

Translate this page into:
Quinoline-pyrimidine hybrid compounds from 3-acetyl-4-hydroxy-1-methylquinolin-2(1H)-one: Study on synthesis, cytotoxicity, ADMET and molecular docking
⁎Corresponding author.
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Some novel 2-amino-6-aryl-4-(4′-hydroxy-N-methylquinolin-2′-on-3′-yl)pyrimidines have been synthesized from α,β-unsaturated ketones of 3-acetyl-4-hydroxy-N-methylquinolin-2-one by reaction of corresponding α,β-unsaturated ketones with guanidine hydrochloride. The purity and structure of the obtained products have been confirmed by thin layer chromatography, IR, 1H NMR, 13C NMR, HSQC, HMBC and MS spectra. All the synthesized of 3-(2-amino-6-arylpyrimidin-4-yl)-4-hydroxy-1-methylquinolin-2(1H)-ones 6a-i were screened for their in vitro cytotoxic activity against human hepatocellular carcinoma HepG2 and squamous cell carcinoma KB cancer lines. Compounds 6b and 6e had the best activity in the series, with IC50 values equal to 1.33 μM. Compounds 6a-g exhibited weak or insignificant activity with liver cancer cell lines HepG2, while compounds 6a and 6g had more powerful activity in this sequence, with IC50 values equal to 47.99 and 89.38 μM, respectively. ADMET properties showed that compounds 6b, 6e, and 6f possessed the drug-likeness behavior. Cross-docking results indicated that two hydrogen bonding interactions in the binding pocket, as potential ligand binding hot-spot residues for compounds 6b and 6e, may be one of the mechanisms of action responsible for the higher cytotoxic effect on HepG2 and KB cells.
Keywords
3-Acetyl-4-hydroxyquinolin-2(1H)-one
Cytotoxicity
2-Aminopyrimidine
α,β-Unsaturated ketones

1 Introduction
Pyrimidines (1,3-diazines) and their fused analogues formed a large group of heterocyclic compounds and could impart diverse pharmacological properties (Sanjiv et al., 2019). They had been described in a wide range of biological potential agents, such as anticancer (Huang et al., 2019), antiviral (Meneghesso et al., 2012), antimicrobial (Mallikarjunaswamy et al., 2017), and antioxidant (Bhalgat et al., 2014), etc. Numerous compounds having pyrimidine ring and pyrimidine-fused heterocyclic compounds were reported about their anticancer activity via multiple different mechanisms and targets (Sanjiv et al., 2019). These such compounds exhibited antitumor activity against different cancer cell lines, such as human liver cancer HepG2 and Huh-7 (Huang et al., 2019, Abbass et al., 2020), prostate cancer PC-3 (Abbass et al., 2020), CNS cancer SNB-75 (Kassab and Gedawy, 2013), colorectal carcinoma SW480 and HCT-116 (Abbass et al., 2020), cervical carcinoma HeLa and large-cell lung carcinoma NCI-H460 (Gaonkar et al., 2018), etc. For examples, cytarabine (A, Fig. 1) was used in the treatment of acute myeloid leukemia, acute lymphocytic leukemia and in lymphomas, imatinib (B) was the first kinase inhibitor approved for the treatment of chronic myeloid leukemia (Carroll et al., 1997). Another analogous derivative, nilotinib, was used for the treatment of imatinib-resistant chronic myelogenous leukemia (Kantarjian et al., 2006).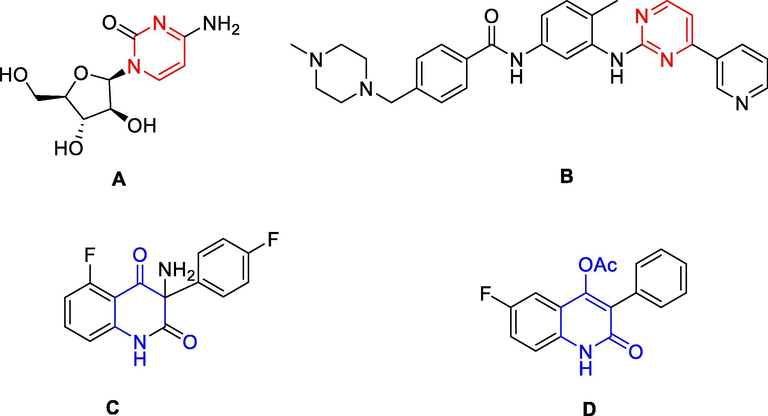
Some bioactive pyrimidine and quinoline against cancer cell lines.
Quinolones had attracted increased attention in both synthetic organic and medicinal chemistry, as recent advancements in the use of this family of important compounds highlight their considerable value (Jain et al., 2019). 4-Hydroxy-2(1H)-quinolones represented the key structure of several natural products with strong bioactivity profiles (Abdou, 2017). Compounds with a quinoline nucleus exhibited various pharmacological properties, including such as anticancer (Kumar et al., 2014), antimicrobial (Ferretti et al., 2014), antimalarial (Romero, 2019) agents, etc. They exhibited potent inhibitory activity against different cancer cell lines (Jain et al., 2019), such as human liver cancer HepG2 (Chrzanowska et al., 2020, Kuang et al., 2018), breast cancer MCF-7 (Chrzanowska et al., 2020), colorectal carcinoma SW480 (Chrzanowska et al., 2020) and HCT-116 (Hassanin et al., 2019), prostate cancer PC-3 (Chrzanowska et al., 2020), cervical carcinoma HeLa (Jin et al., 2019), melanoma SK-MEL-5 (Khusnutdinova et al., 2020), etc. For examples, compound KR-22332, 3-amino-3-(4-fluoro-phenyl)-1H-quinoline-2,4-dione, had significantly inhibited cisplatin-induced apoptosis (Shin et al., 2013). Compound B, 6-fluoro-4-hydroxy-3-phenylquinolin-2(1H)-one, had anticancer activity against MRC-5 cell lines with MIC = 3.2 μM (de Macedo et al., 2017) (Fig. 1).
Two different active pharmacophores could be combined together with or without the help of a linker to get the desired hybrid target molecule (Bargh et al., 2019, Bérubé, 2016). The new molecule was called hybrid compound (Bargh et al., 2019). The linkers could classify into two types: metabolically stable linker (usually a hydrocarbon chain) and metabolically cleavable linker (that is some functional groups, such as ester, amide (Yang et al., 2013). The latter included acid cleavable, reducible disulfides and those cleavable by exogenous stimuli (Yang et al., 2013, Bargh et al., 2019). Hybrid compounds are more effective compared to the multi-component drugs because of the lower occurrence of drug-to-drug adverse effects (Bérubé, 2016, Abbot et al., 2017). Hybridization of other pharmacophores with 4-quinolone moiety has the potential to provide novel candidates with a synergistic effect in terms of efficacy, lowered resistance selection propensity, activity against resistant bacteria, and reduced susceptibility to efflux pumps and toxicity in comparison to a cocktail of the two drugs (Romero, 2019, Butler et al., 2007). Thus, various 4-quinolone hybrid compounds were designed, synthesized and screened for their in vitro and in vivo antibacterial activities, and several hybrids are under pre-clinical or clinical studies (Gao et al., 2019). Quinolone-pyrimidine based molecular hybrids are as potential next generation of anticancer (Gao et al., 2019, Jain et al., 2019), antibacterial (Hu et al., 2017), anti-HIV-1 (Tian-Qi et al., 2016) agents, etc. Some typical cytotoxic activity against cancer cell lines included against breast cancer MCF-7 and lung cancer SK-LU-1 (Vilchis-Reyes et al., 2010), colon cancer cells HCT-116 (Karthikeyan et al., 2015), liver cancer HepG2, etc. For examples, pyrimidine-quinolone hybrid compound 251D (E) was found to be a highly selective potent inhibitor against both B. subtilis topoisomerase IV and gyrase with IC50 of 43.6 and 31 μM, respectively (Butler et al., 2007) (Fig. 2). Hybrid compounds F had antitumor activity against HepG2 cell lines (Al-Issa, 2013).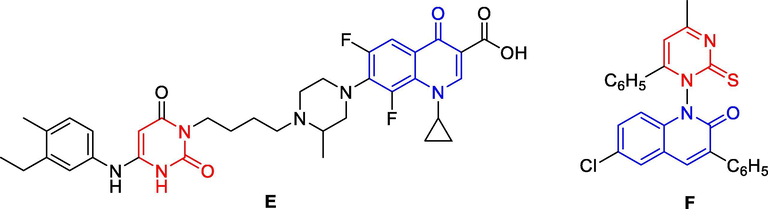
Some quinolone-pyrimidine based molecular hybrids as potential anticancer agents.
Molecular docking studies sometimes were performed to evaluate the anticancer mechanism of compounds action and to endorse with the in vitro results (Mohamady et al., 2020). Several proposed enzyme targets were used in molecular docking for HepG2 cancer activity, such as epidermal growth factor receptor-tyrosine kinase domain with erlotinib (EGFR-TKD) (Abbas et al., 2020), tubulin in complex with colchicine and with the stathmin-like domain (Xavier et al., 2020), human topoisomerase IIα in complex with DNA and etoposide (Mohamady et al., 2020), human topoisomerase IIα in complex with DNA and etoposide (Li et al., 2020, Pedatella et al., 2020), histone acetyltransferase GCN5 (Abulkhair et al., 2020), VEGFR-2 receptor tyrosine kinase complex with a novel 4-amino-furo[2,3-d]pyrimidine (El-Adl et al., 2020), human cholinesterase enzyme (human telomeric G-quadruplex DNA) (Orabi et al., 2019), etc.
Based on above-mentions research works on both moieties, quinoline-4-one and pyrimidine, as well as own research exploration to find new and effective anticancer agents, some quinolone-based pyrimidine hybrid compounds were designed, synthesized and scanned in vitro anticancer activity against human squamous cell carcinoma (KB) and hepatocellular carcinoma (HepG2) cell lines in this study (Scheme 1). Simultaneously, molecular docking study also performed to understanding the interaction mode of these compounds as inhibitors. Molecular docking was performed on human topoisomerase IIα in complex with DNA and etoposide (Li et al., 2020).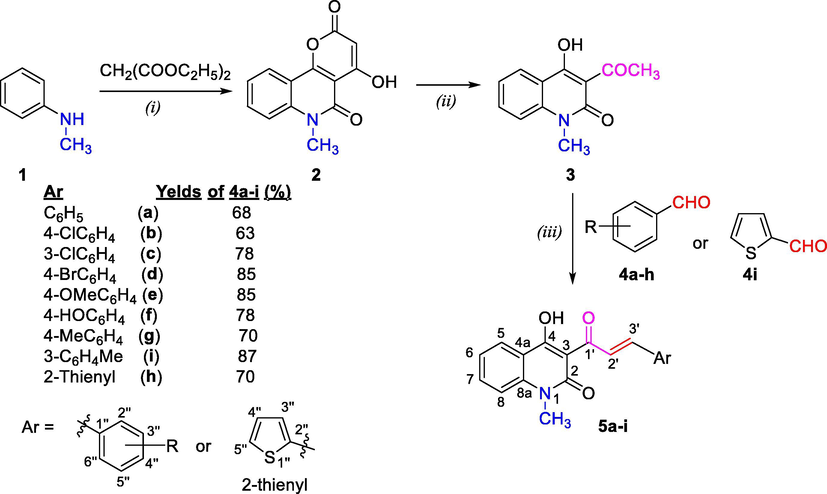
Reaction conditions: (i) Diphenyl ether, 5 h, under reflux; (ii) NaOH 40%, glycerol, 1 h, reflux; (iii) Piperidine, abs. EtOH, under reflux for 32–38 h; where, 4a-i, 5a-i: R = C6H4 (a), 4-ClC6H4 (b), 3-ClC6H4 (c), 4-BrC6H4 (d), 4-MeOC6H4 (e), 4-HOC6H4 (f), 4-MeC6H4 (g), 3-MeC6H4 (h), 2-thienyl (i).
