

Translate this page into:
Potential inhibition of SARS-CoV-2 infection and its mutation with the novel geldanamycin analogue: Ignaciomycin
⁎Corresponding authors. antonystalin@uestc.edu.cn (Antony Stalin), a.staanlin@gmail.com (Antony Stalin), imuthus@hotmail.com (Savarimuthu Ignacimuthu), ignacimuthu@stxavierstn.edu.in (Savarimuthu Ignacimuthu), zouquan@nclab.net (Quan Zou) zouquan@uestc.edu.cn (Quan Zou)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
The impact of novel coronavirus (SARS-CoV-2) is very high; its mutant variants provide a higher transmission rate. Due to many mutations in the omicron-variant, it can evade previously available neutralizing antibodies. The inhibitory effect of the novel compound “Ignaciomycin” was investigated using various computational analyses against the RBD of the SARS-CoV-2 wild-type, Delta, and omicron-variants. Molecular docking revealed the potentially stable interaction of Ignaciomycin with an energy value of −8.65 kcal/mol for the omicron-variant. DFT and Hirshfeld surface calculations showed higher efficiency and reactivity with strong electrostatic potential values. In-silico toxicity studies revealed a significant drug-likeness score (7.5912) with non-toxic properties. MD simulation studies confirmed the stability of Ignaciomycin during ∼ 100 ns simulation. Covariance, PCA, and FEL analyses revealed significant fluctuations in residues and atom mobility in all variants based on the strong interaction of Ignaciomycin. Mutations in the Delta and omicron-variants increased the binding efficiency of the RBD of the SARS-CoV-2 S-glycoprotein with human ACE2. Ignaciomycin had high efficiency in interacting with mutated sites in the RBD, thereby blocking the interaction of the RBD with ACE2. Our findings provide a strong hypothesis for preclinical validation of Ignaciomycin as a potential drug against SARS-CoV-2 and its mutant variants.
Keywords
SARS-CoV-2
Mutations
Ignaciomycin
Molecular docking
Dynamics simulation
Viral inhibitor

1 Introduction
The coronavirus (COVID-19) pandemic caused by SARS-CoV-2 continuously disturbs human health and the world's economic condition. According to the COVID-19 Data Repository developed by Johns Hopkins University (JHU) (Dong et al., 2020), there have been approximately 630 million infected cases and 6 million deaths by 2022. So far, developed vaccines have quietly controlled death rates. However, the virus emerges with various mutations and increased interaction between the human ACE2 receptor, thus evading the neutralizing antibodies (Shang et al., 2020, Yan et al., 2020, Yang et al., 2021, Zhang et al., 2021, Chen et al., 2022, Lupala et al., 2022). The World Health Organization (WHO) continuously monitors all mutant variants and designates some of the essential variants as “variants of concern” (VOC). Of all mutant variants, Delta (B.1.617.2) and Omicron (B.1.1.529) are the crucial mutants. Omicron is the current VOC strong interaction with the ACE2-receptor based on the mutations in the receptor binding domain (RBD) of the spike (S) glycoprotein, which significantly neutralize antibodies and form strong electrostatic and hydrophobic surfaces in the receptor binding motif (RBM) of the spike protein (Cao et al., 2022a, Cheng et al., 2022). There are more than 30 mutations in Omicron, and about 15 mutations are in the RBD region (Cao et al., 2022b, Ren et al., 2022).
The spike protein is the critical hotspot antigenicity site for many drugs and antibodies. Due to the unprecedented mutations and the need to develop novel antibodies and drugs targeting the emerging mutations and their resistances and consider immune escape (Lupala et al., 2022). The main thing is that SARS-CoV-2 is a respiratory disease that majorly affects the lungs; at the same time, it can also affect other organs because of immune suppression (Arokiyaraj et al., 2020, Bajo-Morales et al., 2022, Jacob et al., 2022). Due to the neutralizing antibody evasion by the omicron variant, there is an urgent need to develop novel drugs with significant activity. Numerous research reports revealed the inhibitory properties of various natural products from plants, fungi, bacteria, and nanoparticle-based vaccines and drugs against bacterial and viral infections, including SARS-CoV-2 (Arokiyaraj et al., 2020, Nikaeen et al., 2020, Tam et al., 2021, Yousefi et al., 2021a, Yousefi et al., 2021b, Zulfiqar et al., 2021, Akter et al., 2022, Mahdi et al., 2022, Stalin et al., 2022b, Xu et al., 2023).
Geldanamycin belongs to the group of ansamycins, a class of benzoquinones, and is known for its antimicrobial and anticancer activities. However, the use of geldanamycin is restricted due to its hepatotoxicity and other undesirable effects (Taechowisan et al., 2020, Skrzypczak et al., 2021). In addition, geldanamycin is a major inhibitor of heat shock protein 90 (HSP90) and also has antiviral properties against viruses such as influenza and HIV-1. Therefore, geldanamycin analogues and derivatives are continuously being developed to increase its efficacy and reduce side effects (Connor et al., 2007, Li et al., 2012, Sultan et al., 2020).
In the current study, the newly obtained compound “Ignaciomycin” (an analogue of geldanamycin) from the terrestrial Streptomyces sp. CFR16 was investigated for its inhibitory effect against SARS-CoV-2 infection. The RBD of wild-type SARS-CoV-2 S-glycoprotein (https://www.rcsb.org/structure/6M0J), Delta (https://www.rcsb.org/structure/7V8B) and Omicron (https://www.rcsb.org/structure/7T9L), was chosen for the in silico molecular docking analysis, followed by molecular dynamics simulation and covariance matrix with the principal component analysis. In addition, a molecular mechanics Poisson-Boltzmann Surface Area (MM-PBSA) analysis was performed and the perturbation of the free energy and the decomposition of the energy contribution per residue were calculated to confirm the inhibitory effect of the compound Ignaciomycin.
2 Material and methods
2.1 Ligand preparation
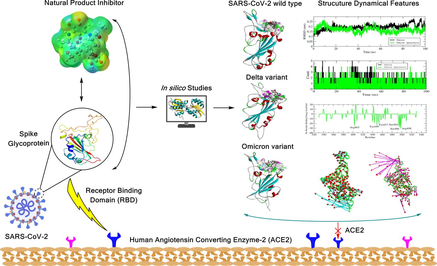
The newly derived geldanamycin analogue compound named Ignaciomycin, which was isolated from terrestrial Streptomyces sp. CFR16 was chosen for computational studies to predict its efficacy against COVID-19. The structural configuration of the Ignaciomycin (CCDC Deposition Number: 2218814) was obtained from single-crystal X-ray diffraction (XRD) analysis (Pachaiyappan, 2022) (Fig. 1). The conformer PDB format structure was derived, and its frontier HOMO, LUMO orbitals energies, and electrostatic potential was computed using the Gaussian09 program with density functional theory (DFT) and the basis set of B3LYP/ 6-31G(d,p) (M. J. Frisch, 2016, R.D. Dennington, 2016). Besides, Hirshfeld surface analysis was calculated to analyze the inter- and intramolecular interactions using CrystalExplorer-3.1 (Spackman et al., 2021).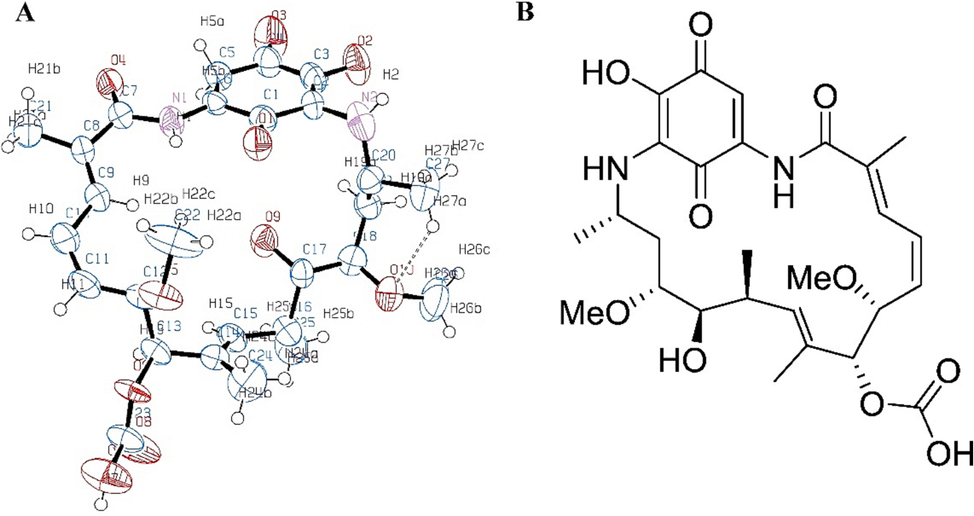
Structure of Ignaciomycin. (A) ORTEP diagram of the newly derived Geldanamycin analogue compound Ignaciomycin. (B) 2D structure of Ignaciomycin.
2.2 Protein preparation and molecular docking
The Receptor binding domain (RBD) of the SARS-CoV-2 spike glycoprotein of the wild-type (PDB ID:6M0J), Delta variant (PDB ID:7V8B), and Omicron variant (PDB ID: 7T9L) was chosen for this study. The structures were loaded into AutoDock software (Morris et al., 2009), water molecules and co-crystalized compounds were removed, and polar and nonpolar hydrogens, Kollman charges, and Gasteiger charges were added. COACH and CASTP servers (Dundas et al., 2006, Yang et al., 2013a, Yang et al., 2013b) were used to define the active sites. Then, the ligand parameters were included, and grid boxes (the number of grid points 100x100x100) were formed to cover the active sites. Further, the coordinates of the XYZ grid dimensions were set for the wild-type (x-33.507, y-30.09, z-8.015), Delta variant (x-186.556, y-193.596, z-278.008), and Omicron variants (x-228.181, y-175.319, z-251.794) with the default distance (0.375 Å) parameters.
