Sagittasine C
[M + H]+
3.52
C33H40O16
693.2354
−5.0
547.1799,385.1280,329.0650
Flavonoids
e
P43
Echinacoside
[M−H]-
3.62
C35H46O20
785.2506
−0.4
623.2188,461.1660,179.0347,161.0240
Phenylethanoid glycosides
a,d
P44
Purpureaside C
[M−H]-
3.62
C35H46O20
785.2506
−0.4
623.2188,179.0347,161.0240,133.0291
Phenylethanoid glycosides
a
P45
Kankanoside F
[M−H]-
3.62
C26H40O17
623.2199
1.1
477.1605,461.1660,179.0347,161.0240
Phenylethanoid glycosides
d
P46
Acuminatoside
[M + H]+
3.69
C45H60O24
985.3565
1.8
831.2673,673.2218,285.0762
Flavonoids
e
P47
Hexandraside F
[M + H]+
[M + HCOO]-
3.72
10.15C39H50O20
839.2960
883.2865−1.0
−1.4677.2450,531.1863,369.1334,313.0706
675.2296,366.1104,351.0870,323.0926Flavonoids
e
P48
Epimedin B
[M + H]+
[M + HCOO]-
3.74
10.20C38H48O19
809.2860
853.2767−0.3
−0.6677.2450,531.1863,369.1334,313.0708
645.2182,366.1104,351.0870,323.0926Flavonoids
e
P49
2-phenylethyl-β-primeveroside
[M−H]-
3.76
C19H28O10
415.1589
−4.9
191.0560
Phenylethanoid glycosides
a
P50
Renifolin
[M + H]+
3.77
C23H34O8
439.2318
−1.9
369.1334,85.0285
Phenolics
b
P51
Epimedin C
[M + H]+
[M + HCOO]-
3.78
10.42C39H50O19
823.3024
867.29230.6
−0.6677.2450,531.1863,369.1334,313.0708
659.2344,513.1762,366.1105,351.0865,323.0925Flavonoids
e
P52
Demethyl syringin
[M−H]-
3.79
C16H22O9
357.1187
−1.1
177.0550
Phenylpropanoids
d
P53
Icariside I
[M + H]+
[M−H]-
3.79
12.13C27H30O11
531.1861
529.17060.0
−1.8369.1334,313.0708
367.1177,352.0929Flavonoids
e
P54
Korepimedoside A
[M + H]+
3.80
C37H44O17
761.2619
−4.3
531.1863,369.1334,313.0708
Flavonoids
e
P55
Sagittatoside A
[M + H]+
[M−H]-
3.80
12.44C33H40O15
677.2441
675.22910.0
−0.5531.1862,369.1334,313.0706
367.1176,352.0938Flavonoids
e
P56
Jionoside A1/A2
[M−H]-
4.03
C36H48O20
799.2643
−2.9
623.2190,135.0449
Phenylethanoid glycosides
a
P57
Cistanoside A
[M−H]-
4.03
C36H48O20
799.2643
−2.9
623.2190,399.1288,219.0656
Phenylethanoid glycosides
d
P58
Campneoside Ⅱ or its isomer
[M−H]-
4.08
C29H36O16
639.1924
−1.1
621.1798,399.1288,219.0656
Phenylethanoid glycosides
a,d
P59
Rehmapicrogenin
[M−H]-
4.19
C10H16O3
183.1026
−0.1
139.1136
Other compounds
a
P60
Kankanoside K1/K2
[M−H]-
4.29
C36H48O21
815.2621
0.7
784.2413,769.2551
Phenylethanoid glycosides
d
P61
8-Prenylkaempferol
[M + H]+
[M−H]-
4.32
12.57C20H18O6
355.1178
353.10250.5
−1.7299.0553,208.0328,191.0011
323.0921,295.0614Flavonoids
e
P62
Ligupurpuroside A
[M−H]-
4.33
C35H46O19
769.2555
−0.7
623.2181,595.1662,459.1136
Phenylethanoid glycosides
a
P63
Poliumoside
[M−H]-
4.33
C35H46O19
769.2555
−0.7
623.2181,595.1662,459.1136
Phenylethanoid glycosides
d
P64
Baohuoside Ⅱ
[M + H]+
[M−H]-
4.33
11.65C26H28O10
501.1760
499.15990.9
−2.2355.1174,299.0553,191.0011
352.0946,297.0392Flavonoids
e
P65
Dunnisinoside
[M + HCOO]-
4.39
C26H30O13
595.1659
−1.6
235.0247,151.0028
Iridoid glycosides
a
P66
Jionoside A1/A2
[M−H]-
4.42
C36H48O20
799.2648
−2.3
623.2184,595.1665,135.0449
Phenylethanoid glycosides
a
P67
Arenarioside
[M−H]-
4.63
C34H44O19
755.2378
−3.5
579.1307
Phenylethanoid glycosides
d
P68
Pinoresinol-β-D-glucoside
[M−H]-
4.75
C26H32O11
519.1866
−1.2
461.1649,357.1335,161.0241
Phenylpropanoids
d
P69
Acteoside
[M−H]-
4.81
C29H36O15
623.1976
−0.9
461.1649,315.1048,179.0355,161.0241
Phenylethanoid glycosides
a,d
P70
Tubuloside A
[M−H]-
4.84
C37H48O21
827.2607
−1.0
665.2281,623.1976,461.1649,161.0241
Phenylethanoid glycosides
d
P71
Hyperoside
[M−H]-
4.89
C21H20O12
463.0876
−1.4
300.0255,271.0232
Flavonoids
b,e
P72
Isoquercitrin
[M−H]-
4.89
C21H20O12
463.0876
−1.4
300.0255,285.0392,271.0232
Flavonoids
b,e,f
P73
Sagittatoside B
[M + H]+
5.06
C32H38O14
647.2339
0.7
563.3768,248.1323
Flavonoids
e
P74
2-O-Galloylhyperin
[M−H]-
5.06
C28H24O16
615.0978
−2.2
609.1450
Flavonoids
b
P75
2-O-Rhamnosylicariside Ⅱ or its isomer
[M + H]+
[M−H]-
5.19
10.42C33H40O14
661.2493
659.23390.3
−0.9369.1333,313.0709
513.1759,367.1175,351.0866,323.0920,296.0332Flavonoids
e
P76
Syringaresinol-4-O-β-D-glucopyranoside
[M−H]-
5.20
C28H36O13
579.2085
0.3
417.1546
Phenylpropanoids
d
P77
Icaritin
[M + H]+
[M−H]-
5.22
13.65C21H20O6
369.1335
367.11810.7
−1.5313.0709,135.0441
352.0958,309.0399,297.0407Flavonoids
e
P78
Jionoside B1/B2
[M−H]-
5.29
C37H50O20
813.2818
−0.6
579.1715,459.1140
Phenylethanoid glycosides
a
P79
Rehmapicroside
[M−H]-
5.45
C16H26O8
345.1543
−3.4
161.0238,135.0446
Other compounds
a
P80
Kankanoside A
[M−H]-
5.45
C16H26O8
345.1543
−3.4
161.0242
Iridoid glycosides
d
P81
Isomer of acteoside
[M−H]-
5.54
C29H36O15
623.1973
−1.3
461.1660,315.1009,179,0326,161.0242
Phenylethanoid glycosides
a,d
P82
Forsythiaside A
[M−H]-
5.54
C29H36O15
623.1973
−1.3
461.1660,315.1078,161.0242
Phenylethanoid glycosides
a,d
P83
3, 6-Disinapoyl sucrose
[M−H]-
5.59
C34H42O19
753.2248
0.0
223.0604,205.0503,190.0266
Other compounds
g
P84
Campneoside Ⅰ or its isomer
[M−H]-
5.60
C30H38O16
653.2104
2.6
623.1973,621.1834,461.1660,161.0242
Phenylethanoid glycosides
a,d
P85
Afrormosin
[M + H]+
5.63
C17H14O5
299.0912