2 Experimental
2.1 General
Melting points were determined by open capillary method on STUART SMP3 instrument (BIBBY STERILIN, UK) and are uncorrected. IR spectra (KBr disc) were recorded on an Impact 410 FT-IR Spectrometer (Nicolet, USA). 1H and 13C NMR spectra were recorded on Bruker Avance Spectrometer AV500 (Bruker, Germany) at 500 MHz and 125 MHz, respectively, using DMSO‑d6 as solvent and TMS as an internal standard. ESI-mass spectra were recorded on LC-MSD-Trap-SL (Agilent Technologies, Inc., USA) mass spectrometer. All reactions were monitored by thin layer chromatography, carried out on silica gel 60 WF254S aluminum sheets (Merck, Germany) and were visualized with UV light. Chemical reagents in high purity were purchased from the Merck Chemical Company (in Viet Nam). All materials were of reagent grade for organic synthesis. 4-Hydroxy-6-methyl-2H-pyrano[3,2-c]quinoline-2,5(6H)-dione (2) was prepared from N-methylaniline by known procedure (Roschger and Stadlbauer, 1990, Faber and Kappe, 1984).
2.2 Synthesis of 3-acetyl-4-hydroxy-1-methylquinolin-2(1H)-one (3)
This compound was prepared by modified procedure (Faber and Kappe, 1984). To suspension of pyronoquinoline 2 (0.01 mol, 2.5 g) in ethylene glycol (32 mL) a sodium hydroxide solution (40%, 0.052 mol, 3.2 mL) was added. Reaction mixture was boiled within 1 h with stirring, cooled to room temperature and then in ice bath. The cold solution was poured into cold water (65 mL), and neutralized by concentrated HCl (5.5 mL) until the precipitate completely separated (to acidic medium, pH 3). The precipitate was filtered and washed several times with water, dried at temperature of 80 °C, crystallized from 96% ethanol. Yield: 2.19 g (90%). M.p. 141–142 °C, ref. (Roschger and Stadlbauer, 1990): 141–142.5 °C. IR (KBr, ν, cm−1): 3488, 1650, 1620, 1594, 1564, 1501, 764, 687; 1H NMR (500 MHz, DMSO‑d6) δ (ppm): 8.08 (dd, J = 7.75, 1.5 Hz, 1H, H-5), 7.80 (td, J = 8.5, 1.5 Hz, 1H, H-7), 7.52 (d, J = 8.5 Hz, 1H, H-8), 7.31 (t, J = 7.75 Hz, 1H, H-6), 3.53 (s, 3H, N-CH3), 2.70 (s, 3H, 3-COCH3).
2.3 General procedure for synthesis of (E)-4-hydroxy-3-(3-(aryl)acryloyl)-1-methylquinolin-2(1H)-ones (5a-i)
To a mixture of 3-acetyl-4-hydroxy-N-methyl-2(1H)-quinolone (3, 5 mmol, 1.085 g) and appropriate (un)substituted benzaldehydes 4a-h or thiophene-2-carbaldehyde (4i) (5 mmol) in absolute ethanol (25 mL) was added piperidine (0.1 mL). The reaction mixture was heated under reflux for 32–38 h. After reaction, solvent was led to evaporate to half a volume. Separated solid product was filtered, washed with a little of 96% ethanol (2 × 2 mL), crystallized from appropriate solvents to afford the titled compounds 5a-i.
2.3.1 (E)-4-Hydroxy-3-(3-(phenyl)acryloyl)-1-methylquinolin-2(1H)-one (5a)
From 3 (5 mmol, 1.085 g) and 4a (R = H, 5 mmol, 0.53 g, 0, 51 mL), under reflux for 35 h. Yield: 1.037 g (68%) of 5a as yellow crystals. M.p.: 160–161 °C (96% ethanol/DMF, 5:1 by volume); Rf = 0.74 (n-hexane/ethyl acetate = 5:3 by volume). IR (KBr), ν (cm−1): 3436 (νOH), 1655 (νC⚌O lactam), 1622 (conj. ketone), 1540 (νC⚌C arene), 978 (δCH⚌ trans-alkene); 1H NMR (500 MHz, DMSO‑d6), δ (ppm): 8.59 (d, J = 15.75 Hz, 1H, H-3ʹ), 8.14 (d, J = 7.5 Hz, 1H, H-5), 7.93 (d, J = 15.75 Hz, 1H, H-2ʹ), 7.82 (t, J = 7.5 Hz, 1H, H-7), 7.75–7.74 (m, 2H, H-2″ & H-6″), 7.55 (d, J = 7.5 Hz, 1H, H-8), 7.50–7.49 (m, 3H, H-3″, H-4″ & H-5″), 7.34 (t, J = 7.5 Hz, 1H, H-6), 3.59 (s, 3H, N-CH3); ESI-MS(+): calcd. for C19H15NO3, M+H = 306.1 Da, found: m/z 305.9 [M+H]+.
2.3.2 (E)-4-Hydroxy-3-(3-(4-chlorophenyl)acryloyl)-1-methylquinolin-2(1H)-one (5b)
From 3 (5 mmol, 1.085 g) and 4b (R = 4ʹ-Cl, 5 mmol, 0.702 g), under reflux for 32 h. Yield: 1.069 g (63%) of 5b as yellow–brown crystals. M.p.: 165–166 °C (96% ethanol/5:1 by volume); Rf = 0.74 (n-hexane/ethyl acetate = 5:3 by volume). IR (KBr), ν (cm−1): 3427 (νOH), 1645 (νC⚌O lactam), 1620 (conj. ketone), 1593, 1546 (νC⚌C arene), 974 (δCH⚌ trans-alkene); 1H NMR (500 MHz, DMSO‑d6), δ (ppm): 8.56 (d, J = 16.0 Hz, 1H, H-3′), 8.12 (dd, J = 8.0, 1.0 Hz, 1H, H-5), 7.87 (d, J = 16.0 Hz, 1H, H-2′), 7.80 (t, J = 7.5 Hz, 1H, H-7), 7.75 (d, J = 9.0 Hz, 2H, H-2″ & H-6″), 7.55–7.53 (m, 3H, H-8, H-3″ & H-5″), 7.33 (t, J = 7.5 Hz, 1H, H-6), 3.58 (s, 3H, N-CH3); ESI-MS(+): calcd. for C19H1435ClNO3/C19H1437ClNO3, (M+H)/((M+2)+H) = 340.1//342.1 Da, found: m/z 339.9 [M+H]+, 341.8 [(M+2)+H]+.
2.3.3 (E)-4-Hydroxy-3-(3-(3-chlorophenyl)acryloyl)-1-methylquinolin-2(1H)-one (5c)
From 3 (5 mmol, 1.085 g) and 3c (R = 3ʹ-Cl, 5 mmol, 0.702 g), under reflux for 35 h. Yield: 1.324 g (78%) of 5c as pale-yellow crystals. M.p.: 168–169 °C (96% ethanol/5:1 by volume); Rf = 0.70 (n-hexane/ethyl acetate = 5:3 by volume). IR (KBr), ν (cm−1): 3102 (νC—H aryl), 1650 (νC⚌O lactam), 1623 (conj. ketone), 1597, 1542 (νC⚌C arene), 984 (δCH⚌ trans-alkene); 1H NMR (500 MHZ, DMSO‑d6), δ (ppm): 8.56 (d, J = 16.0 Hz, 1H, H-3′), 8.16 (dd, J = 8.0, 1.5 Hz, 1H, H-5), 7.86 (d, J = 16.0 Hz, 1H, H-2′), 7.81 (td, J = 8.5, 1.5 Hz, 1H, H-7), 7.76 (s, 1H, H-2″), 7.71–7.69 (m, 1H, H-6″), 7.56 (d, J = 8.5 Hz, 1H, H-8), 7.52–7.51 (m, 2H, H-4″ & H-5″), 7.35 (t J = 8.0 Hz, 1H, H-6), 3.62 (s, 3H, N-CH3); ESI-MS(+): calcd. for C19H1435ClNO3/C19H1437ClNO3, (M+H)/((M+2)+H) = 340.1/342.1 Da, found: m/z 399.9 [M+H]+, 341.9 [(M+2)+H]+.
2.3.4 (E)-4-Hydroxy-3-(3-(4-bromophenyl)acryloyl)-1-methylquinolin-2(1H)-one (5d)
From 3 (5 mmol, 1.085 g) and 4d (R = 4ʹ-Br, 5 mmol, 0.925 g), under reflux for 36 h. Yield: 1.632 g (85%) of 5d as pale-yellow crystals. M.p.: 200–201 °C (96% ethanol/5:1 by volume); Rf = 0.68 (n-hexane/ethyl acetate = 5:3 by volume). IR (KBr), ν (cm−1): 3423 (νOH), 1688 (νC⚌O lactam), 1616 (conj. ketone), 1535 (νC⚌C arene), 980 (δCH⚌ trans-alkene); 1H NMR (500 MHZ, DMSO‑d6), δ (ppm): 1H NMR (500 MHz, DMSO‑d6), δ (ppm): 8.58 (d, J = 16.0 Hz, 1H, H-3′), 8.12 (dd, J = 7.75, 1.25 Hz, 1H, H-5), 7.86 (d, J = 16.0 Hz, 1H, H-2′), 7.81 (td, J = 8.5, 1.25 Hz, 1H, H-7), 7.72–7.67 (m, 4H, H-2″ & H-6″, H-3″ & H-5″), 7.55 (t, J = 8.5 Hz, 1H, H-8), 7.33 (t, J = 7.75 Hz, 1H, H-6), 3.89 (s, 3H, N-CH3); ESI-MS(+): calcd. for C19H1479BrNO3/ C19H1481BrNO3, (M+H)/((M+2)+H) = 384.0/386.0 Da, found: m/z 383.8 [M+H]+, 385.8 ([(M+2)+H]+).
2.3.5 (E)-4-Hydroxy-3-(3-(4-methoxyphenyl)acryloyl)-1-methylquinolin-2(1H)-one (5e)
From 3 (5 mmol, 1.085 g) and 4e (R = 4ʹ-OMe, 5 mmol, 0.68 g), under reflux for 36 h. Yield: 1.423 g (85%) of 5e as yellow crystals. M.p.: 178–179 °C (96% ethanol/5:1 by volume); Rf = 0.81 (n-hexane/ethyl acetate = 5:3 by volume). IR (KBr), ν (cm−1): 3103 (νC—H aryl), 1643 (νC⚌O lactam), 1599 (conj. ketone), 1532, 1504 (νC⚌C arene), 980 (δCH⚌ trans-alkene); 1H NMR (500 MHz, DMSO‑d6), δ (ppm): 8.51 (d, J = 15.5 Hz, 1H, H-3′), 8.13 (d, J = 7.5 Hz, 1H, H-5), 7.93 (d, J = 15.5 Hz, 1H, H-2′), 7.81 (t, J = 8.0 Hz, 1H, H-7), 7.72 (d, J = 7.5 Hz, 2H, H-2″ & H-6″), 7.55 (d, J = 8.0 Hz, 1H, H-8), 7.33 (t, J = 7.5 Hz, 1H, H-6), 7.06 (d, 7.5 Hz, 2H, H-3″ & H-5″), 3.83 (s, 3H, 4″–OCH3), 3.59 (s, 3H, N-CH3); ESI-MS(+): calcd. for C20H17NO4, M = 335.1 Da, found: m/z 335.9 [M+H]+.