The prepared ligand and proteins were used for the molecular docking analysis using autoDock4 (Morris et al., 2008, Stalin et al., 2020, Stalin et al., 2022b). The protein molecule’s rigidity was maintained, and the ligand molecule was treated flexibly. The default docking parameters and Lamarck's Genetic Algorithm were used to run the docking for 250,000 evaluations. The least energy-docked confirmations of ligand–protein complexes were screened, and their interactions were analyzed.
2.3 Molecular dynamics simulation
After the docking analysis, the finalized complex of the compound Ignaciomycin with the RBD of the SARS-CoV-2 spike glycoprotein of the wild-type, Delta variant, and Omicron variant was used for the molecular dynamics simulation (GROMACS-2018.6 package) (Van Der Spoel et al., 2005, Abraham et al., 2015, Stalin et al., 2022a) to determine the confirmational trajectory changes in the proteins.
The ligand topology was generated using the CHARMM General Force Field (CGenFF) server 4.6 (Vanommeslaeghe et al., 2010) and the protein topology was developed with the CHARMM36-jul2021 forcefield (Huang et al., 2017). The system was neutralized with 0.15 M sodium chloride ions. Then, the system was energy minimized with 50000 ns steps using the steepest descent minimization algorithm. Then, the system was equilibrated by the NVT (substance amount, volume, and temperature) and the NPT (substance amount, pressure, and temperature) ensembles at 300 K and 1.0 bar pressure for 50000 ns steps each with the Particle Mesh Ewald (PME) coulomb type and Verlet cut-off scheme (Darden et al., 1993). Then, the system performed a ∼ 100 ns simulation in the ubuntu server 20.0.4 LTS with an Intel(R) Xeon(R) CPU E5-2620 v3 @ 2.40 GHz processor. Grace Plotting Tool (https://plasma-gate.weizmann.ac.il/Grace/) was used to analyze the simulation data and develop the figures for RMSD (root mean square deviation), RMSF (root mean square fluctuation), the average number of H-bonds, the solvent-accessible surface area, and the radius of gyration. Data analysis of the covariance matrix and free energy landscape (FEL) was developed using the gmx anaeig and sham tools, and the associated eigenvectors and positions of the lowest energy conformations were determined. Discovery Studio 2021 client, PyMol, and UCSF Chimera 1.16 tool were used to visualize the binding information of the docked and simulated structures and Mathematica software was used to generate the FEL plots.
2.4 Molecular mechanics Poisson-Boltzmann surface area (MMPBSA) analysis
To determine the free energy binding affinity, the last 2000 frames of the 100 ns simulated complex of the compound Ignaciomycin and the three RBD of the SARS-CoV-2 spike glycoprotein of the wild-type, Delta variant, and Omicron variant were analyzed by g_MMPBSA software (Kumari et al., 2014) using Molecular mechanics Poisson − Boltzmann surface area (MM-PBSA) calculations.
3 Results
3.1 Hirshfeld surface analysis
The structural configuration of the Ignaciomycin (CCDC Deposition Number: 2218814) was obtained from single-crystal X-ray diffraction (XRD) analysis (Pachaiyappan, 2022) (Fig. 1). The surface and close contacts of atoms in Ignaciomycin were studied by Hirshfeld surface analysis. The points on the molecular surface ranged from red to white to blue, showing intermolecular contacts with the corresponding internuclear distance smaller, equal, and larger than the Van Der Waal radii. The dnorm, shape index, and curvedness of Hirshfeld molecular surface analysis for the compound Ignaciomycin are shown in Figure (Fig. 2). These parameters are shown in ranges from −0.592 to 6.208, −1.0000 to 1.0000, and −4.0000 to 0.4000, respectively.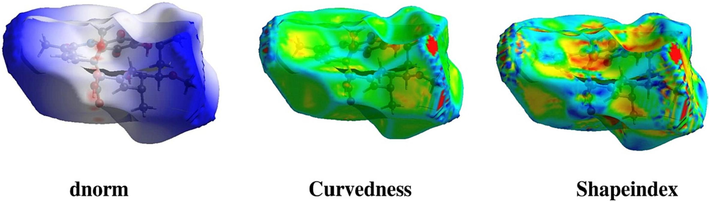
Physiochemical structural analysis. Hirshfeld diagram over dnorm, Curvedness, and Shape index of Ignaciomycin.
Two-dimensional (2D) fingerprint diagrams were also constructed to analyze the intermolecular atomic contacts in the Ignaciomycin crystal structure. The distance between the Hirshfeld surface and the nearest atomic nucleus within the surface is called the internal distance (di). The distance between the Hirshfeld surface and the outside of the surface is called the external distance (de). These two terms, di and de, in the 2D fingerprint diagram reveal more information about the molecular surface based on the interatomic contacts.
The 2D fingerprint diagrams show that intermolecular interactions such as H…H, H…O, O…O, C…H, N…H, and C…C clearly dominate the graphical Hirshfeld surfaces (Fig. 3). In addition to a 2D fingerprint map of Ignaciomycin, Fig. 3 illustrates the percentage contribution of selected interatomic contacts to the Hirshfeld surface region for Ignaciomycin. Using the most accurate 2D fingerprint diagrams available, the contribution of the intermolecular interactions H…H (40.9 %), H…O (20.7 %), O…O (2.9 %), C…H (2.5 %), C…O (1.8 %), H…N (0.9 %), and C…C (1.4 %) that make up the entire Hirshfeld surface is carefully examined. The entire Hirshfeld surface of the molecule was covered by the intermolecular interactions of H…H and H…O in proportions of 40.9 % and 20.7 %, respectively. The deep red spots on the dnorm Hirshfeld surfaces indicate that the main intermolecular hydrogen bonding is primarily due to H…O/O…H close-contact interactions.
Intramolecular atomic structural interaction analysis. Two-dimensional (2D) Fingerprint plot overall interactions and individual interactions in crystal packing of Ignaciomycin.
The molecule contains an intramolecular hydrogen bond between O10 and H27a (2.552 Å). H…O/O…H contacts contribution is 20.7 %, which plays a substantial role in crystal packing. The set of 60 potential intermolecular hydrogen bonds, including long range of force of attractions, are responsible for packing the crystal, and the interactions are shown in the Supplementary section (SI Fig. 1).
3.2 HOMO-LUMO analysis
The energies of the frontier orbitals can be easily correlated with various parameters of the molecules. The larger the EHOMO value, the greater the molecule's ability to donate electrons, and the smaller the ELUMO value, the greater the ability to accept electrons. The considerable ΔE value indicates moderate kinetic stability and lowers chemical reactivity. The electronic transition absorption corresponds to the transition from HOMO to LUMO by electron transfer (Fig. 4A).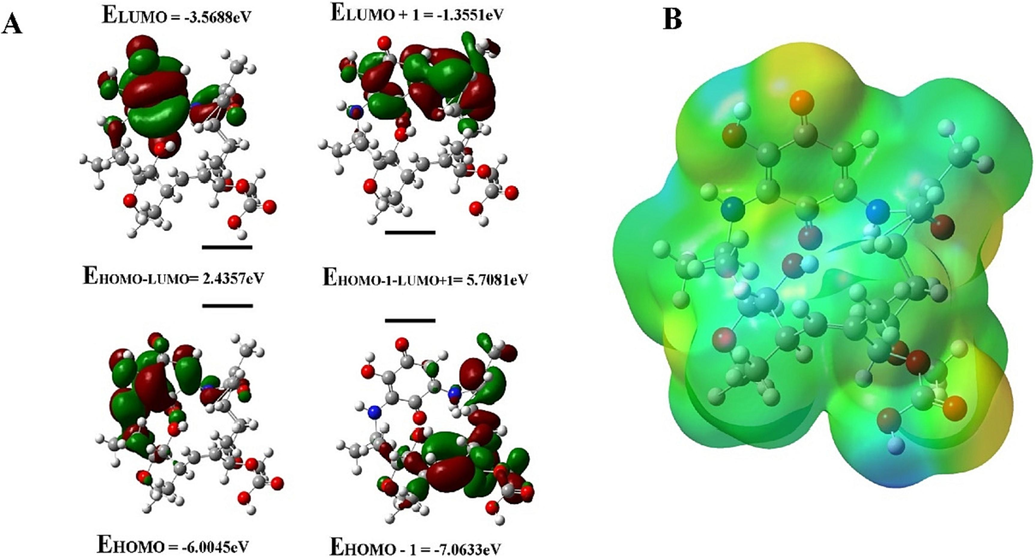
HOMO and LUMO energy calculation. (A) Electron density distribution and the energies (eV) of HOMOs and LUMOs of Ignaciomycin (The red and green colors represent the positive and negative phases of the molecule, respectively). (B) The 3D molecular electrostatic potential surface identifies the active sites for electrophilic and nucleophilic attack for Ignaciomycin in optimized geometry.
In the compound Ignaciomycin, the electron density of HOMO (-6.0045 eV) is distributed over the quinone and the adjacent side chain. At the same time, HOMO −1 (-7.0633 eV) of Ignaciomycin is mainly distributed over the carbonyl group involved in conjugation with the double bonds of the side chain. The LUMO (-3.5688 eV) is primarily located on the quinone ring and is weakly distributed over a close side chain with an amide bond. The LUMO + 1 (-1.3551 eV) is distributed over the quinone ring and is partially distributed over the close side chain. The calculated HOMO-LUMO energy gap is 2.4357 eV (Fig. 4A).
3.3 Chemical reactivity
The frontier orbital energies clearly show various properties, such as chemical potential (μ), chemical hardness (η), and electrophilicity index (ω), which are listed in Table 1. The above properties are calculated using HOMO and LUMO energies with the help of the Koopmans theorem and Parr approximation.
