

Translate this page into:
Potential bioactive compounds as SARS-CoV-2 inhibitors from extracts of the marine red alga Halymenia durvillei (Rhodophyta) – A computational study
⁎Corresponding author.
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
The respiratory infection COVID-19 caused by the virus SARS CoV-2 has continued to be a major health problem worldwide and has caused more than a million mortalities. Even if the development of COVID-19 vaccines has shown much progress, efforts to find novel, natural anti-viral drugs should be pursued. Halymenia durvillei is a marine red alga widely distributed around Southeast Asia. This study aimed to develop new anti SARS CoV-2 compounds from ethanolic and ethyl acetate extracts of H. durvillei via a computational approach, focusing onthe inhibitory action against the main protease (3CL-Mpro). In this study, 37 compounds were extracted and identified by GC–MS analysis. The potentials of compounds 1–2 tetradecandiol and E,E,Z-1,3,12-nonadecatriene-5,14-diol were identified for therapeutic purposes based on our pharmacophore study, while cholest-5-En-3-Ol (3.Beta.)- had a high fitness score in molecular docking studies both in monomer and dimer state compared to the N3 inhibitor and remdesivir affinity scores. As these compounds show competitive affinity scores against the 3CL-Mpro, these natural compounds may be effective for the treatment of COVID-19 infection. The ADME and pharmacokinetic studies should also be employed to assess the ability of the natural compounds as oral drugs. These promising results have shown the potentials of H. durvillei as an alternative drug in addressing COVID-19 infection. Accordingly, further studies should explore the effectiveness of these active compounds.
Keywords
COVID-19
Natural products chemistry
Molecular docking
Pharmacophore

1 Introduction
In December 2019, a major cluster of atypical pneumonia caused by a novel coronavirus was reported in Wuhan city, China (Wang et al., 2020; Zhou et al., 2020). This coronavirus was initially named 2019 novel coronavirus (2019-nCoV) by the World Health Organization (WHO) and later renamed severe acute respiratory syndrome coronavirus 2 (SARS- CoV-2) (Gorbalenya et al., 2020; Meng et al., 2020). Despite efforts to contain the outbreak within the initial area, the dissemination of SARS-CoV-2 has continued to spread widely to other nations. In March 2020, the WHO declared the uncontrollable spread of SARS-CoV-2 as a pandemic (Cucinotta and Vanelli, 2020). Now, almost 2 years later, this unprecedented pandemic remains a global health threat with a total of 205,338,159 confirmed cases worldwide and 4,333,094 fatalities (WHO, 2021a) causing enormous economic upheaval.
Global collaborations are necessary to find effective way to control SARS-CoV-2 (Kaur and Gupta, 2020). The development of potential drugs and vaccines usually takes a sizable amount of time but, considering the high morbidity and mortality of the SARS-CoV-2, the pace of development had to go at a very fast pace (Belete, 2021). At least 294 vaccine candidates are in various stages of clinical development to date (WHO, 2021b) with some already in use by countries for mass vaccinations (Jabal et al., 2021). Vaccines reduce the risks of getting infected but questions remain regarding the duration of protection and protection response in natural infection (Long et al., 2020), as well to reduce the number of symptomatic cases for those infected (Olliaro et al., 2021). However, concerns for the protection provided by vaccines are raised related to the multiple SARS-CoV-2 variants that have been circulating globally (Iacobucci, 2021; Kirby, 2021).
The use of vaccines is for the precautionary use for the healthy, while the sick requires drugs to recover. The current drugs used include those with appropriate pharmacological and therapeutic efficacies such as remdesivir, hydroxychloroquine, chloroquine, lopinavir, umifenovir, favipiravir, and oseltamivir (Ita, 2020; Jean and Hsueh, 2020). To date, no specific therapeutic drugs have been reported to specifically target SARS-CoV-2 (Wu et al., 2020). Therefore, the search for potential therapeutic regimens to treat the sick is still ongoing by considering the available alternatives. Furthermore, the use of synthetic compounds has shown several adverse effects in the patients. On the other hand, the development of new drugs from natural sources was found to be less toxic and involved lower financial cost (Permana et al., 2020). Furthermore, to the best of our knowledge, there has been no report regarding the extreme toxicity of antiviral compounds obtained from natural product. Therefore, it is crucial to develop an alternative treatment of COVID-19 from natural products, including those from the marine environment such as algae-derived compounds (Hans et al., 2021).
Seaweeds have been recognized as rich and valuable sources of biological compounds that may provide potential solutions to the SARS-CoV-2 pandemic (Pereira and Critchley, 2020). Seaweed compounds showed activity against a broad spectrum of viruses (Jha et al., 2020; Hans et al., 2021). A group from Morocco found one seaweed compound that inhibits the Herpes simplex virus type 1 (HSV-1) by cell viability method (Rhimo et al., 2010). Cirne-Santos et al. (2019) also revealed that the brown seaweed Dictyota menstrualis (Hoyt) Schnetter, Hörnig & Weber-Peukert can inhibit the viral replication of the Zika virus (ZIKV). Moreover, the capability of seaweeds could be an alternative in combating coronaviruses through several mechanisms: penetration, replication, or targeting the host‘s cellular targets (Chen et al., 2020; Zaporozhets and Besednova, 2020).
The inhibitory effect of the natural compounds of seaweeds is related to their ability to inhibit the virus from attaching to the main protease, i.e., 3 chymotrypsin-like protease Main Protease (3CL-Mpro), which is fundamental for the replication of SARS-CoV-2 (Muteeb et al., 2020; Palese, 2020). The 3CL-Mpro is a key enzyme that has an important role in SARS-CoV-2 replication mechanism. It has been identified by researchers as a protein target in anti-coronavirus drugs (Qamar et al., 2020). X-ray crystallography of the 3CL-Mpro structure was obtained from PDB with 6LU7 code. The structure is comprised of three domains, i.e., domain I (residues from 8 to 100), domain II (residues from 101 to 176) and domain III (residues from 1200 to 306) where domain I and domain II are called as N-terminal domains with β-sheets and 13 β-strands, while domain III is a C-terminal domain that consists of 5 α-helices. The N-terminal domain plays a critical role as binding substrate that is located in a cleft between domain I and domain II. Residues number 176 to 199 from N-terminal and C-terminal domains are the loop that joins these domains. The overall molecular architecture of SARS-CoV-2 3CL-Mpro is illustrated in Fig. 1.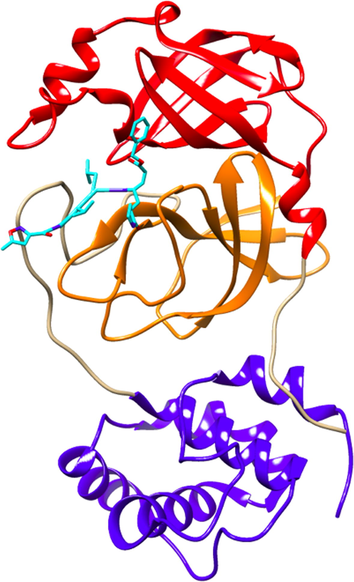
X-ray structure of 3CL-Mpro of SARS-CoV-2 (6LU7 pdb): domain I (red), domain II (orange) and domain II (blue), and its inhibitors NO3 within the active site (between domain I and II).
Nowadays, drug discovery using computational design via molecular docking has become more popular and is becoming an important device used by scientists. Traditional drug discovery methods are expensive and time-consuming. Instead, molecular docking has been proven to be a more robust, effective, low-cost and less time-consuming process (Bharti and Shukla, 2021). Therefore, this study investigated an in silico method that evaluates the inhibitory effect of natural compounds from the red marine alga Halymenia durvillei Bory de Saint-Vincent against SARS-CoV-2. As previously explained, the main protease (3CL-Mpro) is the significant protein in the life cycle of SARS-CoV-2. This enzyme was used as one of the main targets of this computational study. Identifying the potential of seaweed against 3CL-Mpro main protease using computational study approaches has been conducted by Gentile et al. (2020) who showed that some natural compounds from the Marine Natural Product Library could be used as inhibitors with pharmacophore and molecular docking properties. Moreover, an in silico analysis of the marine seaweed database identified some red algae metabolites have a high-affinity against 3CL-Mpro of SARS-CoV-2. However, not all algae from the databases have been studied for their potentials against SARS-CoV-2.
Here, we identify the compounds in Halymenia durvillei that can potentially act as an inhibitor to 3CL-Mpro of SARS-CoV-2 by employing computational approaches. These involved virtual screening, pharmacophore study and molecular docking with 37 compounds of H. durvillei. The findings of this study may provide a new perspective for the development of natural-based drugs that are widely available, with high-efficiency, and low-toxicity for the treatment in the current and future pandemics.
