

Translate this page into:
Recent developments of synthesis and biological activity of sultone scaffolds in medicinal chemistry
⁎Corresponding author. ahmupharm@126.com (Wenjian Tang)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Sulfonyl drugs are widely used to treat various types of diseases, in which sultones have several desirable properties that have led to their increased use, including improving its simple and effective synthetic methods and/or potentially enhancing potency of drugs. This review focuses on the recent progress in typical synthesis of sultones and the latest developments in various therapeutic applications, including carbonic anhydrase inhibitory activity, HIV-I and HIV-II inhibitory activity, human cytomegalovirus inhibitory activity, and cholinesterase inhibitory activity and their structure–activity relationship. We hope that this review will encourage medicinal chemists to consider the potential benefits to incorporating sultone scaffold into future drug discovery.
Keywords
Sultone
Sulfonyl drug
Anti-carbonic anhydrase activity
Anti-HIV activity
Cholinesterase activity
Structure–activity relationship

- HIV
-
human immunodeficiency virus
- BuChE
-
butyrylcholinesterase
- HCMV
-
human cytomegalovirus
- VZV
-
varicella virus
- CA
-
carbonic anhydrase
- BVDV
-
bovine viral diarrhea virus
- SuFEx
-
sulfur (VI) fluoride exchange
- ESF
-
ethenesulfonyl fluoride
- CCCC
-
carbene cyclization cycloaddition cascade
- RCM
-
ring-closing metathesis
- AIDS
-
acquired immunodeficiency syndrome
- RT
-
reverse transcriptase
- NNRTIs
-
non-nucleoside reverse transcriptase inhibitors
- HCV
-
hepatitis C virus
- AD
-
alzheimer's disease
- ChE
-
cholinesterase
- AChE
-
acetylcholinesterase
- CAS
-
catalytic active site
- PAS
-
peripheral anion site
- CAM
-
chick chorioallantoic membrane
Abbreviations
1 Introduction
Sultone moieties (Fig. 1), the internal esters of sulfonic acids introduced by Erdmann in 1888, have emerged as significantly valuable heterocycles that has been widely used as a general building block in the field of medicinal chemistry (Erdmann, 1988; Pustenko and Žalubovskis, 2017). Novel sultone-functionalized heterocyclic compounds will be particularly useful and desirable in the fields of diversity-oriented chemistry, medicinal chemistry, chemical biology, drug discovery and pharmaceutical industry (Fang et al., 2019; Revathi et al., 2018; Zhang et al., 2018). The early synthesis methods involved carbanion-mediated sulfonate intermolecular or intramolecular coupling reaction (CSIC reaction) (Postel et al., 2003) or sulfonation of olefins with dioxane-sulfur trioxide (Tin and Durst, 1970). Mondal (Mondal, 2012) and Pustenko (Pustenko and Žalubovskis, 2017) further summarized the synthesis methods of sultones in 2012 and 2017, including cycloaddition reaction (Ghandi et al., 2012; Tian et al., 2003), intramolecular Diels-Alder reaction (Bovenschulte et al., 1989; Chan et al., 1997; Doye et al., 1997; Ghandi et al., 2013; Metz et al., 1992; Metz et al., 1994; Metz et al., 1996; Moghaddam et al., 2013; Plietker et al., 2000), ring closure translocation (Karsch et al., 2004; Le-Flohic et al., 2003), Pd-catalyzed reaction (Tamaru et al., 1990), Rh-catalyzed C-H insertion (Wolckenhauer et al., 2007), Heck reaction (Wolckenhauer et al., 2008). Currently, the simple and effective synthesis method of sultones have received an increasing attention to synthetic organic chemists and biologists.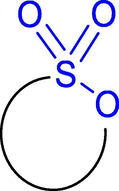
Structure of sultone.
Besides their synthetic application, Molecules containing sultone scaffolds are widely used to treat various types of diseases, for example, phenolsulfonphthalein (F1) (Fig. 2) is a non-toxic compound that has been approved for human kidney function testing (Itagaki et al., 2018; Russel et al., 1987). In previous reports, sultones were found to have diverse biological activities, such as skin sensitization (Bolt and Golka, 2004; Meschkat et al., 2001; Ritz et al., 1975), anti-tumor activity, anti-virus (human immunodeficiency virus (HIV-I, II), human cytomegalovirus (HCMV) inhibitory activity, and varicella virus (VZV)) inhibitory activity (Balzarini et al., 1992; Bonache et al., 2005; Bonache et al., 2008; Bonache et al., 2011; Camarasa et al., 1992; Camarasa et al., 2004; Camarasa et al., 2005; Camarasa et al., 2006a; Camarasa et al., 2006b; Das et al., 2011; De-Castro et al., 2003; De-Castro et al., 2005; De-Castro et al., 2006a; De-Castro et al., 2006b; De-Castro et al., 2007; De-Castro et al., 2008; De-Castro et al., 2009; De-Castro et al., 2011; De-Castro and Camarasa, 2018; Lobatón et al., 2002; Perez-Perez et al., 1992; Rodríguez-Barrios et al., 2001; Velazquez et al., 1992; Velázquez et al., 2004; Sluis-Cremer et al., 2006), carbonic anhydrase (CA) inhibitory activity (Grandane et al., 2014; Grandane et al., 2015a; Grandane et al., 2015b; Nakai et al., 2018; Nocentini et al., 2018; Pustenko et al., 2017; Tanc et al., 2013; Tanc et al., 2015; Tars et al., 2012), selective butyrylcholinesterase (BuChE) inhibitory activity (Xu et al., 2019), and so on. This review mainly summarizes the advances in the recent synthesis and pharmacology, and discusses the structure–activity relationship (SAR) of active compounds.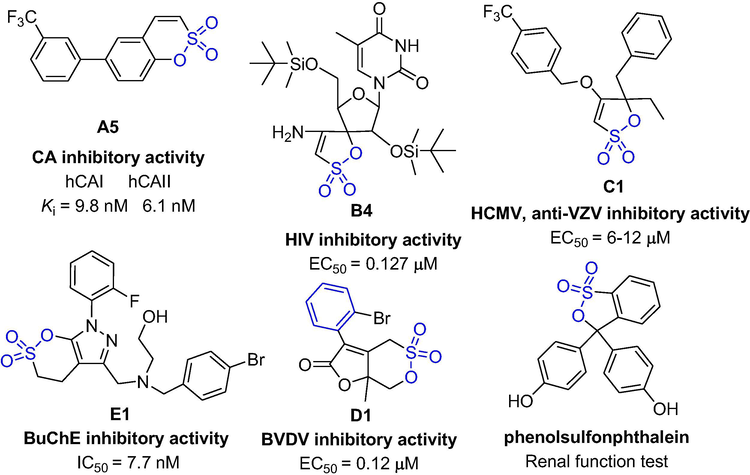
The structures of some active compounds.
2 Synthesis of sultones
Recently, many powerful methods for sultones synthesis were discovered, such as Julia–Kocienski reaction, Diels-Alder reaction, closed-loop metathesis, Rh-catalyzed C-H insertion, Rh-catalyzed carbene cycloaddition cascade, or other metal catalysts reactions using palladium, rhodium, gold, copper, ruthenium.
2.1 Julia–Kocienski reaction
Smith et al. prepared firstly γ-sultones in a stereocontrolled method through the epoxide and the homologous Julia-Kocienski reaction (Scheme 1). In the early experiment, it was expected that alkoxysulfones 1b gave sulfinates 1c through Smiles rearrangement, subsequently 1c lose SO2 and cyclized to produce cyclopropane. But compound 1a and (R)-propylene oxides 2a (R = Me) under the basic conditions of LiN(SiMe3)2 at room temperature gave γ-sultones 2d (R = Me). Both the yield and the ee values were>99%, and no cyclopropane was observed. The yield of γ-sultones obtained from compound 1a with a terminal alkyl-substituted epoxide was excellent. Protected alcohols, amines, ketones, halogen-substituted epoxides, diepoxides, and alkyl-substituted terminal epoxides can all give the corresponding substituent γ-sultones. However, when the substrate is a bis-epoxide, only a mono-sulfonylation product can be obtained, because the formation of the first internal sulphur ring may slow down the formation of the second sultone ring. The disubstituted epoxide reaction may involve a single inversion of stereochemistry (cis-1,2-butene oxide produces only trans-sultone, while trans-1,2-butene oxide produces only cis-sulfonate lactone) (Smith et al., 2015). This pathway provides access to γ-sultones using a one-pot method in a stereo-controlled manner.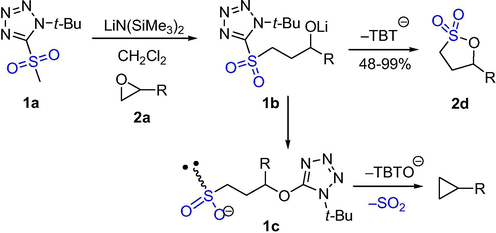
The synthesis of γ-sultones by Julia–Kocienski reaction.
2.2 Sulfur fluoride exchange (SuFEx) click reaction
2.2.1 Nickel-catalyzed SuFEx reaction
Chen et al. have developed a mild and efficient nickel-catalyzed cyclization method by using sulfur (VI) fluoride exchange (SuFEx) click reaction for the synthesis of sultone-functionalized pyridines (Scheme 2). The sultones 3 were obtained by Ni-catalyzed cyclization reaction from substrate (E)-2-phenylethanesulfonyl fluoride (1 eq) and 2-acetylpyridine (2 eq) in acetonitrile. The (hetero)aralkylsulfonyl fluorides with p- or m-substituted groups have moderate to excellent yield. In which, the reaction time for the o-substituted one is longer, but the yield is higher (>90%); the halide group (F, Cl and Br) have better tolerance to the catalytic conditions; the yield of the p-substituted one with stronger electron-withdrawing ester group is reduced (44%); the 2-aralkylsulfonyl fluoride with a disubstituted aromatic ring has a good to excellent yield (73–89%). This method is also applicable to 2-arylsulfonyl fluoride, thiophene, dibenzothienyl, and indole with complex and heterocyclic aromatic substrates (including naphthyl). 2-Phenylethenesulfonyl fluorine can react with substituted 2-acetylpyridine to form sultones in moderate to good yields. However, when the substituent group is adjacent to the N atom of pyridine, the yield becomes low. When 6,7-dihydroquinoline-8(5H)-one was reacted with various 2-substituted ethenesulfonyl fluorides, a novel class of fused-sultones 4 were obtained in a yield of 37–99%. Besides, this method is practical so that sultone-functionalized pyridines can be prepared on a reasonable scale (Chen et al., 2018). An efficient Ni-catalyzed annulative process for the synthesis of sultones were developed using SuFEx chemistry. This method features a wide scope to serve as an irreplaceable asset for medicinal chemistry and medicinal discovery.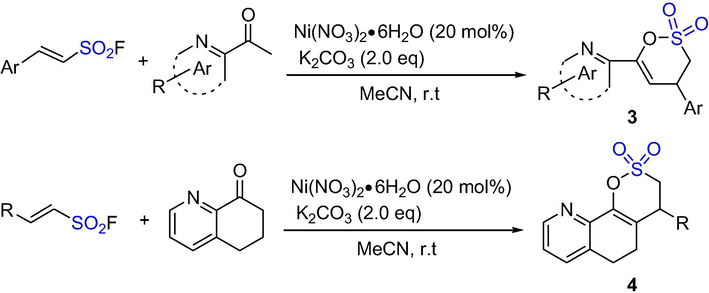
The synthesis of δ-sultones by Ni-catalyzed SuFEx reaction.
2.2.2 DBU-catalyzed SuFEx click reaction
Chen et al. synthesized a new class of δ-sultones through direct cyclization of ethenesulfonyl fluoride (ESF) and pyrazolone or 1,3-dicarbonyl compounds catalyzed by 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) (Scheme 3). The δ-sultones 5 were obtained from pyrazolones or 1,3-dicarbonyl compounds (1.0 eq) with (E)-2-(hetero)arylethenesulfonyl fluoride (1.0 eq), DBU (5–30 mol%) and NaHCO3 (1.0 eq) in CH2Cl2 at room temperature for 3–24 h in excellent yields (>90%) (Dong et al., 2014). It is a simple and effective synthesis method.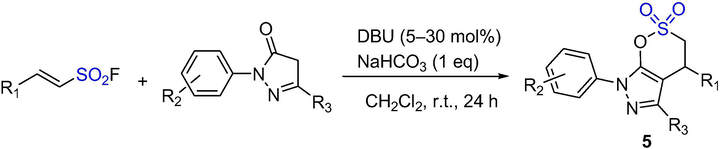
The synthesis of δ-sultones by DBU-catalyzed SuFEx reaction.
2.3 Photocatalytic redox reaction
The γ- and δ-sultones 6 were synthesized from different α,ω-enol and trifluoromethylsulfonyl chloride by one-step synthesis under (Cu(dap)2)Cl photo-redox catalysis (Scheme 4). Focusing on fluorinated sultones, a wide range of substrates can be cleanly converted to the title compounds. The fluorinated sultones are attractive as structural motifs in drug synthesis with being demonstrated with the synthesis of a trifluoroethyl-substituted analogue of a benzoxathiin that has high anti-arrhythmic activity (Rawner et al., 2016). This method uses an inexpensive low load copper catalyst to obtain the corresponding sultones with moderate to excellent yields.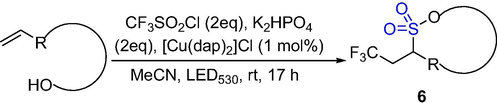
The synthesis of δ-sultones by photocatalytic redox reaction.