Compound
Ignaciomycin
EHOMO (eV)
−6.0045
ELUMO (eV)
−3.5688
ELUMO-EHOMO
2.4357
Chemical Potential μ (eV)
−4.787
Chemical Hardness η (eV)
1.21785
Electrophilicity ω (eV)
9.407
Chemical Softness S (eV)
0.411
3.4 Electrostatic potential analysis
The molecular surface with red markings has a high negative ESP surface (electron-rich centers) and acts as a nucleophilic center. The molecular surface with blue hues has a positive ESP surface (electron-poor centers) and serves as an electrophilic center. Green shades represent neutral sites. Thus, the molecular electrostatic potential calculation was assessed to identify the active sites for electrophilic and nucleophilic attack for Ignaciomycin in optimized geometry. As shown in Fig. 4B, the blue < green < red sequence on the molecular surface of Ignaciomycin shows an increasing potential, indicating various sites of negative nature at O1 and O3 in the quinone ring, O4 of the amide and O6, O7, O8 of the carboxylic acid. Positive nature is easily found at H2 of the amine, H2a of the hydroxyl group, and H7 of the carboxylic acid atoms of butyrolactone ring. These positive centers in systems can interact with nucleophilic centers and are involved in H-bonding with proteins of pathogens.
3.5 Protein-Protein interaction
Here, we investigated the H-bonding interactions between the S-glycoprotein of Delta and Omicron with the ACE2-receptor (Table 2). In the Delta, Tyr449, Gln493, Gly496, and Thr500 showed H-bonds with Asp38, Lys31, Lys353, and Tyr41; in Omicron, Tyr449, Arg493, Arg498, and Gly502 showed H-bonds with Asp38, Glu35, Asp38, and Lys353, with the ACE2-receptor. Interestingly, no hydrophobic interactions, salt-bridges, and unfavorable electrostatic interactions with the ACE2-receptor occurred with Delta (SI-Table 2). The Omicron-ACE2-receptor complex formed the hydrophobic interaction Phe486-Leu79, similar to wild-type, but there was no H-bonding between Asn487-Gln24. This was because of the other mutation that occurred in the RBD of the Omicron and the potential short contacts formed between Gly476-Gln24 and Tyr501-Tyr41 (SI Table 3). The Phe486-Leu79 hydrophobic interaction contributes to the segregation of the nonpolar residues and induces H-bonding between Asn487-Gln24 in the wild-type but not in the Delta. * The interactions between the S-glycoprotein of the wild-type with the ACE2-receptor were already analyzed, and the data are displayed here for reference [15].
Potential Hydrogen Bonds
ACE2
SARS-Cov-2-S RBD–Wild-type*
Type of H-Bond
Distance (D-A)\AA
ResNum
ResName
Chain-1
AtomName
ResNum
ResName
Chain-2
AtomName
24
GLN
A
OE1
487
ASN
E
ND2
SS
2.69
30
ASP
A
OD2
417
LYS
E
NZ
SS
2.90
42
GLN
A
NE2
446
GLY
E
O
SB
3.24
42
GLN
A
NE2
449
TYR
E
OH
SS
2.79
353
LYS
A
O
502
GLY
E
N
BB
2.78
353
LYS
A
NZ
496
GLY
E
O
SB
3.08
ACE2
SARS-Cov-2-S RBD – Delta
Type of H-Bond
Distance (D-A)\AA
ResNum
ResName
Chain-1
AtomName
ResNum
ResName
Chain-2
AtomName
38
ASP
F
OD2
449
TYR
A
OH
SS
3.19
31
LYS
F
NZ
493
GLN
A
OE1
SS
3.26
353
LYS
F
NZ
496
GLY
A
O
BS
2.63
41
TYR
F
OH
500
THR
A
O
BS
2.66
ACE2
SARS-Cov-2-S RBD – Omicron
Type of H-Bond
Distance (D-A)\AA
ResNum
ResName
Chain-1
AtomName
ResNum
ResName
Chain-2
AtomName
38
ASP
D
OD2
449
TYR
A
OH
SS
3.23
35
GLU
D
OE2
493
ARG
A
NH1
SS
2.69
38
ASP
D
OD1
498
ARG
A
NH1
SS
3.17
353
LYS
D
O
502
GLY
A
N
BB
2.99
Ligand
Protein
Interacting residues & atoms in bond (ligand - Receptor)
Interaction type
Distance
Binding Energy(kcal/mol)
Vdw_hb_desolv_energy(kcal/mol)
Inhibition constant
Ligand efficiency
Ignaciomycin
6M0J – wild-type
TYR453 (OH-OAN)GLY496 (N-OAH)TYR505 (OH-OBB)GLN493 (OE1:B-HAL)SER494 (O-HAL)TYR449 (OH-HAI)
H-Bond: Conventional H-BondH-Bond: Conventional H-BondH-Bond: Conventional H-BondH-Bond: Conventional H-BondH-Bond: Conventional H-BondH-Bond: Conventional H-Bond
3.01292.73012.61952.09012.61772.3543
−6.58
−7.20
80.88 (uM)
0.23
Ignaciomycin
7v8b - Delta
GLN493 (HE21-OBG)ASN501 (OD1-HAI)PHE497 (HA-OAG)SER494 (OG-CBH)TYR449 (OH-CAS)GLY496 (O-CAS)TYR505
H-Bond: Conventional H-BondH-Bond: Conventional H-BondH-Bond: Carbon H-BondH-Bond: Carbon H-BondH-Bond: Carbon H-BondH-Bond: Carbon H-BondHydrophobic: Pi-Pi T-shaped
2.59531.85952.16063.07363.10013.67174.8145
−7.02
−6.07
208.33 (uM)
0.26
Ignaciomycin
7t9l-Omicron
SER496 (OG-OBB)ARG498 (NH1-OBB)ARG498 (NH1-OBG)ARG498 (NH2-OBD)TYR501 (OH-OBG)HIS505 (ND1-OAN)SER494 (O-HBS)TYR453 (OH)
H-Bond: Conventional H-BondH-Bond: Conventional H-BondH-Bond: Conventional H-BondH-Bond: Conventional H-BondH-Bond: Conventional H-BondH-Bond: Conventional H-BondH-Bond: Conventional H-BondH-Bond: Pi-Donor H-Bond
3.00663.17783.23412.48872.97472.63692.05173.4505
−8.65
−5.9
72.18 (uM)
0.29
3.6 Docking analysis
The ADMET properties of Ignaciomycin were analyzed using OSIRIS-DataWarrior (SI-Table 4) (Lopez-Lopez et al., 2019). The modification of Geldanamycin at the quinone ring can increase its potency, metabolic stability, and water solubility. The hydroxyl (OH) group was added to the benzoquinone moiety and carboxylic acid groups in the 10th position of Ignaciomycin, which may result in lower metabolic toxicity and higher potency and water solubility compared to Geldanamycins (SI Table 4) and could lead to an improvement in metabolic potency. Ignaciomycin has 5H-bonding donors and 12H-bonding acceptors and has good drug-likeness (7.5912) compared to Geldanamycin. Both cLogP (1.4933) and cLogS (-3.502) indicate that the hydrophilicity and water solubility of Ignaciomycin were improved. Moreover, it shows no adverse effects such as mutagenic, tumorigenic, reproductive, or irritant effects. However, experimental studies are needed to confirm its stability and toxicity.
Complex
Binding free energy(kJ/mol)
Electrostatic energy (kJ/mol)
Van der Waal energy(kJ/mol)
Polar solvation energy(kJ/mol)
SASA energy(kJ/mol)
Wild-type-Ignaciomycin
−75.599+/-0.541
−284.686+/- 0.402
−1.662+/-0.282
215.114+/-0.623
−4.358+/-0.025
Delta-Ignaciomycin
−178.494+/-0.858
−395.904+/- 0.673
−9.806+/-0.391
232.623+/-1.022
−5.375+/-0.034
Omicron-Ignaciomycin
−237.658+/-0.375
−560.012+/- 0.490
−24.975+/-0.414
356.902+/-0.500
−9.575+/-0.028
Upon docking at the RBD-ACE2 interface, Ignaciomycin formed a strong conventional and carbon H-bonding with amino acid residues Tyr453, Gly496, Tyr505, Gln493, Ser494, and Tyr449 in the wild-type (Fig. 5A), Gln493, Asn501, Phe497, Ser494, Tyr449, and Gly496 in the Delta (Fig. 5B) and Ser496, Arg498, Tyr501, His505, and Ser494 in the Omicron (Fig. 5C), respectively. The least energy-confirmed poses of docked complexes were selected for further analysis and all interactions are listed in Table 3.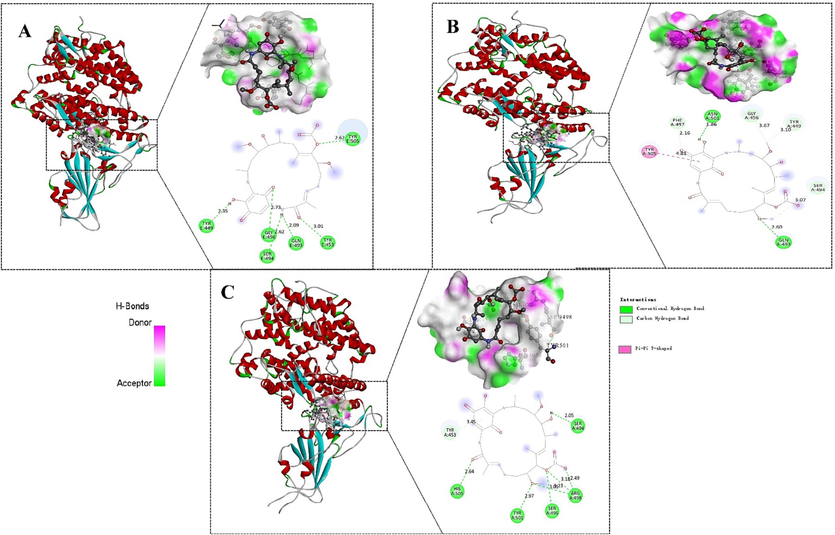
Molecular docking (Ligand-protein) interaction. The interaction with the least energy mode of Ignaciomycin with the RBD of SARS-CoV-2 S-glycoprotein placed in the cavity of the surface region of the (A) wild-type, (B) delta-variant, (C) omicron-variant.