−0.5
193.0494
Flavonoids
f
P86
Kankanoside G
[M−H]-
5.66
C29H36O14
607.2024
−1.4
461.1660
Phenylethanoid glycosides
d
P87
Kankanoside H1/H2
[M−H]-
5.70
C37H48O20
811.2663
−0.4
665.2288
Phenylethanoid glycosides
d
P88
Cistanoside C
[M−H]-
5.97
C30H38O15
637.2122
−2.5
564.4120,511.3497
Phenylethanoid glycosides
d
P89
Campneoside Ⅰ or its isomer
[M−H]-
6.02
C30H38O16
653.2080
−1.1
637.2144,621.1840,461.1664
Phenylethanoid glycosides
a,d
P90
Kaempferol-3,7-O-rhamnoside
[M−H]-
6.23
C27H30O14
577.1557
−1.0
431.0953
Flavonoids
e
P91
Kankanoside I
[M−H]-
6.41
C35H46O18
753.2606
−0.7
607.2021
Phenylethanoid glycosides
d
P92
Leucosceptoside A
[M−H]-
6.95
C30H38O15
637.2127
−1.8
564.4120,315.1096
Phenylethanoid glycosides
a
P93
Syringaresinol
[M−H]-
6.99
C22H26O8
417.1545
−2.3
387.1112,181.0510
Phenylpropanoids
d,f
P94
Tubuloside B
[M−H]-
7.23
C31H38O16
665.2084
−0.4
461.1655,161.0240
Phenylethanoid glycosides
d
P95
2′-Acetylacteoside
[M−H]-
7.26
C31H38O16
665.2081
−0.9
503.1782,461.1654,161.0241
Phenylethanoid glycosides
a,d
P96
Jiocarotenoside A1/A2
[M−H]-
7.32
C21H34O9
429.2121
−2.1
174.9559,161.0241,112.9860
Other compounds
a
P97
Martynoside or its isomer
[M−H]-
7.73
C31H40O15
651.2289
−0.8
591.2077,175.0398
Phenylethanoid glycosides
a
P98
Cistanoside D or its isomer
[M−H]-
7.73
C31H40O15
651.2289
−0.8
591.2077
Phenylethanoid glycosides
d
P99
Ikarisoside B or its isomer
[M−H]-
8.15
C32H38O15
661.2134
−0.6
352.0946,297.0394
Flavonoids
e
P100
Rouhuoside
[M−H]-
8.20
C38H48O20
823.2663
−0.4
661.2130,631.2026
Flavonoids
e
P101
Epimedoside E
[M−H]-
8.25
C37H46O19
793.2561
0.0
631.2026
Flavonoids
e
P102
Epimedoside A
[M−H]-
8.43
C32H38O15
661.2132
−1.0
514.1472,499.1582,353.1021
Flavonoids
e
P103
Diphylloside B
[M−H]-
8.63
C38H48O19
807.2722
0.6
645.2186,514.1480
Flavonoids
e
P104
Cistanoside D or its isomer
[M−H]-
8.64
C31H40O15
651.2304
1.5
529.1719,514.1480
Phenylethanoid glycosides
d
P105
Martynoside or its isomer
[M−H]-
8.67
C31H40O15
651.2281
−2.1
529.1719,514.1480
Phenylethanoid glycosides
a
P106
Elycohol-7-O-glucoside
[M + H]+
8.89
C21H22O11
451.1250
3.3
207.0323
Flavonoids
c
P107
Cistansinenside A
[M−H]-
9.67
C32H40O16
679.2236
−1.1
673.2147,353.1019
Phenylethanoid glycosides
d
P108
Cuhuoside
[M + HCOO]-
9.81
C33H40O16
737.2319
2.8
529.1731
Flavonoids
e
P109
Icariin
[M + HCOO]-
10.37
C33H40O15
721.2343
−0.8
513.1759,367.1175,351.0866,323.0920,296.0332
Flavonoids
e
P110
Curcumin
[M−H]-
10.37
C21H20O6
367.1180
−2.0
338.1142,217.0499
Other compounds
c
P111
Isomer of icariside Ⅱ
[M−H]-
10.38
C27H30O10
513.1763
−0.7
367.1176,352.0934,324.1005
Flavonoids
e
P112
Epicatechin gallate
[M−H]-
[M + H]+
10.59
16.48C22H18O10
441.0817
443.0976−2.2
0.8121.0296
237.0794,207.0325Flavonoids
b
P113
Paulownin
[M−H]-
10.98
C20H18O7
369.0967
−3.4
341.1027
Phenylpropanoids
a
P114
Anhydroicaritin-3-O- rhamnopyranosyl(4 → 1)-furan acid-7-O-glucopyranoside
[M−H]-
11.02
C39H48O19
819.2712
−0.6
657.2176,367.1180
Flavonoids
e
P115
3,4-Dihydroxyacetophenone
[M + H]+
11.09
C8H8O3
153.0547
0.3
135.0438,121.0284,93.0334
Phenolics
a
P116
Dibutyl phthalate
[M + H]+
11.09
C16H22O4
279.1591
0.1
149.0233
Other compounds
c
P117
Ikarisoside B or its isomer
[M−H]-
11.38
C32H38O15
661.2139
0.1
481.1487,352.0946
Flavonoids
e
P118
Rhein
[M + HCOO]-
11.40
C15H8O6
329.0289
−4.1
267.0486,211.1332
Other compounds
f
P119
Ikarisoside F
[M−H]-
11.52
C31H36O14
631.2037
0.7
571.1821,437.1605
Flavonoids
e
P120
2-O-Rhamnosylicariside Ⅱ or its isomer
[M−H]-
12.62
C33H40O14
659.2335
−1.6
513.1760,367.1165,351.0869,323.0920,296.0324
Flavonoids
e
P121
Breviflavone A
[M−H]-
12.66
C25H26O7
437.1595
−2.3
281.0423
Flavonoids
e
P122
Icariside Ⅱ
[M−H]-
12.68
C27H30O10
513.1760
−1.2
367.1164,352.0912,324.0966
Flavonoids
e
P123
Asiatic acid
[M−H]-
12.91
C30H48O5
487.3420
−1.8
471.3105,211.1334,167.1441
Organic acids
b
P124
Anhydroicaritin-3-O- rhamnopyranosyl-furan acid
[M−H]-
13.03
C33H38O14
657.2163
−4.0
367.1184,352.0943
Flavonoids
e
P125
Yinyanghuo B
[M−H]-
13.70
C25H26O6
421.1661
1.0
366.1085,311.1668,227.1074,211.0756
Flavonoids
e
P126
Spasuberoside A
[M + HCOO]-
14.74
C22H25O9
478.1508
5.8
433.1522
Flavonoids
f
P127
Linolenic acid
[M−H]-
15.08
C18H30O2
277.2171
−0.7
271.2276,165.1188
Organic acids
g
P128
Palmitic acid
[M−H]-
16.48
C16H32O2
255.2328
−0.8
223.0281
Organic acids
a,b,d,f,g
P129
Oleic acid
[M−H]-
16.86
C18H34O2
281.2478
−2.7
183.0115
Organic acids
b,g
P130
Juglanin
[M + H]+
17.03
C20H18O10
419.0956
−4.0
355.0701,221.0843
Flavonoids
b
P131
Stigmasterol
[M + H]+
17.51
C29H48O
413.3779
0.3
360.3229,109.0645
Other compounds
c
P132
Stearic acid
[M−H]-
18.43
C18H36O2
283.2639
−1.2
231.9680
Organic acids
a,g
P133
Eleutheroside A
[M + HCOO]-
21.08
C35H60O6
621.4372
0.0
369.3371,323.3314
Other compounds
b,c,d,f
P134
Behenic acid
[M−H]-
22.17
C22H44O2
339.3267
−0.4
322.2127
Organic acids
a
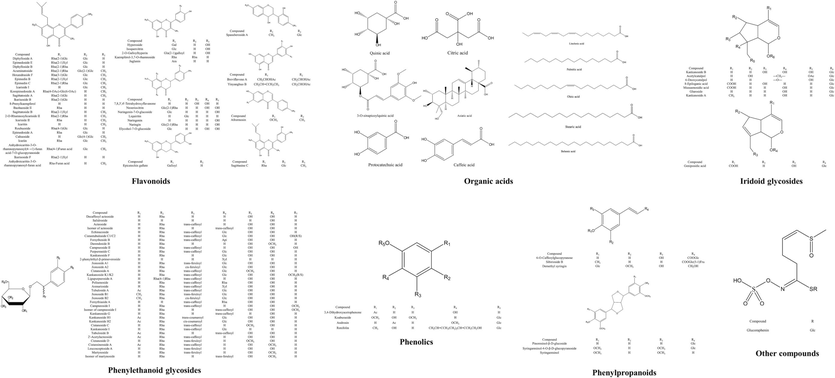
Chemical structure of the main constitutions found in THJGW.