2 Material and methods
2.1 Collection of plant materials and extract preparation
Sample of the red seaweed was collected from Kayangan Island, Makassar, Indonesia. Morphometric approaches were used to identify the samples. The fresh samples were washed with water or distilled water to remove particles, then dried by Herbs Dryer. To get optimal result in phytochemical extraction,maceration was used with two different solvents, ethanol and ethyl acetate in 3x30 min with sonication. Samples of 126.32 g and 127.15 g dried algae were added with 600 mL of 96% ethanol and 600 mL of ethyl acetate, respectively. Afterwards, the mixtures were filtered to get the extract which was further subjected to rotary evaporation to remove the organic solvents.
2.2 Gas Chromatography-Mass Spectrometry (GC–MS) analysis of Halymenia durvillei extracts
GC–MS analysis was done using the Ultra Shimadzu QP2010 Gas Chromatograph Mass Spectrometer coupled with AOC-20i Autosampler to identify the phytochemicals of Halymenia durvillei extracts. The following conditions were used: 1 µL injection volume; SH-Rxi-5Sil capillary column MS 30 m column length with 0.25 mm inner diameter; vacuum fractionation distillation used a Vigreux column with 20 cm long and 2.4 cm inner diameter. About 5 g H. durvillei was inserted into a 250 mL round bottom flask connected to a fractionation column and vacuum pump. A bath jacket heated the flask at 200 °C and a pressure of 96 kPa. Furthermore, distillates obtained at each change in steam temperature during the distillation process were carried out in 50 mL round bottom flasks, afterwhich H. durvillei alcohol levels of each fraction were measured. In addition, GC–MS analysis was conducted under these specific conditions: helium carrier gas, injector temperature of 250 °C with splitless mode, pressure at 76.9 kPa, and carrier gas flow rate at 14 mL/min and a ratio of 1:10. Ion source temperature and interfaces are 200 °C and 280 °C. Solvent cut time was 3 min, with range of mass spectrum of 400–700 m/z. The initial temperature of the column was 110 °C with a hold time of 2 min, and the temperature was increased to 200 °C at a rate of 10 °C / min with the final temperature of 280 °C with a holding time of 9 min at a rate of 5 °C/ min so that the total analysis time was 36 min. Determination of compounds used the NIST and Wiley libraries (Rifai et al., 2019).
2.3 Ligand praparation
All of the 37 three-dimensional structures of compounds from GC/MS results were collected from PubChem ( https://pubchem.ncbi.nlm.nih.gov/). Moreover, the ligand N3 that are from co-crystalized protein of 6LU7 PDB was extracted, while several drugs such as remdesivir, lopinavrin, ritonavir and hydroxychloroquine that are repurposed against the 3CL-Mpro were also collected from Pubchem. A dataset of natural compounds and other drugs had been prepared by UCSF Chimera 1.14 ( https://www.cgl.ucsf.edu/chimera/)with adding hydrogens and charges, and minimizing into their 3D structure by Gasteiger Force Field in UCSF Chimera and then continued to be screened in the pharmacophore study and molecular docking study.
2.4 Protein preparation
The protein structure of 3-CL Main Protease or 3CL-Mpro that was responsible for severe acute respiratory syndrome-2 of COVID-19 was obtained from Protein Data Bank ( https://www.rcsb.org/) with latest version in 6LU7 PDB. The protein of 3CL-Mpo was obtained from X-ray diffraction method with 2.16 Å resolution. It was consisted of 306 residues in monomer state and 612 residues in dimer state which was divided into 2 subunits, they are subunit 1 (from residue 1 – residue 302) and subunit 2 (from residue 303 – residue 612). Both monomeric and dimeric state of 3CM Main Protease were prepared by PyMOL 2.5. The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC and UCSF Chimera 1.14 removing water molecules, inhibitor and other heteroatoms.
2.5 Validation and virtual screening of pharmacophore-based approach
The pharmacophore study has been one of the important methods in drug discovery. This method gives information of the interaction patterns of molecules that are represented by three-dimensional (3D) features. The features such as the formation of hydrogen bonds (donor and acceptor), charged interaction (ions), hydrophobic interactions and aromatic contacts (Kaserer et al., 2015) determine the type of interactions between ligand and protein target. LigandScout version 4.4 was used for pharmacophore elucidation and established hit compound in virtual screening. In establishing a pharmacophore model, four antiviral drugs that are used for the treatment of COVID-19 were analyzed as training sets for the ligand-based pharmacophore model: remdesivir, lopinavir, ritonavir and hydroxychloroquine. The following conditions were used: fragment screening mode; retrieval mode with get best matching conformation; three as the minimum number of required features and checking exclusion volumes. The generated pharmacophore was then used as the model template in generating hits compounds from the dataset compounds that should have at least one match in every feature. The compound hits resulting from this study were then analyzed in the molecular docking study through their molecular interactions.
2.6 Molecular docking
Molecular docking studies were done using GOLD software version 1.10.5 developed by The Cambridge Crystallographic Data Centre (CCDC) and AutoDock Tools version 1.5.6 developed by The Scripps Research Institute (Morris et al., 2009). All ligands that were used in previous study were used in this current approach as ligands. This step employed allosteric binding site and active site into monomeric and dimeric form of SARS-CoV-2 3CL-Mpro. In monomer state, the protein chain A was docked with GOLD and AutoDock software into active site docking and allosteric site docking. In GOLD software, the docking study was only run with specific active site because GOLD is not capable of allosteric site searching. Therefore, in establishing the active site of the protein, the position of N3 was pointed as the active site position. Meanwhile, the protein monomer-ligand docking at the the active site with AutoDock software was configured with grid size, 20x30x30 and center –13x11x74 (x,y,z), while in allosteric site, the grid box of allosteric docking in monomeric state is 50x60x55 with grid center –26x14x59 (x,y,z)
On the other hand, in dimeric state docking, AutoDock software was employed to calculate the affinity of each subunit of 3CL-Mpro and compared the results with monomeric state. For subunit 1, the grid was configured with grid size and center are the same as with monomeric's configuration and subunit 2 was configured with grid size 20x30x30 and center –32x14x30 (x,y,z). This approach was aimed to analyze the binding affinity of each ligand from Halymenia durvillei against SARS-CoV-2 3CL-Mpro. The most remarkable affinities of ligands were continuously analyzed for their molecular interactions with protein PyMOL 2.5 and UCSF Chimera 1.14.
2.7 Docking validation and ligand efficiency
The docking study was validated by doing re-docking of the co-crystalized ligand from 3CL-Mpro (6LU7 PDB), N3. The ligand was removed from the active site of protein and re-docked again at the same site using AutoDock. The re-docked complex was then superimposed with the real co-crystalized 3CL-Mpro with its inhibitor (6LU7) and calculated the root mean square deviation using PyMOL between these proteins and its inhibitor. The docking result was in acceptable result if the N3 as the ligand and the protein has the same position at the active site with similar pose mode. These were done to validate the docking procedure to ensure the validation of docking (Shivanika et al, 2020). Furthermore, the ligands were calculated of their Ligand Efficiency (LE) with this equation: Where −ΔG is the free energy of binding and HA is the number of non-hydrogen atoms of the ligand. This study aims to consider the binding affinity of the whole compound with the receptor per non-hydrogen atom. This gives an information about the affinity and the size of molecules which very useful to lead a compound in further optimization (Schultes et al, 2010).
2.8 Lipinski’s rule of 5 and pharmacokinetic
The importance of lipophilicity and solubility are the key molecular properties in the absorption of a drug. The prediction of the oral availability of a drug is divided to the several characteristics in the Lipsinki’s rule of five which was done by SwissAdme ( http://www.swissadme.ch/) by Swiss of Bioinformatics website: logP (≤5), Molecular weight (MW ≤ 500 g/mol), number of hydrogen bond acceptors (HBA ≤ 10), number of hydrogen bond donors (HBD ≤ 5), rotatable bond(nRotb ≤ 10) and polar surface area (PSA ≤ 140 Å2). Meanwhile, the evaluation of ADME-Tox properties of a drug is essential to select candidate compounds that have possibilities to be approved and not being discarded in the clinical phase. The method was done by using ADMETLab 2.0 ( https://admetmesh.scbdd.com/) developed by CBDD Team of Medicine of Zhejiang University. This study analyzed the solubility, absorption, permeability and metabolites of a drug (Brito, 2011).
3 Results and discussion
In this study, the extraction of phytochemicals from Halymenia durvillei was conducted using two different solvents, ethanol and ethyl acetate, in order to get compounds under both polar and semi-polar conditions. According to Deepak et al. (2018), ethyl acetate extraction of the brown seaweed Sargassum wightii Greville revealed all the tested phytochemicals while ethanol extraction has been used to obtain compounds from algae, especially the red alga Kappaphycus alvarezii (Doty) L.M. Liao which yielded phytochemicals which show mostly high polarity and solubility in water (Bhuyar et al., 2020). Therefore, in this present study, we used ethanol and ethyl acetate as extraction solvents.