2.4 N-Heterocyclic carbine-catalyzed (3 + 3) annulation
As shown in Scheme 5, a series of unsaturated δ-sultones were obtained from (3 + 3) annulation of α,β-unsaturated sulfonyl azolium intermediates with trimethylsilyl enol ethers of various 1,3-dicarbonyl compounds in modest yields (40–88%). The δ-sultones 7 were synthesized from different trimethylsilyl (TMS) enol ethers, α,β-unsaturated sulfonyl fluoride and Imes (10 mol%) in tetrahydrofuran (THF) for 17 h. The electron-deficient sulfonyl fluorides have more efficient conversion than the electron-rich ones. The yields of 2-Furyl-substituted sultone and ESF-derived sultone are moderate. The TMS enol ether derived from 1,3-cyclohexadione and the acyclic TMS enol ether of acetylacetone gave the δ-sultones in acceptable yields. TMS enol ethers derived from unsymmetric diketones gave the expected δ-sultones in good yields and modest regioselectivity. In contrast, TMS enol ethers derived from β-ketoesters gave the methyl- and isopropyl-substituted δ-sultones in good yields (Ungureanu et al., 2015).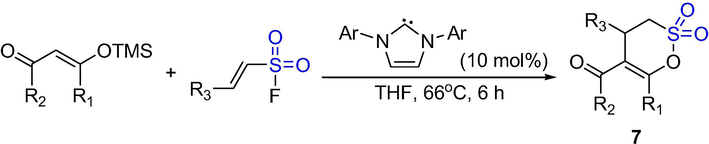
The synthesis of δ-sultones by carbene-catalyzed (3 + 3) annulation.
2.5 C-H activation reaction
2.5.1 Pd-catalyzed C-H activation reaction
Li et al. firstly reported the functionalization with the sulfonic acid group as the directing group in a C–H activation reaction. (60)Fullerene-fused sultones has been employed in the unprecedented Pd-catalyzed C–H activation reaction of arylsulfonic acids to afford (60)fullerene-fused sultones. As shown in Scheme 6, C60-fused sultones 8a were obtained from arylsulfonic acids and C60 in 18–30% yields. C60-fused sultones were treated in o-dichlorbenzene (ODCB) with excess BF3·OEt2 at 150 °C to give arylsulfonate-substituted fullerenes 8b in good yields (Li et al., 2012b).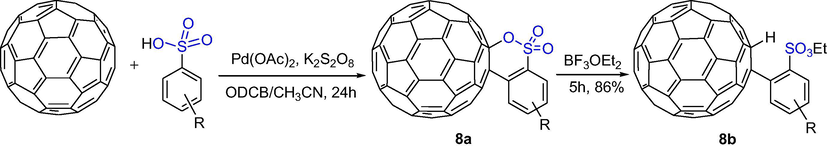
The synthesis of C60-fused sultones by Pd-catalyzed C-H activation reaction.
Li et al. disclosed a Pd-catalyzed cyclization of arylsulfonic acids with simple arenes to give aromatic sultones through sulfonic acid group-directed multiple C-H activation and C-C/C-O formation. As shown in Scheme 7, one isomer aryl sulfonyl lactone 9 can be obtained by reacting aryl sulfonic acid with aromatics (20 eq), Pd(OAc)2 (10 mol%) and (NH4)2S2O8 (3 eq) in ODCB solution at 90 °C for 10 h. except the reaction between toluene and 4-methylbenzenesulfonic acid produced an inseparable regioisomers 9a and 9b at a ratio of 2:1 (Li et al., 2012a).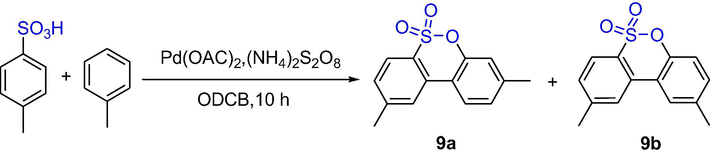
The synthesis of aromatic sultones by Pd-catalyzed C-H activation.
2.5.2 Rh-catalyzed C-H activation reaction
Qi et al. described a new Rh-catalyzed synthesis of sultones via the oxidative coupling of sulfonic acids with internal alkynes. The sultones 10 were obtained from different arylsulfonic acids via aryl C–H activation assisted by a sulfonic acid group in Scheme 8, for example, alkynes with electron-donating groups had good yields, para-alkynes or diarylalkynes with o-OMe had lower yields, asymmetric alkynes showed moderate yields but higher regioselectivity, 1-naphthalenesulfonic acid and diphenylacetylene exhibited moderate reactivity (Qi et al., 2014). A broad scope of substrates has been examined and good functional group compatibility has been realized. This approach provides a new access to sultones.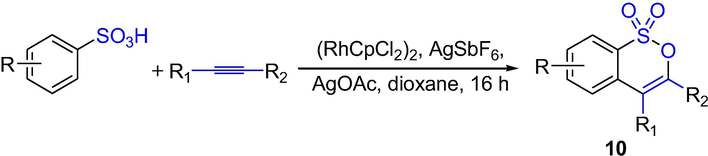
The synthesis of δ-sultones by Rh-catalyzed C-H activation reaction.
2.6 C-H insertion reaction
Liyanage et al. reported that key intermediates can be prepared through intramolecular C-H insertion on diazosulfonate substrates. The δ-sultones 12a–d were synthesized from the diazosulfonate substrates of borneol (11a), 3-methylpentan-1-ol (11b), and isomenthol (11c) in high yields in Scheme 9. This C-H insertion reaction was used to prepare a series of stereoselective sultones from easily available materials (Liyanage et al., 2015). By using C–H insertion on sulfonyl substrates, a series of useful intermediates were prepared from inexpensive and easily available starting materials.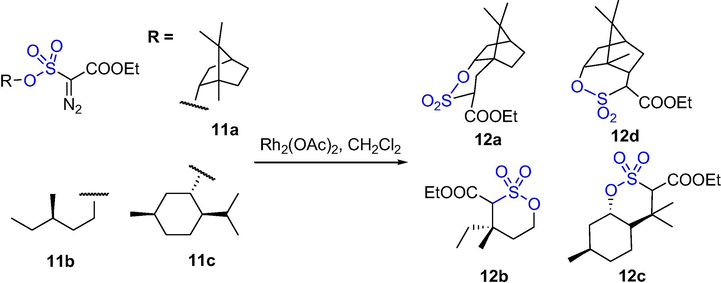
The synthesis of δ-sultones by Rh-catalyzed C-H insertion reaction.
2.7 Baylis-Hillman (BH) reaction
A solvent-dependent method was used to synthesize β-keto-benzo-δ-sultone scaffolds. As shown in Scheme 10, the sultones 13a were obtained in high yields in DMF using a one-pot, DBU-catalyzed condensation of 2-hydroxybenzaldehydes and (E)-2-phenylethenesulfonyl chlorides, on the other hand, the initially prepared 2-formylphenyl-(E)-2-phenylethenesulfonate derivatives underwent DBU-catalyzed reactions to sultones 13b in moderate to good yields. These reactions presumably proceed via DBU-catalyzed o-sulfonylation/intramolecular Baylis–Hillman/1,3-H shift or dehydration tandem sequences, respectively (Ghandi et al., 2011). These broaden the benzo-δ-scaffolds that are accessible through intramolecular BH reactions, and many of them may represent interesting pharmacophores.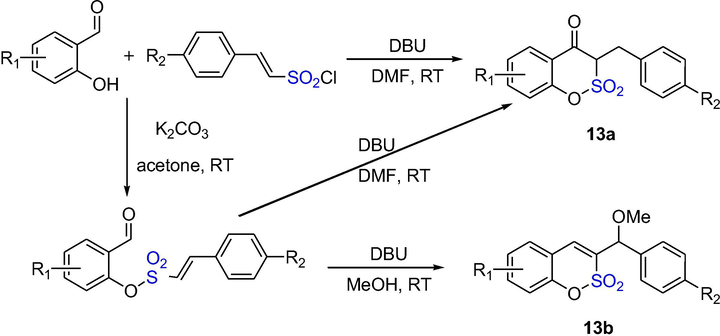
The synthesis of δ-sultones by Baylis-Hillman reaction.
2.8 Asymmetric hydrogenation
Chudasama et al. reported the use of aerobically initiated, metal-free hydroacylation of various C = C and N = N acceptor molecules with a wide range of aldehydes, for example, the hydroacylation of vinyl sulfonate 14b with n-butyraldehyde 14a in 1,4-dioxane gave the pentafluorophenyl (PFP) γ-keto-sulfonate 14c. The sultone 14 could be achieved by reducing the ketone moiety of γ-keto-PFP-sulfonate and subsequently eliminating pentafluorophenol in good yield (Scheme 11) (Chudasama et al., 2013).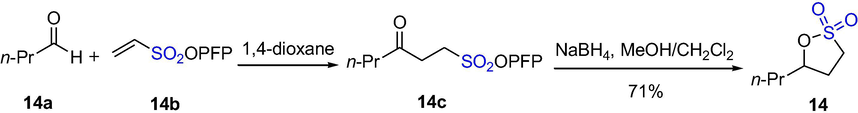
The synthesis of sultones by hydroacylation–elimination.
2.9 Heterocyclization of arylalkynes
Gaitzsch et al. reported the synthesis of 1,3-dienic δ-sultones 15 via a one-step reaction of arylalkynes with sulfotrioxide in dioxane (Scheme 12). It is presumed that alkynes 15a were first sulfonated with SO3 to afford a highly reactive β-sultones existing in the cyclic 15b and zwitterionic 15c form. Subsequent electrophilic addition of alkyne to this β-sultones then gives the stable δ-sultones 15. Thus, a range of monosubstituted arylalkynes were investigated in a heterocyclization with SO3 to give δ-sultones. The resulting sultones were regioselectively brominated with Br2 or NBS (Gaitzsch et al., 2011).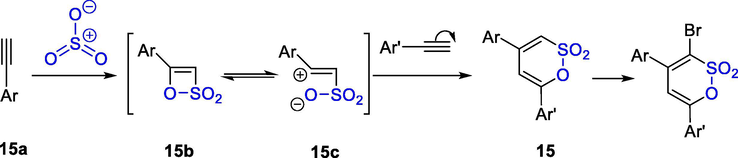
One-step synthesis of 1,3-dienic δ-sultones in the presence of SO3.
2.10 Cycloaddition reaction
2.10.1 Iodine-catalyzed oxidative heterocyclic reaction
Yoshimura et al. has developed an efficient cycloaddition of heterocyclic alkenes with nitrile oxides generated in situ from the corresponding aldoximes using Koser's reagent. The oxidative cyclization of various aldoximes with 1-propene-1,3-sultone affords the respective isoxazoline-ring-fused heterobicyclic products 16 in moderate to good yields (Scheme 13). Furthermore, under similar conditions, the reaction of aldoxime with a cyclic phospholene-oxide produces the corresponding heterobicyclic phospholene oxides in moderate yields. The structures of bicyclic phospholene oxide and two sultones were established by X-ray crystallography (Yoshimura et al., 2017). This reaction generally afforded the corresponding heterobicyclic products in moderate to good yields.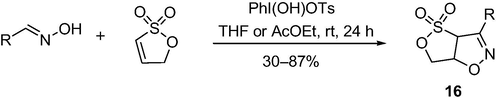
The synthesis of sultones by iodine-catalyzed cycloaddition.
2.10.2 Rh-catalyzed carbene cyclization cycloaddition reaction
Carbene cyclization cycloaddition cascade (CCCC) is a powerful and atomically economical reaction that produces stereogenic oxapolycyclic adducts. Shi et al. reported that vinylsulfonates are excellent dipolarophiles for carbonyl ylides derived from diazoketones in Rh-catalyzed intramolecular cycloadditions (Scheme 14). The cycloaddition of diazoketones 17a–d and 17e–h under the catalysis of 3 mol% Rh2(oct)4 in CH2Cl2 occurred smoothly to give the γ-sultones 18a–d and δ-sultones 18e–h, respectively, in high yields and excellent stereoselectivities. The CCCC reaction of vinyl sulfonate (+)-19 gave sultone-bridged hydroazulene (+)-20 with the only diastereomer under the catalysis of Rh2(OAc)4 in 90% yield (Shi et al., 2009). The multifunctional substrate forms polycyclic sultones with high yield and very good diastereoselectivity under mild reaction conditions.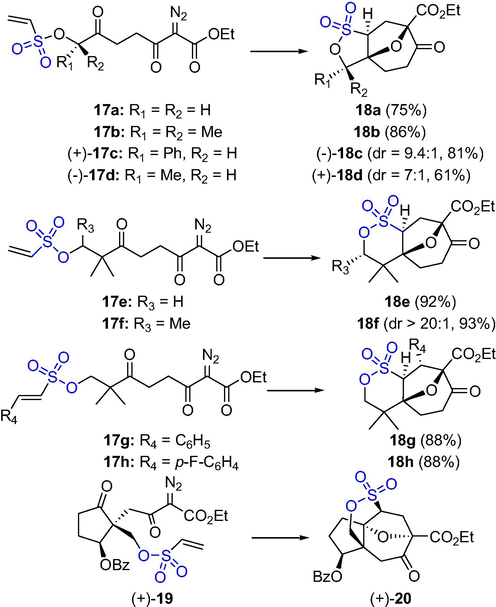
The synthesis of sultones by Rh-catalyzed cycloaddition.