3.7 MD simulation
RMSD plotting of Ignaciomycin-RBD in the wild-type and Delta showed that residues converged and structural stability was maintained throughout the simulation period (Fig. 6A, B). For Delta, there was a structural deviation and upward shift of ∼ 50 ns, which then typically converged the rest of the simulation (Fig. 6B). For Ignaciomycin-Omicron, there was a structural deviation upward at ∼ 20 ns and then a significant structural deviation was observed throughout the simulation. These deviations were due to the mutations and the strong interaction of the Ignaciomycin in the RBD region (Fig. 6C). The backbone RMSD of Ignaciomycin confirmed its stability (Fig. 6D) and the Apo wild-type, Delta, and Omicron confirmed the structural variation due to the mutations (SI Fig. 2).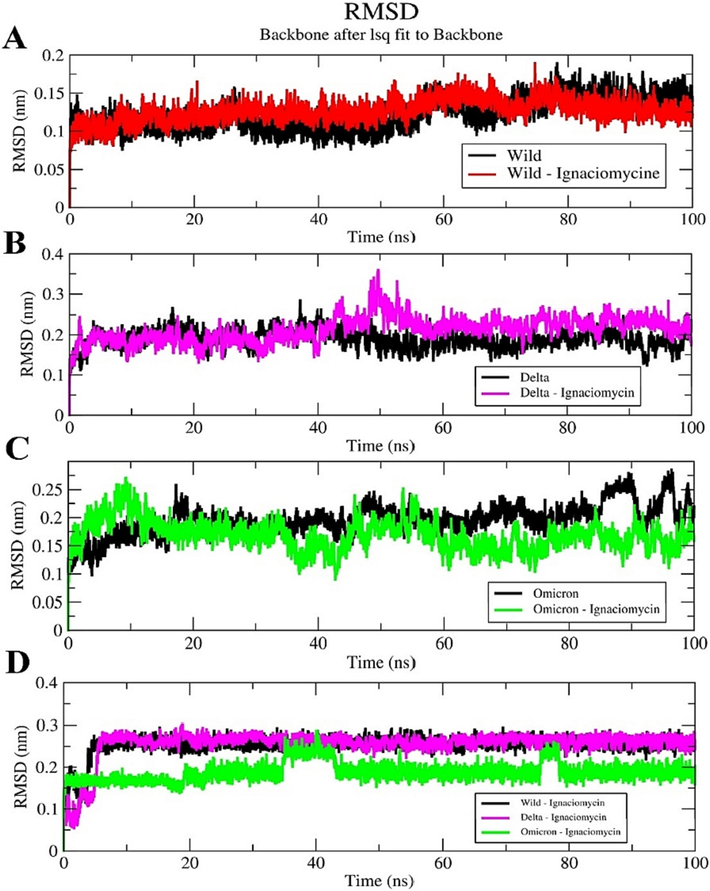
Root Mean Square Deviation (RMSD) plot. (A) Ignaciomycin-wild-type. (B) Ignaciomycin-delta. (C) Ignaciomycin-omicron. (D) Ignaciomycin alone in all the three complexes.
In all three complexes, Ignaciomycin maintained its stability throughout the simulation and retained strong conventional H-bonding and other interactions, similar to docking analysis (SI Figs. 3, 4, 5); these interactions significantly altered the RBD region of wild-type, Delta, and Omicron, and confirmed by secondary structure analysis (SI Figs. 6, 7, 8). The number of intermolecular H-bond contacts of all three complexes is shown in Fig. 7.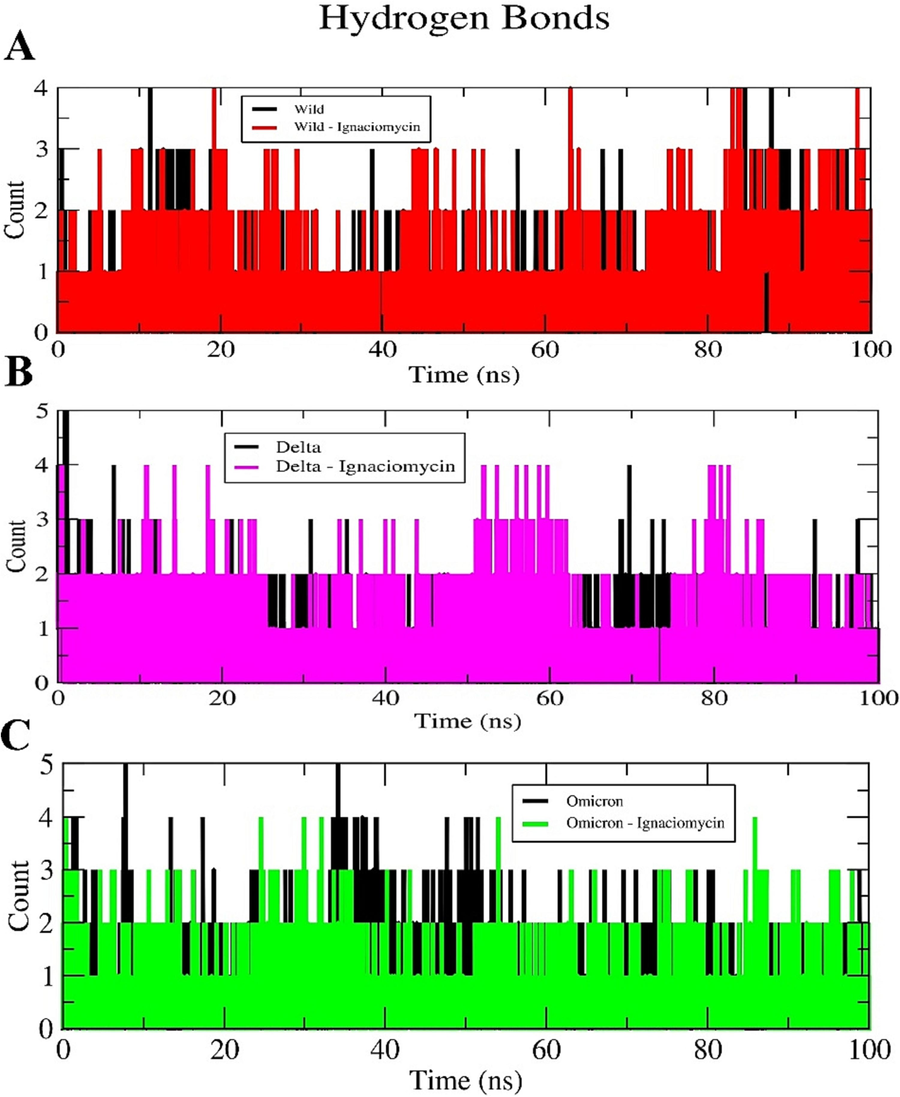
Intermolecular Hydrogen bond plot. (A) Ignaciomycin-wild-type. (B) Ignaciomycin-delta. (C) Ignaciomycin-omicron.
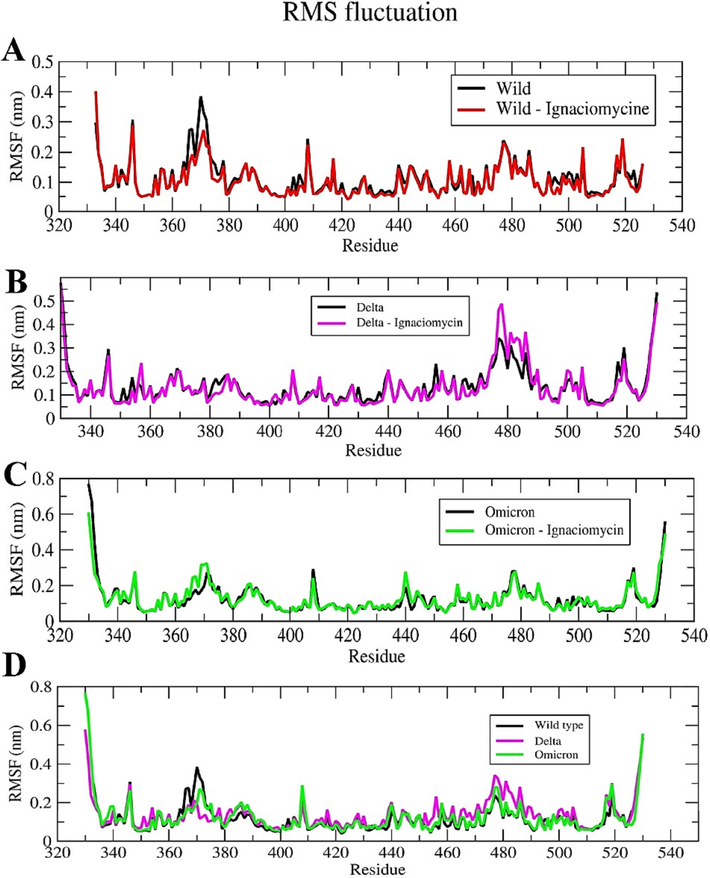
Root Mean Square Fluctuation (RMSF) plot. (A) Ignaciomycin-wild-type. (B) Ignaciomycin-delta. (C) Ignaciomycin-omicron. (D) superimposed RMSF plot for wild-type. (E) delta and omicron-variants alone.