This current investigation revealed 37 compounds (Table 1) from GC–MS analysis of ethanolic and ethyl acetate extracts of H. durvillei. Furthermore, GC–MS chromatograms are presented in Fig. 2. The details of all compounds are listed in Table 1. Three compounds were obtained from ethanol extraction namely caryophyllene, eucalyptol and 1,4,7,-cycloundecatriene, while 1,5,9,9-tetramethyl-, Z, Z, Z- and the rest of the compounds were obtained from ethyl acetate extraction. From Table 1, it is shown that eucalyptol from ethanolic extracts was the most compound with 97.55 area % (154 MW). By contrast, 3,7,11,15-Tetramethyl-2-hexadecen-1-ol was the least compound from the ethyl acetate extract with 0.23 area % (296 MW).
Extraction
Compounds
Area %
Molecular Weight (MW)
Smiles
PubChem ID
1
Ethanol
Eucalyptol
97.55
154
CC1(C2CCC(O1)(CC2)C)C
2758
2
Caryophyllene
1.71
204
CC1 = CCCC(=C)C2CC(C2CC1)(C)C
5281515
3
1,4,7,-Cycloundecatriene, 1,5,9,9-tetramethyl-, Z,Z,Z-
0.74
204
CC1 = CCC = C(CC = CC(CC1)(C)C)C
5368784
4
Ethyl acetate
N-Hexadecanoic Acid
10.07
256
CCCCCCCCCCCCCCCC(=O)O
985
5
1,2-Tetradecanediol
1.49
230
CCCCCCCCCCCCC(CO)O
89436
6
15-Isopropenyloxacyclopentadecan-2-One
0.03
266
CC(=C)C1CCCCCCCCCCCCC(=O)O1
557450
7
15-Methyl-Z-11-Hexadecenal
0.46
252
CC(C)CCC = CCCCCCCCCCC = O
5369022
8
1-Dodecanol
0.61
186
CCCCCCCCCCCCO
8193
9
1-Hepten-1-Ol, Acetate
0.36
156
CCCCCC = COC(=O)C
5463146
10
1-Octadecene
3.09
252
CCCCCCCCCCCCCCCCC = C
8217
11
1-Tetradecanol
1.82
214
CCCCCCCCCCCCCCO
8209
12
2-Pentadecanone, 6,10,14-Trimethyl-
0.47
268
CC(C)CCCC(C)CCCC(C)CCCC(=O)C
10408
13
3,7,11,15-Tetramethyl-2-Hexadecen-1-Ol
0.23
296
CC(C)CCCC(C)CCCC(C)CCCC(=CCO)C
5366244
14
5-Octadecene, (E)-
0.33
252
CCCCCCCCCCCC/C = C/CCCC
5364598
15
9-Octadecenal, (Z)-
0.76
266
CCCCCCCCCCCCC = CCCCC
5364492
16
9-Octadecenamide, (Z)-
0.9
281
CCCCCCCCC = CCCCCCCCC = O
5283454
17
9-Octadecenoic Acid (Z)-
0.7
282
CCCCCCCCC = CCCCCCCCC(=O)N
445639
18
9-Octadecenoic Acid, (E)-
2.56
884
CCCCCCCCC = CCCCCCCCC(=O)O
5364430
19
Benzene, 1,2-Dimethyl-
5.5
106
CCCCCCCCC = CCCCCCCCC(=O)O
7237
20
Benzene, Ethenyl-
4.96
104
C = CC1 = CC = CC = C1
12872668
21
Cholest-5-En-3-Ol (3.Beta.)-
4.1
386
CC(C)CCCC(C)C1CCC2C1(CCC3C2CC = C4C3(CCC(C4)O)C)C
5997
22
Cis-1-Chloro-9-Octadecene
4.68
286
CCCCCCCCC = CCCCCCCCCCl
5367784
23
Cis-9-Hexadecenal
4.24
238
CCCCCCC = CCCCCCCCC = O
5364643
24
Cyclopentadecanone, 2-Hydroxy-
0.66
240
C1CCCCCCC(C(=O)CCCCCC1)O
543400
25
Dodecanal
0.3
184
CCCCCCCCCCCC = O
8194
26
E,E,Z-1,3,12-Nonadecatriene-5,14-Diol
3.55
294
CCCCCC(C = CCCCCCCC(C = CC = C)O)O
5364768
27
Ethylbenzene
6.27
106
CCC1 = CC = CC = C1
7500
28
Heptadecanoic Acid
0.24
270
CCCCCCCCCCCCCCCCC(=O)O
10465
29
Hexadecanal
1.08
240
CCCCCCCCCCCCCCCC = O
984
30
Hexadecanamide
0.33
255
CCCCCCCCCCCCCCCC(=O)N
69421
31
Neophytadiene (2,6,10-Trimethyl,14-Ethylene-14-Pentadecne)
0.24
278
CC(C)CCCC(C)CCCC(C)CCCC(=C)C = C
10446
32
Pentadecanoic Acid
1.14
242
CCCCCCCCCCCCCCC(=O)O
13849
33
Phenol, 4-(2,2,3,3-Tetramethylbutyl)-
0.29
206
CC(C)(C)C(C)(C)CC1 = CC = C(C = C1)O
41234
34
Tetradecanal
3.66
212
CCCCCCCCCCCCCC = O
31291
35
Tetradecanoic Acid
1.06
228
CCCCCCCCCCCCCC(=O)O
11005
36
Z-(13,14-Epoxy)Tetradec-11-En-1-Ol Acetate
0.49
268
CC(=O)OCCCCCCCCCCC = CC1CO1
5363633
37
Z-2-Tridecen-1-Ol
0.54
198
CCCCCCCCCCC = CCO
5364998
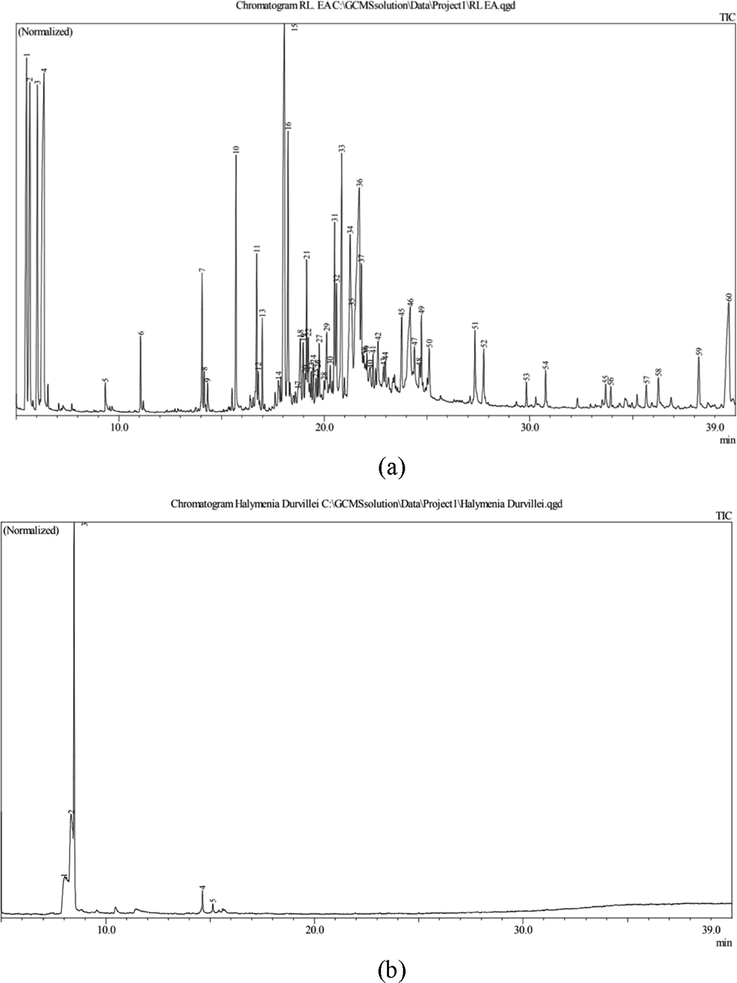
The chromatogram of GC-MS extraction of H. durvillei (a) ethyl acetate and (b) ethanol.
The 2-D chemical structure of natural compounds of H. durvillei.
The 2-D chemical structure of natural compounds of H. durvillei.
Most of the identified compounds are fatty acids or lipid constituting the most prominent phytochemicals from this seaweed as shown at Table 1 (see Fig. 3). This result is supported by the research of Chirasuwan et al. (2009) that showed lipid extracts to be potential antiviral agents and Schoggins and Randall (2013) reported that the roles of lipids in the antiviral response to infection are in two ways with directly inhibiting viral infection and regulating the adaptive and inflammatory responses. Moreover, according to recent study of SARS-CoV-2, the role of bioactive plasmalogens (vinyl ether glycerophospholipids) has the ability to enrich lung surfactant formulations to stimulate the respiratory process in severely infected individuals. Therefore, the role of lipid likely plasmogens can be suggested as an anti-viral prophylactic, a lipid biomarker in SARS-CoV and SARS-CoV-2 infections, and a potential anti-viral therapeutic component of lung surfactant development for COVID-19 patients (Deng and Angelova, 2021). These previous studies have shown the effectiveness of several lipid compounds as antiviral agent. Therefore, to confirm the potential of these compounds against viral activity, they were investigated to analyze their intermolecular interactions with the enzyme 3CL-Mpro of SARS-CoV-2 as ligand and protein through pharmacophore and molecular docking study.