2.10.3 (2 + 2) cycloaddition reaction
Koch et al. reported the catalytic asymmetric synthesis of β-sultones, which has enabled a rapid access to a number of highly enantioenriched biologically interesting sulfonyl and sulfinyl compounds, either two vicinal stereocenters, such as in β-hydroxy-sulfonamides, -sulfonates, -sulfones, -sulfonic acids, -sulfinic acids, γ-sultines, and γ-sultones or a single stereocenter, such as in α-branched alkyl or allyl sulfonic acids (Scheme 15). The inherent ring strain of the four-membered heterocycles 21 was used to produce the sulfene intermediates in asymmetric catalysis. The reactivity of a sulfene as an electrophile could be reverted by the formation of a nucleophilic zwitterionic sulfene-amine adduct. The cooperative catalysis achieves a combination of high enantioselectivity and reactivity (Koch and Peters, 2011). The catalytic asymmetric synthesis of β-sultones have firstly been developed. This methodology enables a rapid access to a number of highly enantioenriched sulfonyl and sulfinyl compounds.
The synthesis of β-sultones by (2 + 2) cycloaddition.
2.11 Closed-ring reaction
2.11.1 Acid-catalyzed cyclization
To facilitate more detailed studies of sultones, a new method was used to synthesize the sulfonic acid lactones and finish its ring-closing reaction. A new sultone, (E)-ethyl 4-oxo-6-styryl-3,4-dihydro-1,2-oxathiine-5-carboxylate 2,2-dioxide (S-CA), was synthesized and identified. Its synthesis extended the method of ring-closing reaction of sulfonic acid lactones. Li et al. synthesized the sultone 22 by a fast ring-closing reaction (Scheme 16). The reaction of cinnamoyl chloride with ethyl acetoacetate under NaH condition gives ethyl 2-cinnamoyl-3-ketobutyrate, subsequently transfers to sultone 22 by ring-closing reaction under Ac2O and H2SO4. Compound 22 selectively suppress small angiogenesis in chorioallantoic membrane, without influencing middle or large angiogenesis (Li et al., 2015).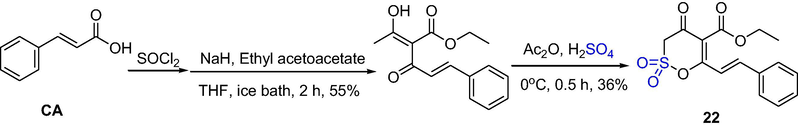
The synthesis of δ-sultone by acid-catalyzed cyclization.
2.11.2 Base-catalyzed cyclization
Xu et al. (Xu et al., 2014) reported the synthesis of a novel series of bicycle δ-sultones containing γ-lactones and their activity against bovine viral diarrhea virus (BVDV). The alkylsulfonate intramolecular cyclization reaction (CSIC reaction) gave the δ-sultones 23 in 32–70% yields (Scheme 17).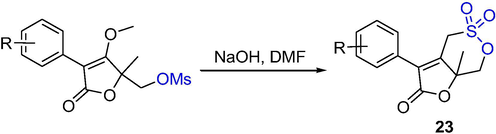
The synthesis of δ-sultones by base-catalyzed cyclization.
2.11.3 DBU-catalyzed intramolecular aldol cyclization
Grandane et al. reported a synthetic method of benzo-δ-sultones as bioisosteres of coumarin through the intramolecular aldol cyclization of mesylsalicyl aldehydes in the presence of DBU (Scheme 18). The benzo-δ-sultones 24 were obtained from mesyl derivatives by cyclization reaction in CH2Cl2 under a catalytic of DBU in good yields (Grandane et al., 2012). This method is reproducible and applicable for a broad scope of substrates with electron-withdrawing and -donating substituents.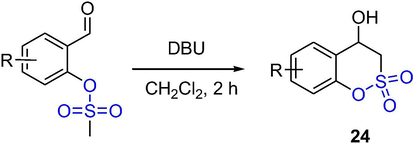
The synthesis of δ-sultones by DBU-catalyzed aldol cyclization.
2.11.4 Pd-catalyzed Heck cyclization
Mondal et al. reported an effective method for the synthesis of seven-membered fused-sultones by intramolecular Heck cyclization catalyzed by Pd(PPh3)4 (Scheme 19). The seven-membered fused-sultones 25 were obtained in excellent yield and regio- and stereo-selectivity from the mixture of raw materials (1.5 eq), 3 mol% Pd(PPh3)4 and HCOONa in DMF-H2O at 100 °C for 1 h. Heck-type cyclization is regio- and stereo-selective, which the stereochemistry of exocyclic double bond in the dibenzo-fused sultone is opposite to that of the benzopyridin-fused sulfonate (Mondal et al., 2015a). The method offers the regio- and stereo-selective synthesis of highly functionalized benzopyrido-fused or dibenzo-fused sultone derivatives in high yield under mild conditions.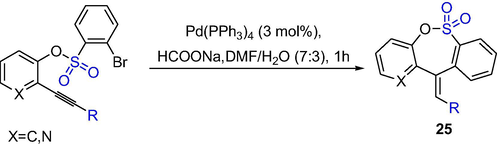
The synthesis of sultones by Heck cyclization.
2.11.5 Pd-catalysed intramolecular cyclization
A Pd-catalyzed intramolecular arylation of 2-bromobenzenesulfonates was reported (Scheme 20). The arylation was affected by the substituents on phenol moiety, for example, electron-donating ones favor the reaction while electron-withdrawing ones are unfavorable. The clization of the intermediate phenyl 2-bromobenzenesulfonates 26a gave sultones 26b in good yields, which effective method generates tricyclic and tetracyclic sultones (Bheeter et al., 2012; Majumdar et al., 2009).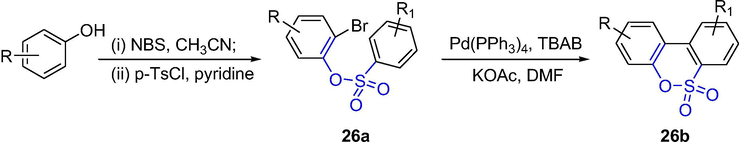
The synthesis of sultones by Pd-catalyzed intramolecular cyclization.
This Pd-catalyzed intramolecular arylation can be used to synthesize the regioselective uracil-, coumarin- and quinolone-fused benzosultams and benzosultones (Scheme 21). Pd-catalyzed cyclization of uracil 2-bromobenzenesulfonates gave uracil-fused benzosultones 27 in high yields (Mondal et al., 2015b).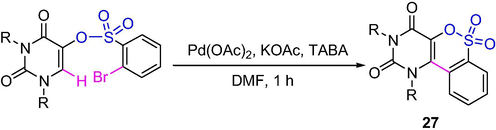
The synthesis of sultones by Pd-catalyzed intramolecular cyclization.
2.11.6 Dimer gold-catalyzed cyclization
Similarly, a dimeric phosphine-gold complex as a photocatalyst can catalyze the photoreductive radical reaction of an aryl bromide to give the biaryl compound 28 (Scheme 22). Sultone 28 was formed in the presence of the dimerized phosphine-gold complexes (Au2(μ-dppm)2)Cl2 (2.5 mol%) under ultraviolet radiation A (UVA) or sunlight (Au2(μ-dppm)2)Cl2 (5.0 mol%) in MeCN in good yields (Revol et al., 2013). This photo-redox process has uniqueness and high synthesis potential.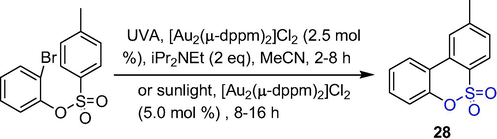
The synthesis of δ-sultones by gold-catalyzed cyclization.
2.11.7 Direct cyclization/desaturation radical cascade reaction
A sulfonylation/rearrangement sequence of allyl ether containing β-lactam or glucofuranoside with 2-(3,3-diethyltriazine-1-enyl)-4-methylbenzene-1-sulfonyl chloride gave the functionalized 1,3-diene-2-yl arenesulfonates, which suffered a direct cyclization/desaturation radical cascade to give title δ-sultones 29a-b in moderate yields (Scheme 23). Experiments and density functional theory (DFT) calculations demonstrated stereoselective cyclization of the readily formed core through intramolecular Diels-Alder reaction. This metal-free C-C cyclization reaction is feasible and universal (Alcaide et al., 2016).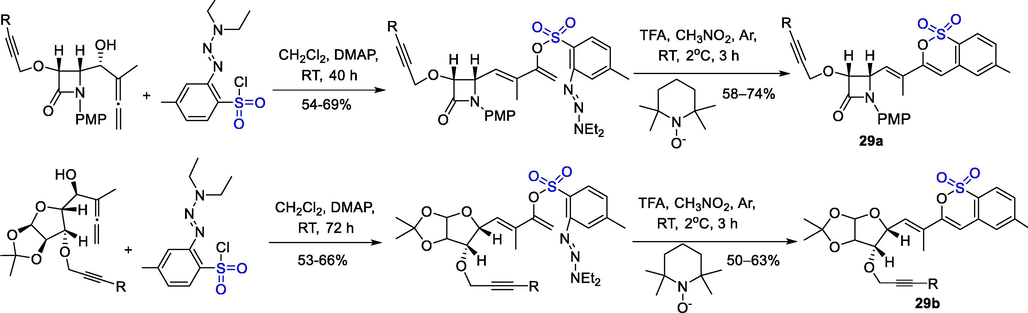
The synthesis of enantiopure polycyclic-fused sultones.
2.11.8 Grubbs-catalyzed ring-closing metathesis
Mondal et al. reported the synthesis of seven-membered sultones fused with different carbo- and heterocycles through ring-closing metathesis (RCM) in good yields (Scheme 24). Sulfonylation reaction of o-allylphenol derivatives with 2-chloroethylsulfonyl chloride gave the vinyl-sulfonates with yields of 81–95%, which were transferred to the sultones 30 under catalysis of Grubbs’ 2nd generation (3 mol %) with yields of 70–80% (Mondal and Debnath, 2014).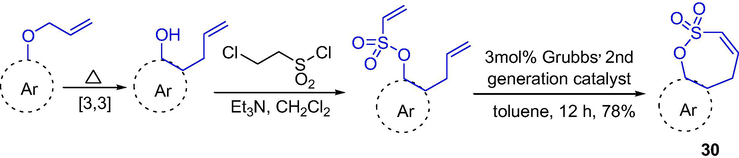
The synthesis of seven-membered sultones by Grubbs-catalyzed RCM.
Walleser et al. reported a twofold ring-closing metathesis under forcing conditions provided unsaturated lactone/unsaturated sultone (Scheme 25). Ethanesulfonate 31 with different functional groups gave monometathesis products 32a (18%), 32b (52%) and cross metathesis products 32c (9%) mixtures under catalysis of 1 mol% Grubbs II, while only sultone 32d was obtained under catalysis of 2 mol% Grubbs II catalyst in 75% yield (Walleser and Brückner, 2014).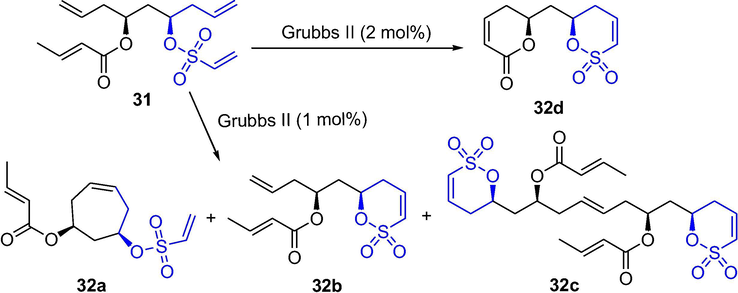
The synthesis of sultone by Grubbs-catalyzed RCM.
2.11.9 Ru-catalyzed olefin metathesis
Pustenko et al. reported the synthesis of sultones using Ru-catalyzed olefin metathesis. The seven-membered fused-sultones 33 were obtained from the reaction of 4-substituted 2-ethenylprop-2-ene-1-sulfonate with Ru-catalyst (tricyclohexyl phosphine(1,3-bis(2,4,6-trimethyl-phenyl)imidazol-2-ylidene)(3-phenyl-1H-inden-1-ylidene)ruthenium(II) dichloride) (0.05 eq) in dry toluene (10 mL/0.2 g) at 70 °C for 4 h in yields of 84–96% (Scheme 26) (Pustenko et al., 2017).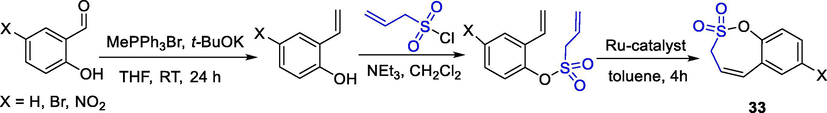
The synthesis of sultones by Ru-catalyzed olefin metathesis.
Although we reviewed the various synthetic methods of the sultone scaffolds, there is an urgent need to develop more effective and mild synthetic methods to obtain plenty of sultone compounds with new scaffolds to enrich sultone compound libraries. In addition, there are few reports on synthesis of sultones based on rational drug design. We hope that researchers can optimize the methods of batch synthesis of sultones to find the potential sultone inhibitors by high-throughput screening.
3 Biological activities of sultones
Sulfonyl compounds are widely used as basic structural frameworks in pharmaceutical chemistry (Zhao et al., 2018; Zhao et al., 2019). Phenolsulfonphthalein (F1) is a synthetic small molecular as various medical diagnostic agents for many years, for example, phenolsulfonphthalein has been used clinically as a diagnostic marker to detect renal function by estimating urinary excretion rate after intravenous injection, in addition to improving the accuracy of gastric secretion studies and oviduct patency assessments (Itagaki et al., 2005; Horhota and Fung, 1978). It was also found that sultone has a variety of biological activities, including antiviral (HIV-1, HIV-II, HCMV and VZV) activity, anticarbonic anhydrase (CA) activity, selective antibutyrylcholine (BuChE) activity (Fig. 2).