2.3.6 (E)-4-Hydroxy-3-(3-(4-hydroxyphenyl)acryloyl)-1-methylquinolin-2(1H)-one (5f)
From 3 (5 mmol, 1.085 g) and 4f (R = 4ʹ-OH, 5 mmol, 0.61 g), under reflux for 38 h. Yield: 1.251 g (78%) of 5f as yellow crystals. M.p.: 254–255 °C (96% ethanol/5:1 by volume); Rf = 0.76 (n-hexane/ethyl acetate = 5:3 by volume). IR (KBr), ν (cm−1): 3216 (νOH), 1627 (νC⚌O lactam), 1602 (conj. ketone), 1537, 1516 (νC⚌C arene), 980 (δCH⚌ trans-alkene); 1H NMR (500 MHz, DMSO‑d6), δ (ppm): 10.24 (s, 1H, 4″–OH), 8.47 (d, J = 15.75 Hz, 1H, H-3ʹ), 8.12 (d, J = 8.0 Hz, 1H, H-5), 7.92 (d, J = 15.75 Hz, 1H, H-2ʹ), 7.79 (t, J = 8.0 Hz, 1H, H-7), 7.62 (d, J = 8.75 Hz, 2H, H-2″ & H-6″), 7.53 (d, J = 8.0 Hz, 1H, H-8), 7.32 (t, J = 8.0 Hz, 1H, H-6), 6.88 (d, J = 8.75 Hz, 2H, H-3″ & H-5″), 3.58 (s, 3H, N-CH3); ESI-MS(+): calcd. for C19H15NO4, M+H = 322.1 Da, found: m/z 321.9 [M+H]+; ESI-MS(−): calcd. for C19H15NO4, M−H = 320.1 Da, found: m/z 319.9 [M−H]−.
2.3.7 (E)-4-Hydroxy-3-(3-(4-methylphenyl)acryloyl)-1-methylquinolin-2(1H)-one (5 g)
From 3 (5 mmol, 1.085 g) and 4 g (R = 4ʹ-Me, 5 mmol, 0.60 g), under reflux for 38 h. Yield: 1.116 g (70%) of 5g as yellow crystals. M.p.: 166–167 °C (96% ethanol/5:1 by volume); Rf = 0.82 (n-hexane/ethyl acetate = 5:3 by volume). IR (KBr), ν (cm−1): 3103 (νC—H aryl), 1649 (νC⚌O lactam), 1618 (conj. ketone), 1535 (νC⚌C arene), 987 (δCH⚌ trans-alkene); 1H NMR (500 MHz, DMSO‑d6), δ (ppm): 8.56 (d, J = 15.75 Hz, 1H, H-3ʹ), 8.11 (d, J = 7.5 Hz, 1H, H-5), 7.89 (d, J = 15.75 Hz, 1H, H-2ʹ), 7.79 (t, J = 8.0 Hz, 1H, H-7), 7.62 (d, J = 7.5 Hz, 2H, H-2″ & H-6″), 7.53 (d, J = 8.0 Hz, 1H, H-8), 7.32 (t, J = 7.5 Hz, 1H, H-6), 7.30 (d, J = 7.5 Hz, 2H, H-3″ & H-5″), 3.57 (s, 3H, N-CH3), 2.36 (s, 3H, 4-CH3); ESI-MS(+): calcd. for C20H17NO3, M+H = 320.1 Da, M+Na = 342.1 Da, found: m/z 319.9 [M+H]+, 341.9 [M+Na]+.
2.3.8 (E)-4-Hydroxy-3-(3-(3-methylphenyl)acryloyl)-1-methylquinolin-2(1H)-one (5h)
From 3 (5 mmol, 1.085 mg) and 4h (Ar = 3ʹʹ-MeC6H4, 5 mmol, 600 mg), under reflux for 32 h. Yield: 1308 mg (87%) of 5h as yellow crystals. M.p.: 172–173 °C (96% ethanol); Rf = 0.82 (n-hexane/ethyl acetate = 5:3 by volume). IR (KBr), ν (cm−1): 3110, 3034 (νC—H aryl), 2917, 2849 (νC—H alkyl), 1633 (νC⚌O lactam), 1625 (conj. ketone), 980 (δCH⚌ trans-alkene). 1H NMR (500 MHz, DMSO‑d6), δ (ppm): 8.57 (d, J = 16.0 Hz, 1H, H-3ʹ), 8.13 (d, J = 8.0 Hz, 1H, H-5), 7.88 (d, J = 16.0 Hz, 1H, H-2ʹ), 7.80 (t, J = 8.0 Hz, 1H, H-7), 7.53–7.51 (m, 3H, H-8, H-2ʹʹ, H-6ʹʹ), 7.38–7.29 (m, 3H, H-6, H-4ʹʹ, H-5ʹʹ), 3.58 (s, 3H, N-CH3), 2.36 (s, 3H, 3″–CH3); ESI-MS(+): calcd. for C20H17NO3, M+H = 320.1 Da, found: m/z 320.3 (100%) [M+H]+.
2.3.9 (E)-4-Hydroxy-3-(3-(2-thienyl)acryloyl)-1-methylquinolin-2(1H)-one (5i)
From 3 (5 mmol, 1.085 mg) and 4i (Ar = 2ʹʹ-thienyl, 5 mmol, 701 mg), under reflux for 50 h. Yield: 1078 mg (70%) of 5i as bright yellow crystals. M.p.: 211–212 °C (96% ethanol); Rf = 0.80 (n-hexane/ethyl acetate = 5:3 by volume). IR (KBr), ν (cm−1): 3106, 3086 (νC—H aryl), 1655 (νC⚌O lactam), 1607 (conj. ketone), 967 (δCH⚌ trans-alkene). 1H NMR (500 MHz, DMSO‑d6), δ (ppm): 8.41 (d, J = 15.5 Hz, 1H, H-3ʹ), 8.12–8.10 (m, 2H, H-5, H-2ʹ), 7.79–7.78 (m, 2H, H-7, H-5ʹʹ), 7.61 (d, 1H, J = 3.5 Hz, H-3ʹʹ), 7.53 (d, J = 8.0 Hz, 1H, H-8), 7.31 (t, J = 8.0 Hz, 1H, H-6), 7.20 (td, 1H, J = 3.5, 1.0 Hz, H-4ʹʹ), 3.58 (s, 3H, N-CH3); ESI-MS(+): calcd. for C17H13NO3S, M+H = 312.1 Da, found: m/z 312.4 (100%) [M+H]+.
2.4 General procedure for synthesis of 3-(2-amino-6-arylpyrimidin-4-yl)-4-hydroxy-1-methylquinolin-2(1H)-ones (6a-i)
A mixture of appropriate α,β-unsaturated ketones 5a-i (1 mmol), guanidine hydrochloride (1 mmol, 95 mg) and NaHCO3 (1.5 mmol, 126 mg) in DMF (30 mL) on water bath at 70 °C for 48–62 h. Cooled reaction mixture to room temperature and poured in crushed ice. Separated solid product was filtered, washed with cold water, and crystallized from appropriate mixture of 96% ethanol and DMF to afford the tilted compounds 6a-i.
2.4.1 3-(2-Amino-6-phenylpyrimidin-4-yl)-4-hydroxy-1-methylquinolin-2(1H)-one (6a)
From 4a (R = H, 1 mmol, 305 mg) and guanidine hydrochloride (1 mmol, 95 mg). Reaction time: 58 h. Yield: 220 mg (64%) of 6a as white crystals. M.p.: 231–232 °C (from 96% ethanol/DMF = 4:1 in volume), Rf = 0.74 (n-hexane/acetone = 5:3). IR (KBr), ν (cm−1): 3450–3152 (νOH hydrogen bonding), 3328, 3152 (νNH2), 3053 (νC—H aryl), 1640 (νC⚌O lactam), 1586, 1494 (νC⚌C arene); 1H NMR (500 MHz, DMSO‑d6), δ (ppm): 8.99 (s, 1H, H-5ʹ), 8.16 (dd, J = 7.5, 1.0 Hz, 1H, H-5), 8.07 (dd, J = 6.75, 2.25 Hz, 2H, H-2ʹʹ & H-6ʹʹ), 7.96 (s, 2H, NH2), 7.66 (td, J = 8.0, 1.5 Hz, 1H, H-7), 7.57 (m, 3H, H-3ʹ′, H-4ʹ′ & H-5″), 7.46 (d, J = 8.0 Hz, 1H, H-8), 7.25 (t, J = 7.5 Hz, 1H, H-6), 3.59 (s, 3H, N-CH3); 13C NMR (125 MHz, DMSO‑d6), δ (ppm): 175.7 (C⚌O lactam), 166.1 (C-2ʹ & C-6′), 159.5 (C-4), 155.3 (C-4′), 140.0 (C-8a), 136.8 (C-1ʹʹ), 132.9 (C-7), 131.2 (C-4ʹʹ), 128.9 (C-3ʹʹ & C-5ʹʹ), 127.1 (C-2ʹʹ & C-6ʹʹ), 125.5 (C-5), 121.3 (C-6), 120.0 (C-4a), 114.5 (C-8), 101.3 (C-5ʹ), 89.2 (C-3), 26.6 (N-CH3); ESI-MS: calcd. for C20H16N4O2, M+H = 345.14 Da, found: m/z 345.07 (100%) [M+H]+.
2.4.2 3-(2-Amino-6-(4-chlorophenyl)pyrimidin-4-yl)-4-hydroxy-1-methylquinolin-2(1H)-one (6b)
From 4b (R = 4-Cl, 1 mmol, 339 mg) and guanidine hydrochloride (1 mmol, 95 mg). Reaction time: 55 h. Yield: 234 mg (62%) of 6b as white crystals. M.p.: 234–235 °C (from 96% ethanol/DMF = 4:1 in volume), Rf = 0.82 (n-hexane/acetone = 5:3). IR (KBr), ν (cm−1): 3450–3207 (νOH hydrogen bonding), 3321, 3207 (νNH2), 3013 (νC—H aryl), 2950, 2862 (νC—H alkyl), 1632 (νC⚌O lactam), 1552, 1495 (νC⚌C arene); 1H NMR (500 MHz, DMSO‑d6), δ (ppm): 8.97 (s, 1H, H-5ʹ), 8.14 (d, J = 7.75 Hz, 1H, H-5), 8.07 (d, J = 8.0 Hz, 2H, H-2ʹʹ & H-6ʹʹ), 7.97 (s, 2H, NH2), 7.66 (d, J = 8.5 Hz, 1H, H-7), 7.63 (d, J = 8.0 Hz, 2H, H-3ʹʹ & H-5ʹʹ), 7.45 (d, J = 8.5 Hz, 1H, H-8), 7.24 (t, J = 7.75 Hz, 1H, H-6), 3.58 (s, 3H, N-CH3); 13C NMR (125 MHz, DMSO‑d6), δ (ppm): 175.6 (C⚌O lactam), 164.6 (C-4, C-2′ & C-6ʹ), 155.3 (C-4′), 140.0 (C-8a), 136.0 (C-4ʹʹ), 135.6 (C-1ʹʹ), 133.0 (C-7), 129.0 (C-3ʹʹ & C-5ʹʹ), 128.8 (C-2ʹʹ & C-6ʹʹ), 125.4 (C-5), 121.2 (C-6), 120.0 (C-4a), 114.5 (C-8), 101.2 (C-5ʹ), 89.2 (C-3), 28.6 (N-CH3); ESI-MS: calcd. for C20H1535ClN4O2/C20H1537ClN4O2, (M+H)/((M+2)+H) = 379.1/381.1 Da, found: m/z 378.9 [M+H]+, 380.9 [(M+2)+H]+.