3.1.1 Flavonoids
In this study, a total of 45 flavonoids including flavonoid O-glycosides and flavonoid aglycones were identified in both positive and negative ion modes in THJGW. The main sources of flavonoids were Epimedium brevicornum, followed by Spatholobus, Rhizoma drynariae, and Pyrola.
37 flavonoid O-glycosides were characterized in the mass spectrum of THJGW. They were divided into two categories: 8-prenyl flavonoid O-glycosides and flavonoid O-glycosides without isopentenyl groups, according to the presence of isopentenyl at the C-8 position in their structures. Glycosidic bonds connected by the oxygen atom in flavonoid O-glycosides were cleaved in both positive and negative ion modes, which were characterized mostly by the neutral losses of 162 Da (Glc), 146 Da (Rha), and 132 Da (Xyl).
3.1.1.1 8-Prenyl flavonoid O-glycosides
8-Prenyl flavonoid O-glycosides derived from Epimedium brevicornum show similar fragmentation patterns in mass spectrometry. First, in positive ion mode, the cleavage pathway typically involves losing the C-3 position of an O-substituted diglycosyl or one of them. Second, the loss of the C-7 position of an O-substituted or the C-3 position of the remaining glycosyl to generate anhydroicaritin. Finally, the prenyl group at the C-8 position was lost. In negative ion mode, firstly, cleavage starts with the loses the C-7 position of the O-substituted glycosyl group, followed by the C-3 position of the O-substituted diglycosyl, and finally the prenyl group at the C-8 position was lost. The following describes the identification process for several representative compounds.
Compounds 47, 48, 51, and 55 separately showed molecular ion peaks at m/z 839.2960, 809.2860, 823.3024, and 677.2441 for [M + H]+ in positive ion mode. The loss of a molecule glycosyl group was observed to generate fragment ions at m/z 677 in compounds 47, 48, and 51, which were corresponding to [M + H-Glc]+, [M + H-Xyl]+, and [M + H-Rha]+, respectively. Compounds 47, 48, 51, and 55 showed similar characteristic fragment ions at m/z 531, 369, and 313. Ions at m/z 531 were separately corresponding to [M + H-Rha-Glc]+, [M + H-Rha-Xyl]+, [M + H-2Rha]+, and [M + H-Rha]+. Then they separately lost a glucosyl group to generate anhydroicaritin at m/z 369. Ions at m/z 313 were the losses of the C4H8 group, corresponding to [M + H-Rha-2Glc-isobutenyl]+, [M + H-Rha-Xyl-Glc-isobutenyl]+, [M + H-2Rha-Glc-isobutenyl]+, and [M + H-Rha-Glc-isobutenyl]+, respectively. Likewise, fragment ions at m/z 367, 366, 352, 351, and 323 with similar cleavage pathways to positive mode for compounds 47, 48, 51, and 55 were observed in negative ion mode, also including losses of neutral molecules such as CH3 and CO2. According to the UNIFI data matching and the cleavage pathways, compounds 47, 48, 51, and 55 were identified as hexandraside F, epimedin B, epimedin C, and sagittatoside A, respectively. Furthermore, compounds 41, 42, and 64 exhibited similar fragmentation pathways with them in the mass spectrum, and in combination with accurate masses, they were identified as diphylloside A, sagittasine C, and baohuoside II respectively.
Compound 109 showed [M + HCOO]- at m/z 721.2343, and the main fragment ions at m/z 513.1759, 367.1175, 351.0866, 323.0920, and 296.0332, which were derived from losses of the glycosyl group and neutral molecules. Fragment ions at m/z 513.1759 and 367.1175 were separately corresponding to [M−H−Glc]- and [M−H−Glc−Rha]-, and ions at m/z 351.0866, 323.0920, and 296.0332 were corresponding to [M−H−Glc−Rha−CH3−H]-, [M−H−Glc−Rha−CH3−H−CO]-, and [M−H−Glc−Rha−CH3−H−C4H7]-, respectively. Based on the literature reported (Chang et al., 2021), comparing the retention time and fragmentation pattern with the reference compound, compound 109 was identified as icariin, and its possible cleavage pathway in mass spectrometry is shown in Fig. 3.
MS/MS spectrum of icariin standard (A) and icariin (B), as well as its possible cleavage pathway (C).
Compounds 111 and 122 gave the [M−H]- ion at m/z 513, and the fragment ions at m/z 367, 352, and 324. The fragment ion at m/z 367 was the loss of a rhamnosyl group, corresponding to [M−H−Rha]-. The ions at m/z 352 and 324 were generated by losing a molecule of methyl and carbon monoxide in a similar pathway, which were corresponding to [M−H−Rha−CH3]- and [M−H−Rha−CH3−CO]-, respectively. By comparing the retention time and fragmentation pattern with the reference compound, compounds 111 and 122 were separately identified as isomer of icariside II and icariside II, and the possible cleavage pathway of icariside II in mass spectrometry is shown in Fig. S1.
3.1.1.2 Flavonoid O-glycosides without isopentenyl groups
The primary cleavage pattern in mass spectrometry of flavonoid O-glycosides without isopentenyl groups is the loss of glycosyl groups to generate a series of deglycosylated fragment ions, followed by a secondary bombardment of their aglycones.
Compound 36 gave a [M + H]+ ion at m/z 435.1285, and the fragment ions at m/z 273.0763 and 153.0181. The fragment ion at m/z 273.0763 was corresponding to the loss of a molecule glucosyl as [M + H-Glc]+. The ion at m/z 153.0181 was generated by the Retro-Diels-Alder (RDA) cleavage reaction of its aglycone. By comparison of the accurate mass and the cleavage pathways, compound 36 was identified as naringenin-7-O-glucoside.
Compound 39 showed [M−H]- at m/z 579.1712, and the fragment ions at m/z 271.0607 and 151.0033, which were separately corresponding to [M−H−Rha−Glc]- and [M−H−Rha−Glc−C8H8O]-. The fragment ion at m/z 151.0033 was generated by the RDA cleavage reaction. Based on the accurate mass and compared with the standard, compound 39 was identified as naringin. In the same way, compound 71, with a similar cleavage pathway, was identified as hyperoside.