3.1 Carbonic anhydrase inhibitors
Carbonic anhydrases (CAs, EC 4.2.1.1) are ubiquitous Zn2+ metallo-enzymes, and 16 isozymes have been identified. The α-CAs are present in vertebrates, protozoa, algae and cytoplasm of green plants and in some bacteria, the β-CAs are predominantly found in bacteria, algae and chloroplasts of both mono- as well as dicotyledons, but also in many fungi and some archaea. The γ-CAs were found in archaea and some bacteria, whereas the δ- and ζ-CAs seem to be present only in marine diatoms. Among those identified, there are eight cytosolic proteins (CA I, CA II, CA III, CA VII, CA VIII, CA X, CA XI, CA XIII), two mitochondrialmatrix proteins (CA VA, CA VB), one secreted protein (CA VI), two glycosylphosphatidylinositol (GPI)-anchored proteins (CA IV and CA XV), and three transmembrane proteins (CA IX, CA XII, CA XIV). Indeed, 16 such isozymes were described in non-primates, CAs I-XV with two V-type isoforms, CA VA and CA VB, and 15 isoforms are known in primates, as CA XV is not expressed in these mammals (Fisher et al., 2012; Lee et al., 2009; McKenna and Supuran, 2014; Savile and Lalonde, 2011; Vullo et al., 2013).
CAs catalyze the hydration of CO2 and the dehydration of bicarbonate, which play an important role in regulating the intracellular and extracellular pH in various physiological processes. Abnormal levels or activities of these enzymes are often associated with different diseases (Alterio et al., 2012; Krishnamurthy et al., 2008). CA II is associated with glaucoma, edema, epilepsy, altitude sickness, et al. Inhibiting CA II can reduce bone loss in osteoporosis. CA IV is a drug target for several pathologies including glaucoma (along with CA II and XII), pigmented retinitis and stroke. CA VA and VB are targets of anti-obesity drugs, while CA IX and XII are targets of anticancer drugs (Alterio et al., 2012; Krishnamurthy et al., 2008). CA IX is a marker of the progression of many types of hypoxic tumors, such as primary tumors and metastatic tumors (Alterio et al., 2012). CA XII over-expression is found in meningiomas, hemangioblastomas, gliomas, brain tumors, and so on. In breast cancer, CA XII expression levels indicate a potential attack of the disease (Alterio et al., 2012; Baig et al., 2019; Ilie et al., 2011; Parkkila et al., 2000). Because CAs with multiple isoforms exist in a variety of tissues and organs, it is important to design selective CA inhibitors.
3.1.1 Mechanism of sulfocoumarin as CA inhibitors
The sulfocoumarin is converted into 2-hydroxyphenyl-vinylsulfonic acid by hydrolysis of the intramolecular sulfonic acid ester mediated by esterase CA, and then isomerized to obtain trans-vinylsulfonic acid. For (E)-2-(5-bromo-2-hydroxyphenyl) ethene-1-sulfonic acid (Fig. 3), the OH moiety in ortho to the ethenylsufonate group participates in a bifurcated hydrogen bond with the hydroxyl of Thr200 and through a bridging water molecule, with the carbonyl oxygen of Pro201. hCA IX and XII have larger active sites than hCA I and II, which may lead to the selective inhibition of CA isoforms by sulfocoumarin (Alterio et al., 2012; Nocentini et al., 2018; Supuran, 2016).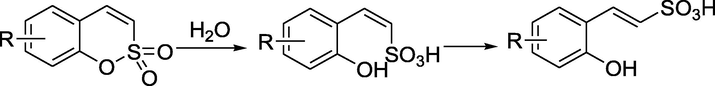
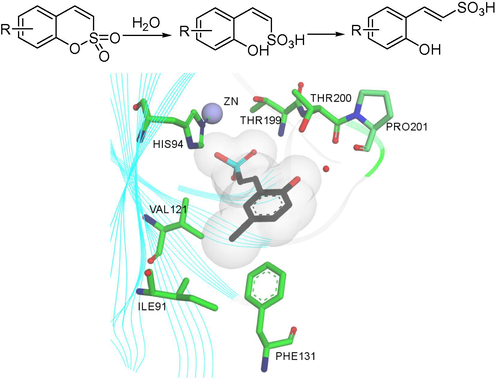
Active site CA-mediated hydrolysis of trans-vinylsulfonic acid. Binding of trans-vinylsulfonic acid within the active site of the CA II/IX mimic (PDB: 4BCW). The Zn (II) ion (larger sphere), His ligand (His94), water molecule (small sphere) coordinated to the zinc are shown by X-ray crystallography (Tars et al., 2012). Discovery Studio Client v18.1.0 was used to present a 3D image.
3.1.2 SAR analysis
Table 1 shows the structure and activity of inhibitors containing sultone scaffold, and discuss their structure–activity relationship as shown in Fig. 4. “nd ” represents the activity to be measured and the activity is poor (Ki > 10 μM). “-” represents the inhibitor activity has not been measured.
Compd
R1
R2
Ki (nM)
Ref.
hCA
IhCA II
hCA VA
hCA IX
hCA XII
A1
4-F-phenyl
H
nd
nd
_
95.3
4.6
(Grandane et al., 2015b)
A2
3-F-phenyl
H
nd
nd
_
10.3
9.4
A3
4-tBu-phenyl
H
nd
nd
_
9.1
6.8
A4
4-CF3-phenyl
H
nd
nd
_
29.7
3.7
A5
3-CF3-phenyl
H
nd
nd
_
9.8
6.1
A6
4-CH3OOC-phenyl
H
nd
nd
_
9.0
9.1
A7
–NO2
H
nd
nd
_
3770
3160
(Tars et al., 2012 Nocentini et al., 2018)
A8
–NH2
H
6780
8890
_
46
23
A9
Cl
H
nd
nd
_
136.3
89.5
(Nocentini et al., 2018)
A10
H
–NO2
nd
nd
_
37.0
81.7
A11
H
–NH2
nd
nd
_
33.5
38.4
A12
H
-Cl
nd
nd
_
33.7
60.9
A13
–OCH2CH2OH
H
nd
nd
60
26.8
10.4
(Tanc et al., 2015)
A14
-OMe
H
nd
nd
nd
42.1
10.9
(Grandane et al., 2015a)
A15
–CN
H
nd
nd
nd
33.8
26.6
A16
H
BnO
nd
2.5
835
nd
nd
(Tanc et al., 2013)
A17
H
4-Cl-phenylacetyloxyl
nd
2.6
3910
nd
nd
A18
H
2-Br-phenylacetyloxyl
nd
1.9
7060
nd
nd
A19
cyclopropyl
H
nd
nd
_
60.6
5.9
(Grandane et al., 2014)
A20
3-Cl-phenyl
H
nd
nd
_
93.1
7.6
A21
4-Cl-phenyl
H
nd
nd
_
7.8
17.7
A22
2-NH2-phenyl
H
nd
nd
_
7.9
6.3
A23
3-NH2-phenyl
H
nd
nd
_
875
9.2
A24
4-CF3-phenyl
H
nd
nd
_
136
5.5
A25
4-F-phenyl
H
nd
nd
_
531
6.7
A26
Ph
H
6860
7760
_
29
32
(Tars et al., 2012)
A27
Et2NCH2-
H
8110
9370
_
25
7
A28
4-CF3O-phenyl
H
8430
9640
_
74
14
A29
H
2-NH2-phenyl
nd
nd
_
9.5
9.2
(Grandane et al., 2014)
A30
H
3-MeO-phenyl
nd
nd
_
9.1
7.6
A31
H
4-MeO-phenyl
nd
nd
_
8.3
44.0
A32
H
3-CF3-phenyl
nd
nd
_
9.6
8.9
A33
H
4-CF3-phenyl
nd
nd
_
7.2
30.1
A34
H
4-F-phenyl
nd
nd
_
8.3
7.8
A35
H
3-Cl-phenyl
nd
nd
_
8.8
9.4
A36
H
4-Cl-phenyl
nd
nd
_
9.2
31.2
A37
4-(4-CF3O-phenyl) −1,2,3-triazole
H
nd
5770
_
340
1720
(Grandane et al., 2015b)
A38
4-(2- NH2-phenyl) −1,2,3-triazole
H
nd
nd
_
460
2320
A39
–NO2
H
nd
nd
_
27
640
A40
H
–OCH3
nd
2.4
91
nd
nd
(Tanc et al., 2013)
A41
H
4-Cl-phenylacetyloxyl
nd
2.2
206
nd
nd
A42
H
2-Br-phenylacetyloxyl
nd
3.4
141
nd
nd
A43
Bn
H
nd
nd
_
81.3
4.0
(Grandane et al., 2015a)
A44
2-furyl
H
nd
nd
_
33.0
6.2
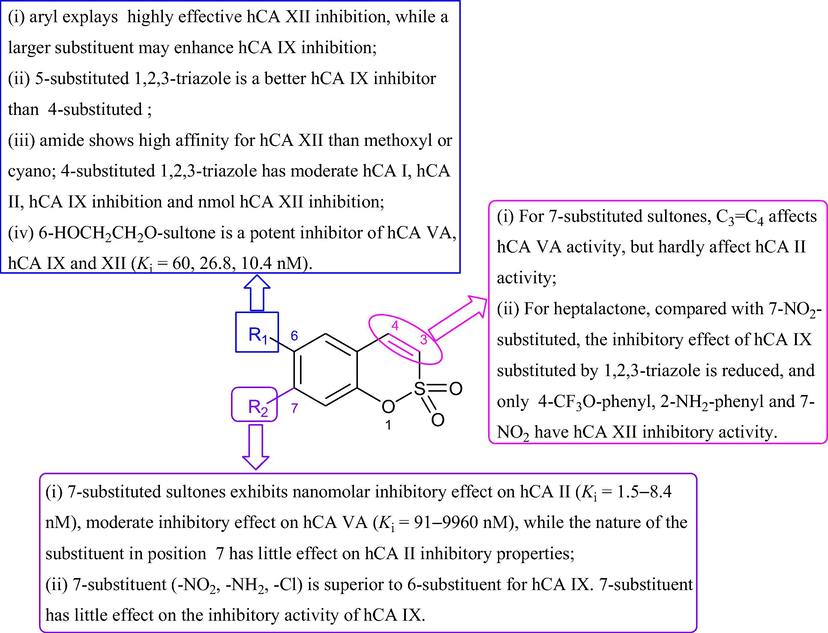
SARs of the sultone-based hCA inhibitors.
6-Substituted sultones:
-
6-Arylsultones are potent and selective inhibitors of tumor-associated hCA IX and XII. The size and position of substituted phenyl significantly affect the inhibitory activity, slightly larger groups may enhance the hCA IX inhibitory activity (t-Bu in A3, COOMe in A6), m-substituted phenyl is more active than the p-substituted (A5 > A4, A2 > A1). Compounds with 6-aryl are highly effective hCA XII inhibitors (Grandane et al., 2015b).
-
Compounds with 1,2,3-triazole scaffold are effective hCA IX and hCA XII inhibitors. 5-Substituted 1,2,3-triazole is better hCA IX inhibitor than 4-substituted one (A33 > A24, A34 > A25, A35 > A20), in which, most substitution patterns, both aliphatic and aromatic R groups, lead to highly effective hCA XII inhibitors (Grandane et al., 2014).
-
6-carboxamido-substituted sultones are selective and effective inhibitor of hCA IX and XII. The introduction of carbon unit spacers or heterocyclic five-membered rings (A43, A44) reduced hCA IX inhibitory effect (Ki = 81.3, 33.0 nM). All 6-amide-substituted sultones exhibited high affinity for hCA XII (Ki = 2.1–8.7 nM), which was higher than that of 6-methoxy (A14) and 6-cyano (A15) sultones (Ki = 10.9, 26.6 nM) (Grandane et al., 2015a).
-
Some of 4-substituted 1,2,3-triazoles have moderate hCA I and hCA II inhibition, effective hCA IX inhibition, and low nanomolar hCA XII inhibition (A26-A28) (Tars et al., 2012).
-
6-Hydroxyethoxylsultone (A13) is a potent inhibitor of hCA VA (Ki = 60 nM), and excellent inhibitory activity on tumor-associated enzymes hCA IX and XII (Ki = 26.8, 10.4 nM) (Grandane et al., 2012; Tanc et al., 2015).
7-Substituted sultones:
-
7-substituted sultones and 3,4-dihydrosultones showed low nanomolar inhibitory effect on hCA II (Ki = 1.5–8.4 nM), moderate inhibitory effect on hCA VA (Ki = 91–9960 nM). The changes of substituents and the C3 = C4 bond have no significant effect on the inhibition of hCA II (A41 ≈ A42 ≈ A17 ≈ A18). Compounds with methoxyl (A40), 4-chlorophenylacetyloxyl (A41) and 2-bromophenylacetyloxyl (A42) for 3,4-dihydrosulfocoumarin skeleton have good hCA VA inhibitory activity (Ki = 91–206 nM) (Tanc et al., 2013).
-
For heptasultones: only A37 is a moderate hCA II inhibitor (Ki = 5.77 µM). A39 (7-NO2) has the best hCA IX inhibitory activity (Ki = 27 nM), and the 7-substituted 1,2,3-triazole does not increase hCA IX inhibitory activity (A39 > A37 > A38). 7-NO2 (A39), 1,2,3-triazole with 4-trifluoromethoxyphenyl (A37) and 2-aminophenyl (A38) have hCA XII inhibitory activity (Ki = 0.64–2.32 µM) (Pustenko et al., 2017).
-
7-Substituent (–NO2, –NH2, -Cl) is superior to 6-substituent for hCA IX (A10 > A7, A11 > A8, A12 > A9). This 7-substituent has little effect on the inhibitory activity of hCA IX (A10 ≈ A11 ≈ A12) (Nocentini et al., 2018).