2.4.3 3-(2-Amino-6-(3-chlorophenyl)pyrimidin-4-yl)-4-hydroxy-1-methylquinolin-2(1H)-one (6c)
From 4c (R = 3-Cl, 1 mmol, 339 mg) and guanidine hydrochloride (1 mmol, 95 mg). Reaction time: 52 h. Yield: 234 mg (62%) of 6c as white crystals. M.p.: 230–231 °C (from 96% ethanol/DMF = 3:1 in volume), Rf = 0.81 (n-hexane/acetone = 5:3). IR (KBr), ν (cm−1): 3450–3150 (νOH hydrogen bonding), 3325, 3232 (νNH2), 3059 (νC—H aryl), 2948, 2836 (νC—H alkyl), 1628 (νC⚌O lactam), 1593, 1550 (νC⚌C arene); 1H NMR (500 MHz, DMSO‑d6), δ (ppm): 8.91 (s, 1H, H-5ʹ), 8.11 (dd, 1H, J = 7.72, 1.25 Hz, H-5), 8.03 (s, 1H, H-2″), 7.96 (d, 1H, J = 7.5 Hz, H-4ʹʹ), 7.94 (s, 2H, NH2), 7.63–7.56 (m, 3H, H-7, H-5ʹ′ & H-6ʹ′), 7.40 (d, 1H, J = 8.5 Hz, H-8), 7.21 (t, 1H, J = 7.75 Hz, H-6), 3.55 (s, 3H, N-CH3); 13C NMR (125 MHz, DMSO‑d6), δ (ppm): 175.4 (C⚌O), 164.0 (C-4, C-2′ & C-6ʹ), 155.4 (C-4′), 140.0 (C-8a), 138.9 (C-3′ʹ), 133.7 (C-1ʹʹ), 132.9 (C-7), 130.8 (C-2ʹ′), 130.7 (C-5ʹʹ), 126.6 (C-4ʹ′), 125.6 (C-6ʹ′), 125.4 (C-5), 121.2 (C-6), 120.4 (C-4a), 114.4 (C-8), 101.4 (C-5ʹ), 89.3 (C-3), 28.6 (N-CH3); ESI-MS: calcd. for C20H1535ClN4O2/C20H1537ClN4O2, (M−H)/((M+2)−H) = 377.08/379.0 Da, found: m/z 377.09 (100%) [M−H]+ and 379.11 (41%) [(M+2)−H]+.
2.4.4 3-(2-Amino-6-(4-bromo)phenylpyrimidin-4-yl)-4-hydroxy-1-methylquinolin-2(1H)-one (6d)
From 4d (R = 4-Br, 1 mmol, 384 mg) and guanidine hydrochloride (1 mmol, 95 mg). Reaction time: 48 h. Yield: 208 mg (74%) of 6d as white crystals. M.p.: 262–263 °C (from 96% ethanol/DMF = 4:1 in volume), Rf = 0.79 (n-hexane/acetone = 5:3). IR (KBr), ν (cm−1): 3450–3207 (νOH hydrogen bonding), 3308, 3207 (νNH2), 3075 (νC—H aryl), 2918 (νC—H alkyl), 1622 (νC⚌O lactam), 1592, 1550 (νC⚌C arene); 1H NMR (500 MHz, DMSO‑d6), δ (ppm): 8.93 (s, 1H, H-5ʹ), 8.12 (dd, 1H, J = 8.0, 1.25 Hz, H-5), 7.96 (d, 2H, J = 8.5 Hz, H-2ʹʹ & H-6ʹʹ), 7.93 (s, 2H, NH2), 7.75 (d, 2H, J = 8.5 Hz, H-3ʹʹ & H-5ʹʹ), 7.62 (td, 1H, J = 8.0, 1.25 Hz, H-7), 7.40 (d, 1H, J = 8.0 Hz, H-8), 7.21 (t, 1H, J = 8.0 Hz, H-6), 3.55 (s, 3H, N-CH3); 13C NMR (125 MHz, DMSO‑d6), δ (ppm): 175.5 (C⚌O), 164.6 (C-2′ & C-6ʹ), 162.3 (C-4), 155.3 (C-4′), 140.0 (C-8a), 135.9 (C-1ʹʹ), 132.9 (C-7), 131.9 (C-3ʹʹ & C-5ʹʹ), 129.0 (C-2ʹʹ & C-6ʹʹ), 125.4 (C-5), 124.9 (C-4ʹʹ), 121.2 (C-6), 120.0 (C-4a), 114.4 (C-8), 101.1 (C-3 & C-5ʹ), 28.6 (N-CH3); ESI-MS: calcd. for C20H1579BrN4O2/C20H1581BrN4O2, (M−H)/((M+2)−H) = 421.03/423.03 Da, found: m/z 421.07 (16%) [M−H]+ and 423.03 (17%) [M+2−H]+.
2.4.5 3-(2-Amino-6-(4-methoxy)phenylpyrimidin-4-yl)-4-hydroxy-1-methylquinolin-2(1H)-one (6e)
From 4e (R = 4-OMe, 1 mmol, 335 mg) and guanidine hydrochloride (1 mmol, 95 mg). Reaction time: 57 h. Yield: 231 mg (62%) of 6e as white crystals. M.p.: 241–242 °C (from 96% ethanol/DMF = 3:1 in volume), Rf = 0.68 (n-hexane/acetone = 5:3). IR (KBr), ν (cm−1): 3462–3207 (νOH hydrogen bonding), 3293, 3207 (νNH2), 2920, 2858 (νC—H alkyl), 1636 (νC⚌O lactam), 1545, 1496 (νC⚌C arene); 1H NMR (500.13 MHz, DMSO‑d6), δ (ppm): 8.94 (s, 1H, H-5ʹ), 8.15 (dd, J = 8.0, 1.25 Hz, 1H, H-5), 8.05 (d, J = 8.0 Hz, 2H, H-2ʹʹ & H-6ʹʹ), 7.87 (s, 2H, NH2), 7.64 (td, J = 8.5, 1.25 Hz, 1H, H-7), 7.44 (d, J = 8.5 Hz, 1H, H-8), 7.24 (td, J = 8.0, 1.0 Hz, 1H, H-6), 7.12 (d, J = 8.0 Hz, 2H, H-3ʹʹ & H-5ʹʹ), 3.85 (s, 3H, N-CH3), 3.58 (s, 3H, 4″–OCH3); 13C NMR (125 MHz, DMSO‑d6), δ (ppm): 175.9 (C⚌O), 165.6 (C-6ʹ), 162.0 (C-2′), 158.9 (C-4ʹʹ), 155.0 (C-4′), 140.0 (C-8a), 132.9 (C-7), 130.0 (C-1″), 129.9 (C-2ʹʹ & C-6ʹʹ), 125.5 (C-5), 121.2 (C-6), 120.0 (C-4a), 114.5 (C-8), 114.4 (C-3ʹʹ & C-5ʹʹ), 100.3 (C-5ʹ), 55.5 (4″–OCH3), 28.7 (N-CH3); ESI-MS: calcd. for C21H18N4O3, M = 374.14 Da, found: m/z 375.0 (100%) [M+H]+.
2.4.6 3-(2-Amino-6-(4-hydroxyphenyl)pyrimidin-4-yl)-4-hydroxy-1-methylquinolin-2(1H)-one (6f)
From 4f (R = 4-OH, 1 mmol, 321 mg) and guanidine hydrochloride (1 mmol, 95 mg). Reaction time: 60 h. Yield: 208 mg (58%) of 6f as white crystals. M.p.: 250–251 °C (from 96% ethanol/DMF = 3:1 in volume), Rf = 0.78 (n-hexane/acetone = 5:2). IR (KBr), ν (cm−1): 3453–3197 (νOH hydrogen bonding), 3322, 3197 (νNH2), 2918, 2858 (νC—H alkyl), 1645 (νC⚌O lactam), 1597, 1487(νC⚌C arene); 1H NMR (500 MHz, DMSO‑d6), δ (ppm): 10.15 (s, 1H, 4″–OH), 8.91 (s, 1H, H-5ʹ), 8.14 (dd, J = 7.75, 1.5 Hz, 1H, H-5), 7.96 (d, J = 8.5 Hz, 2H, H-2ʹʹ & H-6ʹʹ), 7.86 (s, 2H, NH2), 7.63 (td, J = 8.0, 1.5 Hz, 1H, H-7), 7.42 (d, J = 8.0 Hz, 1H, H-8), 7.22 (t, J = 7.75 Hz, 1H, H-6), 6.92 (d, J = 8.5 Hz, 2H, H-3ʹʹ & H-5ʹʹ), 3.57 (s, 3H, N-CH3); 13C NMR (125 MHz, DMSO‑d6), δ (ppm): 175.9 (C⚌O), 165.9 (C-6ʹ), 160.7 (C-2′), 158.6 (C-4ʹʹ), 154.8 (C-4′), 140.0 (C-8a), 132.7 (C-7), 129.1 (C-2ʹʹ & C-6ʹʹ), 127.4 (C-1ʹʹ), 125.5 (C-5), 121.1 (C-6), 120.0 (C-4a), 115.7 (C-3ʹʹ & C-5ʹʹ), 114.4 (C-8), 99.8 (C-5ʹ), 95.9 (C-3), 28.5 (N-CH3); ESI-MS: calcd. for C20H16N4O3, M−H = 359.11 Da, found: m/z 359.12 (17%) [M−H]+.