A pharmacophore study to determine the biological features of inhibitors of the SARS-CoV-2 was applied and found to generate ligand-based pharmacophore models from the well-known drugs of SARS-CoV-2: remdesivir, lopinavir, ritonavir and hydroxychloroquine. Throughout the 10 generated models, the highest score was chosen as the template with 0.7886 scores using Ligand Scout calculation. The template model has at least two hydrogen donor and hydrogen acceptor features which were used in virtual screening approach. As a result, from 37 compounds of H. durvillei, only two passed the virtual screening accession: 1–2 tetradecandiol and E, E, Z-1,3,12-nonadecatrienome-5,14-diol with their pharmacophore fit at 38.95 and 38.99, respectively. The pharmacophore features (Fig. 4) of 1–2 tetradecandiol are two hydrogen donors and one hydrogen acceptor which interact with hydroxyl groups, respectively, while E, E, Z-1,3,12-nonadecatriene-5,14-diol has two hydrogen acceptors and one hydrogen donor. Furthermore, these two compounds as well as other compounds are the subject of continued study for their molecular interactions between protein and its inhibitors (ligands).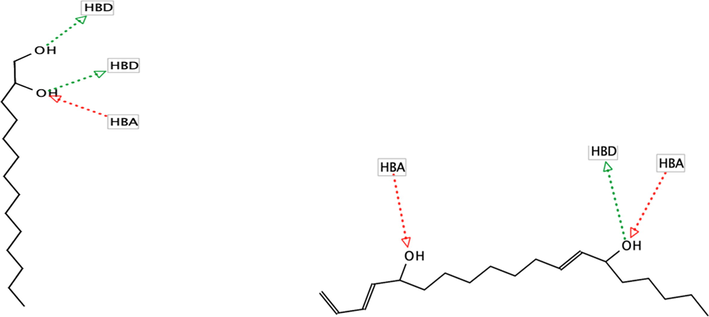
Pharmacophore features of (a) 1–2 tetradecandiol and (b) E,E,Z-1,3,12-nonadecatriene-5,14-diol.
Although only two compounds passed the pharmacophore-based virtual screening approach, the docking result of main protease 3CL-Mpro with the natural compounds of H. durvillei showed considerable fitness scores that are shown in Table 2. It appears that all the natural molecules in monomer docking showed good binding energy and have competitive fitness scores ranging from 27,43 to 17,73 in GOLD and binding affinity ranging from 2.9 to 5.0 in AutoDock, except the positive controls. In both calculation of GOLD and AutoDock, Cholest-5-En-3-Ol (3.Beta.)- is the strongest ligand with a fitness score of 27,43 (GOLD) and 5.0 binding affnity (AutoDock), while the hits compound from the screening approach, 1–2 Tetradecanadiol and E, E, Z-1,3,12-Nonadecatriene-5,14-diol have 21,13 and 25,65 fitness scores in GOLD and −3.6 and −3.8 binding affinity in AutoDock, respectively. On the other hand, N3 and remdesivir that are co-crystallized ligands from X-ray crystallography protein structure (PDB: 6LU7) and well-known drugs of SARS-Cov-2have fitness scores of 24,44 and 16,78 in GOLD, then –6.1 and –5.9 in AutoDock, respectively. This gives information that in GOLD calculation, they are below those from other natural compounds while their scores are just over from the others compounds at AutoDock calculation. The ligand efficiency (LE) calculation showed that the Cholest-5-En-3-Ol (3.Beta.)- is −0.18 kcal/mol, while N3 and remdesivir are −0.12 and −0.14, respectively. According to Schultes et al (2010), the LE calculation showed that some compounds are competitive with the positive controls, which means that the compound are prioritized as the hits compound according to their LE with smaller low affinity for further optimization step. Furthermore, to examine the interaction of these potential compounds in monomeric docking with their competitors (N3 and remsidivir) with active site residues, Fig. 5 illustrates different binding modes of protein–ligand complexes and residues type in Table 3.
Compounds
Monomer
Dimer
Ligand Efficiency
(kcal/mol_
GOLD
Autodock
Subunit1
Subunit2
Cholest-5-En-3-Ol (3.Beta.)-
27,43
−5.0
−7.3
−7.3
−0,18
E,E,Z-1,3,12-Nonadecatriene-5,14-Diol
25,65
−3.6
−5.1
−4.8
−0,28
Neophytadiene (2,6,10-Trimethyl,14-Ethylene-14-Pentadecne)
25,48
−3.4
−4.8
−5.1
−0,19
3,7,11,15-Tetramethyl-2-Hexadecen-1-Ol
25,25
−4.6
−5.1
−4.8
−0,22
1-Octadecene
24,84
−3.0
−3.6
−3.8
−0,17
5-Octadecene, (E)-
24,52
−3.0
−4.0
−3.7
−0,17
Cis-1-Chloro-9-Octadecene
24,52
−3.3
−4.1
−4.1
−0.17
N3 (positive control)
24,44
−6.1
−7.7
−7.6
−0,12
2-Pentadecanone, 6,10,14-Trimethyl-
23,92
−4.6
−4.7
−4.5
−0,24
15-Isopropenyloxacyclopentadecan-2-One
22,50
−5.0
−6.0
−6.0
−0,26
Z-(13,14-Epoxy)Tetradec-11-En-1-Ol Acetate
22,18
−4.8
−5.2
−5.0
−0,25
9-Octadecenal, (Z)-
22,02
−3.1
−4.6
−4.1
−0,16
9-Octadecenamide, (Z)-
21,90
−4.0
−5.0
−4.6
−0,18
Z-2-Tridecen-1-Ol
21,80
−3.8
−4.5
−4.0
−0,27
1,2-Tetradecanediol
21,70
−3.8
−4.6
−4.8
−0,24
9-Octadecenoic Acid, (E)-
21,68
−3.6
−4.8
−4.2
−0,16
15-Methyl-Z-11-Hexadecenal
21,65
−3.1
−4.6
−4.1
−0,17
Caryophyllene
21,57
−4.4
−5.1
−5.1
−0,29
9-Octadecenoic Acid (Z)-
21,36
−4.1
−4.3
−4.2
−0,21
Cis-9-Hexadecenal
21,23
−3.5
−4.2
−4.0
−0,18
1-Tetradecanol
21,13
−3.2
−4.5
−3.5
−0,21
1-Dodecanol
21,02
−3.7
−4.1
−3.8
−0,28
Heptadecanoic Acid
20,75
−3.7
−4.6
−4.2
−0,46
Hexadecanal
20,42
−4.0
−4.2
−4.3
−0,19
Phenol, 4-(2,2,3,3-Tetramethylbutyl)-
20,32
−4.5
−5.0
−5.6
−0,30
Hexadecanamide
20,30
−3.4
−4.5
−4.5
−0,24
Cyclopentadecanone, 2-Hydroxy-
19,99
−4.6
−5.8
−5.8
−0,27
1,4,7,-Cycloundecatriene, 1,5,9,9-Tetramethyl-, Z,Z,Z-
19,93
−4.4
−5.3
−5.3
−0,29
Benzene, Ethenyl-
19,90
−3.8
−3.9
−3.8
−0,35
N-Hexadecanoic Acid
19,65
−3.7
−4.4
−4.4
−0,21
Tetradecanal
19,44
−2.9
−3.8
−3.9
−0,19
Pentadecanoic Acid
19,31
−4.3
−4.3
−4.6
−0,25
Benzene, 1,2-Dimethyl-
19
−3.4
−3.9
−4.2
−0,43
Tetradecanoic Acid
18,96
−3.8
−4.5
−4.0
−0,24
Eucalyptol
18,72
−4.0
−4.4
−4.5
−0,36
1-Hepten-1-Ol, Acetate
17,94
−3.7
−4.4
−4.1
−0,34
Dodecanal
17,73
−3.0
−4.2
−3.6
−0,18
Ethylbenzene
16,82
−3.8
−3.9
−3.7
−0,18
Remdesivir
16,78
−5.9
−8.4
−7.9
−0,14
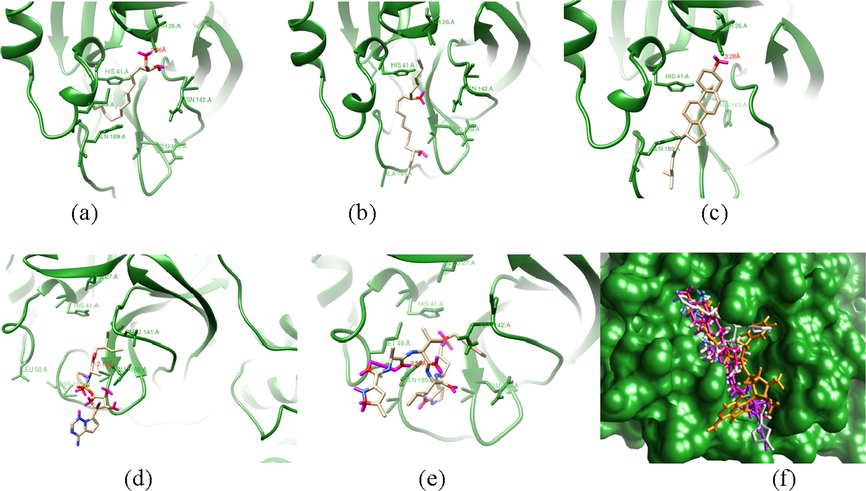
The binding modes of selected potential compounds within the active site of X-ray structure main protease (6LU7 pdb). (a) 1–2 tetradecandiol, (b) E,E,Z-1,3,12-nonadecatriene-5,14-diol, (c) Cholest-5-En-3-Ol (3.Beta.)-, (d) remdesivir , (e) NO3 and (f) superimposed image of the selected potential compounds.