The dynamic behavior of the individual residues was investigated based on the RMSF of all complexes. The combined H-bonds and hydrophobic interactions were reflected as residual fluctuations in the RMSF plots of all simulated complexes, especially in the corresponding active and mutant residues of the RBD region (Fig. 8). Compared to the wild-type, the residues at both the N- and C-terminus of the Delta and Omicron variants showed flexibility during the simulation period. In particular, the flexibility is greater in the RBD region due to the mutation and it was even greater during the interaction of Ignaciomycin in the apo and complex structures. These fluctuations can be seen in the RMSF of the mutant residues of the Delta (Arg452 and Lys478) (Fig. 8B, D) and Omicron (Leu371, Phe373, Lys440, Arg493, Ser496, Arg498, Tyr501 and His505) (Fig. 8C, D) variants.
3.8 Covariance and PCA analysis
The diagonal matrix of five eigenvectors and their eigenvalues for each complex and the apo state were summarized based on the RMSD and RMSF projections (SI Figs. 9, 10, 11), and the atomic fluctuations in the collective motions of each particle in the S-glycoprotein and RBD regions were analyzed. Each of the collected eigenvectors defined the corresponding atom contributing to the collective motion of particles per atom (Figs. 9, 10, 11).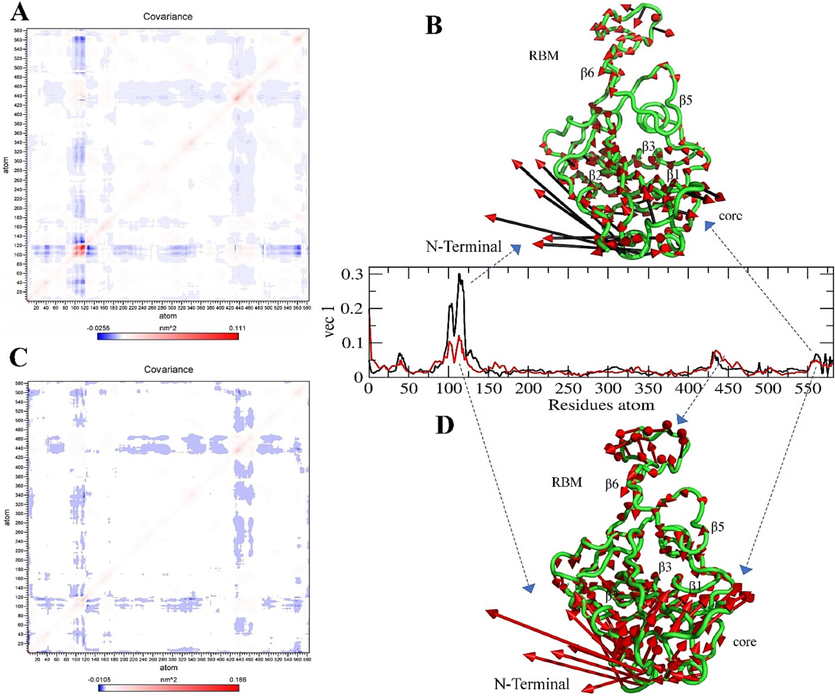
Covariance and Principal Component Analysis of Wild-type. (A, B) Total atom fluctuation and motility analyzed by covariance matrix and PCA for the apo wild-type and (C, D) Ignaciomycin-wild-type; (B) the black and red arrow marks indicate the atomic motion in the wild-type; (D) the full red color indicates the atomic motion in the Ignaciomycin-wild-type; in the covariance matrix, (A, C) the red color indicates the interacting two atoms are moving together, and blue indicates atoms moving on the opposite side.
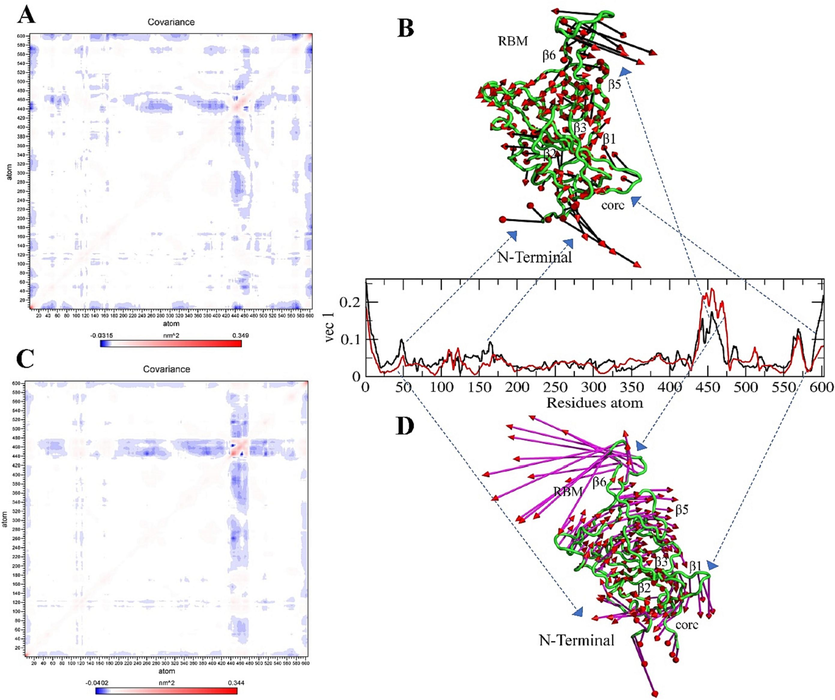
Covariance and Principal Component Analysis of Delta variant. (A, B) Total atom fluctuation and motility analyzed by covariance matrix and PCA for the apo delta and (C, D) Ignaciomycin-delta; (B) the black and red arrow marks indicate the atomic motion in the delta; (D) the full magenta with red color indicates the atomic motion in the Ignaciomycin-delta; in the covariance matrix, (A, C) the red color indicates the interacting two atoms are moving together, and blue indicates atoms moving on the opposite side.
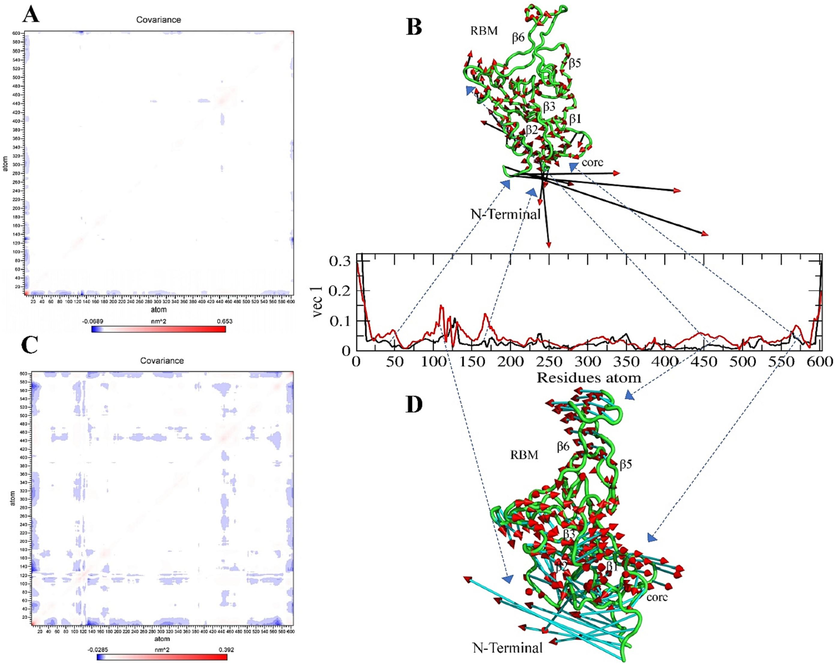
Covariance and Principal Component Analysis of Omicron variant. (A, B) Total atom fluctuation and motility analyzed by covariance matrix and PCA for the apo omicron and (C, D) Ignaciomycin-omicron; (B) the black and red arrow marks indicate the atomic motion in the omicron; (D) the full cyan with red color indicates the atomic motion in the Ignaciomycin-omicron; in the covariance matrix (A, C) the red color indicates the interacting two atoms are moving together, and blue indicates atoms moving on the opposite side.
In the wild-type, based on the collective motion particles, there was atomic motion in the core region of the N-terminal and the β1- and β2-sheets. Similarly, a slight particle fluctuation was observed in the β6 sheets of the RBM region (Fig. 9A, B). After interaction with Ignaciomycin, particle motion was slightly altered in the core region of N-terminus. Atomic fluctuations were observed in the β6-, β3- and β5- sheets and opposite manner in the β1-, and β2-sheets (Fig. 9C, D).
The Delta-variant showed large collective motion particles in β5- and β6-sheets in the RBM region. These fluctuations probably occurred due to mutation in residues Leu452Arg and Thr478Lys. Moreover, there were fluctuations and opposing movements in the core region of N-terminus and residues in most β-sheets (Fig. 10A, B). A contradictory large atomic rotational drift was observed at the RBM, the core region of N-terminus, and most β-sheets after Ignaciomycin interaction (Fig. 10C, D).
In the Omicron-variant, slight particle motions were observed in the RBM region, due to the mutated non-polar and positively charged amino acids and their enhanced hydrophobic surface in the RBM regions. Simultaneously, opposite atomic motions and fluctuations were observed in the core region of the N-terminus and a slight drift across residues in most β-sheets similar to the Delta-variant (Fig. 11A, B). After interaction with Ignaciomycin, there were many atomic rotational drifts and significant fluctuations in the collective motion particles in an anticorrelated manner in the RBM region and the core region of N-terminal, respectively. Moreover, most of the existing atoms in β-sheets were moved in a partially correlated and anti-correlated manner. This large number of collective motions is due to the strong interaction of Ignaciomycin with the non-polar and positively charged mutant amino acids in the RBM region of the Omicron-variant (Fig. 11C, D).