2.4.7 3-(2-Amino-6-(4-methylphenyl)pyrimidin-4-yl)-4-hydroxy-1-methylquinolin-2(1H)-one (6g)
From 4 g (R = 4-Me, 1 mmol, 319 mg) and guanidine hydrochloride (1 mmol, 95 mg). Reaction time: 62 h. Yield: 257 mg (72%) of 6g as white crystals. M.p.: 245–246 °C (from 96% ethanol/DMF = 4:1 in volume), Rf = 0.82 (n-hexane/acetone = 5:3). IR (KBr), ν (cm−1): 3405–3205 (νOH hydrogen bonding), 3318, 3205 (νNH2), 2918, 2858 (νC—H alkyl), 1630 (νC⚌O lactam), 1553, 1462 (νC⚌C arene). 8.98 (s, 1H, H-5ʹ), 8.15 (dd, J = 7.5, 1.25 Hz, 1H, H-5), 7.98 (d, J = 8.0 Hz, 2H, H-2ʹʹ & H-6ʹʹ), 7.93 (s, 2H, NH2), 7.66 (td, J = 8.25, 1.0 Hz, 1H, H-7), 7.45 (d, J = 8.25 Hz, 1H, H-8), 7.38 (d, J = 8.0 Hz, 2H, H-3ʹʹ & H-5ʹʹ), 7.25 (t, J = 7.5 Hz, 1H, H-6), 3.58 (s, 3H, N-CH3), 2.40 (s, 3H, CH3); 13C NMR (125 MHz, DMSO‑d6), δ (ppm): 175.7 (C⚌O lactam), 165.9 (C-2ʹ & C-6′), 163.0 (C-4), 155.1 (C-4′), 141.3 (C-4ʹʹ), 140.0 (C-8a), 134.0 (C-1ʹʹ), 132.9 (C-7), 129.5 (C-3ʹʹ & C-5ʹʹ), 127.1 (C-2ʹʹ & C-6ʹʹ), 125.5 (C-5), 121.2 (C-6), 120.0 (C-4a), 114.5 (C-8), 100.8 (C-5ʹ), 89.2 (C-3), 28.6 (N-CH3), 21.0 (4″–CH3); ESI-MS: calcd. for C21H18N4O2, M+H = 359.15 Da, found: m/z 359.05 (100%) [M+H]+.
2.4.8 3-(2-Amino-6-(3-methylphenyl)pyrimidin-4-yl)-4-hydroxy-1-methylquinolin-2(1H)-one (6h)
From 4h (R = 3-CH3, 1 mmol, 319 mg) and guanidine hydrochloride (1 mmol, 95 mg). Reaction time: 62 h. Yield: 247 mg (68%) of 6h as white crystals. M.p.: 233–234 °C (from 96% ethanol/DMF = 5:1 in volume), Rf = 0.76 (n-hexane/acetone = 5:3). IR (KBr), ν (cm−1): 3442 (νOH hydrogen bonding), 1630 (νC⚌O lactam), 1586, 1494 (νC⚌C arene); 1H NMR (500 MHz, DMSO‑d6), δ (ppm): 8.96 (s, 1H, H-5ʹ), 8.16 (dd, J = 8.0, 1.0 Hz, 1H, H-5), 7.93 (s, 2H, NH2), 7.88 (s, 1H, H-2ʹʹ), 7.86 (d, J = 8.0 Hz, 1H, H-6ʹʹ), 7.65 (td, J = 8.0, 1.5 Hz, 1H, H-7), 7.47–7.44 (m, 2H, H-8, H-5ʹʹ), 7.39 (t, J = 7.5 Hz, 1H, H-4ʹʹ), 7.24 (t, J = 7.5 Hz, 1H, H-6), 3.58 (s, 3H, N-CH3), 2.42 (s, 3H, CH3); 13C NMR (125 MHz, DMSO‑d6), δ (ppm): 175.6 (C⚌O lactam), 166.1 (C-2ʹ & C-6′), 159.5 (C-4), 155.1 (C-4′), 139.9 (C-8a), 138.0 (C-5ʹʹ), 136.8 (C-1ʹʹ), 132.8 (C-7), 131.8 (C-4ʹʹ), 128.7 (C-3ʹʹ), 127.5 (C-2ʹʹ), 125.4 (C-5), 124.3 (C-6ʹʹ), 121.2 (C-6), 114.4 (C-8), 101.2 (C-5ʹ), 88.5 (C-3), 28.6 (N-CH3), 21.0 (3ʹʹ-CH3); ESI-MS: calcd. for C21H18N4O2, M+H = 359.1 Da, found: m/z 359.0 (100%) [M+H]+.
2.4.9 3-(2-Amino-6-(2-thienyl)pyrimidin-4-yl)-4-hydroxy-1-methylquinolin-2(1H)-one (6i)
From 4i (Ar: 2ʹʹ-thienyl, 1 mmol, 311 mg) and guanidine hydrochloride (1 mmol, 95 mg). Reaction time: 70 h. Yield: 246 mg (70%) of 6i as light-yellow crystals. M.p.: 241–242 °C (from 96% ethanol/DMF = 3:1 in volume), Rf = 0.78 (n-hexane/acetone = 5:3). IR (KBr), ν (cm−1): 3450 (νOH hydrogen bonding), 3315, 3195 (νNH2), 1641 (νC⚌O lactam), 1590, 1528 (νC⚌C arene); 1H NMR (500 MHz, DMSO‑d6), δ (ppm): 8.86 (s, 1H, H-5ʹ), 8.15 (dd, J = 8.0, 1.0 Hz, 1H, H-5), 7.94 (s, 2H, NH2), 7.87–7.84 (m, 2H, H-4ʹʹ, H-5ʹʹ), 7.65 (td, J = 8.0 Hz, 1H, J = 8.0, 1.5 Hz, 1H, H-7), 7.45 (d, J = 8.0 Hz, 1H, H-8), 7.27–7.22 (m, 2H, H-6, H-3ʹʹ), 3.58 (s, 3H, N-CH3); 13C NMR (125 MHz, DMSO‑d6), δ (ppm): 175.7 (C⚌O lactam), 160.8 (C-6′), 159.0 (C-4), 154.9 (C-4′), 142.3 (C-5ʹʹ), 140.0 (C-8a), 132.8 (C-7), 131.7 (C-5ʹʹ), 128.9 (C-3ʹʹ), 128.5 (C-4ʹʹ), 125.4 (C-5), 121.2 (C-6), 114.4 (C-8), 99.58 (C-5ʹ), 28.6 (N-CH3); ESI-MS: calcd. for C19H16N4O2S, M+H = 351.1 Da, found: m/z 350.9 (100%) [M+H]+.
2.5 Cytotoxicity assay
Dilution series (128, 32, 8, 2, and 0.5 µg/mL of each compound 6a-g) were prepared and used for 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay (Scudiero et al., 1988). Two cancer cell lines were seeded at a density of 3 × 104 cells/well and treated with a range of concentrations in triplicate in 96-well cell culture plates, whereupon cell proliferation was assessed using a standard MTT assay. Specifically, the growth inhibitory activity of pyrimidines was determined using MTT, which correlates the cell number with the mitochondrial reduction of MTT to a blue formazan precipitate. In brief, the cells were plated in 96-well plates and allowed to attach overnight. The medium was then replaced with serum-free medium containing the test compounds and cells were incubated at 37 °C for 72 h. The medium was then replaced with fresh medium containing 1 mg/mL MTT. Following incubation at 37 °C for 2–4 h, the wells were aspirated, the dye was solubilized in DMSO and the absorbance was measured at 540 nm using a Tecan™ GENios® Microplate Reader (Conquer Scientific, USA). The viability of cells was compared with that of the control cells. The slope of the absorbance change was used for calculating the reaction rate. Negative controls were performed in the absence of enzyme and compound, and positive controls in the presence of enzyme and 100% DMSO. The percentage of residual activity was calculated as the difference in absorbance between the time 6 and 2 min, obtained by the average of two experiments carried out in triplicate. The obtained rate were related to the rate when the inhibitor was absent. IC50 values were calculated from linear extrapolations of reaction rate (as a function of the logarithm of the concentration). The IC50 values were determined with increasing concentrations of inhibitor (128, 32, 8, 2, and 0.5 µg/mL) versus % of inhibition, in triplicate in two independent experiments. The experimental data were analyzed with TableCurve 2D Software (Systat Software, Inc.) and the IC50 values determined by linear regression. It is important to stress the fact that all compounds are soluble in the assay mixtures at the described experimental conditions.
2.6 In silico physicochemical property calculation and druglikeness evaluation
SwissADME online (http://www.swissadme.ch/) (Daina et al., 2017) prediction tools were applied for determination of physicochemical properties, lipophilicity, water solubility, pharmacokinetics, druglikeness and medicinal chemistry parameters, and also Topological Polar Surface Area (TPSA), number of rotatable bonds, LogP, violations of Veber's rule (Lagorce et al., 2011), and violations of Lipinski's rule of five (Lagorce et al., 2011). The human intestinal absorption (%ABS) was calculated using the Zhao's approach as follows (Zhao et al., 2002):
2.7 Molecular docking studies
The two-dimensional structures (.mae) of the best bioactive compounds 6b,6e and 6f (ligands) and ellipticine, were drawn and the structure was analyzed by using 2D sketcher and 3D builder of Maestro 11.8 (Schrödinger, LLC, New York, NY, USA) (Schrödinger, 2018). The three-dimensional structures of these compounds (ligands) were generated from two-dimensional structures prepared first using LigPrep 3.6 using OPLS-2005 force field. The tautomeric isomers for the ligands were searched and energy minimizations were carries out by applying the OPLS 2005 force fields, at pH 7.0 ± 2.0 The Epik v.4.3 methodology was used when preparing the ligands. Then geometrically minimized with MacroModel 12.2 followed by conformational analysis using MMFFs force field. Monte Carlo Multiple Minimum (MCMM) conformational search was used with 2500 iterations and convergence threshold of 0.05 kJ/mol. Water was chosen as solvent. Truncated Newton Conjugate Gradient minimization was used with 2500 iterations and convergence threshold of 0.05 kJ/mol. Other parameters were used as default. Crystal structure of human topoisomerase IIα in complex with DNA and etoposide was retrieved from the RCSB Protein Data Bank with PDB ID of 5GWK (https://www.rcsb.org/structure/5GWK) (Wang et al., 2017). This structure was solved by X-ray crystallography at 3.15 Å resolution. Coordinates of the protein-ligand complex were fixed for errors in atomic representations and optimized using Protein Preparation Wizard Maestro v. 11.5 (Maestro, v. 11.5: Schrödinger, LLC, New York, NY, USA), using OPLS-2005 force field for structural optimization and minimization and for Receptor Grid Generation tool of Glide v.8.1. The Glide HTVS 8.1 algorithm (High-Throughput Virtual Screening Mode) was employed using a grid box volume of 10 × 10 × 10 Å. Briefly, Glide approximates a systematic search of positions, orientations and conformations of the ligand in the receptor binding site using a series of hierarchical filters. The bond orders were assigned to residues, hydrogen atoms were added at pH 7.0 ± 2.0. The restrained minimizations were carried out using the OPLS 2005 force field with an RMSD cut-off value of 0.3 Å for heavy atom convergences.
3 Results and discussion
3.1 Chemistry
3.1.1 Synthesis of (E)-4-hydroxy-3-(3-(aryl)acryloyl)-1-methylquinolin-2(1H)-ones
Starting material for our research, 3-acetyl-4-hydroxy-1-methylquinolin-2(1H)-one (3), was obtained from 4-hydroxy-6-methyl-2H-pyrano[3,2-c]quinoline-2,5(6H)-dione (2). The latter was prepared from N-methylaniline according to known method of Stadlbauer et al. (Roschger and Stadlbauer, 1990). N-Methylaniline is a readily available reagent and procedure for synthesis of 2 from this reagent was easy to handle. Based on literatures and above-mentioned methods, we have prepared 4-hydroxy-6-methyl-2H-pyrano[3,2-c]quinoline-2,5(6H)-dione (2, a pyronoquinoline). Ring opening and subsequent spontaneous decarboxylation processes of this pyronoquinoline with sodium hydroxide gave compound 3. These processes performed in ethylene glycol as solvent instead glycerol (Roschger and Stadlbauer, 1990) under reflux for 1 h and subsequent spontaneous decarboxylation, and gave good yields (90%).