3.1.2 Iridoid glycosides
A total of 10 iridoid glycosides were inferred and identified from THJGW in the negative ion mode of mass spectrometry. Radix Rehmanniae Praeparata and Desertliving Cistanche were the major sources of iridoid glycosides. These compounds shared a similar cleavage pathway in mass spectrometry, primarily cleavage of the glycosidic bond and the aglycone ring in negative ion mode, which was also associated with the loss of neutral molecules. Compound 12 was selected as an example to analyze and discuss the cleavage pathway of iridoid glycosides in mass spectrometry. Compound 12 showed [M−H]- at m/z 373.1133 and the fragment ions at m/z 211.0568, 193.0480, and 149.0628. The ion at m/z 211.0568 was generated by losing the glucosyl group, which was corresponding to [M−H−Glc]-. The ions at m/z 193.0480 and 149.0628 were corresponding to the loss of a molecule water and carbon dioxide as [M−H−Glc−H2O]- and [M−H−Glc−H2O−CO2]-, respectively. According to the fragmentation pathways and standard comparison, compound 12 was identified as geniposidic acid, and its possible cleavage pathway in mass spectrometry is shown in Fig. S2.
3.1.3 Phenylethanoid glycosides
A total of 39 phenylethanoid glycosides were identified from THJGW in both positive and negative ion modes, with the majority coming from Radix Rehmanniae Praeparata and Desertliving Cistanche. In negative ion mode, these compounds typically cleave glycosidic and ester bonds, lose neutral glycosyl groups and phenylethanoid aglycones, or lose phenylpropene acyl groups and other small molecules. Positive ion mode typically results in the loss of neutral glycosyl groups, hydroxytyrosol (HT), and some neutral molecules usually occurs.
Compounds 69 and 81 showed the [M−H]- ion at m/z 623, and the fragment ions at m/z 461, 315, 179, and 161. Similar cleavage pathways were found in their mass spectrometry. The ions at m/z 461 and 315 were separately corresponding to [M−H−caffeoyl]- and [M−H−caffeoyl−Rha]-. The ion at m/z 179 was corresponding to the loss of deprotonated caffeic acid as [caffeic acid-H]-, and the ion at m/z 161 was caused by the loss of a molecule water, which was corresponding to [caffeic acid-H –H2O]-. By comparing the retention time and fragmentation pattern with the reference compound, compounds 69 and 81 were separately identified as acteoside and isomer of acteoside, and the possible cleavage pathway of acteoside in mass spectrometry is shown in Fig. 4.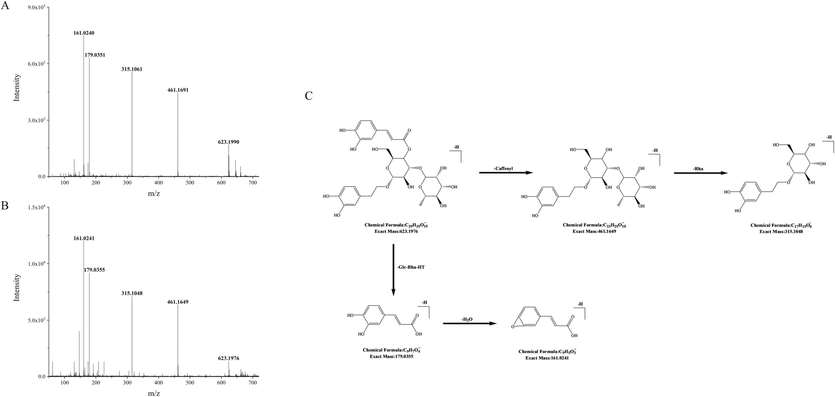
MS/MS spectrum of acteoside standard (A) and acteoside (B), as well as its possible cleavage pathway (C).
Compound 43 showed [M−H]- at m/z 785.2506, and the fragment ions at m/z 623.2188, 461.1660, 179.0347, and 161.0240. The ions at m/z 623.2188 and 461.1660 were separately corresponding to [M−H−caffeoyl]- and [M−H−caffeoyl−Glc]-. The ion at m/z 179.0347 was corresponding to the loss of deprotonated caffeic acid as [caffeic acid-H]-, and the ion at m/z 161.0240 was corresponding to [caffeic acid-H-H2O]-. Based on the accurate mass and compared with the standard, compound 43 was identified as echinacoside. Consistently, compounds 45 and 95 shared similar cleavage patterns with it and were identified as kankanoside F and 2′-acetylacteoside, respectively.
3.1.4 Phenylpropanoids
A total of 9 phenylpropanoids, consisting of simple phenylpropanoids, coumarins, and lignans, were analyzed in negative ion mode. Radix Rehmanniae Praeparata and Desertliving Cistanche were the primary sources of phenylpropanoids. These compounds primarily lose glycosyl groups and neutral molecules, with compound 16 serving as a brief example of their fragmentation pathways in mass spectrometry. Compound 16 showed the [M−H]- ion at m/z 341.0871, and its fragment ions at m/z 179.0347 and m/z 135.0449 were detected. The ion at m/z 179.0347 was generated by the loss of a glucosyl group, which was corresponding to [M−H−Glc]-, and the ion at m/z 135.0449 was corresponding to the loss of a molecule carbon dioxide as [M−H−Glc−CO2]-. Based on the accurate mass and comparison with the cleavage pattern, compound 16 was identified as 6-O-caffeoylglucopyranose.
3.1.5 Organic acids
11 organic acids were discovered in positive and negative ion modes, which existed in the 7 herbs of THJGW. The structure of these compounds with a strong signal in negative ion mode contains carboxyl groups, which typically lose some neutral molecules. Considering compound 4 as an example, it gave a [M−H]- ion at m/z 191.0558, and the fragment ions at m/z 173.0448, 128.0434, 85.0287, 71.0133, and 59.0141. The ions at m/z 173.0448 and 128.0434 were separately corresponding to [M−H−H2O]- and [M−H−H2O−HCOO]-, and the ions at m/z 85.0287, 71.0133, and 59.0141 were corresponding to the cleavage of cyclohexane. Based on the accurate mass and compared with the standard, compound 4 was identified as quinic acid.
3.1.6 Phenolics
4 phenolics were identified in both positive and negative ion modes, the majority of which came from Pyrola. The cleavage behaviors of these compounds in mass spectrometry were primarily due to the loss of substituents on the benzene ring. As an example, compound 115 showed a [M + H]+ ion at m/z 153.0547, and the fragment ions at m/z 135.0438, 121.0284, and 93.0334, which were corresponding to [M + H-H2O]+, [M + H-H2O-CH2]+, and [M + H-H2O-CH2-CO]+, respectively. Based on the accurate mass and comparison with the cleavage pattern, compound 115 was identified as 3,4-dihydroxyacetophenone.
3.1.7 Other compounds
Besides the above major compounds, 16 other compounds such as sugars, monoterpenoids, sterols, and glucosinoltes were identified from THJGW, and their mass spectrum information is shown in Table 1.
3.2 Target network pharmacology analysis
3.2.1 Analysis of absorbed components in serum
The pharmacokinetic parameters of major components among seven medicinal materials herbs in the formula were pre-searched and determined. Most of them had half-lives within 0.4–4 h and peak times within 0.16–2 h. Therefore, serum samples were collected in the 0–2 h range at 0.25, 0.5, 1, and 2 h points after the last dose in order to access more absorbed components efficiently. The serum samples from the blank group and various administration time groups were analyzed using the UPLC-Q-TOF-MSE method, and their BPI chromatograms in positive and negative ion modes are shown in Fig. S3. A total of 38 prototype components were identified in serum samples using rapid matches with the UNIFI platform, as well as a comparison of retention times and fragment ions of each component from the chemical constituent analysis of THJGW. These components include 12 flavonoids, 8 organic acids, 6 iridoid glycosides, 2 phenolics, 2 phenylpropanoids, 1 phenylethanoid glycoside, and 7 other compounds. Most of them could be detected within 0.25 h after administration, while only 2 components remaining detectable at 2 h after administration. The mass spectrometry information for these compounds is detailed in Table S1.