Compounds
Residues within 5 Å
NO3
Thr24, Thr25, Thr26, Leu27, His41, Met49, Tyr54, Phe140, Leu141, Asn142, Gly143, Ser144, Cys145, His163, His164, Met165, Glu166, Leu167, Pro168, His172, Asp187, Arg188, Gln189, Thr190, Ala191, Gln192
CHOLEST-5-EN-3-OL (3.BETA.)-
Thr 24, Thr25, Thr26, Leu27, His41, Val42, Met49, Leu141, Asn142, Gly143, Ser144, Cys145, His163, His164, Met165, Glu166, Leu167, Pro168, Phe185, Val186, Asp187, Arg188, Gln189, Thr190, Ala191, Gln192, Ala193
1–2 Tetradecandiol
Thr24, Thr25, Thr26, Leu27, His41, Val42, Cys44, Asp48, Met49, Leu50, Pro51, Tyr54, Asn142, Gly143, Ser144, Cys145, His164, Met165, Glu166, Val186, Asp187, Arg188, Gln189
E,E,Z-1,3,12-Nonadecatriene-5,14-diol
Thr24, Thr25, Thr26, Leu27, His41, Val42, Met49, Gly143, Cys145, Tyr154, His164, Met165, Glu166, Leu167, Pro168, Val186, Asp187, Arg188, Gln189
Remdesivir
Thr24, Thr25, Thr26, Leu27, His41, Met49, Leu50, Phe140, Leu141, Asn142, Gly143, Ser144, Cys145, Gly146, His163, His164, Met165, Glu166, Leu167, Pro168, His172, Gln189, Thr190, Ala191
Allosteric Site
Residue within 5 Å
A
Arg131, Lys137, Thr198, Thr199, Ile200, Tyr237, Tyr239, Leu271, Leu272, Gly275, Met276, Ala285, Leu286, Leu287, Asp289,
B
Trp218, Phe219, Asn221, Phe223, Ser267, Glu270, Leu271, Asn274, Gly275, Asn277, Arg279
C
Phe3, Arg4, Lys5, Trp207, Ile281, Leu282, Gly283, Ser284, Glu288, Phe291
D
Phe8, Lys102, Val104, Arg105, Ile106, Gln107, Gln110, Thr111, Asn151, Ile152, Asp153, Ser158, Thr292, Phe294
E
Val13, Glu14, Gly15, Cys16, Met17, Val18, Trp31, Ala70, Gly71, Val73, Leu75, Asn95, Pro96, Lys97
F
Thr24, Thr25, Thr26, Leu27, His41, Ser46, Met49, Asn119, Asn142, Gly143, Cys145,
G
Phe140, Leu141, Asn142, Gly143, Ser144, Cys145, His163, Met165, Glu166, His172
Compounds
Monomer
Allosteric Site
1-Dodecanol
−4.0
D
1-Hepten-1-Ol, Acetate
−4.2
D
1-Octadecene
−3.5
D
1-Tetradecanol
−4.0
D
1,2-Tetradecanediol
−4.3
D
1,4,7,-Cycloundecatriene, 1,5,9,9-Tetramethyl-, Z,Z,Z-
−5.9
D
15-Isopropenyloxacyclopentadecan-2-One
−6.2
D
15-Methyl-Z-11-Hexadecenal
−4.6
D
2-Pentadecanone, 6,10,14-Trimethyl-
−4.7
A
3,7,11,15-Tetramethyl-2-Hexadecen-1-Ol
−4.4
D
5-Octadecene, (E)-
−3.9
D
9-Octadecenal, (Z)-
-4.3
D
9-Octadecenamide, (Z)-
−4.4
D
9-Octadecenoic Acid (Z)-
−4.6
D
9-Octadecenoic Acid, (E)-
−4.7
D
Benzene, 1,2-Dimethyl-
−4.6
D
Benzene, Ethenyl-
−4.2
D
Caryophyllene
−6.1
D
Cholest-5-En-3-Ol (3.Beta.)-
−7.8
A
Cis-1-Chloro-9-Octadecene
−4.0
D
Cis-9-Hexadecenal
−4.3
D
Cyclopentadecanone, 2-Hydroxy-
−5.9
A
Dodecanal
−3.8
D
E,E,Z-1,3,12-Nonadecatriene-5,14-Diol
−5.4
D
Ethylbenzene
−4.3
D
Eucalyptol
−5.0
D
Heptadecanoic Acid
−4.5
D
Hexadecanal
−4.4
D
Hexadecanamide
−4.8
D
N-Hexadecanoic Acid
−4.2
D
Neophytadiene (2,6,10-Trimethyl,14-Ethylene-14-Pentadecne)
−4.6
D
N3 (positive control)
−6.0
D
Pentadecanoic Acid
−4.6
D
Phenol, 4-(2,2,3,3-Tetramethylbutyl)-
−5.6
D
Remdesivir
−5.6
A
Tetradecanal
−4.5
D
Tetradecanoic Acid
−4.3
D
Z-(13,14-Epoxy)Tetradec-11-En-1-Ol Acetate
−5.2
D
Z-2-Tridecen-1-Ol
−4.1
D
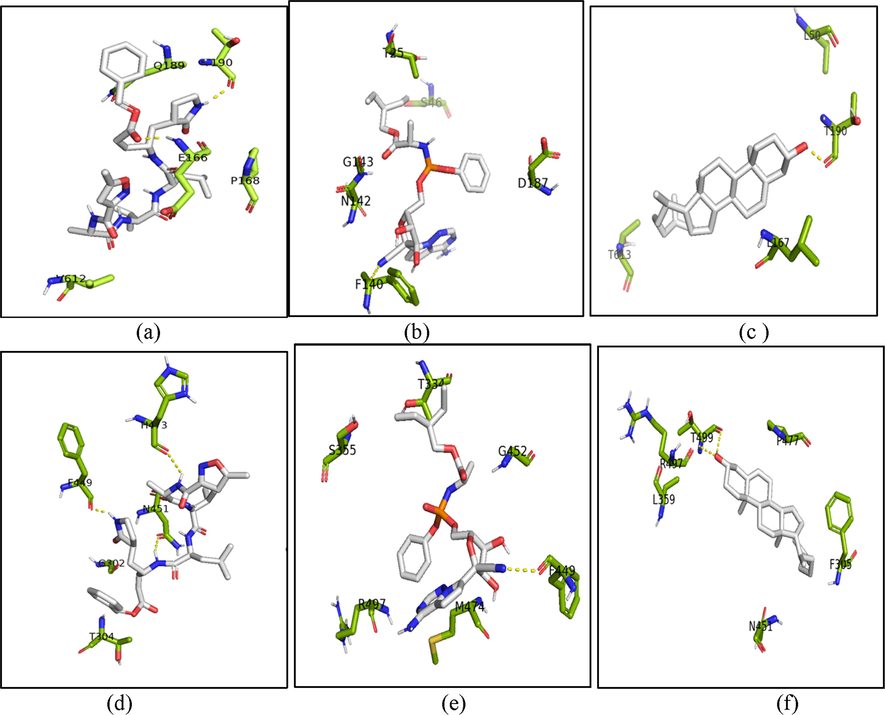
Interactions between complexes comprised of ligands and dimeric state of the 3CL-Mpro. The co-crystalized ligand N3 bound to subunit 1 (a) and 2 (d), remdesivir bound to subunit 1 (b) and 2 (e), and Cholest-5-En-3-Ol (3.Beta.)- bound to subunit 1 (c) and 2 (f) of dimer state of the 3CL-Mpro (6LU7 PDB). The active site residues of protein are shown in the green sticks visalization, the ligands are shown with grey stick representations and the interacting residues of hydrogen bonds within 3 Å are shown in yellow dashes. The figures were constructed with PyMOL 2.5.