Condensation of compound 3 with different (un)substituted benzaldehydes 4a-h and thiophene-2-carbaldehyde 4i yielded corresponding α,β-unsaturated ketones 5a-i (Scheme 1). Piperidine was used as a catalyst for condensation reaction. Reaction took place within 32–38 h. These α,β-unsaturated ketones were obtained with yields of 68–87%.
IR spectra of these α,β-unsaturated ketones 5a-i had characteristic absorption bands of trans-vinyl group in range of 987–974 cm−1 (Donald L. Pavia et al., 2015). The configuration trans was confirmed by two resonance signals at δ = 7.93–7.86 ppm and δ = 8.59–7.47 ppm belonged protons H-2′ and H-3′, respectively. These signals had the roof effects that showed the coupling constants J = 15.75–16.0 Hz. These values indicated that the resulting alkene had trans configuration. Other absorption bands appeared at 3436–3103 cm−1 (for phenol OH group), 1688–1627 cm−1 (for conjugated ketone carbonyl group), and 1622–1602 cm−1 (for conjugated lactam and alkene groups). Benzene ring was specified by absorption bands in region at 1540–1504 cm−1. N-methyl group on quinolinone had chemical shift at δ = 3.57–3.89 ppm, p-methyl group on benzene ring had resonance signal at δ = 2.36 ppm, p-methoxy at 3.83 ppm. Generally, protons on benzene ring of phenyl acryloyl moiety had upfield chemical shifts, at δ = 7.75–7.62 ppm (for protons H-2″ and H-6″), 7.55–6.88 (for protons H-3″ and H-5″), with coupling constants of J = 7.5–9.0 Hz in cases of p-substituted benzene ring, whereas protons on benzene moiety of quinoline ring had both downfield and upfield chemical shifts, in order of δ = 8.16–8.11 ppm (for H-5), 7.93–7.53 (for H-8), 7.82–7.79 ppm (for H-7), and 7.35–7.32 ppm (for H-6). In compounds 5a-i the phenolic hydroxyl group on position 4 of quinolone ring had no chemical shift downfield in DMSO‑d6 solvent, possibly due to keto-enol tautomerism of this group and hydrogen-bonding formation to ketone carbonyl group (Fig. 3) (Abdou, 2017). Compound 5f had signal at δ = 9.81 ppm in its 1H NMR spectrum that belonged to 4″-hydroxyl group on benzene ring.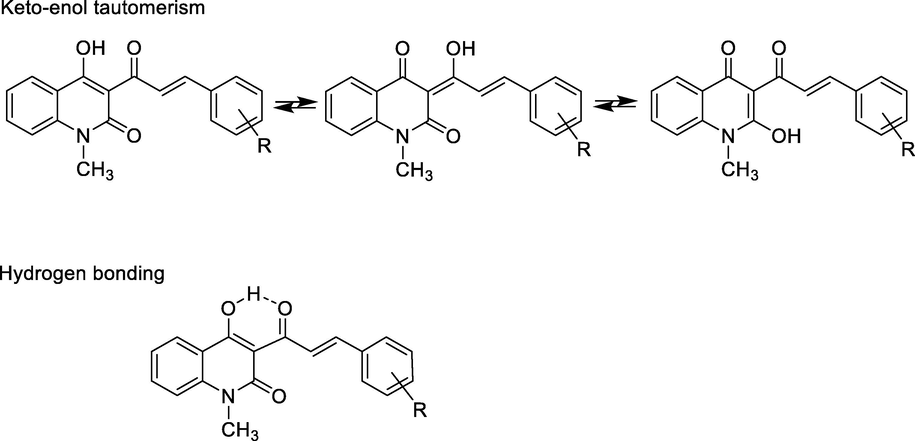
Keto-enol tautomerism and hydrogen bonding of compounds 5a-i.
3.1.2 Synthesis of 3-(2-amino-6-arylpyrimidin-4-yl)-4-hydroxy-1-methylquinolin-2(1H)-ones
Substituted 2-aminopyrimidines 6a-i were synthesized by ring-closure condensation reaction of obtained (un)substituted α,β-unsaturated ketones 5a-i with guanidine hydrochloride under reflux in dried DMF. Sodium bicarbonate was used as base in order to remove HCl from guanidine hydrochloride (Scheme 2). In our research DMF was used as a solvent because ketones 5a-i were dissolved in methanol or ethanol. Ring-closure condensation process occurred during the period 48–62 h with product yields of 58–74%.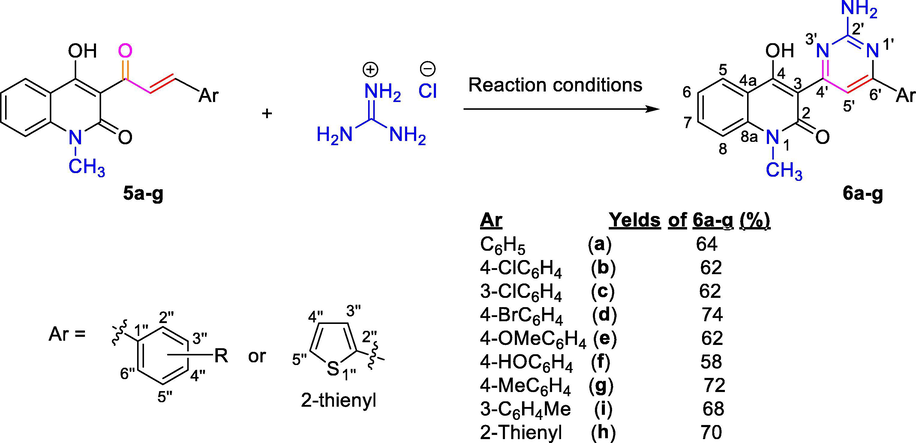
Reaction conditions: NaHCO3, dried DMF, 70 °C, 48–62 h; where, 5a-i, 6a-i: R = C6H4 (a), 4-ClC6H4 (b), 3-ClC6H4 (c), 4-BrC6H4 (d), 4-MeOC6H4 (e), 4-HOC6H4 (f), 4-MeC6H4 (g), 3-MeC6H4 (h), 2-thienyl (i).
Structurally, the formation of 2-aminopyrimidines 6a-i from α,β-unsaturated ketones 5a-i of 3-acetyl-4-hydroxy-1-methylquinolin-2(1H)-one 3 could be confirmed by spectral data (IR, NMR, and MS). The presence of the amino group (primary amine group) on pyrimidine ring was specified by both IR and NMR spectra. In IR spectra of these compounds, characteristic absorption band for out-of-plane bending vibration of C—H of trans-alkene group in the region at 987–974 cm−1 disappeared, simultaneously, new absorption bands appeared in regions at 3328–3293 and 3232–3197 cm−1; these bands characterized the stretching vibrations for N—H bond in 2-aminopyrimidine ring. In 1H NMR spectra of these 2-aminopyrimidines, the disappearance of signals at δ = 7.93–7.86 ppm and δ = 8.59–7.47 ppm for protons H-2′ and H-3′, respectively, and the appearance of proton chemical shift at δ = 8.98–8.91 ppm, which was assigned to pyrimidine proton H-5′, suggested that the pyrimidine ring was formed in the reaction of α,β-unsaturated ketones 5a-i with guanidine (Scheme 2). This signal assigned to proton H-5′ on pyrimidine ring base on HSQC and HMBC spectra of a representative compound 6g (see Section 2.5, Supplementary Data in online version of this article). Carbon-13 chemical shift of carbon atom C-5′ was at δ = 101.4–99.8 ppm (Thanh and Mai, 2009). Additionally, a resonance signal that appeared at δ = 7.97–7.86 ppm as singlet belonged to NH2 protons in 2-aminopyrimidine ring. The phenolic hydroxyl group in position 4 on quinolinone ring had no resonance signal in 1H NMR spectra because strong hydrogen-bonding formation to nitrogen atom in 2-aminopyrimidine ring and/or its tautomerism took place (Fig. 4). In 1H NMR spectrum of pyrimidine 6f, chemical shift appeared at δ = 10.15 ppm as singlet belonged to 4″-hydroxyl group on benzene ring. Generally, protons H-2″ and H-6″ on benzene ring at position 6 of pyrimidine ring had downfield chemical shifts, at δ = 8.07–7.76 ppm, and protons H-3″ and H-5″ had upfield resonance signals at δ = 7.75–6.57 ppm, with J = 8.0–8.5 Hz in cases of p-substituted benzene ring, whereas protons on benzene moiety of quinoline ring had both downfield and upfield chemical shifts, in order of H-5 (8.16–8.11 ppm), H-7 (7.66–7.56 ppm), H-8 (7.46–7.40 ppm), and H-6 (δ = 7.31–7.21 ppm).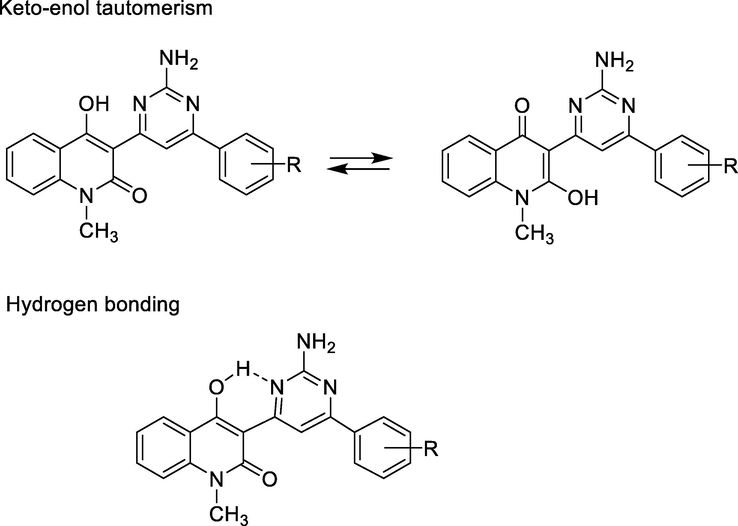
Keto-enol tautomerism of compounds 6a-i.
3.2 Cytotoxic activity
All the synthesized of 3-(2-amino-6-arylpyrimidin-4-yl)-4-hydroxy-1-methylquinolin-2(1H)-ones 6a-i were screened for their in vitro cytotoxic activity against two representatives: human squamous cell carcinoma (KB) and hepatocellular carcinoma (HepG2) cell lines. Ellipticine was used as reference. Evaluated cytotoxic results for 6a-i were given in Table 1. Figs. 5 and 6 displayed dose-dependent cell growth inhibition percentages against KB and HepG2 cell lines by the synthesized compounds 6a-i. Note: N.E. = not evaluated.
Entry
IC50 (μM)
KB
HepG2
6a
9.72 ± 2.33
1.99 ± 0.06
6b
1.32 ± 0.05
7.73 ± 7.34
6c
7.73 ± 7.25
7.13 ± 7.35
6d
13.61 ± 5.43
12.60 ± 5.95
6e
1.33 ± 0.05
8.99 ± 6.68
6f
5.06 ± 0.19
5.56 ± 7.68
6g
23.46 ± 4.20
1.38 ± 0.09
6h
N.E.
N.E.
6i
N.E.
N.E.