3.2 Anti-HIV activity
HIV is the causative agent of acquired immunodeficiency syndrome (AIDS), which has caused significant damage worldwide over the past 40 years. Side effects and resistance to long-term chemotherapy may be the most important factor in the failure of treating and eradicating HIV infection (Barre-Sinoussi et al., 1983; Nanfack et al., 2017; Trivedi et al., 2020). To date, 20 drugs have been approved for the treatment of AIDS. However, viral rebound during therapy, the emergence of drug-resistant and the need for long-term treatment modalities are the main causes for the failure of current antiretroviral therapy. There is still a need for the development of new drugs with less toxic, active against growing drug-resistant or novel targets in the viral life cycle. Eleven of anti-HIV drugs target the reverse transcriptase (RT). The non-nucleoside RT inhibitors (NNRTIs), (2′,5′-bis-O-(tert-butyldimethylsilyl)-β-D-ribofuranosyl)-3′-spiro-5′'-(4′'-amino-1′',2′'-oxathiole-2′',2′'-dioxide) nucleosides (TSAO) derivatives, are an unusual class of compounds that exert their unique selectivity for HIV-1 through a specific interaction with the p51 subunit of HIV-1 RT (Camarasa et al., 2005).
TSAO compounds are the only NNRTIs for which amino acids at both HIV-1 RT subunits (p66 and p51) are needed for optimal interaction with the enzyme. In fact, it has been demonstrated that the heterodimeric enzyme shows a reduced DNA binding ability in the presence of the 5-fold molar excess of TSAO-T. TSAO-e3T can induce the dissociation of RT into inactive monomers under certain conditions (Velazquez et al., 1992). SAR in Fig. 5 showed that the 3′-spironucleosides with S stereospecificity (B1-B3) didn’t have anti-HIV-1 activity, while those with R configuration (B4-B6) and tert-butyldimethylsilyl (TBDMS) at C-5′ and C-2′ had significant anti-HIV-1 activity. The replacement of 2′-TBDMS group led to reduce twice to 20-fold anti-HIV-1 activity while that of the 5′-position showed inactivity. The base part of TSAO molecule is structurally less stringent, which plays a modulatory role in its activity/cytoxicity (Camarasa et al., 1992; Perez-Perez et al., 1992; Rodríguez-Barrios et al., 2001).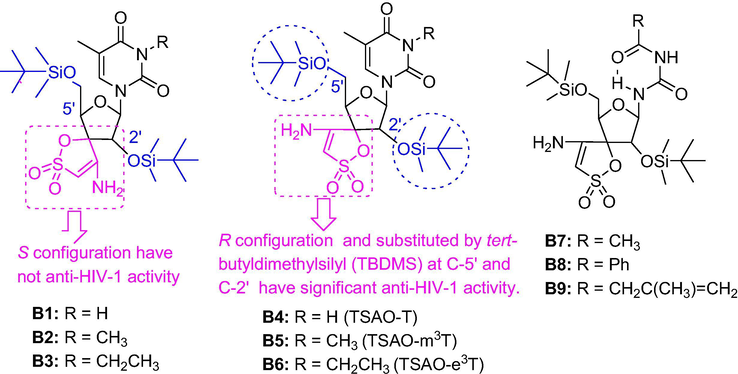
SARs of base-modified TSAO compounds.
As first small non-peptide molecule, TSAO compounds interfere with the HIV-1 RT dimerization process (Bonache et al., 2011; De-Castro et al., 2011; De-Castro and Camarasa, 2018). As shown in Fig. 6, crystal analysis of TSAO-T into HIV-1 RT showed that TSAO-T binding morphology is similar to the “dragon” configuration, in which the thymine base, ribose ring and helix are similar to the head, body and tail, respectively. The “head” thymidine is between the aromatic side chains of Tyr188 and Phe227. One sulfonyloxy group of the sultone group forms a hydrogen bond with Lys103, and the other S = O group interacts with RT through water. As two wings of dragon NNRTI of 2′ and 5′ two TBDMS moieties, 5′-TBDMS (I wing) part interacts with Trp229, Pro95, Tyr183 and Leu100, while 2′-TBDMS (II wing) part interacts with Tyr318 (Das et al., 2011).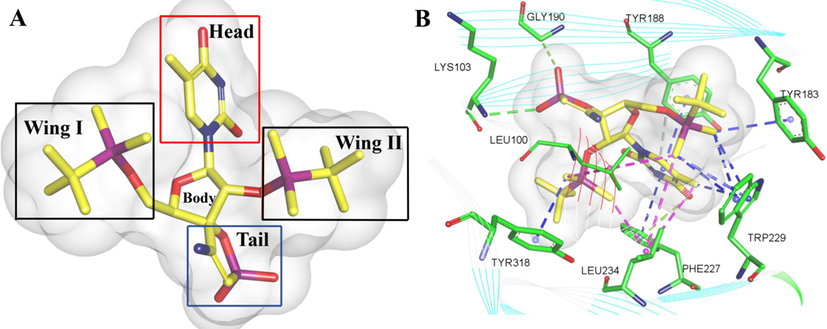
3D conformations of compound B4 (A) (TSAO-T) docked in HIV-1 RT (PDB: 3QO9) (Das et al., 2011). Discovery Studio Client v18.1.0 were used to present a 3D images.
Replacement of the thymine base of TSAO-T by other pyrimidines, purines or 1,2,4-substituted triazoles didn’t significantly influence the activity or toxicity, for example, the abasic TSAO analogue B9 showed only moderate loss of activity, which may be due to the energy penalty involved in breaking the intramolecular H-bond in order to adopt a suitable conformation for binding into the enzyme (Camarasa et al., 2005). Further modifications at the N-3 position of thymine (introducing alkyl, alkenyl and alkynyl; polar groups such as acids, amides, alcohols and amines; lipophilic groups; aromatic groups; amino acids and others) led to interesting results. As shown in Fig. 7, some compounds exhibited attenuated cytotoxicity and higher selectivity indices (SI = CC50/EC50), such as the SIs of compounds B10 and B11 increased two orders of magnitude (200 and 12500, respectively), that of compound B13 increased twice to 6-fold. Moreover, several compounds with polar groups exhibited more effective in inhibiting/disrupting dimerization of HIV-1 RT than TSAO-T (Bonache et al., 2005; Bonache et al., 2008; Camarasa et al., 2004; Sluis-Cremer et al., 2006).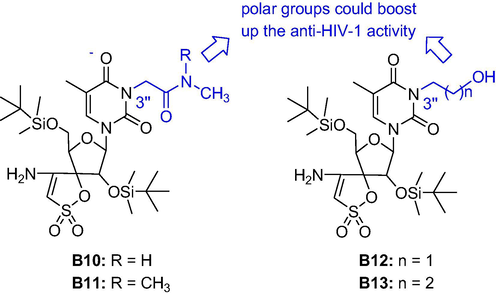
Structures of TSAO with polar groups at the N-3 position of thymine.
The 4′'-amino group at the spiro moiety was substituted by different carbonyl functionalities (including keto, carboxylic acid, ester, amide and urea groups), which may interfere with hydrogen bond formation of the residue Lys138 in TSAO-resistant HIV-1 strains (De-Castro et al., 2005). Surprisingly, some compounds in Fig. 8 were firstly observed activity against HCMV replication at sub-toxic concentrations. In particular, compound B21 represents the first TSAO derivative with dual anti-HIV-1 and HCMV activity, which is important because HCMV infection is common for AID patients. Moreover, compounds B17-B19 only gained anti-HCMV activity, resulting in the first TSAO molecules specific for HCMV replication.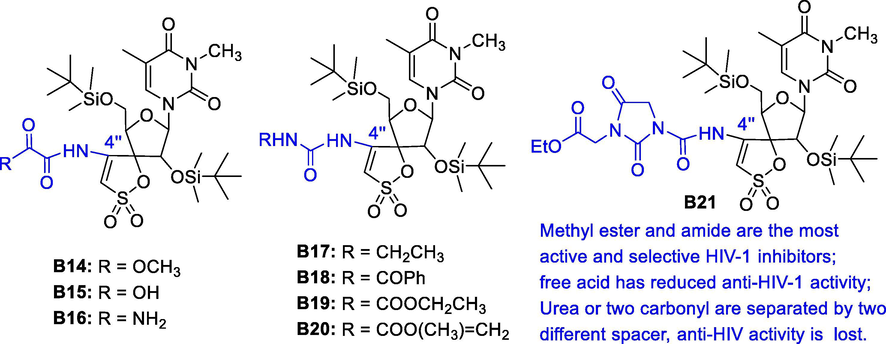
Potent anti-HIV-1 and HCMV activity of TSAO derivatives B14-B21.
The 3′'-postion of spiro moiety was substituted by different functional groups (halogen, alkenyl, alkynyl, allyl, aromatic and heteroaromatic groups), which may give additional interactions with residues adjacent to Glu138 of p51 subunit (Lobatón et al., 2002). Amongst them, the 4′'-amino group of the sultone was maintained to allow the crucial interaction of TSAO derivatives with residue Glu138 of HIV-1 RT p51 subunit. As shown in Fig. 9, the brief SAR of compounds B22-B27 on HIV-1 and HIV-2 replication activities was discussed, and the inhibitory activity of B10-B27 against HIV is shown in Table 2. “-” represents the inhibitor activity has not been measured.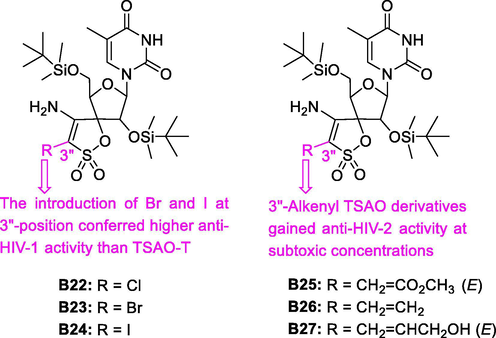
Potent anti-HIV-1 and anti-HIV-2 activity of TSAO derivatives B22-B27.
Compd
EC50 (μM)
SI
IC50 (μM)
EC50 (μM)
Ref.
MT-4
CEM
CEM
HCMV
(AD-169)HCMV
(Davis)
HIV-1
HIV-2
HIV-1
HIV-2
HIV-1 RT
-
-
-
B10
0.03
>250
0.02
>50
>12500
3.1
–
–
(Bonache et al., 2005)
B11
0.04
>250
0.02
>250
12,250
3.5
–
–
B12
0.06
>10
0.04
>10
282
3.1
–
–
B13
0.03
>2
0.01
>2
386
3.0
–
–
B14
0.08
> 4
0.06
>4
–
–
253
179
(De-Castro et al., 2005)
B15
1.30
> 10
0.2
>50
–
–
>80
36
B16
0.0057
> 2
0.7
>10
–
–
6.5
6.5
B17
>2
>2
>2
> 2
–
–
1.8
2.0
B18
>2
>10
>2
>2
–
–
1.2
0.8
B19
>2
>2
>2
>2
–
–
2.3
1.8
B20
>0.8
>0.8
>0.8
>0.8
–
–
>20
>4
B21
0.88
>2
0.24
>2
–
–
0.29
0.32
B22
> 10
>10
> 10
> 10
–
–
–
–
(Lobatón et al., 2002)
B23
0.93
>10
0.85
>10
–
–
–
–
B24
0.12
>2
0.06
>2
–
–
–
–
B25
3.7
9.9
3.0
7.3
–
–
–
–
B26
3.2
3.3
4.0
4.0
–
–
–
–
B27
6.6
5.4
> 2
1.2
–
–
–
–
TSAO-T
0.06
>20
0.06
>20
200
–
–
–
(Bonache et al., 2005)
TSAO-m3T
0.06
>50
0.04
>250
2875
3.1
–
–
Among the human immunodeficiency virus (HIV) reverse transcriptase (RT) inhibitors, the so called nonnucleoside RT inhibitors (NNRTIs) have gained a definitive place in the treatment of HIV infections in combination with nucleoside analogue RT inhibitors (NRTIs) and HIV protease inhibitors (PIs). The virus can be markedly suppressed for a relatively long period of time when exposed to multiple drug combination therapy (highly active antiretroviral therapy, HAART). TSAO derivatives are a peculiar group of highly functionalized nucleosides that belong to the so-called NNRTIs. They exert their unique selectivity for HIV-1 through a specific interaction with the p51 subunit of HIV-1 RT. They are the first small molecules that seem to interfere with the dimerization process of the enzyme. This review covers the work carried out with this unique class of specific inhibitors of HIV-1 reverse transcriptase, including SAR studies, its mechanism of action, resistance studies, model of interaction with the enzyme, etc.
Emergence of drug-resistant viral strains is one of the major milestones and the main cause for the failure of antiretroviral therapy. Combination of different anti-HIV agents has become the standard clinical practice to prevent emergence of virus-drug resistance. The Glu138Lys RT mutant virus had the most marked resistance to TSAOs, followed by the Glu138Gln, Glu138Phe, Glu138Gly, Glu138Tyr, and Glu138Ala virus mutants. In the mutant Glu138Lys RT HIV-1 strains, virus replication can be completely inhibited by NNRTIs (i.e., TIBO, BHAP, nevirapine). TSAO derivatives have been used in several double and triple drug combination studies with both NNRTIs. Individually used 3TC, TSAO-m3T and the thiocarboxanilide UC10 rapidly led to the emergence of drug-resistant HIV-1 mutants (Glu138Lys for TSAO-m3T, Met184Val for 3TC, and Lys103Thr/Asn for UC10). When 3TC was combined with either TSAO-m3T, UC10 or UC42, emergence of drug-resistant virus was markedly delayed or even fully suppressed (Balzarini et al., 1993; Balzarini et al., 1995; Balzarini et al., 1996; Perez-Perez et al., 1992).