In addition to analyzing the importance of the residues in the 3CL-Mpro Table 3 shows the classification of the residues in the active site from the different ligands. The importance of residues describes their contribution to the strength of the ligand in the protein cavity where they are significantly matched. Moreover, this list of active residues from candidate compounds and the competitors confirmed that they are also in the same cleft with similar active residues within 5 Å. The residues of the 3CL-Mpro are from number 24 to 192: THR24, THR25, THR26, LEU27, HIS41, MET49, GLY143, SER144, CYS145, HIS163, HIS164, MET165, GLU166, LEU167, PRO168, GLN189, THR190, ALA191 and GLN192 (see Fig. 5).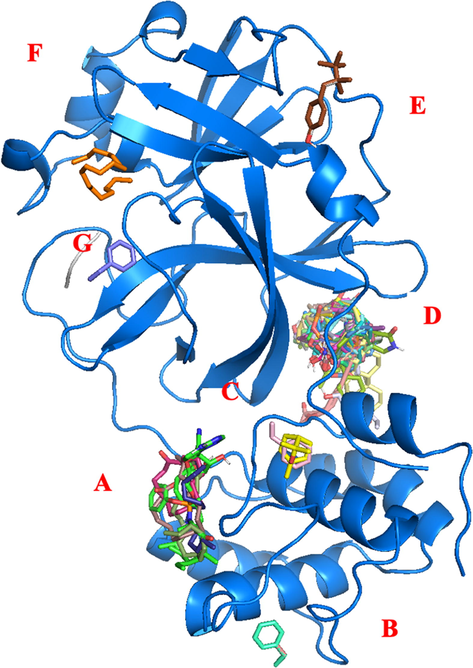
Allosteric site in monomer state.
Since the monomeric docking showed the potential result for natural compounds, this study also had done to calculate the data set of H. durvillei compounds in biological conformation of the 3CL-Mpro in dimer state. This study was purposed to identifying the binding affinity and molecular interaction of each compound bound to each subunit of 3CL-Mpro. In the Table 2, dimer docking calculation was done by AutoDock which was divided into subunit 1 and subunit 2. Docking studies between compounds, positive controls (N3 and remdesivir) and 3CL-Mpro (dimer state) showed that all ligands bound precisely into the active site into each subunit. The highest affinity score between natural compounds is Cholest-5-En-3-Ol (3.Beta.)- with −7.3 for each subunit. These scores compared with the positive controls of N3 and remdesivir are significantly competitive due to the scores of N3 in subunit 1 and subunit 2 are −7.7 and –7.6, while remdesivir showed the higher scores than other ligand with –8.4 and –7.9 at each subunit. These calculations were depicted in the Fig. 6 to analyze the molecular interaction between ligands and the protein.
Most of compounds are bound precisely into the active site, however, there are some compounds which found its site with low energy affinity score near the active site. In subunit 1, 1,4,7,-Cycloundecatriene, 1,5,9,9-Tetramethyl-, Z,Z,Z- and Benzene Ethenyl have another active site which 1,5,9,9-Tetramethyl-, Z,Z,Z- attach into the cleft of α3 and α4, while Benzene Ethenyl has the allosteric site near to the active site that is located between L14 and L15. Meanwhile, Phenol, 4-(2,2,3,3-Tetramethylbutyl)- and Benzene,1,2-Dimethyl- in subunit 2 were bound between domain II of subunit 2 and domain III of subunit 1. The states of both compounds are quite far from the active site with ± 14 Å distance.
N3 in subunit 1 was bound by residues in the active site and formed hydrogen bonds in 3 Å with backbone Thr190 and Glu166. On subunit 2, N3 has non-polar interaction with residues at the active site and polar interactions with the side chains Phe449, Asn451 and His473. Remdesivir has polar interaction with backbone atoms with Phe140 in subunit 1 and Phe449 in subunit 2. Moreover, for the highest score among other natural compounds, Cholest-5-En-3-Ol (3.Beta.)- bound to the backbone of Thr190 with hydrogen bond in subunit 1 and this compound formed two hydrogen bond atoms with backbone atoms O and N of Thr499.
On the other hand, to identify candidate inhibitors of 3CL-Mpro activity, this study performed a virtual screen by docking of natural compound from H. durvillei targetting the putative allosteric site of the protein in monomer state. The data set of natural compounds and other positive controls (N3 and remdesivir) were docked to the protein receptor. Eventually, the docking calculations of AutoDock present seven allosteric sites, including the active site. In Fig. 7, it is shown that most of ligands in the first conformation or low energy bound to the allosteric site A and D, while other sites B, C, E, F (active site) and G only bound to the ligands in others conformations (see Table 5). Nb: For the classification endpoints, the prediction probability values are transformed into six symbols: 0–0.1( − − −), 0.1–0.3(--), 0.3–0.5(-), 0.5–0.7(+), 0.7–0.9(++), and 0.9–1.0(+++).
Compounds
Mass (Da)
Rbond
HBA
HBD
PSA (Å 2)
LOGP
Acute Toxicity
Carcinogenicity
Eucalyptol
154
0
1
0
9.23
2.67
–
+
Caryophyllene
204
0
0
0
0.00
4.24
–
–
1,4,7,-Cycloundecatriene, 1,5,9,9-tetramethyl-, Z,Z,Z-
204
0
0
0
0.00
4.32
–
+ + +
N-Hexadecanoic Acid
256
14
2
1
37.30
5.20
–
- - -
1,2-Tetradecanediol
230
12
2
2
40.46
3.85
–
- - -
15-Isopropenyloxacyclopentadecan-2-One
266
1
2
0
26.30
4.71
–
–
15-Methyl-Z-11-Hexadecenal
252
13
1
0
17.07
5.35
–
–
1-Dodecanol
186
10
1
1
20.23
3.94
–
+ + +
1-Hepten-1-Ol, Acetate
156
6
2
0
26.30
2.54
–
+ + +
1-Octadecene
252
15
0
0
0.00
7.20
–
- - -
1-Tetradecanol
214
12
1
1
20.23
4.67
–
- - -
2-Pentadecanone,6,10,14-Trimethyl-
268
12
1
0
17.07
5.66
–
- - -
3,7,11,15-Tetramethyl-2-Hexadecen-1-Ol
296
13
1
1
20.23
6.22
–
–
5-Octadecene, (E)-
252
14
0
0
0.00
6.97
–
- - -
9-Octadecenal, (Z)-
266
14
0
0
0.00
6.97
–
- - -
9-Octadecenamide, (Z)-
281
15
1
0
17.07
5.94
–
–
9-Octadecenoic Acid (Z)-
282
15
1
1
43.09
5.32
–
- - -
9-Octadecenoic Acid, (E)-
884
15
2
1
37.3
5.71
–
- - -
Benzene, 1,2-Dimethyl-
106
15
2
1
37.3
5.71
–
- - -
Benzene, Ethenyl-
104
1
0
0
0.00
2.72
–
- - -
Cholest-5-En-3-Ol (3.Beta.)-
386
5
1
1
20.23
6.76
–
- - -
Cis-1-Chloro-9-Octadecene
286
15
0
0
0.00
7.03
–
–
Cis-9-Hexadecenal
238
13
1
0
17.07
5.18
–
–
Cyclopentadecanone,2-Hydroxy-
240
0
2
1
37.3
3.58
–
- - -
Dodecanal
184
10
1
0
17.07
3.94
–
–
E,E,Z-1,3,12-Nonadecatriene-5,14-Diol
294
14
2
2
40.46
4.94
–
+
Ethylbenzene
106
1
0
0
0.00
2.8
–
+
Heptadecanoic Acid
270
15
2
1
37.3
5.57
–
- - -
Hexadecanal
240
14
1
0
17.07
5.43
–
–
Hexadecanamide
255
14
1
1
43.09
4.83
–
- - -
Neophytadiene(2,6,10-Trimethyl,14-Ethylene-14-Pentadecne)
278
13
0
0
0.00
7.07
–
- - -
Pentadecanoic Acid
242
14
2
1
37.3
4.43
–
- - -
Phenol,4-(2,2,3,3-Tetramethylbutyl)-
206
3
1
1
20.23
3.79
–
–
Tetradecanal
212
12
1
0
17.07
4.67
–
–
Tetradecanoic Acid
228
12
2
1
37.3
4.45
–
- - -
Z-(13,14-Epoxy)Tetradec-11-En-1-Ol Acetate
268
13
3
0
38.83
4.03
–
–
Z-2-Tridecen-1-Ol
198
10
1
1
20.23
4.11
–
–
The residues of each allosteric sites of 3CL-Mpro are shown in Table 4. The identification was done by UCSF Chimera calculation of amino acids within 5 Å. Allosteric site A and B located at the domain III of receptor, allosteric site C and D located between domain II and III, and allosteric site E, F and G are between domain I and II. Allosteric site A bound with ligands in a cleft that is surrounded with α-helices 6 (residue 201–214) , 7 (residue 227–237), and 9 (261–274) with loop number L17 (residue 176–200), L19 (238–243) and L21 (275–292). Allosteric site B consists of L18 and α9. Allosteric site C is located between N-terminal loop (residue 1–10), L21 and β-sheets 9 (residue 121–131) and α6. Allosteric site D consists of L1, L11, L21, β7, β8, β9, β11. β12 and α10. Allosteric site E consists of α1, β1- β4, L8 and L10. Allosteric site F was included by α1, β1, β2, β4, β5, β7, β11, β12, L3, L10 and L17. The last allosteric site G consists L14, L16, L17 and β12- β13 (see Figure 7)
Compound
GI
BBB
P-glycoprotein substrate
CYP1A2 inhibitor
CYP2C19 inhibitor
CYP2C9 inhibitor
CYP2D6 inhibitor
CYP3A4 inhibitor
Cholest-5-En-3-Ol (3.Beta.)-
Low
No
No
No
No
Yes
No
No
E,E,Z-1,3,12-Nonadecatriene-5,14-Diol
High
Yes
Yes
Yes
No
Yes
Yes
Yes
Neophytadiene (2,6,10-Trimethyl,14-Ethylene-14-Pentadecne)
Low
No
Yes
No
No
Yes
No
No
3,7,11,15-Tetramethyl-2-Hexadecen-1-Ol
Low
No
Yes
No
No
Yes
No
No
1-Octadecene
Low
No
No
Yes
No
No
No
No
5-Octadecene, (E)-
Low
No
No
Yes
No
No
No
No
Cis-1-Chloro-9-Octadecene
Low
No
No
Yes
No
No
No
No
2-Pentadecanone, 6,10,14-Trimethyl-
High
No
Yes
No
No
Yes
No
No
15-Isopropenyloxacyclopentadecan-2-One
High
Yes
No
No
Yes
Yes
No
No
Z-(13,14-Epoxy)Tetradec-11-En-1-Ol Acetate
High
Yes
No
Yes
No
Yes
No
The allosteric site docking revealed that all ligands bound to the several sites of the receptors. However, most of the compounds showed that the other lowest energy was found at the allosteric site D rather than the other site. This phenomenon also has been shown by N3 that has the allosteric site in the D point and remdesivir was in the A point. Even though, most of compounds has the allosteric site to bound, the comparison of the lowest energy between allosteric sites and initial active site are competitive.