3.2.2 Targets of active components and OA
3636 targets of 38 identified prototype absorbed components were predicted via the Swiss Target Prediction database platform, and 751 remained after removal of duplicates. A total of 3849 disease targets were acquired from the GeneCards database platform. Targets with a relevance score > 1 were screened and combined with the predicted targets from the OMIM and TTD database platforms. After removing duplicates, 1172 OA targets were obtained. Furthermore, 178 potential targets of THJGW for OA therapy were identified, and the venn diagram is shown in Fig. S4.
3.2.3 PPI network analysis
The PPI network of intersection targets was created with STRING and then imported into Cytoscape software for visualization, as shown in Fig. S5. The network consists of 142 nodes and 493 edges. The nodes represented targets, and the edges represented interactions between targets. Node size and color correspond to degree value, with smaller nodes and blue color indicating lower values and larger nodes and orange color indicating higher values. The degree value represents the importance of a node within the network, with a higher value indicating greater significance. The top 10 core targets ranked by MCC scores were VEGFA, STAT3, RELA/NF-κB p65, PIK3R1, PIK3CA, MMP9, MMP1, IL-6, HDAC1, and FGF2, respectively, as shown in Fig. S6.
3.2.4 Network topology analysis
Cytoscape was utilized to analyze and visualize the topology structure of the “herbs-components-targets-disease” network, as depicted in Fig. 5. The entire network includes 224 nodes and 1062 edges. The network topology was analyzed using CytoNCA and the top-ranked 11 key components were screened with high values of degree centrality (DC) and between centrality (BC), as shown in Table S2. Due to the large number of absorbed components, only these 11 components were selected and showed DC values ranging from 31.0 to 40.0.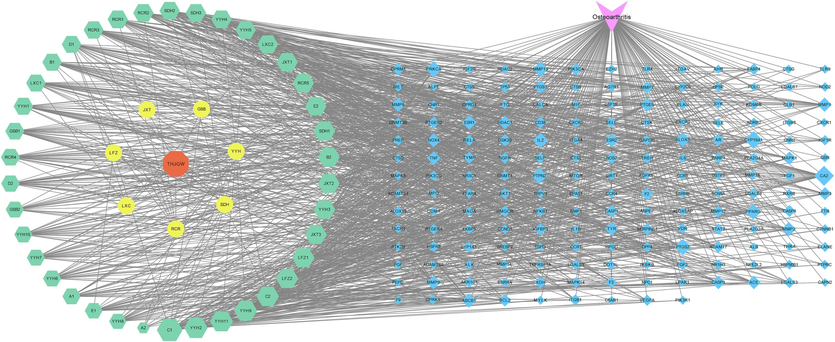
“herbs-components-targets-disease” network of THJGW.
3.2.5 GO enrichment and KEGG pathway analysis
GO function enrichment analysis was performed using Metascape, and the top 10 items were selected based on their p-value as shown in Fig. S7. The molecular functions (MF) category includes activities such as endopeptidase activity, protease binding, nuclear receptor activity, chemokine binding, and fibronectin binding. The biological processes (BP) category includes positive regulation of response to external stimulus, inflammatory response, response to hormone, response to molecule of bacterial origin, positive regulation of locomotion, and so on. The cellular components (CC) category includes the extracellular matrix, membrane raft, side of membrane, secretory granule lumen, lytic vacuole, and so on. KEGG pathway enrichment analysis resulted in Fig. S8, which displays the top 20 representative pathways as pathways in cancer, lipid and atherosclerosis, prostate cancer, microRNAs in cancer, transcriptional misregulation in cancer, and so on. This suggests that THJGW may have a role in the treatment of OA via these pathways. Further research focused on the lipid and atherosclerosis pathway due to its lower p-value and higher rich factor.
3.3 Molecular docking verification
It is widely assumed that binding energies less than −5.0 kcal/mol indicate favorable binding interactions between the compounds and targets. Lower the binding energy indicates stronger docking affinity. The molecular docking results among caffeic acid, acteoside, 8-prenylkaempferol, icaritin, rhein, naringenin, icariside Ⅱ, baohuoside Ⅱ, 5-(Glucosyl-α-1–6-glucose)-hydroxymethylfurfural (5-GGMF), 8-epiloganic acid, glucoraphenin and 10 core targets are shown in Fig. 6A. The binding energies of all docking results were in the range of −5.3 to −9.7 kcal/mol, indicating stable binding of these key components and core targets. This binding stability is closely correlated to the therapeutic effect of THJGW in treating OA disease. Notably, the core target proteins IL-6, STAT3, and RELA/NF-κB p65 had binding energies below −6.5 kcal/mol with strong binding affinity to most key components. It suggests that they are key targets of THJGW in the treatment of OA. Four of docking results with lower binding energies were visualized in Fig. 6B-E using the Discovery Studio software, and their complexes were primarily stabilized by hydrogen bonds and hydrophobic interactions.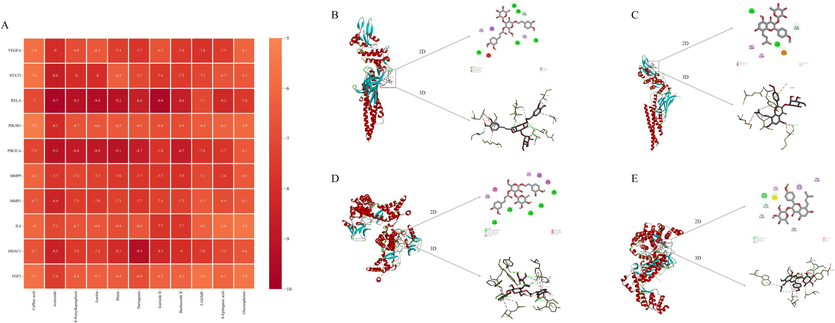
Mollecular docking evaluation on the binding affinity of key components and core targets. (A) Heatmap of the molecular docking of 11 key components with 10 core targets. (B) Molecular docking model of STAT3 and acteoside; (C) Molecular docking model of STAT3 and icariside Ⅱ; (D) Molecular docking model of RELA and acteoside; (E) Molecular docking model of RELA and icariside Ⅱ.
3.4 Effects of THJGW on LPS-induced RAW264.7 macrophages
The MTT assay results, shown in Fig. S9, revealed that LPS at various concentrations reduced viability compared to untreated control cells. Among the tested concentrations, 1 µg/mL of LPS was chosen for the subsequent studies as it exhibited the lowest cell viability (Fig. S9A). THJGW exhibited no significant cytotoxicity at concentrations of 6.25–200 µg/mL in RAW264.7 cells, but significantly increased cell viability at a concentration of 50 µg/mL (Fig. S9B). Furthermore, THJGW had no significant cytotoxic effect on RAW264.7 cells stimulated by LPS (1 µg/mL), as shown in Fig. S9C. Pre-treatment with varying concentrations of THJGW had a distinct protective effects on cells compared to the LPS-induced group. However, this effect gradually attenuated with the increasing concentrations, particularly at 100 μg/mL, while cell viability was still higher than in the LPS-treated group. Consequently, concentrations of 6.25–50 μg/mL of THJGW were chosen for further experiments.