3.3 Anti-HCMV and anti-VZV activity
Human cytomegalovirus (HCMV) belongs to a widely distributed member of the herpesvirus family that generally remains unnoticed in the human body, but can be severely pathogenic in immunocompromised patients (Kenneson and Cannon, 2007; Lilleri and Gerna, 2016; Plotkin and Boppana, 2019). HCMV is found to lead to a variety of serious diseases (mainly pneumonia and gastroenteritis) that can severely cause to death (Boppana and Britt, 1995; Perotti and Perez, 2019; Sandhu and Buchkovich, 2020), and is the main viral cause of congenital diseases affecting children by causing deafness, learning disabilities, and mental retardation (Gupta et al., 2014). In despite of tremendous efforts in vaccine and treatment, HCMV infections still threaten the lives of individuals with poor immune function and newborns, but currently available compounds for HCMV have poor drug-like properties such as oral bioavailability, toxicity, and so on. Therefore, it is needed for anti-HCMV compounds with effective safety and novel mechanisms of action. 4′'-Benzoylurea-TSAO derivatives showed pronounced antiviral activity against replication of HCMV at micromolar concentrations (0.29–2.0 µM) that is below the toxicity threshold. In particular, compound C1 showed the highest anti-HCMV activity. The brief SAR was discussed in Fig. 10 (De-Castro et al., 2007; De-Castro et al., 2008).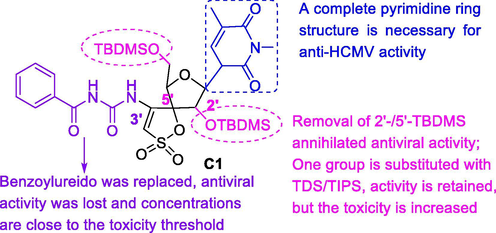
Anti-HCMV activity of 4′'-benzoylurea-TSAO derivatives.
Herpesvirus infections are the most frequent causes of viral infections in immunocompetent as well as in immunocompromised patients. Several 4-benzyloxy-γ-sultone derivatives were found to have a selective inhibitory activity against HCMV and Varicella zoster virus (VZV) replication in vitro. These novel compounds don’t show cross-resistance against mutant drug-resistant HCMV strains, pointing to a novel antiviral mechanism. The brief SAR was discussed in Fig. 11 (De-Castro et al., 2009).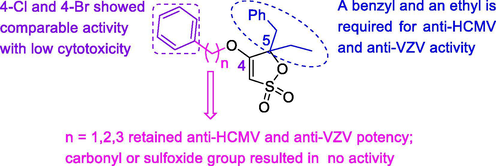
Anti-HCMV and anti-VZV activity of 4-benzyloxy-γ-sultone derivatives.
3.4 Anti-BVDV activity
Hepatitis C virus (HCV) is a major health problem worldwide. The most common complications are fibrosis (20–30%) and cirrhosis (10–20%), in which 1–4% has developed hepatocellular carcinoma and 350,000 people each year die from HCV-related advanced liver disease (Alkharsah et al., 2020; Darweesh et al., 2015; Lapa et al., 2019; Rau et al., 2013). There is an urgent need for efficient and selective HCV replication inhibitors. HCV and BVDV belong to the Flavivirus genus with similar genomic structure and replication pathways. BVDV infection is able to affect most of the organ system, which acute infection may lead to peripheral blood lymphocyte reduction, apoptosis and immunosuppression. Antiviral agents targeting BVDV replication may inhibit HCV replication. As shown in Fig. 12, novel δ-sultones containing γ-lactone may be developed as antiviral agents against HCV infection (Lanyon et al., 2014; Liu et al., 2018; Tabarrini et al., 2006; Villalba et al., 2016; Xu et al., 2014).
Anti-BVDV activity of δ-sultone.
3.5 Selective BuChE inhibitors
Alzheimer's disease (AD) is a chronic neurodegenerative disease. The most effective strategy of improving the symptoms of AD is to use cholinesterase (ChE), acetylcholinesterase (AChE) and butyrylcholine (BuChE), inhibitors to restore acetylcholine (ACh) levels (Kumar et al., 2018; Liu et al., 2017; Zhou et al., 2016). Clinical studies have shown that BuChE inhibition in cerebrospinal fluid is correlated with cognitive function in AD patients. Selective BuChE inhibitors infused into the cerebral cortex of rats can increase extracellular ACh levels by 15 times, and no choline-like adverse reactions have been found. In advanced AD patients, BuChE took over the role of AChE, reaching almost 80% of the overall ChE activity (Contestabile, 2011). Therefore, selective BuChE inhibitors have great potential to develop drug candidates.
Recent, a class of novel δ-sultone-fused pyrazole scaffold was discovered as highly selective sub-micromolar BuChE inhibitors, which is an effective selective BuChE inhibitor and has non-toxic, lipophilic and significant neuroprotective activity. Moreover, this scaffold showed reversible, mixed-type BuChE inhibitory activity, which may be used to improve disease symptoms in progressive AD. Further research found that there are two flexible amino acids (Leu286 and Val288) in the acyl pocket of hBuChE compared to hAChE, which allows binding of bulkier ligands. Simultaneously, a ChE inhibitor (reversible or pseudo-irreversible) contains a basic nitrogen atom crucial for the interaction with the ChE binding site. Therefore, the structure-based optimization of δ-sultone-fused pyrazoles was conducted to further improve the potency and selectivity of BuChE inhibitor (Zhang et al., 2020). Fig. 13 showed SAR of δ-sultone-fused pyrazoles as BuChE inhibitors.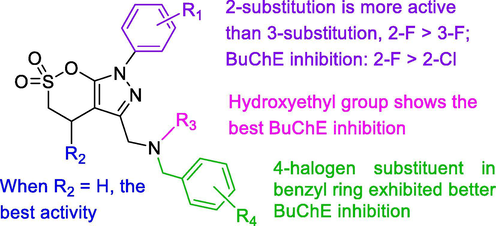
SARs of δ-sultones as BuChE inhibitors.
The amino acid sequence of AChE and BuChE is 65% homologous, and its protein structure contains a catalytic active site (CAS), a deep canyon and a peripheral anion site (PAS). The two aromatic residues of hAChE burst into the canyon to occupy the valley space, while the replacement of hBuChE with smaller residues provided a wider space, allowing larger substrates to bind and be hydrolyzed (Brus et al., 2014; Cavallaro et al., 2018; Chen et al., 2018). The different structural characteristics of the two enzymes lead to substrate specificity: AChE is more selective for small acetylcholine molecules, and BuChE has greater affinity for various neuroactive peptides (Chierrito et al., 2018; Košak et al., 2018; Panek et al., 2018). Molecular docking (Fig. 14) showed that compound E1 is nicely bound into hBuChE via a strong hydrogen bond interaction between the hydroxyl unit with Asn68, p–π interaction between the benzyl ring with Leu286, Val288 and Phe329 in the acyl sub-pocket, and a π–anion interaction between the S atom of sultone ring with His438 in the hydrolysis sub-pocket (Zhang et al., 2020).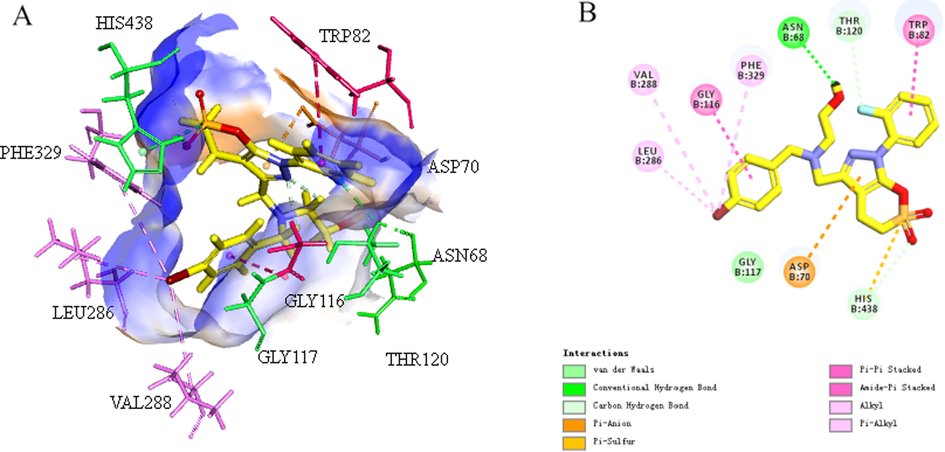
(A) 3D mode of the pocket surface of compound E1 with receptor hBuChE (PDB ID: 5LKR), (B) 2D mode of the interaction of active compound with receptor hBuChE (conventional H bond and C–H bond, halogen, π–anion, alkyl, and π–alkyl are represented by green, light green, brown, pink and light pink lines, respectively). Discovery Studio Client v18.1.0 were used to present images.
3.6 β-Receptor antagonists
As shown in Figs. 15, 8-(3-amino-2-hydroxypropoxy)benzoxathiin was developed as an ideal remedy for cardiovescular disorders such as arrhythmie, angina pectoris, hypertension, and so on. Compounds with N-alkyl or an aralkyl substitution showed clearly its effect as a β-blocker which may be applied to angina pectoris and hypertension in addition to arrhythmia, wherein R1 represents hydrogen atom or a lower alkyl group, and R2 represents a lower alkyl group or an aralkyl group. Among them, N-t-Bu substitution substitution showed excellent anti-arrhythmic effect with 96.3% of rat brain synaptosome membrane β-receptor inhibitory effect and 0.03 mg/kg (ED50) activity at 1 µM, while toxicity and side-effects are very low (Hori, 1985).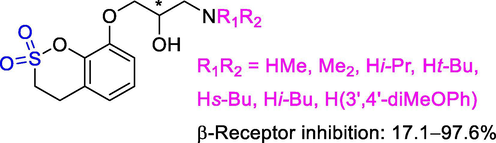
δ-Sultones as β-receptor inhibitors.
3.7 Anti-angiogenic effect and skin sensitization, carcinogenic effect
The angiogenesis activity of S-CA was evaluated by the chick chorioallantoic membrane (CAM) model. As shown in Fig. 16, In vivo a murine sarcoma S180 model, reduction of the tumor weight and tumor HE staining regions demonstrated that S-CA (iv, 10 mg/kg) had potent inhibition effects (44.7% inhibitory rate in S180 mice) and low acute toxicity (LD50: 25.6 mg/kg) (Li et al., 2015). Alk-1-ene-γ-sultone has been proven to be a particularly strong skin sensitizer, while alkane-γ-sulfolactone has been found to be a medium sensitizer (Fig. 16). In rats of either weekly or a single intravenous injecting 1,3-propanelactone, tumors were observed at various tissues including the brain and nervous system. Skin exposure to 1,3-propane sultone can cause tumors in the skin and lymph reticulum of bisexual mice, while subcutaneous injection can cause lung cancer (regenerative adenocarcinoma) in male rats (Meschkat et al., 2001).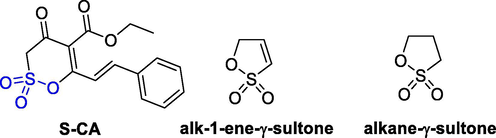
The anti-angiogenesis and skin sensitization, carcinogenic effects of δ-sultone.
Sultone stents show a wide range of pharmacological activities, but reports on preclinical trials of sultone are rarely a matter to be solved. Future medicinal chemists should increase the relevance of sultone pharmacokinetic studies to provide a theoretical basis for the subsequent application of sultone structured drugs as clinical candidate compounds. CA inhibitors are used to treat different diseases. Based on the difference of CA isoforms, rational drug design can be used to improve selectivity and activity of CA inhibitors. SARs of TSAO as anti-HIV agents can be used to optimize the base part of TSAO, which plays a modulatory role in its activity/cytoxicity. In the future, these compounds with benign activity and a clear mechanism of action can be regarded as valuable candidate prototypes for the design and development of candidate drugs. It is interesting to explore the role of sultones in the field of medicine by studying the SAR, the toxicity level of active compounds and their mechanism of action, as well as discussing those binding conformations in the active site.
4 Conclusion
This review aims to provide a wide range of information on sultone scaffolds, the unique sultone scaffold has multiple pharmacological activities in medicinal chemistry. In this review, we discuss in detail various strategies for the simple and effective synthesis of sultone derivatives from readily available raw materials. In addition, sultone-based drugs have great potential therapeutic value for a variety of drug targets, such as anti-CA, anti-viral, anti-tumor, et al. We have disclosed present role of diverse sultone in the field of medicinal chemistry by discussing insights of the SAR studies, their mechanism action and binding conformation of these heterocycles with various targets. Additionally, we hope that SARs can be further used for structural optimization. Target-based sultone synthesis should be designed to obtain novel compounds with target selectivity. Synthetic chemists can develop the methods of batch synthesis of sultones to build up sultone compound libraries find potent lead compounds containing sultone in the future.
Acknowledgments
Financial support was provided by the Natural Science Foundation of Anhui Province (2008085MH272) and Natural Science Fundation of Anhui Provincial Department of Education (KJ2019ZD21) and Anhui Provincial Training Programs of Innovation and Entrepreneurship for Undergraduates (20810366046).
Notes.
The authors declare no competing financial interest.
Declaration of Competing Interest
The authors declared that there is no conflict of interest.
References
- Metal-free allene-based synthesis of enantiopure fused polycyclic sultones. Chem. Eur. J.. 2016;22(1):285-294.