In addition, free energy landscape analysis was used to analyze the confirmations of the lowest energy of wild-type, delta and omicron along with the binding of Ignaciomycin based on the PC1 and PC2 plots with the comparison of RMSD and Radius of Gyration against Gibbs free energy. The overall FEL plots of all variants are shown in the figure (Fig. 12). There are large and small confirmations of the lowest energy obtained in the wild type, whereas, in the wild-type Ignaciomycin complex, there is a large energy distribution obtained in the minimal cluster (Fig. 12A). In contrast, two differentials’ confirmations of lowest energy were obtained in both the delta and delta-Ignaciomycin complexes, but the delta-Ignaciomycin complex showed the lowest energy intense to the minimal cluster (Fig. 12B). The omicron and omicron-Ignaciomycin complexes showed a single minimal energy cluster, but the omicron-Ignaciomycin complex showed a strong large distribution in the minimal cluster (Fig. 12C). These particular free energy confirmations distributed different energy clusters due to the mutations and interaction of Ignaciomycin reduced the residual fluctuation and developed the minimal energy distribution in the principal components of all complexes. These results are also confirmed by the analysis of the eigenvectors associated with the covariance matrix.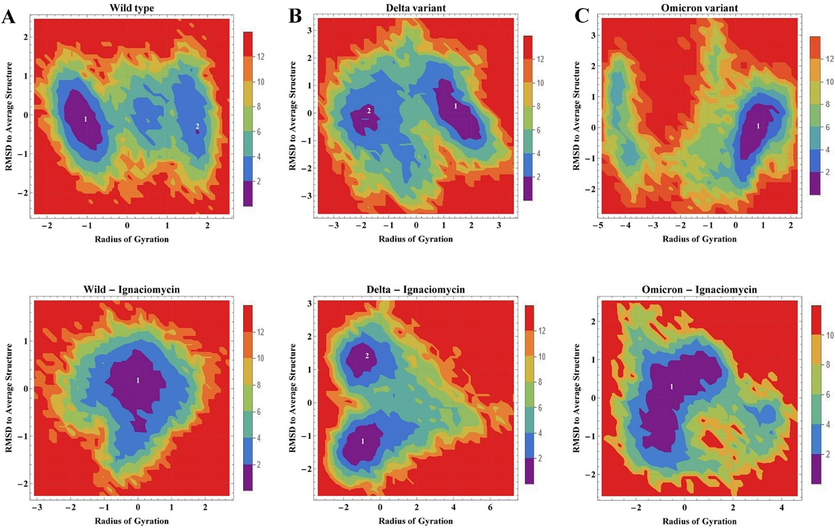
Free energy landscape (FEL) plot. 2D projection of FEL of (A) wild-type, (B) Delta and (C) Omicron variant along with Ignaciomycin interaction based on the PC1 and PC2 in comparison with RMSD and Radius of gyration. The energy minima of the lowest free energy formation shown in violet and blue.
Furthermore, changes in the solvent-accessible surface area (SASA) and the average area per residue in the overall simulation were calculated and confirmed the stability of Ignaciomycin (SI Fig. 12, 13, 14).
3.9 Free energy binding analysis
The last 2000 frames of ∼ 100 ns simulated complexes were used to determine the contribution of binding free energy and decomposition of each residue in the interaction of Ignaciomycin with all the three variants (Table 4). The G-binding energy of the complexes Ignaciomycin with the wild-type, delta, and omicron variants was determined to be −75.602 +/- 24.417 kJ/mol, −178.487 +/- 37.302 kJ/mol and −237.671 +/- 16.456 kJ/mol, respectively.
The free
G-binding energy of Ignaciomycin with Delta and Omicron showed comparatively and significantly the highest binding value, confirming the strong interaction of Ignaciomycin and blocking of RBD-ACE2 interaction due to the mutation (Fig. 13B, C). Simultaneously, Ignaciomycin also showed a better
G-binding free energy for the wild-type (Fig. 13A). Moreover, Ignaciomycin showed significant electrostatic and polar solvation energy with all variants.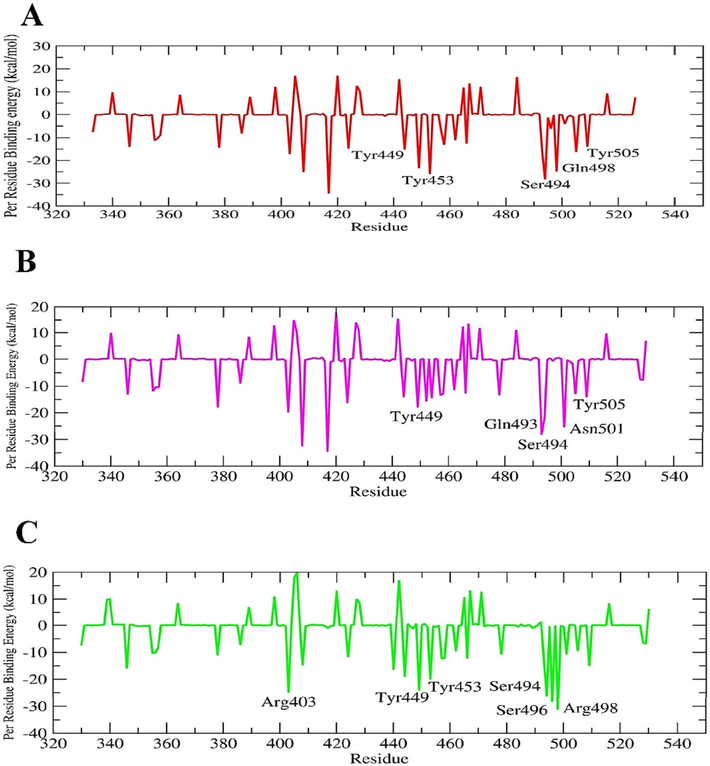
The favorable per-residue energy contribution plot. Interaction energy contribution of Ignaciomycin complex with (A) wild-type, (B) delta variant, and (C) Omicron variant.
4 Discussion
The severity of viral infections is usually determined by where mutations occur in the virus’s genetic material and affect its functional properties. Because the natural variant of SARS-CoV-2 has infected a larger number of people, the mutant variants have now also caused significant problems. The new mutant variants of SARS-CoV-2 are highly contagious compared to the native virus, and the Omicron variant, in particular, is one of the most important (Lupala et al., 2022). The N-terminal S-glycoprotein, especially the RBD of SARS-CoV-2, has strong H-bonds and electrostatic interactions with the binding domain of the human ACE2-receptor (Tortorici and Veesler, 2019).
In this current study, we investigated the interactions between the SARS-CoV-2 S-glycoprotein of Delta and Omicron with the ACE2-receptor and the findings were compared with the previously published wild-type data (Lan et al., 2020, Stalin et al., 2022b). The Omicron showed significant conformational changes in the RBD compared to the wild-type, based on the critical mutations at residues in the RBD, such as Asn417, Asp339, Lys440, Leu371, Phe373, Phe375, Ser446, Asn477, Lys478, Ala484, Arg493, Ser496, Arg498, Tyr501, and His505, which significantly altered the electrostatic surface of the RBD-ACE2 interface due to the large side chains of mutant amino acids such as Lys440 and Lys478 (Yang et al., 2021). These mutants Arg493, Lys478, and Arg498, generated positive charges in the RBD interface of the Omicron and strongly interacted with ACE2, which had negatively charged amino acids such as Glu35 and Asp38. Besides, the RBD of the Delta exhibited mutations in Arg452 and Lys478, which are also generated positive charges (Li et al., 2021, Lupala et al., 2022).
Moreover, Omicron formed the salt-bridge between Arg493-Glu35 and Arg498-Asp38 and also formed the unfavorable electrostatic interactions between Arg403-Lys353, Arg493-Lys31, Arg493-His34, Arg498/His505-Lys353. Besides, some favorable electrostatic interactions and short contacts in Delta and Omicron were also analyzed, which differed from the wild-type (SI Table 3). In our previous alanine scanning mutagenesis study (Stalin et al., 2022b), we predicted that important residues such as Lys417, Tyr449, Tyr453, Leu455, Phe456, Gln493, Gln498, Thr500, Asn501, and Tyr505 were responsible for stabilizing the S-glycoprotein-ACE2-receptor complex in wild-type. Some predicted residues were confirmed by Omicron, such as Lys417Asn, Gln493Lys, Gln498Arg, Asn501Tyr and Tyr505His. The mutants Asn417, Lys440, Arg493, and Arg498 altered the electrostatic surface in the RBD of Omicron and increased ACE2 interaction affinity and decreased vaccine affinity (Yang et al., 2021). However, the mutant residues Arg452 and Lys478 in the Delta did not interact with the ACE2-receptor but caused conformational changes in the protein (Goher et al., 2021, Zhao et al., 2022).
The above-mentioned amino acids present in the RBD of wild-type, Delta and Omicron are the main active sites for inhibiting SARS-CoV-2 inhibition. Therefore, it is important to check the interaction of drugs or natural compounds with these active sites. Geldanamycin is a benzoquinone and belongs to the ansamycins group with known antimicrobial and anticancer activities. Since Geldanamycin exhibits hepatotoxicity due to the benzoquinone moiety, effective doses and uses are limited (Taechowisan et al., 2020, Skrzypczak et al., 2021). Geldanamycin and its derivatives are potent inhibitors of heat shock protein 90 (HSP90) and inhibit most viruses such as influenza virus, HIV-1, and herpes by altering the host response (Connor et al., 2007, Li et al., 2012, Sultan et al., 2020). For viral replication, HSP90 plays an important role in viral protein synthesis. Recent research has shown that geldanamycin blocks viral replication by inhibiting HSP90 (Connor et al., 2007, Li et al., 2012, Sultan et al., 2020, Kasperkiewicz, 2021). Some geldanamycin derivatives modified at positions C17 and C19 showed their antiviral activity against the influenza virus, HIV-1, hepatitis C and B, and herpes (Connor et al., 2007, Qu et al., 2011, Kousara et al., 2017, Taechowisan et al., 2020). Therefore, we speculate that Ignaciomycin, the analogue of geldanamycin, may also inhibit HSP90 and block viral replication and also act as an anticancer agent. However, we focused on determining the efficacy of Ignaciomycin in inhibiting SARS-CoV-2 and its mutations through molecular docking and dynamics simulation analyses and confirmed its inhibitory activity. Based on this docking analysis, Ignaciomycin was found to form interactions with the major amino acids in the RBD and RBM of the SARS-CoV-2 spike glycoprotein in the wild-type, delta, and omicron variants. The binding affinities are more or less similar, and especially in the omicron variant, Ignaciomycin formed the interactions with the mutant residues Ser496, Arg498, Tyr501, His505, and also with Ser494 as in the wild-type and delta variants.