To investigate the anti-inflammatory effect of THJGW, LPS was used to stimulate RAW264.7 macrophages to imitate the chronic inflammatory environment. The ELISA results (Fig. 7A-B) showed a significant upregulation in the concentration of pro-inflammatory cytokines after LPS treatment for 24 h. The secretion of TNF-α and IL-6 in cell supernatants was visibly enhanced. However, pre-treatment with varying concentrations of THJGW prior to stimulation significantly diminished the increase of their secretion, and treatment with high concentrations (50 μg/mL) of THJGW notably suppressed the LPS-induced levels of TNF-α and IL-6.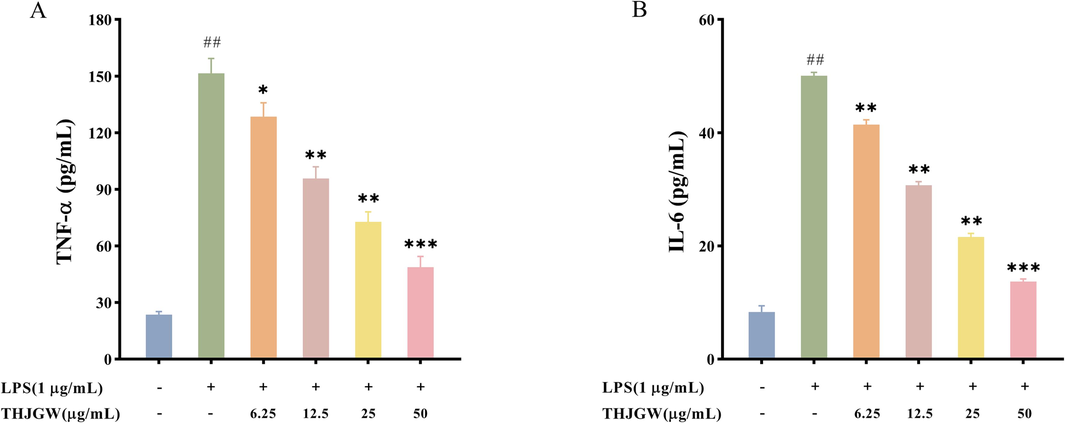
Effects of THJGW on the secretion of TNF-α and IL-6 in LPS-induced RAW264.7 macrophages. (A-B) Concentrations of TNF-α and IL-6. All data are expressed as the mean ± SD. vs. control group ## p < 0.01. vs. LPS-treated group * p < 0.05, ** p < 0.01, *** p < 0.001.
3.5 THJGW inhibits the expression of phosphorylated STAT3 and NF-κB p65 in LPS-induced RAW264.7 cells
According to the results of target network pharmacology and molecular docking, STAT3 and NF-κB p65 were identified as key factors in the lipid and atherosclerosis pathway, playing a significant role in the treatment of THJGW on OA disease. Thus, to further investigate the effects of THJGW, we examined its impact on the expression levels of STAT3 and NF-κB p65 in LPS-stimulated RAW264.7 cells. According to the Western blot results in Fig. 8, STAT3 and NF-κB p65 proteins were slightly phosphorylated in the control group cells. However, inducing macrophages with LPS significantly increased the phosphorylation expression of these two proteins. The protein expression levels of P-STAT3 and P-p65 were significantly reduced with THJGW pre-treatment, while it did not influence the expression of total STAT3 and NF-κB p65 proteins.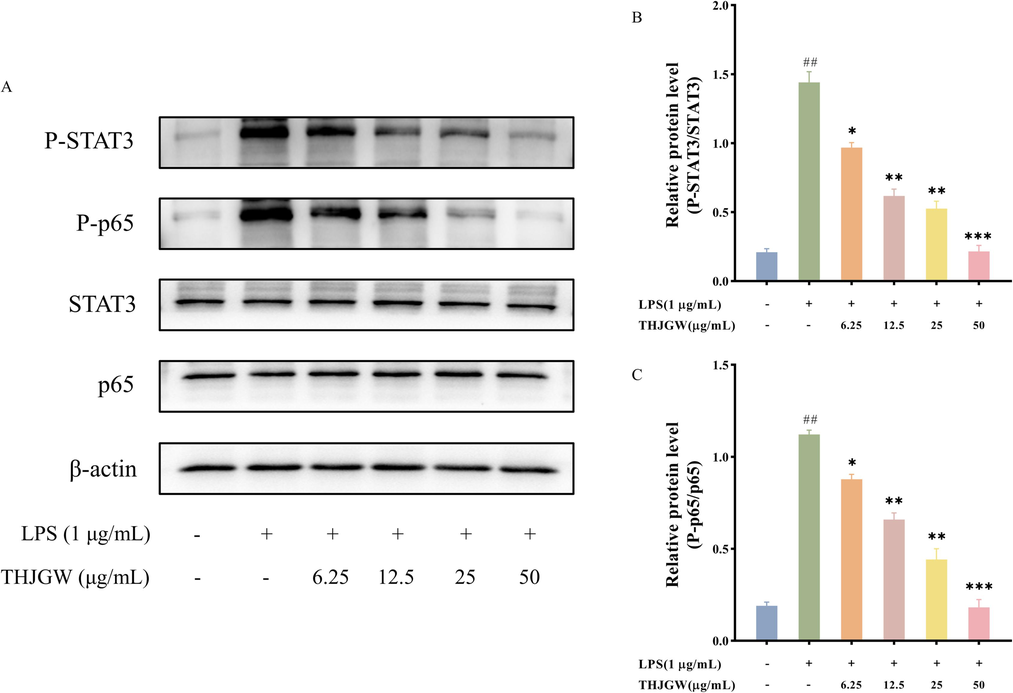
Suppressive effects of THJGW on LPS-stimulated STAT3 and p65 phosphorylation in RAW264.7 macrophages. (A) Western blot protein bands; (B-C) Relative protein levels. The data are presented as means ± SD (n = 3). vs. control group ## p < 0.01. vs. LPS-treated group * p < 0.05, ** p < 0.01, *** p < 0.001.
4 Discussion
In this study, a total of 134 components were identified from THJGW using the UPLC-Q-TOF-MSE technology, and the possible cleavage pathways and chemical structures of its main compounds were determined. There were two major categories of compounds: 45 flavonoids and 39 phenylethanoid glycosides. The flavonoids mostly consist of 8-prenyl flavonoid O-glycosides, such as icariin and icariside II, which were typically loss glycosyl and neutral molecules in mass spectrometry. The phenylethanoid glycosides, such as echinacoside and acteoside, primarily cleave glycosyl, HT, and small molecules. Six major constituents in the THJG formula have been characterized by the UPLC method in a previous study (Li et al., 2021). Compared to the previous study, the in vitro chemical constituents of THJGW were characterized with greater accuracy, sensitivity, rapidity, and comprehensiveness using high-resolution mass spectrometry. This advanced characterization technique provided a solid foundation for subsequent research on the pharmacodynamic materials of THJGW in the current study.
Network pharmacology and molecular docking technology provided an effective tool for analyzing the active ingredients, targets, and pathways of TCM. In this study, target network pharmacology was utilized, which largely avoided the shortcomings of false positive target sites observed in traditional network pharmacology (Xu et al., 2017). A total of 38 prototype absorbed components from serum were identified using the UPLC-Q-TOF-MSE method and were selected as candidates for the target network pharmacology analysis. The results showed that caffeic acid, 8-prenylkaempferol, icaritin, icariside Ⅱ, acteoside, glucoraphenin, rhein, baohuoside Ⅱ, naringenin, 8-epiloganic acid, and 5-GGMF were the key components of THJGW for treating OA. Previous studies suggested that caffeic acid may promote osteoblast proliferation, differentiation, and maturation, resulting in improved bone formation (Melguizo-Rodríguez et al., 2019). Icaritin, the metabolite of icariin, was found to have significant anti-inflammatory activity. It may reduce LPS-induced inflammatory responses in mouse chondrocytes by lowering nitric oxide and matrix metalloproteinases levels (Liu et al., 2010). Icariside II likely suppressed the progression of osteoporosis in rats by promoting osteoblast differentiation, inhibiting adipocyte formation, and modulating immune function (Xi et al., 2019). Acteoside was investigated to prevent bone loss in osteoporotic mice by inhibiting NF-κB and activating the PI3K/AKT signaling pathway (Yang et al., 2019). Naringenin has been proven in vivo and in vitro to be effective in treating iron overload-induced OA by alleviating cartilage damage and reducing oxidative stress levels (Pan et al., 2022). These reports mostly agreed with our predictions. However, there were also a few studies that held the opposite opinion. Folwarczna et al. discovered that caffeic acid did not minimize bone resorption in osteoporotic rats and may have altered bone mechanical properties in normal rats (Folwarczna et al., 2009), which could be attributed to oral administration. The PPI network analysis identified VEGFA, STAT3, RELA/NF-κB p65, PIK3R1, PIK3CA, MMP9, MMP1, IL-6, HDAC1, and FGF2 as key targets for THJGW in treating OA.