Ellipticine
1.24 ± 0.08
1.14 ± 0.06
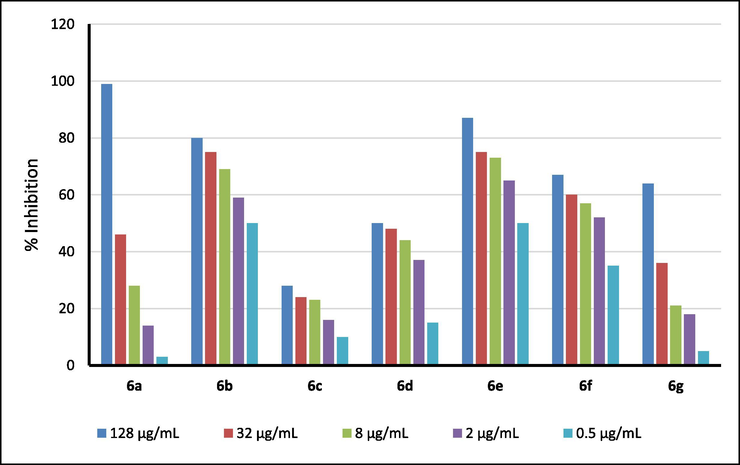
Dose-dependent cell growth inhibition percentages against KB cell lines by the synthesized compounds 6a-i.
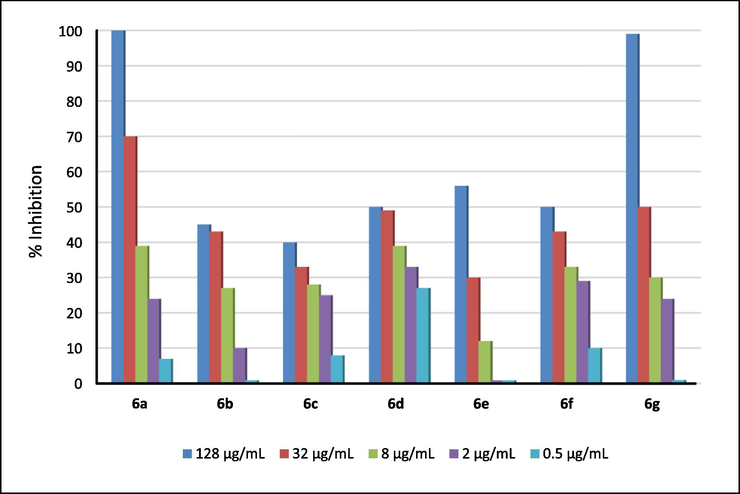
Dose-dependent cell growth inhibition percentages against HepG2 cell lines by the synthesized compounds 6a-1.
With the exception of compounds 6a, 6d and 6g that displayed negligible inhibitory effect on the KB carcinoma cell lines, the remaining compounds had a good to medium inhibitory effect. The order of the inhibitory effect of these compounds was 6b > 6e > 6f > 6c > 6a > 6d > 6g, of which compounds 6b and 6e had the best activity in the series with IC50 values of 1.32 and 1.33 μM, respectively, when compared to IC50 value with 1.24 μM of a positive reference drug ellipticine. Compound 6f had significant inhibitory activity with IC50 of 5.06 μM, and compounds 6a and 6c exhibited weak-medium cytotoxic activity with IC50 values of 9.72 and 7.73 μM against this cell lines, respectively.
Compounds 6a-g also exhibited significant activity with liver cancer cell lines HepG2, and the order of inhibitory activity against HepG2 cell lines was as follows: 6g > 6a > 6f > 6c > 6b > 6e > 6d. Two compounds 6a and 6g showed the highest activity in this sequence, with IC50 values equal to 1.99 and 1.38 μM, respectively, when compared to the IC50 value of ellipticine (with IC50 = 1.14 μM). The remaining compounds in the series had a negligible activity for liver cancer cell lines HepG2. Compound 6f also had significant inhibitory activity with IC50 of 5.56 μM. Compounds 6b,6c, and 6e exhibited medium cytotoxic activity with IC50 values of 7.13–8.99 μM. The substituents (electron-donating and withdrawing) on benzene ring had complex influences to cytotoxic activity of compounds 6a-g against both KB and HepG2 cell lines. For examples, compounds 6b (with electron-withdrawing chloro group) and 6e (with electron-donating methoxy group) had similar cytotoxicity (IC50 values of 1.32 and 1.33 μM against KB cell lines, or IC50 values of 7.73 and 8.99 μM against HepG2 cell lines). Compound 6g with weak electron-donating methyl group exhibited excellent cytotoxicity against HepG2 cell lines (IC50 = 1.38 μM), but had the worst cytotoxic activity against KB cell lines (IC50 = 23.46 μM). Compound 6a with unsubstituted benzene ring also expressed similar cytotoxicity when compared with cytotoxic activity of compound 6g with IC50 values of 9.72 and 1.99 μM, respectively.
3.3 ADMET studies
To estimate the drug-likeness of the compounds, in silico absorption, distribution, metabolism, excretion, and toxicity (ADMET) (Lagorce et al., 2011) predictions were carried out for selected synthesized compounds 6a-g were screened using SwissADME software (Daina et al., 2017). From Table 2, it could be observed that all the synthesized compounds have shown promising percentage absorption (69.58–76.56%). The most active compounds 6b and 6c showed 76.56% and 73.38% absorption, respectively. The designed structures were tested for compliance with rules evaluating bioavailability of a compound after oral administration Lipinski’s rule of five and Veber filter. The first one assumes that compounds having LogPo/w (octanol/water partition coefficient) lower than 5, molecular weight (MW) below 500, less than 10H-bond acceptors (HBA), and less than 5H-bond donors (HBD) are more likely to show favorable bioavailability. The Veber rule extends the range of parameters by rotatable bonds (preferably RB < 10) and topological polar surface area (preferably TPSA ≤ 140 Å2). The Egan rule considers good bioavailability for compounds with TPSA ≤ 132 Å2 and −1 < LogP < 6. Note. a MW: molecular weight (expressed as Dalton); b TPSA: topological polar surface area (Å2); %ABS: human intestinal absorption (percentage absorption according to Zhao(Zhao et al., 2002)); n-ROTB: number of rotatable bonds; n-ON: number of hydrogen bond acceptors; n-OHNH: number of hydrogen bond donors; LogP: logarithm of partition coefficient of compound between n-octanol and water; LV: Lipinski's violations; Veber’s violation.
Entry
MWa
TPSAb
%ABSc
n-ROTBd
n-ONe
n-OHNHf
LogPg
L.V.h
V.V.i
6a
344.37
94.03
76.56
2
4
2
2.04
0
0
6b
378.81
94.03
76.56
2
4
2
2.53
0
0
6c
378.81
94.03
76.56
2
4
2
2.53
0
0
6d
423.26
94.03
76.56
2
4
2
2.64
0
0
6e
374.39
103.26
73.38
3
5
2
1.73
0
0
6f
360.37
114.26
69.58
2
5
3
1.50
0
0
6g
358.39
94.03
76.56
2
4
2
2.26
0
0
From Table 2, it can be observed that all the synthesized compounds are fulfilled the criteria for orally active drug. Compound 6b was predicted to adhere to the Lipinski’s rule of five, suggesting that the structure would serve as a druggable lead structure. ADMET properties and target predictions of other compound 6a,d-g and reference drug ellipticine were represented in the figures in Section 3 in Supplementary Data (see online version of this article).
Obtained ADMET revealed that compounds possessed fairly high lethal dose and can be considered suitable for the druggable point of view. All synthesized compounds exhibited good predicted values of percentage human oral absorption and none of the compound possessed mutagenicity (predicted in qualitative terms). Therefore, based upon the values of predicted drug likeness parameters, all obtained compounds possessed the drug-likeness behavior.
3.4 In silico studies
It is known that the main targets of quinolones are DNA topoisomerases, which are essential for DNA replication, transcription, recombination and condensed DNA remodeling, and function by carrying out transient single- and double-strand breaks (Aldred et al., 2014). The enzyme human topoisomerase IIα in complex with DNA and etoposide (hTopoII) is an important anticancer drug target (Xiao et al., 2014, El-Metwally et al., 2020, Mohamady et al., 2020). Due to the availability of multiple inhibitor‐binding sites in this enzyme, the anti‐hTopoII agents possess high chemical diversity. Molecular docking simulations were performed in order to better understand the molecular basis for the inhibition of topoisomerases by ellipticine (reference drug) and compounds (ligands, herein) 6b,6e, and 6f against above-mentioned cancer cell lines. Based on the results obtained from the enzymatic assays, molecular modeling studies were performed as a step toward understanding the interaction mode of these compounds as inhibitors. In this study, a Topo IIβ-DNA cleavage complex was obtained from the Brookhaven Protein Data Bank (PDB code: 5GWK) (Wu et al., 2011) and used as the target protein in molecular docking for virtual screening (Costa et al., 2018, El-Metwally et al., 2020). Three best active compounds 6b, 6e, and 6f, including ellipticine, were examined docking on these enzymes. The favorably docked molecules were ranked according to the HTVS Glide Score. Obtained docking results (glide score, in kcal/mol) were represented in Table 3.
Compds.
Glide score (kcal/mol)
Hydrogen bonding
π-π Stacking interactions
π-Cation interactions
6b
−8.291
C⚌O quinolone and NH2 with DC8 (chain C)
no
no
6e
−8.326
C⚌O with ARG487 (chain A), NH2 with ASP463 (chain A)
DA12 (three, chain F)
no
6f
−7.441
NH2 with DC8 (chain C), OH with GLU461 (chain A)
DG13 (two, chain F)
no
Fig. 7 displayed the docking pockets of ligands 6b,6e,6f and ellipticine. Ligand 6b had two hydrogen bonding interactions, between C⚌O group of quinolone ring and NH2 group with DC8 (chain C), it had not any π-π stacking and π-cation interactions. Ligand 6e had two hydrogen bonding interactions, between C⚌O group of quinolone ring with residue ARG487 on chain A, and another one between NH2 group with residue ASP463 on chain A, it had three π-π stacking interactions with residue DA12 on chain F. Ligand 6f, which exhibited medium cytotoxic activity, had two hydrogen bonding interactions, between NH2 group with DC8 on chain C and between OH group with residue GLU461 on chain A, it had two π-π stacking interactions with residue DG13 on chain F. This suggested that two hydrogen bonding interactions of C⚌O group of quinolone ring and NH2 group, improved the docking score in case of ligands 6b and 6e, and maybe, make their cytotoxic activity became higher.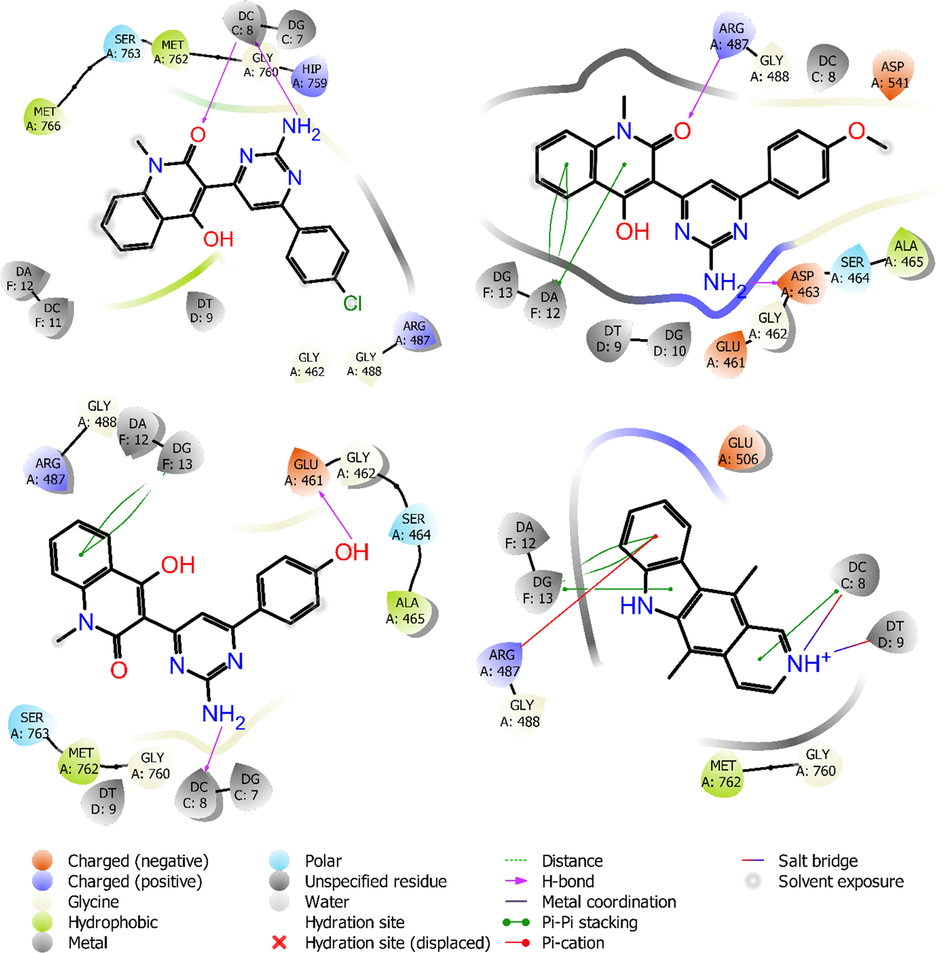
Amino acids involved in intermolecular interactions of ligands 6b (top-left), 6e (top-right) 6f (bottom-left), and ellipticine (bottom-right) with human topoisomerase IIα in complex with DNA (5GWK).