Regarding the ADME properties (see Table 6), the Lipsinki’s rule of five showed that most of natural compound of H. durvillei do not fulfill the Lipsinki properties. Even though molecular weight of each compound is not more than 500 kDa, number of HBA, HBD and PSA has fulfilled the standard of Lispinki’s rule of five. LogP and Rbond of some of compounds have higher value than the standard of Lipsinki’s rule of five which are not acceptable into the Lipsinki’s rule. This also happens to the Cholest-5-En-3-Ol (3.Beta.)- which is the most potential inhibitor of 3CL-Mpro with LogP which is little higher than the standard of LogP with more than 5.
Based on pharmacokinetics prediction result of the natural compound of H.durvillei in Table 7, some of the compounds have high ability in intestinal absorption. In gastrointestinal (GI) and blood–brain-barrier penetration properties only E,E,Z-1,3,12-Nonadecatriene-5,14-Diol, 2-Pentadecanone, 6,10,14-Trimethyl-, 15-Isopropenyloxacyclopentadecan-2-One and Z-(13,14-Epoxy)Tetradec-11-En-1-Ol Acetate have high absorption in digestion tract and penetration into the central nervous systems among the ten potential compounds, which would be delivered to the organ target blood barrier (blood-barrier of lung). Moreover, E,E,Z-1,3,12-Nonadecatriene-5,14-Diol, Neophytadiene (2,6,10-Trimethyl,14-Ethylene-14-Pentadecne), 3,7,11,15-Tetramethyl2-Hexadecen-1-Ol, and 2-Pentadecanone, 6,10,14-Trimethyl- have shown that these compounds has interaction with P-glycoprotein substrate which can cause drug-drug interactions and limiting celullar uptake of drugs from blood into brain and from digestion tract (intestinal lumen) to epithelial cells (Lin and Yamazaki, 2003). Meanwhile, most of compounds had no potential to inhibit P450 protein, however, there are still some compounds from the top ten candidate compounds that could inhibit each inhibitor of P450 protein. According to Bibi (2008), some of the prominent pharmacokinetic interactions between drugs are due to the P450 protein. The inhibitors of these types of protein can reduce metabolism, especially in drug-drug interactions.
Prior work has shown multiple advantages and health benefits of human consumption of red algae (Rhodophyta) such as cardiovascular, cancer, diabetes, anti-bacterial and anti-viral effects (Gamero-Vega et al., 2020) while another study also showed that marine red algae species have crucial anti-viral roles such as in HHV-1 viral DNA synthesis (Montanha et al., 2009). However, there are still lots of red algae with unknown potentials. This study has been conducted to examine one of the most common and largest tropical seaweed, H. durvillei by in silico approaches.
The study found from GC–MS results that ethanolic and ethyl acetate extraction yielded significantly different 3 and 34 compounds, respectively. Belkacami et al. (2020) showed different extraction results using methanol (polar) with 32,6 DC (dielectric constant) and chloroform (semi-polar) with 4,81 DC in the marine green alga Caulerpa racemosa (Forsskål) J. Agardh with methanolic extracts fewer than chloroform extracts. The same differences in polarity and DC were also observed in the current study which indicates that the phytochemicals extracted using semi-polar solvents like ethyl acetate (6 DC) and chloroform were higher than those obtained with polar solvent such as methanol and ethanol with 24,6 DC (Joshi and Adhikari, 2019).
In addition, the pharmacophore study revealed that inhibitor pharmacophore features should have a hydrogen bond donor as well as a hydrogen bond acceptor. Consequently, the virtual screening result of this study established two compounds: 1–2 tetradecandiol and E, E, Z-1,3,12-nonadecatriene-5,14-diol. Most notably, this study is the first to investigate the potential of Indonesian H. durvillei against SARS-CoV-2 through computational study. Our results provide a molecular docking approach in monomer and dimer state of 3CL-Mprotease by analyzing the compounds' molecular interactions. Throughout the result, our study revealed that H. durvillei compounds have a more remarkable fitness score than NO3 and remdesivir whether in monomeric or the biological state (dimeric) state of protein. The allosteric docking study identified that the protein has seven allosteric binding sites. Moreover, the fact that the highest abending affinity scores are from lipid has supported a previous study that lipid could be the potential drug against viruses, especially SARS-CoV-2. Based on the study of Han and Mallampalli (2015) lipid could act as lung surfactant by contributing 70% of its molecular weight. It has important role to stimulate the local pulmonary host defence mechanisms by playing as a barrier against adhesion of microorganisms and could improve the phagocytosis process by alveolar macrophages (inflammation).
Based on ADMET and Pharmacokinetic study, some compounds have not fulfilled the Lipinski's rule of five, while in pharmacokinetics identification, most of compounds are acceptable for absorption in digestion tract. Although our hypothesis is supported numerically, the number of compound types from this alga still needs to be identified and analyzed with more advanced molecular approaches. Future work should therefore include extraction using another solvent, such as non-polar ones and follow-up work in molecular dynamics simulation to assessing molecular behaviors between inhibitors and 3CL-Mpro protein. Moreover, following these promising results, some studies under in vitro and in vivo conditions must now be carried out to further investigate the effectiveness of active compounds in H. durvillei in the treatment of COVID-19. Importantly, the molecular dynamics study and MMGBSA or MMPBSA calculations should be performed for further advanced investigation in in silico study.
4 Conclusions
As noted earlier, the 37 phytochemicals derived from Halymenia durvillei that target the 3CL-Mpro of SARS-CoV-2 are able to interact with binding sites of the target protein effectively as well as exhibit competitive fitness scores. Therefore, the outcomes from computational-based drug discovery could be made a reference for the molecular interaction of the main protease with its ligand inhibitors against SARS-CoV-2. These initial promising results should be assessed further and validated using in vitro and in vivo approaches to better define their treatment mechanism of COVID-19.
Acknowledgments
This research was funded by the COVID-19 Research and Innovation Consortium, Ministry of Research and Technology/National for Research and Innovation Agency (Kemenristek/BRIN) and the Fundamental Research, The Ministry of Education, Culture, Research, and Technology (Kemdikbudristek). The authors want to thank Professor Nicholas Paul (University of the Sunshine Coast, Australia) for his constructive inputs on an earlier version of this manuscript.
Reference
- Review on up-to-date status of candidate vaccines for COVID-19 disease. Infect. Drug Resist.. 2021;14:151-161.
- [CrossRef] [Google Scholar]
- Antioxidant and antibacterial activities and identification of bioactive compounds of various extracts of Caulerpa racemosa from Algerian coast, Asian Pac. J. Trop. Biomed.. 2020;10:87-94.
- [CrossRef] [Google Scholar]
- Molecules against Covid-19: An in silico approach for drug development. J. Electron. Sci. Technol.. 2021;19(1):100095.