KEGG pathway analysis revealed that the top 10 core targets were significantly enriched in signaling pathways associated with anti-OA, including pathways in cancer, lipid and atherosclerosis, prostate cancer, microRNAs in cancer, etc. Especially the lipid and atherosclerosis pathway, which was highly enriched by all ten core targets. Early observations revealed significant amounts of lipid deposition in osteoarthritic chondrocytes, implying a role for lipid metabolism in OA progression (Gkretsi et al., 2011). A majority of proteins related to lipid metabolism, such as peroxisome proliferator-activated receptor and apolipoproteins, were observed to be differentially expressed in osteoarthritic tissues based on several proteomic studies (Wu et al., 2007). Triantaphyllidou et al. have proposed that decreased content or disordered metabolism of high-density lipoprotein contributed to the OA development in metabolic syndrome mice, suggesting that this could be a key factor in OA pathogenesis (Triantaphyllidou et al., 2013). Moreover, studies have found that lipoproteins, which transport lipids in blood circulation, are the primary risk factor for atherosclerosis (Björkegren and Lusis, 2022). For instance, Han et al. have indicated that high-density lipoprotein produced by the intestine protects not only the liver but possibly also the vessel wall to avoid atherosclerosis (Han et al., 2021). However, certain triglyceride-rich lipoproteins directly contribute to the progression of atherosclerosis (Liu et al., 2017). Macêdo et al. have also discovered a link between atherosclerosis and OA pathogenesis (Macêdo et al., 2022). These studies support the predictions, indicating that the lipid and atherosclerosis pathway may be the primary approach for THJGW in the treatment of OA.
The molecular docking results suggested that 5 key components, including acteoside, icaritin, naringenin, icariside Ⅱ, and baohuoside Ⅱ, had higher binding affinities to most of the core targets. Additionally, three core targets, which were IL-6, STAT3, and RELA/NF-κB p65, had higher binding energies for most key components. Previous studies have shown that IL-6 has multiple catabolic functions in cartilage, which were primarily mediated by STAT3, and the IL-6 pathway may be useful as a targeted therapy for OA to alleviate cartilage damage, synovial inflammation, and subchondral bone pathology (Latourte et al., 2016). In vivo and in vitro experiments have shown that the STAT3 pathway may regulate cartilage degradation, with activation leading to cartilage damage and osteophyte formation in OA (Latourte et al., 2017). Kobayashi et al. have proven that NF-κB family member p65 may inhibit chondrocyte apoptosis and participate in the regulation of articular cartilage homeostasis in OA, but canonical activation of the NF-κB signaling pathway promotes cartilage catabolism (Kobayashi et al., 2016). Molecular docking results support the accuracy of target network pharmacology predictions.
The therapeutic effects of herbs in the formula have been reported in several studies. Jhun et al. found that an extract of Radix Rehmanniae Praeparata alleviated inflammatory symptoms and articular cartilage degradation in monosodium iodoacetate-induced OA rats by inhibiting NF-κB and HIF-2α activation (Jhun et al., 2018). Dai et al. discovered that the combination of Rhizoma Drynariae and Epimedium brevicornum inhibited the phosphorylation expression of p38, ERK, and JNK proteins in rat cartilage tissue, thereby preventing OA development via the MAPK signaling pathway (Dai, 2022). Spatholobus extract was observed to prevent bone loss through the inhibition of osteoclast differentiation, indicating that it may have therapeutic potential to attenuate cartilage damage in OA (Im et al., 2014). Likewise, Zhang et al. have demonstrated that Desertliving Cistanche may suppress osteoclast differentiation and corresponding bone resorption in an osteoporosis rat model (Zhang et al., 2019). However, the molecular mechanisms of THJGW in OA treatment need to be determined and thoroughly investigated. Recent clinical studies have provided substantial evidence that inflammation is key driver of OA disease (Knights et al., 2023). Excessive levels of inflammatory mediators such as IL-6, IL-1β, and TNF have been detected in chondrocytes and synovial cells in OA, thus targeting inflammatory therapy might be an effective treatment to prevent or slow OA progression (Robinson et al., 2016). Previous studies have suggested that macrophages contributed substantially to the progression of OA through various secreted mediators that may regulate joint inflammation (Wu et al., 2020a). Therefore, RAW264.7 macrophages were used to mimic an inflammatory environment in vitro to verify the mechanism of THJGW in treating OA. Inflammatory cytokine levels in cell supernatants were substantially increased by LPS (1 µg/mL)-treated RAW264.7 cells, which was significantly reversed by pre-treatment of THJGW at a high concentration (50 μg/mL). The results indicated that the anti-inflammatory effect of THJGW was attributed to inhibiting the pro-inflammatory cytokines, namely TNF-α and IL-6, in LPS-induced RAW264.7 cells. Furthermore, molecular docking results identified STAT3 and RELA/NF-κB p65 as key targets associated with the lipid and atherosclerosis pathway. Wu et al. have demonstrated that inhibition of the phosphorylated expression of these two proteins in macrophages is beneficial for the treatment of inflammatory diseases (Wu et al., 2020b). Our results indicated that expression of phosphorylated STAT3 and NF-κB p65 proteins was visibly increased in LPS-induced cells, suggesting activation of the lipid and atherosclerosis pathway. THJGW pre-treatment significantly restored the phosphorylation levels of STAT3 and NF-κB p65 proteins without affecting total proteins. These findings implied that THJGW effectively inhibits the phosphorylation of STAT3 and NF-κB p65 proteins in LPS-induced macrophages, thereby mediating the lipid and atherosclerosis pathway to exert an anti-inflammatory effect in OA.
5 Conclusion
In the current study, the pharmacodynamic material and mechanism of action of THJGW in OA treatment were investigated. A total of 134 chemical constituents of THJGW in vitro were analyzed by UPLC-Q-TOF-MS technology, and the majority of them were flavonoids and phenylethanoid glycosides. Similarly, 38 prototype absorbed components were identified in serum and considered candidates. The key components acteoside, icaritin, naringenin, icariside Ⅱ, and baohuoside Ⅱ, as well as the core targets IL-6, STAT3, and RELA/NF-κB p65, which were closely associated with therapeutic effects on OA for THJGW, were predicted and validated by targeted network pharmacology and molecular docking technique. Moreover, LPS-induced RAW264.7 macrophages were used in in vitro assays for inflammation. The outcomes further confirmed and indicated that THJGW, possibly through inhibition of the lipid and atherosclerosis pathway, has a potent anti-inflammatory effect in OA. This suppression was achieved through a reduction in the expression levels of inflammatory cytokines TNF-α and IL-6, as well as phosphorylation of STAT3 and NF-κB p65 proteins. In conclusion, this research strategy offers a fresh perspective on how to examine the pharmacodynamic material basis and its mechanism in Chinese medicine. The research findings provide scientific evidence for the scientific application and improvement of quality standards for THJGW. However, this study also has certain limitations. The screened active compounds were not further validated and deeply investigated in terms of their activities and mechanisms of action, and part of this work will be continued in our subsequent studies.