Superimposed poses displayed in Fig. 8 showed that ligands ellipticine (red-orange), 6b (violet), 6e (magenta), 6f (green) all bound at the same position on molecular pocket of human topoisomerase IIα in complex with DNA. Ligands 6b and 6e gave better glide scores when compared with compound 6f for the target protein, with binding score of −8.291 and −8.326 kcal/mol, respectively. The important intermolecular protein-ligand interactions of 6b,6e,6f and ellipticine showed that two highest active compounds 6b and 6e were similar to docking position of ellipticine.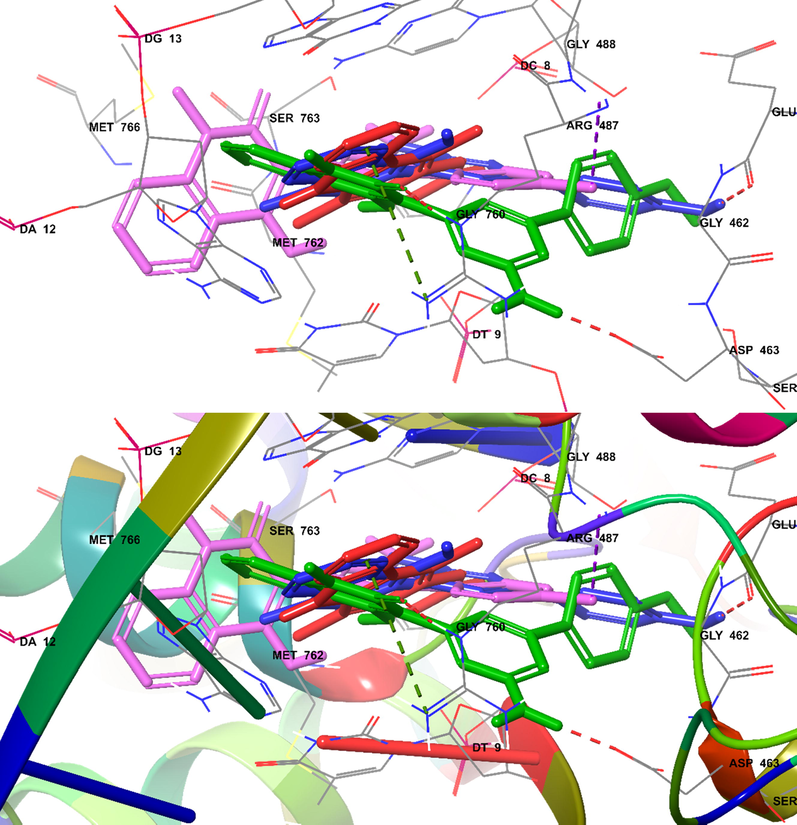
Docked (superimposed) poses showed ellipticine (red), 6b (magenta), 6e (green), 6f (blue).
4 Conclusions
A series of (E)-4-hydroxy-3-(3-(aryl)acryloyl)-1-methylquinolin-2(1H)-ones (5a-i), α,β-unsaturated ketones, were prepared from 3-acetyl-4-hydroxy-1-methylquinolin-2(1H)-one and (un)substituted benzaldehydes with yields of 68–85%. These α,β-unsaturated ketones were converted into 3-(2-amino-6-arylpyrimidin-4-yl)-4-hydroxy-1-methylquinolin-2(1H)-ones (6a-i) by reaction with guanidine hydrochloride in the presence of sodium hydroxide. The yields of quinoline-2-one 6a-were 58–74%. Synthesized compounds 6a-i were evaluated for in vitro cytotoxic activity against two representatives, human squamous cell carcinoma (KB) and hepatocellular carcinoma (HepG2) cell lines. Compounds 6b, 6e, and 6f had remarkable inhibitory activity against the tested cancer cell lines with MIC values of 1.32–5.06 μM. ADMET properties showed that compounds 6b, 6e, and 6f possessed drug-likeness behavior. Cross-docking results indicated that two hydrogen bonding interactions in the binding pocket, as potential ligand binding hot-spot residues for compounds 6b and 6e, may be one of the mechanisms of action responsible for the higher cytotoxic effect on HepG2 and KB cells.
This research was funded by the Scientific Research Project of Ministry of Education and Training (Viet Nam), code: B2019-TNA-11.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Eur. J. Med. Chem.. 2020;198:112363
- [CrossRef]
- J. Heterocycl. Chem.. 2020;57:1154-1164.
- [CrossRef]
- RSC Adv.. 2017;7:28313-28349.
- [CrossRef]
- Arabian J. Chem.. 2017;10:S3324-S3337.
- [CrossRef]
- Bioorg. Chem.. 2020;100:103899
- [CrossRef]
- Saudi Pharm. J.. 2013;21:305-316.
- [CrossRef]
- Biochemistry. 2014;53:1565-1574.
- [CrossRef]
- Chem. Soc. Rev.. 2019;48:4361-4374.
- [CrossRef]
- Expert Opin. Drug Discov.. 2016;11:281-305.
- [CrossRef]
- Arabian J. Chem.. 2014;7:986-993.
- [CrossRef]
- Antimicrob Agents Chemother.. 2007;51:119-127.
- [CrossRef]
- Blood.. 1997;90:4947-4952.
- [CrossRef]
- Eur. J. Med. Chem.. 2020;185:111810
- [CrossRef]
- Costa, D.C.S., De Almeida, G.S., Rabelo, V.W.-H., Cabral, L.M., Sathler, P.C., Alvarez Abreu, P., Ferreira, V.F., Cláudio Rodrigues Pereira Da Silva, L., and Da Silva, F.D.C., 2018. Europ. J. Med. Chem. 156, 524-533. https://doi.org/10.1016/j.ejmech.2018.07.018.
- Sci. Rep.. 2017;7:42717.
- [CrossRef]
- Eur. J. Med. Chem.. 2017;138:491-500.
- [CrossRef]
- Introduction to Spectroscopy. Stamford (USA): Cengage Learning; 2015.
- Bioorg. Chem.. 2020;102:104059
- [CrossRef]
- Bioorg. Chem.. 2020;94:103492
- [CrossRef]
- J. Heterocycl. Chem.. 1984;21:1881-1883.
- [CrossRef]
- Eur. J. Med. Chem.. 2014;81:253-266.
- [CrossRef]
- Eur. J. Med. Chem.. 2019;165:59-79.
- [CrossRef]
- New J. Chem.. 2018;42:2790-2803.
- [CrossRef]
- Med. Chem. Res.. 2019;28:28-38.
- [CrossRef]
- Eur. J. Med. Chem.. 2017;141:335-345.
- [CrossRef]
- Med. Chem. Res.. 2019;28:580-590.
- [CrossRef]
- Arabian J. Chem.. 2019;12:4920-4946.
- [CrossRef]
- J. Enzyme Inhib. Med. Chem.. 2019;34:955-972.
- [CrossRef]
- N. Engl. J. Med.. 2006;354:2542-2551.
- [CrossRef]
- Bioorg. Med. Chem.. 2015;23:602-611.
- [CrossRef]
- Eur. J. Med. Chem.. 2013;63:224-230.
- [CrossRef]
- Chem. Nat. Compd.. 2020;56:465-471.
- [CrossRef]
- RSC Adv.. 2018;8:24376-24385.
- [CrossRef]
- Arabian J. Chem.. 2014;7:409-417.
- [CrossRef]
- Lagorce, D., Reynes, C., Camproux, A.C., Miteva, M.A., Sperandio, O., and Villoutreix, B.O., 2011. In silico adme/tox predictions.
- Int. J. Mol. Sci.. 2020;21:2876.
- Arabian J. Chem.. 2017;10:S484-S490.
- [CrossRef]
- Antiviral Res.. 2012;94:35-43.
- [CrossRef]
- Bioorg. Chem.. 2020;96:103641
- [CrossRef]
- J. Mol. Liq.. 2019;273:559-575.
- [CrossRef]
- Eur. J. Med. Chem.. 2020;187:111960
- [CrossRef]
- Top. Curr. Chem.. 2019;377:9.
- [CrossRef]
- Liebigs Ann. Chem.. 1990;1990:821-823.
- [CrossRef]
- Curr. Bioact. Compd.. 2019;15:289-303.
- [CrossRef]
- Schrödinger, 2018. http://www.schrodinger.com.
- Cancer Res.. 1988;48:4827-4833.
- Neuroscience.. 2013;232:1-12.
- [CrossRef]
- Carbohydr. Res.. 2009;344:2399-2405.
- [CrossRef]
- Curr. Pharm. Des.. 2016;22:6982-6987.
- [CrossRef]
- Eur. J. Med. Chem.. 2010;45:379-386.
- [CrossRef]
- Nucleic Acids Res.. 2017;45:10861-10871.
- [CrossRef]
- Science.. 2011;333:459-462.
- [CrossRef]
- Bioorg. Chem.. 2020;102:104081
- [CrossRef]
- Eur. J. Med. Chem.. 2014;80:267-277.
- [CrossRef]
- Mol Cell Proteomics.. 2013;12:237-244.
- [CrossRef]
- Pharm. Res.. 2002;19:1446-1457.
- [CrossRef]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2020.09.018.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary Data 1
Supplementary Data 1