- [CrossRef] [Google Scholar]
- Association between hepatitis C virus viremia and the rs12979860, rs2228145 and rs1800795 SNP (CT/AC/GG) genotype in Saudi kidney transplant recipients. Saudi. J. Med. Med. Sci.. 2020;8(1):46-52.
- [CrossRef] [Google Scholar]
- Multiple binding modes of inhibitors to carbonic anhydrases: How to design specific drugs targeting 15 different isoforms? Chem. Rev.. 2012;112(8):4421-4468.
- [CrossRef] [Google Scholar]
- Enzyme targeting strategies for prevention and treatment of cancer: Implications for cancer therapy. Semin. Cancer Biol.. 2019;56:1-11.
- [CrossRef] [Google Scholar]
- Treatment of human immunodeficiency virus type 1 (HIV-1)-infected cells with combinations of HIV-1-specific inhibitors results in a different resistance pattern than does treatment with single-drug therapy. J. Virol.. 1993;67(9):5353-5359.
- [CrossRef] [Google Scholar]
- Marked inhibitory activity of non-nucleoside reverse transcriptase inhibitors against human immunodeficiency virus type 1 when combined with (-)2',3'-dideoxy-3'-thiacytidine. Mol. Pharmacol.. 1996;49(5):882-890. PMID:8622638
- [Google Scholar]
- 2',5'-Bis-O-(tert-butyldimethylsilyl) -3'-spiro-5“-(4”-amino-1“,2”-oxathiole-2'',2'-dioxide) pyrimidine (TSAO) nucleo- side analogues: highlyselective inhibitors of human immunodeficiency virus type 1 that are targeted at the viral reverse transcriptase. Proc. Natd. Acad. Sci.. 1992;89(10):4392-4396.
- [CrossRef] [Google Scholar]
- Suppression of the breakthrough of human immunodeficiency virus type 1 (HIV-1) in cell culture by thiocarboxanilide derivatives when used individually or in combination with other HIV-1-specific inhibitors (i.e., TSAO derivatives) PNAS. 1995;92(12):5470-5474.
- [CrossRef] [Google Scholar]
- Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS) Science. 1983;220(4599):868-871.
- [CrossRef] [Google Scholar]
- Palladium-catalysed intramolecular direct arylation of 2-bromobenzenesulfonic acid derivatives. Adv. Synth. Catal.. 2012;354(18):3533-3538.
- [CrossRef] [Google Scholar]
- 1,3-Propane sultone, an extremely potent experimental carcinogen: what should be expected in humans? Toxicol. Lett.. 2004;151(1):251-254.
- [CrossRef] [Google Scholar]
- Improving the antiviral efficacy and selectivity of HIV-1 reverse transcriptase inhibitor TSAO-T by the introduction of functional groups at the N-3 position. J. Med. Chem.. 2005;48(21):6653-6660.
- [CrossRef] [Google Scholar]
- Selective inhibition of human immunodeficiency virus type 1 (HIV-1) by a novel family of tricyclic nucleosides. Antivir. Res.. 2011;92(1):37-44.
- [CrossRef] [Google Scholar]
- Novel N-3 substituted TSAO-T derivatives: synthesis and anti-HIV-evaluation. Nucleos. Nucleot. Nucl.. 2008;27(4):351-367.
- [CrossRef] [Google Scholar]
- Antiviral antibody responses and intrauterine transmission after primary maternal cytomegalovirus infection. J. Infect. Dis.. 1995;171(5):1115-1121.
- [CrossRef] [Google Scholar]
- Intramolekulare Diels-Alder-reaktion von vinylsulfonsäureestern. Angew. Chem.. 1989;101(2):204-206.
- [CrossRef] [Google Scholar]
- Discovery, biological evaluation, and crystal structure of a novel nanomolar selective butyrylcholinesterase inhibitor. J. Med. Chem.. 2014;57(19):8167-8179.
- [CrossRef] [Google Scholar]
- TSAO compounds: the comprehensive story of a unique family of HIV-1 specific inhibitors of reverse transcriptase. Curr. Top. Med. Chem.. 2004;4(9):945-963.
- [CrossRef] [Google Scholar]
- 3'-Spiro nucleosides, a new class of specific human immunodeficiency virus type 1 inhibitors: synthesis and antiviral activity of (2',5'-bis-O-(tert-butyldimethylsilyl) -β-D-xylo- and -ribofuranose)-3'-spiro-5“-(4”-amino-1“, 2”-oxathiole 2“, 2”- dioxide) (TSAO) pyrimidine nucleosides. J. Med. Chem.. 1992;35(15):2721-2727.
- [CrossRef] [Google Scholar]
- TSAO derivatives the first non-peptide inhibitors of HIV-1 RT dimerization. Antivir. Chem. Chemother.. 2005;16(3):147-153.
- [CrossRef] [Google Scholar]
- Dimerization inhibitors of HIV-1 reverse transcriptase, protease and integrase: A single mode of inhibition for the three HIV enzymes? Antivir. Res.. 2006;71(2–3):260-267.
- [CrossRef] [Google Scholar]
- TSAO derivatives, inhibitors of HIV-1 reverse transcriptase dimerization: recent progress. Curr. Pharm. Des.. 2006;12(15):1895-1907.
- [CrossRef] [Google Scholar]
- Alkynyl and β-ketophosphonates: Selective and potent butyrylcholinesterase inhibitors. Bioorg. Chem.. 2018;77:420-428.
- [CrossRef] [Google Scholar]
- New chiral sultam auxiliaries: preparation and their application in asymmetric Diels-Alder reactions. Tetrahedron. 1997;8(15):2501-2504.
- [CrossRef] [Google Scholar]
- Tricyclic pyrazolo (1,5-d) (1,4) benzoxazepin-5 (6H)-one scaffold derivatives: Synthesis and biological evaluation as selective BuChE inhibitors. Eur. J. Med. Chem.. 2018;147:194-204.
- [CrossRef] [Google Scholar]
- A portal to a class of novel sultone-functionalized pyridinesvia an annulative SuFEx process employing earth abundant nickel catalysts. Chem. Commun.. 2018;54(65):9011-9014.
- [CrossRef] [Google Scholar]
- Chameleon-like behavior of indolylpiperidines in complex with cholinesterases targets: potent butyrylcholinesterase inhibitors. Eur. J. Med. Chem.. 2018;145:431-444.
- [CrossRef] [Google Scholar]
- Metal-free, hydroacylation of C-C and N-N bonds via aerobic C-H activation of aldehydes, and reaction of the products thereof. Org. Biomol. Chem.. 2013;11(42):7301-7317.
- [CrossRef] [Google Scholar]
- The history of the cholinergic hypothesis. Behav. Brain Res.. 2011;221(2):334-340.
- [CrossRef] [Google Scholar]
- Characterization of the cytopathic BVDV strains isolated from 13 mucosal disease cases arising in a cattle herd. Virus Res.. 2015;195:141-147.
- [CrossRef] [Google Scholar]
- Crystal structure of tert-butyldimethylsilyl- spiroaminooxathioledioxide-thymine (TSAO-T) in complex with HIV-1 Reverse Transcriptase (RT) redefines the elastic limits of the non-nucleoside inhibitor-binding pocket. J. Med. Chem.. 2011;54(8):2727-2737.
- [CrossRef] [Google Scholar]
- Novel non-nucleoside human cytomegalovirus inhibitors based upon TSAO nucleoside derivatives: structure-activity relationships. Nucleos. Nucleot. Nucl.. 2007;26(6–7):625-628.
- [CrossRef] [Google Scholar]
- Polypharmacology in HIV inhibition: Can a drug with simultaneous action against two relevant targets be an alternative to combination therapy? Eur. J. Med. Chem.. 2018;150:206-227.
- [CrossRef] [Google Scholar]
- 4-Benzyloxy-γ-sultone derivatives: Discovery of a novel family of non-nucleoside inhibitors of human cytomegalovirus and varicella zoster virus. J. Med. Chem.. 2009;52(6):1582-1591.
- [CrossRef] [Google Scholar]
- Discovery of TSAO derivatives with an unusual HIV-1 activity/resistance profile. Antivir. Res.. 2006;71(1):15-23.
- [CrossRef] [Google Scholar]
- Novel (2',5'-Bis-O-(tert-butyldimethylsilyl)-β-D-ribofuranosyl)-3'-spiro-5“-(4”-amino-1“, 2”-oxathiole-2“,2”-dioxide) derivatives with anti-HIV-1 and anti-Human-cytome -galovirus activity. J. Med. Chem.. 2005;48(4):1158-1168.
- [CrossRef] [Google Scholar]
- Unprecedented lability of the 5'-O-tert-butyldimethylsilyl group from 3'-Spiro-5“-(4”-acylamino-1“,2”-oxathiole-2“,2”-dioxide) nucleoside derivatives via neighboring group participation of the 4''-acylamino residue. J. Org. Chem.. 2006;71(4):1407-1415.
- [CrossRef] [Google Scholar]
- Unusual lability of 5'-O-tert-butyldimethylsilyl group on 4''-acyl TSAO derivatives. Nucleos. Nucleot. Nucl.. 2003;22(5–8):959-961.
- [CrossRef] [Google Scholar]
- 4''-Benzoylureido-TSAO derivatives as potent and selective non-nucleoside HCMV inhibitors. Structure-activity relationship and mechanism of antiviral action. J. Med. Chem.. 2008;51(18):5823-5832.
- [CrossRef] [Google Scholar]
- From β-Amino-γ-sultone to unusual bicyclic pyridine and pyrazine heterocyclic systems: synthesis and cytostatic and antiviral activities. ChemMedChem. 2011;6(4):686-697.
- [CrossRef] [Google Scholar]
- Sulfur (VI) fluoride exchange (SuFEx): Another good reaction for click chemistry. Angew. Chem. Int. Ed.. 2014;53(36):9430-9448.
- [CrossRef] [Google Scholar]
- The enantioselective total synthesis of (−)-myltaylenol. Chem. Commun.. 1997;16:1491-1492.
- [CrossRef] [Google Scholar]
- Die Constitution der isomeren Naphtalinderivate. I. Die α-α-disubstituirten Verbindungen. Ann. Chem.. 1888;247(3):306-366.
- [CrossRef] [Google Scholar]
- Synthetic approaches and pharmaceutical applications of chloro-containing molecules for drug discovery: A critical review. Eur. J. Med. Chem.. 2019;173:117-153.
- [CrossRef] [Google Scholar]
- Simple and practical one-step synthesis of new 1,3-dienic δ-sultones from terminal alkynes and some synthetic applications of these compounds. J. Sulfur Chem.. 2011;32(1):3-16.
- [CrossRef] [Google Scholar]
- Solvent-dependent reactions for the synthesis of β-Keto-Benzo-δ-Sultone scaffolds via DBU-catalyzed O-sulfonylation/intramolecular Baylis–Hillman/1,3-H shift or dehydration tandem sequences. J. Org. Chem.. 2011;76(24):9975-9982.
- [CrossRef] [Google Scholar]
- An efficient one-pot, regio- and stereoselective synthesis of novel pentacyclic-fused pyrano(3,2, c)chromenone or quinolinone benzosultone derivatives in water. Tetrahedron. 2013;69(24):4979-4989.
- [CrossRef] [Google Scholar]
- Synthesis of novel tricyclic and tetracyclic sultone scaffolds via intramolecular 1,3-dipolar cycloaddition reactions. Tetrahedron. 2012;68(18):3641-3648.
- [CrossRef] [Google Scholar]
- Facile synthesis of coumarin bioisosteres–1,2-benzoxathiine 2,2-dioxides. Tetrahedron. 2012;68(27–28):5541-5546.
- [CrossRef] [Google Scholar]
- 6-Triazolyl-substituted sulfocoumarins are potent, selective inhibitors of the tumor-associated carbonic anhydrases IX and XII. Bioorg. Med. Chem. Lett.. 2014;24(5):1256-1260.
- [CrossRef] [Google Scholar]
- 6-Substituted sulfocoumarins are selective carbonic anhdydrase IX and XII inhibitors with significant cytotoxicity against colorectal cancer cells. J. Med. Chem.. 2015;58(9):3975-3983.
- [CrossRef] [Google Scholar]
- Synthesis of 6-aryl-substituted sulfocoumarins and investigation of their carbonic anhydrase inhibitory action. Bioorg. Med. Chem.. 2015;23(7):1430-1436.
- [CrossRef] [Google Scholar]
- Hepatitis C virus: Screening, diagnosis, and interpretation of laboratory assays. Asian. J. Transfus. Sci.. 2014;8(1):19-25.
- [CrossRef] [Google Scholar]
- Validity of oral bioavailability estimates of phenolsulfonphthalein based on total urinary excretion from rats. J. Pharm. Sci.. 1978;67(2):267-268.
- [CrossRef] [Google Scholar]
- Novel benzoxathiin derivative, process for preparing them and pharmaceutical composition. Suntory Limited. 1985 EP19850102644
- [Google Scholar]
- Overexpression of carbonic anhydrase XII in tissues from resectable non-small cell lung cancers is a biomarker of good prognosis. Int. J. Cancer. 2011;128(7):1614-1623.
- [CrossRef] [Google Scholar]
- Phenolsulfonphthalein transport by potential-sensitive urate transport system. Eur. J. Pharmacol.. 2005;518(2–3):83-89.
- [CrossRef] [Google Scholar]
- Major role of organic anion transporters in the uptake of phenolsulfonphthalein in the kidney. Eur. J. Pharmacol.. 2018;475(1–3):85-92.
- [CrossRef] [Google Scholar]
- Ring closing metathesis in the synthesis of sultones and sultams. Synthesis. 2004;10:1696-1712.
- [CrossRef] [Google Scholar]
- Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev. Med. Virol.. 2007;17(4):253-276.