- [CrossRef] [Google Scholar]
- Antioxidant and antibacterial activity of red seaweed; Kappaphycus alvarezii against pathogenic bacteria. Global J. Environ. Sci. Manage.. 2020;6(1):47-58.
- [Google Scholar]
- Role of cytochrome P450 in drug interactions. Nutrition Metabol.. 2008;5(1):27.
- [CrossRef] [Google Scholar]
- Pharmacokinetic study with computational tools in the medicinal chemistry course. Brazilian J. Pharm. Sci.. 2011;47(4):797-805.
- [CrossRef] [Google Scholar]
- Application prospect of polysaccharides in the development of anti-novel coronavirus drugs and vaccines. Int. J. Biol. Macromol.. 2020;164:331-343.
- [Google Scholar]
- Anti HSV-1 activity of sulphoquinovosyl diaclyglycerol isolated from Spirulina platensis. ScienceAsia. 2009;35(2):137.
- [CrossRef] [Google Scholar]
- In vitro antiviral activity against Zika virus from a natural product of the Brazilian brown seaweed Dictyota menstrualis, Natural Prod . Comm.. 2019;14(7)
- [CrossRef] [Google Scholar]
- WHO declares COVID-19 a pandemic. Acta Bio Medica: Atenei Parmensis.. 2020;91(1):157.
- [Google Scholar]
- Deepak, P., Sowmiya, R., Kamaraj, C., Josebin, M.P.D., Aiswarya, D., Balasubramani, G., Amutha, V., Perumal, P., 2018, GC-MS profiling, chemical characterization, antioxidant, alpha-amylase and alpha- glucosidase inhibition of selected seaweeds from southeast coast of India : An in vitro approach, J.Drug Delivery Therapeutics, 8(2), 60-72. https://doi.org/10.22270/jddt.v8i2.1665
- Coronavirus-induced host cubic membranes and lipid-related antiviral therapies: A focus on bioactive plasmalogens. Front. Cell Dev. Biol 2021
- [CrossRef] [Google Scholar]
- Gamero-Vega, G., Palacios-Palacios, M., Quitral, V., 2020 Nutritional composition and bioactivecompounds of red seaweed: A mini-review., J. Food Nutr. Res. 8(8), 431-440.
- Putative inhibitors of SARS-CoV-2 main protease from a library of marine natural products: A virtual screening and molecular modeling study. Mar. Drugs. 2020;18(4):225.
- [CrossRef] [Google Scholar]
- Gorbalenya, A.E., Baker, S.C., Baric, R.S., Groot, R.J. de, Drosten, C , Gulyaeva, A.A. , Haagmans, B.L. , Lauber, C. , Leontovich, A.M., Neuman, B.W., 2020. Severe acute respiratory syndrome-related coronavirus: The species and its viruses–a statement of the Coronavirus Study Group. .
- Hans, N., Malik, A., Naik, S. 2021. Bioresource Technology Reports Antiviral activity of sulfated polysaccharides from marine algae and its application in combating COVID-19: Mini review. Bioresource Technol. Rept. 13(October 2020),100623.
- The role of surfactant in lung disease and host defense against pulmonary infections. Annals of the American Thoracic Society. 2015;12(5):765-774.
- [CrossRef] [Google Scholar]
- Covid-19: New UK variant may be linked to increased death rate, early data indicate. BMJ. 2021;372(230):n230
- [Google Scholar]
- Coronavirus disease (COVID-19): Current status and prospects for drug and vaccine development. Arch. Med. Res.. 2020;52(1):15-24.
- [Google Scholar]
- Impact of age, ethnicity, sex and prior infection status on immunogenicity following a single dose of the BNT162b2 mRNA COVID-19 vaccine: Real-world evidence from healthcare workers, Israel, December 2020 to January 2021. Eurosurveillance.. 2021;26(6):1-5.
- [Google Scholar]
- Jean, S., Hsueh, P. 2020. Old and re-purposed drugs for the treatment of COVID-19. Expert Rev. Anti Infect. Ther. 18(9), 843–848. 2020.1771181
- Can sulphated polysaccharides from seaweed provide prophylactic and/or therapeutic solution to COVID-19 pandemic? Current Sci.. 2020;119(2):1-2.
- [Google Scholar]
- An overview on common organic solvents and their toxicity. J. Pharm. Res. Int.. 2019;28(3):1-18.
- [CrossRef] [Google Scholar]
- Kaserer, T., Beck, K.R., Akram, M., Odermatt, A:, & Schuster, D., 2015, Pharmacop models and pharmacophore-based virtual screening: Concepts and applications exemplified on hydroxysteroid dehydrogenases, Molecules, 20 : 22799–22832; doi:10.3390/molecules201219880
- COVID-19 Vaccine: A comprehensive status report. Virus Res.. 2020;288:198114.
- [CrossRef] [Google Scholar]
- New variant of SARS-CoV-2 in UK causes surge of COVID-19. Lancet Respiratory Med.. 2021;9(2):e20-e21.
- [Google Scholar]
- Role of P-glycoprotein in pharmacokinetics: clinical implications. Clin. Pharmacokinet.. 2003;42(1):59-98.
- [CrossRef] [Google Scholar]
- Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections. Nature Med.. 2020;26(8):1200-1204.
- [Google Scholar]
- Coronavirus disease 2019 (COVID-19): emerging and future challenges for dental and oral medicine. J. Dental. Res.. 2020;99(5):481-487.
- [Google Scholar]
- Antiviral activity of carrageenans from marine red algae. Lat. Am. J. Pharm.. 2009;28(3):443-448.
- [Google Scholar]
- Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal.. 2020;10(4):313-319.
- [CrossRef] [Google Scholar]
- Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J.Comput.Chem. 2009;30(16):2785-2791.
- [Google Scholar]
- Screening marine algae metabolites as high – affinity inhibitors of SARS – CoV – 2 main protease (3CLpro): an in silico analysis to identify novel drug candidates to combat COVID – 19 pandemic. Applied. Biolog. Chem.. 2020;63(1)
- [CrossRef] [Google Scholar]
- Olliaro, P., Torreele, E. and Vaillant, M., 2021. COVID-19 vaccine efficacy and effectiveness—the elephant (not) in the room. The Lancet Microbe.
- Palese, L.L. 2020. The structural landscape of SARS-CoV-2 main protease: hints for inhibitor search. ChemRxiv.
- The COVID 19 novel coronavirus pandemic 2020: seaweeds to the rescue? Why does substantial, supporting research about the antiviral properties of seaweed polysaccharides seem to go unrecognized by the pharmaceutical community in these desperate times? J. Appl. Phycol.. 2020;32:1875-1877.
- [Google Scholar]
- Phytosomal nanocarriers as platforms for improved delivery of natural antioxidant and photoprotective compounds in propolis: An approach for enhanced both dissolution behaviour in biorelevant media and skin retention profiles. J. Photochem. Photobiol. B Biol.. 2020;205:111846.
- [CrossRef] [Google Scholar]
- Antiviral activity of the extracts of Rhodophyceae from Morocco. African J. Biotechnol.. 2010;9(46):7968-7975.
- [CrossRef] [Google Scholar]
- Purification and analysis of patchouli alcohol from patchouli oil by vacuum fractionation distillation. J. Phys. Conf. Series. 2019;1341:052016.
- [CrossRef] [Google Scholar]
- Lipids in innate antiviral defense. Cell Host Microbe. 2013;14(4):379-385.
- [CrossRef] [Google Scholar]
- Ligand efficiency as a guide in fragment hit selection and optimization. Drug Discovery Today Technol.. 2010;7(3):e157-e162.
- [CrossRef] [Google Scholar]
- Molecular docking, validation, dynamics simulations, and pharmacokinetic prediction of natural compounds against the SARS-CoV-2 main-protease. J. Biomol. Struct. Dyn. 2020
- [CrossRef] [Google Scholar]
- Unique epidemiological and clinical features of the emerging 2019 novel coronavirus pneumonia (COVID-19) implicatespecial control measures. J. Med. Virol.. 2020;92(6):568-576.
- [Google Scholar]
- WHO coronavirus disease (COVID-19) dashboard. Geneva: World Health Organization; 2021. Available: https://covid19.who.int/.
- WHO coronavirus disease (COVID-19) Draft landscape of COVID-19 candidate vaccines. Geneva: World Health Organization; 2021. Available: https://www.who.int/publications/m/item/ draft-landscape-of-covid-19-candidate-vaccines.
- Wu, R. , Wang, L. , Kuo, H.D. , Shannar, A. , Peter, R. , Chou, P.J. , Li, S. , Hudlikar, R. , Liu, X. , Liu, Z. , Poiani, G.J. , Amorosa, L. , Brunetti, L. & Kong, A.-N. 2020. An update on current therapeutic drugs treating COVID-19. Current Pharmacology Reports (2020). (6):56–70.
- Biologically active compounds from marine organisms in the strategies for combating coronaviruses. AIMS Microbiology. 2020;6(4):470-494.
- [CrossRef] [Google Scholar]
- A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270-273.
- [Google Scholar]