CRediT authorship contribution statement
Dong Xie: Formal analysis, Investigation, Visualization, Writing – original draft. Guangfu Lv: Methodology. Yuchen Wang: Methodology. Wenjing Zhang: Software. Nian Li: Software. Yao Duan: Software. Qing Huang: Resources. Ge Chen: Conceptualization, Writing – review & editing. Zifeng Pi: Conceptualization, Writing – review & editing. Hao Yue: Conceptualization, Writing – review & editing.
Acknowledgements
This research was supported by Science and Technology Development Project of Jilin Province (20210401064YY and 20210508001RQ).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Identification and Analysis of Chemical Constituents and Rat Serum Metabolites in Gushuling Using UPLC-Q-TOF/MS Coupled with Novel Informatics UNIFI Platform. Evid-Based. Compl. Alt.. 2021;2021:2894306.
- [CrossRef] [Google Scholar]
- Molecular Mechanism of the Guizhi Decoction on Osteoarthritis based on an Integrated Network Pharmacology and RNA-sequencing Approach with Experimental Validation. Front. Genet.. 2023;14:1079631.
- [CrossRef] [Google Scholar]
- Network-based approach to prediction and population-based validation of in silico drug repurposing. Nat. Commun.. 2018;9:2691.
- [CrossRef] [Google Scholar]
- Study on the Protective effect and mechanism of the rhizoma drynariae-Epimedium Formula on osteoarthritis in rats. Contrast. Media. Mol. i.. 2022;2022:2869707.
- [CrossRef] [Google Scholar]
- Effects of natural phenolic acids on the skeletal system of ovariectomized rats. Planta Med.. 2009;75:1567-1572.
- [CrossRef] [Google Scholar]
- Lipid metabolism and osteoarthritis: lessons from atherosclerosis. Prog. Lipid. Res.. 2011;50:133-140.
- [CrossRef] [Google Scholar]
- Enterically derived high-density lipoprotein restrains liver injury through the portal vein. Science. 2021;373:eabe6729.
- [CrossRef] [Google Scholar]
- Spatholobus suberectus inhibits osteoclastogenesis and stimulates chondrogenesis. Am. J. Chin. Med.. 2014;42:1123-1138.
- [CrossRef] [Google Scholar]
- Notoginseng radix and rehmanniae radix preparata extract combination (YH23537) reduces pain and cartilage degeneration in rats with monosodium iodoacetate-induced osteoarthritis. J. Med. Food. 2018;21:745-754.
- [CrossRef] [Google Scholar]
- Characterization of steroidal glycosides from the extract of Paris Polyphylla var. Yunnanensis by UPLC/Q-TOF MSE. J. Pharm. Biomed. Anal.. 2012;62:235-249.
- [CrossRef] [Google Scholar]
- Inflammation in osteoarthritis: the latest progress and ongoing challenges. Curr. Opin. Rheumatol.. 2023;35:128-134.
- [CrossRef] [Google Scholar]
- Biphasic regulation of chondrocytes by Rela through induction of anti-apoptotic and catabolic target genes. Nat. Commun.. 2016;7:13336.
- [CrossRef] [Google Scholar]
- Call for standardized definitions of osteoarthritis and risk stratification for clinical trials and clinical use. Osteoarthritis and Cartilage. 2015;23:1233-1241.
- [CrossRef] [Google Scholar]
- IL-6/STAT3 signalling blockade protects against experimental osteoarthritis in mice. Osteoarthritis and Cartilage. 2016;24:S378.
- [CrossRef] [Google Scholar]
- Systemic inhibition of IL-6/Stat3 signalling protects against experimental osteoarthritis. Ann. Rheum. Dis.. 2017;76:748-755.
- [CrossRef] [Google Scholar]
- In vivo effect of the Chinese Tenghuang Jiangu Formula on cartilage destruction and subchondral bone sclerosis. Phytomedicine plus. 2021;1:100121.
- [CrossRef] [Google Scholar]
- Pharmacokinetics integrated with network pharmacology to clarify effective components and mechanism of Wendan decoction for the intervention of coronary heart disease. J. Ethnopharmacol.. 2023;314:116669.
- [CrossRef] [Google Scholar]
- Network pharmacology strategy for revealing the pharmacological mechanism of pharmacokinetic target components of San-Ye-Tang-Zhi-Qing formula for the treatment of type 2 diabetes mellitus. J. Ethnopharmacol.. 2020;260:113044.
- [CrossRef] [Google Scholar]
- Exome-wide association study of plasma lipids in> 300,000 individuals. Nat. Genet.. 2017;49:1758-1766.
- [CrossRef] [Google Scholar]
- Icariin protects murine chondrocytes from lipopolysaccharide-induced inflammatory responses and extracellular matrix degradation. Nutr. Res.. 2010;30:57-65.
- [CrossRef] [Google Scholar]
- Network pharmacology and pharmacokinetics integrated strategy to investigate the pharmacological mechanism of Xianglian pill on ulcerative colitis. Phytomedicine. 2021;82:153458.
- [CrossRef] [Google Scholar]
- Association between osteoarthritis and atherosclerosis: A systematic review and meta-analysis. Exp. Gerontol.. 2022;161:111734.
- [CrossRef] [Google Scholar]
- Bone protective effect of extra-virgin olive oil phenolic compounds by modulating osteoblast gene expression. Nutrients. 2019;11:1722.
- [CrossRef] [Google Scholar]
- Naringenin protects against iron overload-induced osteoarthritis by suppressing oxidative stress. Phytomedicine. 2022;105:154330.
- [CrossRef] [Google Scholar]
- Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol.. 2016;12:580-592.
- [CrossRef] [Google Scholar]
- Perturbations in the HDL metabolic pathway predispose to the development of osteoarthritis in mice following long-term exposure to western-type diet. Osteoarthritis and Cartilage. 2013;21:322-330.
- [CrossRef] [Google Scholar]
- Using network pharmacology and molecular docking to explore the mechanism of Shan Ci Gu (Cremastra appendiculata) against non-small cell lung cancer. Front. Chem.. 2021;9:682862.
- [CrossRef] [Google Scholar]
- Chrysoeriol ameliorates TPA-induced acute skin inflammation in mice and inhibits NF-κB and STAT3 pathways. Phytomedicine. 2020;68:153173.
- [CrossRef] [Google Scholar]
- The role of macrophages in osteoarthritis and cartilage repair. Osteoarthritis and Cartilage. 2020;28:544-554.
- [CrossRef] [Google Scholar]
- Comparative proteomic characterization of articular cartilage tissue from normal donors and patients with osteoarthritis. Arthritis. Rheum.. 2007;56:3675-3684.
- [CrossRef] [Google Scholar]
- Preliminary studies on the anti-osteoporosis activity of Baohuoside I. Biomed. Pharmacother.. 2019;115:108850.
- [CrossRef] [Google Scholar]
- Systematically characterize the absorbed effective substances of Wutou Decoction and their metabolic pathways in rat plasma using UHPLC-Q-TOF-MS combined with a target network pharmacological analysis. J. Pharm. Biomed. Anal.. 2017;141:95-107.
- [CrossRef] [Google Scholar]
- Effect of Tenghuangjiangu capsule on the expression of blood cell and inflammatory cytokines in rats with knee osteoarthritis. J. Biol.. 2020;37:84-87.
- [CrossRef] [Google Scholar]
- Protective effect of acteoside on ovariectomy-induced bone loss in mice. Int. J. Mol. Sci.. 2019;20:2974.
- [CrossRef] [Google Scholar]
- Anti-osteoporotic activity of an edible traditional Chinese medicine Cistanche deserticola on bone metabolism of ovariectomized rats through RANKL/RANK/TRAF6-mediated signaling pathways. Front. Pharmacol.. 2019;10:1412.
- [CrossRef] [Google Scholar]
- Potential molecular mechanisms of Erlongjiaonang action in idiopathic sudden hearing loss: A network pharmacology and molecular docking analyses. Front. Neurol.. 2023;14:1121738.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2024.105711.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