The HOMO and LUMO energies play a crucial role in detecting the reactivity of molecules used for drug design and deciding the capacities of electron-donating and attracting capabilities. The energy gap (ΔE) between HOMO and LUMO indicates the stability of the molecular surface (Houchi and Messasma, 2022b, Missioui et al., 2022). Similarly, the study of molecular ESP provides information about the electrostatic properties of molecules, such as electrophilic and nucleophilic reactions, the possibility of hydrogen bonding, and biological properties and understanding of various interactions, especially non-covalent interactions with complex biological systems (Luo et al., 2006, Spackman and Jayatilaka, 2009, Hazra et al., 2019, Singh et al., 2022). According to ESP mapping (Fig. 4B), Ignaciomycin shows electrostatic negative charges at O1 and O3 in the quinone ring, O4 of the amide, and O6, O7, and O8 of the carboxylic acid, which is involved in a strong interaction with one of the mutant residues Arg498, Ser496 and His505, which has a positive charge in Omicron. Our results are consistent with previous reports (Abian et al., 2020, Arokiyaraj et al., 2020, Oany et al., 2020, Prateeksha et al., 2021, Houchi and Messasma, 2022a, Stalin et al., 2022b).
Structural conformation and stability are important for inhibitors, especially in infectious diseases caused by viruses, bacteria, and fungi. In the Ignaciomycin-omicron complex, the backbone RMSD equilibrated and converged throughout the simulation with moderate fluctuation around ∼ 40 ns and ∼ 80 ns. Even though Ignaciomycin was repositioning its structure and maintained its interaction with residues in the RBD region (Fig. 6C). H-bonds play an important role in determining the molecular stability of proteins. The secondary structure of the protein changes due to ligand interactions, and the stability of the interactions is enhanced by inter and intramolecular H-bonds (Bepari and Reza, 2021, Linani et al., 2022).
After the ∼ 100 ns simulation for the wild-type, the compound Ignaciomycin maintained its stability and retained the strong conventional hydrogen bond with Tyr453, Ser494, and Tyr449 and formed a new carbon-hydrogen bond with Gln498. In addition, Ignaciomycin developed a Pi-Alkyl interaction with Tyr505. Residues Gln493, Gly496, and Asn501 were also located in the hydrophobic surface surrounding them (SI Fig. 3). Like the wild-type, Ignaciomycin in the delta and omicron variants also retained its stability. Ignaciomycin retained strong conventional hydrogen bonds in the delta variant with Gln493 and Asn501 and carbon-hydrogen bonds with Tyr449 and Ser494. In addition, Ignaciomycin maintained the Pi-Pi interaction with Tyr505 (SI Fig. 4). In the omicron variant, Ignaciomycin retained the strong conventional hydrogen bonding with Arg498, Tyr449, and Ser494 and formed a van der Walls interaction with Tyr453. In addition, Ignaciomycin formed a carbon-hydrogen bond with Ser496 and Arg403. Residues Tyr501, His505, Tyr453, and Arg493 were also located in the hydrophobic surface region surrounded by them (SI Fig. 5).
Covariance and PCA were also performed to investigate the flexibility of atoms based on diagonalization and atomic fluctuation in the wild-type, delta, and omicron variants and their docked complexes with Ignaciomycin. Based on the molecular dynamics simulation data, the atomic fluctuation was correlated depending on the atomic interaction between each particle in the atoms of the residues. Based on the atomic fluctuation, the degree of correlation can diverge, and especially when the particles are directly coupled with bonds, they are moved collectively, or else the particle moves in the opposite direction. These correlations between the atomic motions of the particles are related to the fluctuations in structures in the dynamic system and are related to its biological properties and functions (Fenwick et al., 2014, Chen et al., 2021).
In our study, the atom motions and the degree of collinearities for each atom pair of dynamic complexes of the Ignaciomycin with wild-type, delta, and omicron variants were analyzed from the PCA analysis. The diagonal matrix of eigenvectors and eigenvalues summarized and confirmed the atomic fluctuations in collective motions of each particle in the spike glycoprotein and the RBD region of all complexes based on the strong interaction of Ignaciomycin. FEL analysis also confirmed the lowest energy distribution of wild-type, delta, and omicron and the minimal energy changes after the binding of Ignaciomycin. The G-binding energy and decomposition analysis for the contribution of each residue per energy also confirmed the strong interaction of Ignaciomycin. The overall MD simulation analysis proved the structural stability of Ignaciomycin and the strong interaction with the RBD-ACE2 interface of SARS-CoV-2.
5 Conclusion
Docking studies revealed the strong interaction of the novel compound Ignaciomycin with RBD of the wild-type, Delta, and Omicron. Mutations in Delta and Omicron significantly increased RBD-ACE2 binding affinity by altering their electrostatic surface and significantly increasing transmission rate and pathogenesis compared with wild-type. Ignaciomycin exhibited strong efficiency in the mode of H-bonding interaction with the mutated sites in the RBD of Delta (TYR449, TYR453, GLN493, SER494 and ASN501) and Omicron (TYR449, ARG498, SER494, TYR501 and his505), thereby blocking the interaction between RBD and ACE2. Confirmation of the binding stability and free energy of binding of the Ignaciomycin-RBD complex of wild-type, Delta and Omicron was also supported by ∼ 100 ns dynamics simulations and MM-PBSA studies. Therefore, Ignaciomycin could be a potential drug for treating SARS-CoV-2 mutations with resistance to existing drugs. Further experimental studies are required to confirm the mode of action of Ignaciomycin.
Declaration of competing interest.
The author(s) declare(s) that there is no conflict of interest regarding the publication of this article.
CRediT authorship contribution statement
Antony Stalin: Conceptualization, Methodology, Software, Writing – original draft. Pachaiyappan Saravana Kumar: Formal analysis, Writing – review & editing. Balakrishnan Senthamarai Kannan: Writing – review & editing, Software, Validation. Rajamanikam Saravanan: Software, Validation. Savarimuthu Ignacimuthu: Conceptualization. Quan Zou: Conceptualization, Funding acquisition, Project administration.
Acknowledgements
The work was supported by the National Natural Science Foundation of China (No. 62131004).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Structural stability of SARS-CoV-2 3CLpro and identification of quercetin as an inhibitor by experimental screening. Int J Biol Macromol.. 2020;164:1693-1703.
- [CrossRef] [Google Scholar]
- GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX.. 2015;1–2:19-25.
- [CrossRef] [Google Scholar]
- Bioactive potentiality of secondary metabolites from endophytic bacteria against SARS-COV-2: An in-silico approach. PLoS One.. 2022;17:e0269962.
- [Google Scholar]
- Geranii Herba as a Potential Inhibitor of SARS-CoV-2 Main 3CL(pro), Spike RBD, and Regulation of Unfolded Protein Response: An In Silico Approach. Antibiotics (basel).. 2020;9
- [CrossRef] [Google Scholar]
- COVID-19 Biomarkers Recognition & Classification Using Intelligent Systems. Current Bioinformatics.. 2022;17:426-439.
- [CrossRef] [Google Scholar]
- Identification of a novel inhibitor of SARS-CoV-2 3CL-PRO through virtual screening and molecular dynamics simulation. PeerJ.. 2021;9:e11261.
- [Google Scholar]
- Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature.. 2022;602:657-663.
- [CrossRef] [Google Scholar]
- BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature.. 2022;608:593-602.
- [CrossRef] [Google Scholar]
- Conformational transformation of switch domains in GDP/K-Ras induced by G13 mutants: An investigation through Gaussian accelerated molecular dynamics simulations and principal component analysis. Comput Biol Med.. 2021;135:104639
- [CrossRef] [Google Scholar]
- Omicron Variant (B.1.1.529): Infectivity, Vaccine Breakthrough, and Antibody Resistance. J Chem Inf Model.. 2022;62:412-422.
- [CrossRef] [Google Scholar]
- Plaque-neutralizing antibody to BA.2.12.1, BA.4 and BA.5 in individuals with three doses of BioNTech or CoronaVac vaccines, natural infection and breakthrough infection. J Clin Virol.. 2022;156:105273
- [CrossRef] [Google Scholar]
- Antiviral activity and RNA polymerase degradation following Hsp90 inhibition in a range of negative strand viruses. Virology.. 2007;362:109-119.
- [CrossRef] [Google Scholar]
- Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. The Journal of Chemical Physics.. 1993;98:10089-10092.
- [CrossRef] [Google Scholar]
- R.D. Dennington, T. A. K., J.M. Millam, 2016. Gauss View 6.0.16, Gaussian Inc,.
- An interactive web-based dashboard to track COVID-19 in real time. The Lancet. Infectious Diseases.. 2020;20:533-534.
- [CrossRef] [Google Scholar]
- CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res.. 2006;34:W116-W118.
- [CrossRef] [Google Scholar]
- Correlated motions are a fundamental property of beta-sheets. Nat Commun.. 2014;5:4070.
- [CrossRef] [Google Scholar]
- M. J. Frisch, G. W. T., H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, 2016. Gaussian 09, Revision A.02. Gaussian, Inc., Wallingford CT.
- The Delta Variant Mutations in the Receptor Binding Domain of SARS-CoV-2 Show Enhanced Electrostatic Interactions with the ACE2. Med Drug Discov.. 2021;100114
- [CrossRef] [Google Scholar]
- A super-Gaussian Poisson-Boltzmann model for electrostatic free energy calculation: smooth dielectric distribution for protein cavities and in both water and vacuum states. J Math Biol.. 2019;79:631-672.
- [CrossRef] [Google Scholar]
- Exploring the inhibitory potential of Saussurea costus and Saussurea involucrata phytoconstituents against the Spike glycoprotein receptor binding domain of SARS-CoV-2 Delta (B.1.617.2) variant and the main protease (M(pro)) as therapeutic candidates, using Molecular docking, DFT, and ADME/Tox studies. J Mol Struct.. 2022;1263:133032
- [CrossRef] [Google Scholar]
- Exploring the inhibitory potential of Saussurea costus and Saussurea involucrata phytoconstituents against the Spike glycoprotein receptor binding domain of SARS-CoV-2 Delta (B.1.617.2) variant and the main protease (Mpro) as therapeutic candidates, using Molecular docking, DFT, and ADME/Tox studies. J Mol Struct.. 2022;1263:133032
- [CrossRef] [Google Scholar]
- CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods.. 2017;14:71-73.
- [CrossRef] [Google Scholar]
- Deep Reinforcement Learning Framework for COVID Therapy: A Research Perspective. Current Bioinformatics.. 2022;17:393-395.
- [CrossRef] [Google Scholar]
- Covid-19, heat shock proteins, and autoimmune bullous diseases: a potential link deserving further attention. Cell Stress Chaperones.. 2021;26:1-2.
- [CrossRef] [Google Scholar]
- Biomedical Significance of Tryptamine: A Review. Journal of Pharmacovigilance.. 2017;5
- [CrossRef] [Google Scholar]
- g_mmpbsa–a GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model.. 2014;54:1951-1962.
- [CrossRef] [Google Scholar]
- Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature.. 2020;581:215-220.
- [CrossRef] [Google Scholar]
- Geldanamycin, a ligand of heat shock protein 90, inhibits herpes simplex virus type 2 replication both in vitro and in vivo. J Antibiot (tokyo).. 2012;65:509-512.
- [CrossRef] [Google Scholar]
- The impact of receptor-binding domain natural mutations on antibody recognition of SARS-CoV-2. Signal Transduct Target Ther.. 2021;6:132.
- [CrossRef] [Google Scholar]
- Exploring Structural Mechanism of COVID-19 Treatment with Glutathione as a Potential Peptide Inhibitor to the Main Protease: Molecular Dynamics Simulation and MM/PBSA Free Energy Calculations Study. Int J Pept Res Ther.. 2022;28:55.
- [CrossRef] [Google Scholar]
- DataWarrior: an evaluation of the open-source drug discovery tool. Expert Opin Drug Discov.. 2019;14:335-341.
- [CrossRef] [Google Scholar]
- Koopmans' theorem for large molecular systems within density functional theory. J Phys Chem a.. 2006;110:12005-12009.
- [CrossRef] [Google Scholar]
- Mutations on RBD of SARS-CoV-2 Omicron variant result in stronger binding to human ACE2 receptor. Biochem Biophys Res Commun.. 2022;590:34-41.
- [CrossRef] [Google Scholar]
- Green synthesis of DyBa2Fe3O7.988/DyFeO3 nanocomposites using almond extract with dual eco-friendly applications: Photocatalytic and antibacterial activities. International Journal of Hydrogen Energy.. 2022;47:14319-14330.
- [CrossRef] [Google Scholar]
- A possible potential COVID-19 drug candidate: Diethyl 2-(2-(2-(3-methyl-2-oxoquinoxalin-1(2H)-yl)acetyl)hydrazono)malonate: Docking of disordered independent molecules of a novel crystal structure, HSA/DFT/XRD and cytotoxicity. Arabian Journal of Chemistry.. 2022;15:103595
- [CrossRef] [Google Scholar]
- Morris, G. M., R. Huey and A. J. Olson, 2008. Using AutoDock for ligand-receptor docking. Curr Protoc Bioinformatics. Chapter 8, Unit 8 14. https://doi.org/10.1002/0471250953.bi0814s24.
- AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem.. 2009;30:2785-2791.
- [CrossRef] [Google Scholar]
- Application of nanomaterials in treatment, anti-infection and detection of coronaviruses. Nanomedicine (lond).. 2020;15:1501-1512.
- [CrossRef] [Google Scholar]
- Design of novel viral attachment inhibitors of the spike glycoprotein (S) of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) through virtual screening and dynamics. International Journal of Antimicrobial Agents.. 2020;56:106177
- [CrossRef] [Google Scholar]
- CCDC 2218814: Experimental Crystal Structure Determination. CSD Communication 2022
- [CrossRef] [Google Scholar]
- Screening of cryptogamic secondary metabolites as putative inhibitors of SARS-CoV-2 main protease and ribosomal binding domain of spike glycoprotein by molecular docking and molecular dynamics approaches. J Mol Struct.. 2021;1240:130506
- [CrossRef] [Google Scholar]
- Tryptamine derivatives as novel non-nucleosidic inhibitors against hepatitis B virus. Bioorg Med Chem.. 2011;19:3120-3127.
- [CrossRef] [Google Scholar]
- Omicron variant (B.1.1.529) of SARS-CoV-2: Mutation, infectivity, transmission, and vaccine resistance. World J Clin Cases.. 2022;10:1-11.
- [CrossRef] [Google Scholar]
- Structural basis of receptor recognition by SARS-CoV-2. Nature.. 2020;581:221-224.
- [CrossRef] [Google Scholar]
- A DFT study of vibrational spectra of 5-chlorouracil with molecular structure, HOMO-LUMO, MEPs/ESPs and thermodynamic properties. Polym Bull (berl).. 2022;1–29
- [CrossRef] [Google Scholar]
- Anticancer activity and toxicity of new quaternary ammonium geldanamycin derivative salts and their mixtures with potentiators. J Enzyme Inhib Med Chem.. 2021;36:1898-1904.
- [CrossRef] [Google Scholar]
- CrystalExplorer: a program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. Journal of Applied Crystallography.. 2021;54:1006-1011.
- [CrossRef] [Google Scholar]
- Stalin, A., A. Daniel Reegan, M. Rajiv Gandhi, et al., 2022. Mosquitocidal efficacy of embelin and its derivatives against Aedes aegypti L. and Culex quinquefasciatus Say. (Diptera: Culicidae) and computational analysis of acetylcholinesterase 1 (AChE1) inhibition. Comput Biol Med. 146, 105535. https://doi.org/10.1016/j.compbiomed.2022.105535.
- Synthesis of a 1,2,3-bistriazole derivative of embelin and evaluation of its effect on high-fat diet fed-streptozotocin-induced type 2 diabetes in rats and molecular docking studies. Bioorg Chem.. 2020;96:103579
- [CrossRef] [Google Scholar]
- An in-silico approach to identify the potential hot spots in SARS-CoV-2 spike RBD to block the interaction with ACE2 receptor. J Biomol Struct Dyn.. 2022;40:7408-7423.
- [CrossRef] [Google Scholar]
- Sultan, I., S. Howard and A. Tbakhi, 2020. Drug Repositioning Suggests a Role for the Heat Shock Protein 90 Inhibitor Geldanamycin in Treating COVID-19 Infection. https://doi.org/10.21203/rs.3.rs-18714/v1.
- Antiviral activity of geldanamycin and its derivatives against influenza virus. Journal of Applied Pharmaceutical Science.. 2020;10:113-120.
- [CrossRef] [Google Scholar]
- Potential inhibitors for SARS-CoV-2 Mpro from marine compounds. RSC Adv.. 2021;11:22206-22213.
- [CrossRef] [Google Scholar]
- Structural insights into coronavirus entry. Adv Virus Res.. 2019;105:93-116.
- [CrossRef] [Google Scholar]
- CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem.. 2010;31:671-690.
- [CrossRef] [Google Scholar]
- Screening compounds for treating the diabetes and COVID-19 from Miao medicine by molecular docking and bioinformatics. Arabian Journal of Chemistry.. 2023;16:105001
- [CrossRef] [Google Scholar]
- Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science.. 2020;367:1444-1448.
- [CrossRef] [Google Scholar]
- BioLiP: a semi-manually curated database for biologically relevant ligand-protein interactions. Nucleic Acids Res.. 2013;41:D1096-D1103.
- [CrossRef] [Google Scholar]
- Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics.. 2013;29:2588-2595.
- [CrossRef] [Google Scholar]
- Structural Analysis of the SARS-CoV-2 Omicron Variant Proteins. Research (wash D c).. 2021;2021:9769586.
- [CrossRef] [Google Scholar]
- Synthesis, characterization and application of Co/Co3O4 nanocomposites as an effective photocatalyst for discoloration of organic dye contaminants in wastewater and antibacterial properties. Journal of Molecular Liquids.. 2021;337:116405
- [CrossRef] [Google Scholar]
- Dy2BaCuO5/Ba4DyCu3O9.09 S-scheme heterojunction nanocomposite with enhanced photocatalytic and antibacterial activities. Journal of the American Ceramic Society.. 2021;104:2952-2965.
- [CrossRef] [Google Scholar]
- Single-cell RNA analysis reveals the potential risk of organ-specific cell types vulnerable to SARS-CoV-2 infections. Comput Biol Med.. 2021;140:105092
- [CrossRef] [Google Scholar]
- SARS-CoV-2 Omicron variant shows less efficient replication and fusion activity when compared with Delta variant in TMPRSS2-expressed cells. Emerg Microbes Infect.. 2022;11:277-283.
- [CrossRef] [Google Scholar]
- Identification of Potential Inhibitors Against SARS-CoV-2 Using Computational Drug Repurposing Study. Current Bioinformatics.. 2021;16:1320-1327.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Supplementary material to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.105493.
Appendix A
Supplementary material
The following are the Supplementary material to this article:Supplementary material 1
Supplementary material 1