- [CrossRef] [Google Scholar]
- Lewis acid/base catalyzed (2+2)-cycloaddition of sulfenes and aldehydes: A versatile entry to chiral sulfonyl and sulfinyl derivatives. Chem. Eur. J.. 2011;17(13):3679-3692.
- [CrossRef] [Google Scholar]
- The magic of crystal structure-based inhibitor optimization: Development of a butyrylcholinesterase inhibitor with picomolar affinity and in vivo activity. J. Med. Chem.. 2018;61(1):119-139.
- [CrossRef] [Google Scholar]
- Carbonic anhydrase as a model for biophysical and physical-organic studies of proteins and protein-ligand binding. Chem. Rev.. 2008;108(3):946-1051.
- [CrossRef] [Google Scholar]
- Highly functionalized 2-amino-4H-pyrans as potent cholinesterase inhibitors. Bioorg. Chem.. 2018;81:134-143.
- [CrossRef] [Google Scholar]
- Brownlie J., Bovine viral diarrhoea: pathogenesis and diagnosis. Vet. J.. 2014;199(2):201-209.
- [CrossRef] [Google Scholar]
- Hepatitis C virus genetic variability, human immune response, and genome polymorphisms: Which is the interplay? Cells.. 2019;8(4):305.
- [CrossRef] [Google Scholar]
- On carbon dioxide storage based on biomineralization strategies. Micron.. 2009;41(4):273-282.
- [CrossRef] [Google Scholar]
- Neutron diffraction of acetazolamide-bound human carbonic anhydrase II reveals atomic details of drug binding. J. Am. Chem.. 2012;134(36):14726-14729.
- [CrossRef] [Google Scholar]
- Unsaturated sultones from unsaturated sulfonates: Synthesis by Ring-closing metathesis and reactivity. Synlett. 2003;5:0667-0670.
- [CrossRef] [Google Scholar]
- New synthesis method for sultone derivatives: Synthesis, crystal structure and biological evaluation of S-CA. Molecules. 2015;20(3):4307-4318.
- [CrossRef] [Google Scholar]
- Palladium-catalyzed synthesis of aromatic sultones via sulfonic acid group-directed C-H activation. Chinese J. Chem.. 2012;30(9):2041-2046.
- [CrossRef] [Google Scholar]
- Synthesis of (60)fullerene-fused sultones via sulfonic acid group-directed C-H bond activation. Org. Lett.. 2012;14(8):2176-2179.
- [CrossRef] [Google Scholar]
- Strategies to control human cytomegalovirus infection in adult hematopoietic stem cell transplant recipients. Immunotherapy.. 2016;8(9):1135-1149.
- [CrossRef] [Google Scholar]
- Novel ferulic amide derivatives with tertiary amine side chain as acetylcholinesterase and butyrylcholinesterase inhibitors: The influence of carbon spacer length, alkylamine and aromatic group. Eur. J. Med. Chem.. 2017;126:810-822.
- [CrossRef] [Google Scholar]
- PD-1 blockade inhibits lymphocyte apoptosis and restores proliferation and anti-viral immune functions of lymphocyte after CP and NCP BVDV infection in vitro. Vet. Microbiol.. 2018;226:74-80.
- [CrossRef] [Google Scholar]
- Synthetically useful intermediates by diazosulfone and sulfonate C-H insertion. Synth. Commun.. 2015;45(2):226-231.
- [CrossRef] [Google Scholar]
- Synthesis of 3''-substituted TSAO derivatives with anti-HIV-1 and anti-HIV-2 activity through an efficient Palladium-catalyzed cross-coupling approach. J. Med. Chem.. 2002;45(18):3934-3945.
- [CrossRef] [Google Scholar]
- Synthesis of tricyclic and tetracyclic sultones by Pd-catalyzed intramolecular cyclization. Tetrahedron Lett.. 2009;50(33):4781-4784.
- [CrossRef] [Google Scholar]
- Carbonic anhydrase inhibitors drug design. Subcell. Biochem.. 2014;75:291-323.
- [CrossRef] [Google Scholar]
- Studies of the chemical selectivity of hapten, reactivity, and skin sensitization potency. 1. Synthesis and studies on the reactivity toward model nucleophiles of the 13C-labeled skin sensitizers Hex-1-ene- and Hexane-1,3-sultones. Chem. Res. Toxicol.. 2001;14(1):110-117.
- [CrossRef] [Google Scholar]
- Vinylsulfonyl Chloride, a ketene equivalent for the intramolecular Diels-Alder Reaction. Synlett. 1992;1992(12):985-987.
- [CrossRef] [Google Scholar]
- Stereoselective synthesis of substiuted cyclohexenols from a furan-derived sultone via tandem elimination/1,6-addition. J. Org. Chem.. 1994;59(13):3687-3689.
- [CrossRef] [Google Scholar]
- Synthesis of novel pentacyclic thiopyrano indole-annulated benzo-δ-sultone derivatives via a domino Knoevenagel-Hetero-Diels-Alder reaction in water. Tetrahedron Lett.. 2013;54(21):2685-2689.
- [CrossRef] [Google Scholar]
- Recent Developments in the Synthesis and Application of Sultones. Chem. Rev.. 2012;112(10):5339-5355.
- [CrossRef] [Google Scholar]
- Ring-closing metathesis in the synthesis of fused sultones. Tetrahedron Lett.. 2014;55(9):1577-1580.
- [CrossRef] [Google Scholar]
- Synthesis of seven-membered fused sultones by reductive Heck cyclization: an investigation for stereochemistry through DFT study. Tetrahedron. 2015;71(3):476-486.
- [CrossRef] [Google Scholar]
- Synthesis of uracil-, coumarin- and quinolone-fused benzosultams and benzosultones. Synthesis. 2015;47(21):3423-3433.
- [CrossRef] [Google Scholar]
- Evaluation of 99mTc-sulfonamide and sulfocoumarin derivatives for imaging carbonic anhydrase IX expression. J. Inorg. Biochem.. 2018;185:63-70.
- [CrossRef] [Google Scholar]
- Multimethod longitudinal HIV drug resistance analysis in antiretroviral-therapy-naive patients. J. Clin. Microbiol.. 2017;55(9):2785-2800.
- [CrossRef] [Google Scholar]
- Deciphering the mechanism of human carbonic anhydrases inhibition with sulfocoumarins: computational and experimental studies. Chemistry. 2018;24(31):7840-7844.
- [CrossRef] [Google Scholar]
- Design, synthesis, and biological evaluation of 1-benzylamino-2-hydroxyalkyl derivatives as new potential disease-modifying multifunctional anti-Alzheimer’s agents. ACS Chem. Neurosci.. 2018;9(5):1074-1094.
- [CrossRef] [Google Scholar]
- Expression of the membrane-associated carbonic anhydrase isozyme XII in the human kidney and renal tumors. J. Histochem. Cytochem.. 2000;48(12):1601-1608.
- [CrossRef] [Google Scholar]
- Stereospecific synthesis and anti-HIV-1 activity of 1-(2',5'-bis-O- (tert-butyldimethylsilyl)-β-D-ribofuranosyl)-3'-spiro-5“-(4”-amino-1“,2”-oxathiole -2“,2”-dioxide) pyrimidine and pyrimidine-modified nucleosides. J. Med. Chem.. 1992;35(16):2988-2995.
- [CrossRef] [Google Scholar]
- Virus-like particles and nanoparticles for vaccine development against HCMV. Viruses.. 2019;12(1):35.
- [CrossRef] [Google Scholar]
- High Pressure Intramolecular Diels-Alder Reactions of Vinylsulfonates and Vinylsulfonamides. Tetrahedron. 2000;56(6):873-879.
- [CrossRef] [Google Scholar]
- Vaccination against the human cytomegalovirus. Vaccine.. 2019;37(50):7437-7442.
- [CrossRef] [Google Scholar]
- Chemistry of Sulfonate- and Sulfonamide-Stabilized Carbanions-The CSICR eactions. European Asian J. Org Chem.. 2003;2003(19):3713-3726.
- [CrossRef] [Google Scholar]
- 3H–1,2-benzoxathiepine 2,2-dioxides: a new class of isoform-selective carbonic anhydrase inhibitors. J. Enzym. Inhib. Med. Chem.. 2017;32(1):767-775.
- [CrossRef] [Google Scholar]
- Recent advances in sultone synthesis (microreview) Chem. Heterocycl. Compds.. 2017;53(12):1283-1285.
- [CrossRef] [Google Scholar]
- Rh (III)-Catalyzed synthesis of sultones through C-H activation directed by a sulfonic acid group. Chem. Commun.. 2014;50(68):9776-9778.
- [CrossRef] [Google Scholar]
- Impact of genetic SLC28 transporter and ITPA variants on ribavirin serum level, hemoglobin drop and therapeutic response in patients with HCV infection. J. Hepatol.. 2013;58(4):669-675.
- [CrossRef] [Google Scholar]
- Synthesis of trifluoromethylated sultones from alkenols using a copper photoredox catalyst. J. Org. Chem.. 2016;81(16):7139-7147.
- [CrossRef] [Google Scholar]
- Synthesis and chemical transformations of fluorosulfates. Asian J. Org. Chem.. 2018;7(4):662-682.
- [CrossRef] [Google Scholar]
- Photoredox transformations with dimeric gold complexes. Angew. Chem. Int. Ed.. 2013;52(50):13342-13345.
- [CrossRef] [Google Scholar]
- Contact sensitization of guinea-pigs with unsaturated and halogenated sultones. Contact Dermatitis.. 1975;1(6):349-358.
- [CrossRef] [Google Scholar]
- Identification of a putative binding site for (2',5'-Bis-O-(tert-butyldimethylsilyl)-β-D-ribofuranosyl)-3'-spiro-5“-(4”-amino-1“,2”-oxathiole-2“, 2”-dioxide) thymine (TSAO) derivatives at the p51–p66 interface of HIV-1 reverse transcriptase. J. Med. Chem.. 2001;44(12):1853-1865.
- [CrossRef] [Google Scholar]
- Physiologically based pharmacokinetic model for the renal clearance of phenolsulfonphthalein and the interaction with probenecid and salicyluric acid in the dog. J. Pharmacokinet. Biopharm.. 1987;15(4):349-368.
- [CrossRef] [Google Scholar]
- Sandhu, P., Buchkovich, N.J., 2020. HCMV decreases MHC class II by regulating CIITA transcript levels in a myeloid cell line. J. Virol. 901-919. https://doi.org/ 10.1128/JVI.01901-19.
- Biotechnology for the acceleration of carbon dioxide capture and sequestration. Curr. Opin. Biotechnol.. 2011;22(6):818-823.
- [CrossRef] [Google Scholar]
- The rhodium-catalyzed carbene cyclization cycloaddition cascade reaction of vinylsulfonates. Adv. Synth. Catal.. 2009;351(18):3128-3132.
- [CrossRef] [Google Scholar]
- Structure-activity relationships of (2',5'-Bis-O-(tert-butyldimethylsilyl)-β-D-ribofuranosyl)-3'-spiro-5“-(4”-amino-1“,2”-oxathiole-2“, 2”-dioxide) thymine derivatives as inhibitors of HIV-1 reverse transcriptase dimerization. J. Med. Chem.. 2006;49(16):4834-4841.
- [CrossRef] [Google Scholar]
- Sultones and sultines via a Julia-Kocienski reaction of epoxides. Angew. Chem.. 2015;54(50):15236-15240.
- [CrossRef] [Google Scholar]
- Structure and function of carbonic anhydrases. Biochem. J.. 2016;473(14):2023-2032.
- [CrossRef] [Google Scholar]
- Synthesis and anti-BVDV activity of acridones as new potential antiviral agents. J. Med. Chem.. 2006;49(8):2621-2627.
- [CrossRef] [Google Scholar]
- Palladium-catalyzed (2,3) rearrangement of alkyl allyl sulfites to alkyl allylsulfonates. J. Org. Chem.. 1990;55(6):1823-1829.
- [CrossRef] [Google Scholar]
- 7-Substituted-sulfocoumarins are isoform-selective, potent carbonic anhydrase II inhibitors. Bioorg. Med. Chem.. 2013;21(15):4502-4510.
- [CrossRef] [Google Scholar]
- 6-Substituted 1,2-benzoxathiine-2,2-dioxides are isoform-selective inhibitors of human carbonic anhydrases IX. XII and VA. Org. Biomol. Chem.. 2015;13(1):77-80.
- [CrossRef] [Google Scholar]
- Sulfocoumarins (1,2-Benzoxathiine-2,2-dioxides): A class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J. Med. Chem.. 2012;56(1):293-300.
- [CrossRef] [Google Scholar]
- 1,3-Dipolar Cycloaddition Reactionsof Nitrones to Prop-1-ene-1,3-sultone. Synthesis. 2003;9:1329-1334.
- [CrossRef] [Google Scholar]
- A new route to 5- and 6-membered ring sultones. Can. J. Chem.. 1970;48(5):845-851.
- [CrossRef] [Google Scholar]
- Recent advances in the development of integrase inhibitors for HIV treatment. Curr. Hiv. Aids Rep.. 2020;17(1):63-75.
- [CrossRef] [Google Scholar]
- N-Heterocyclic carbene catalyzed synthesis of δ-sultones via α, β-unsaturated sulfonyl azolium intermediates. Angew. Chem. Int. Ed.. 2015;54(40):11780-11784.
- [CrossRef] [Google Scholar]
- Hybrids of (TSAO-T)-(Foscarnet): The first conjugate of foscarnet with a non-nucleoside reverse transcriptase inhibitor through a labile covalent ester bond. J. Med. Chem.. 2004;47(13):3418-3426.
- [CrossRef] [Google Scholar]
- TSAO analogues. Synthesis and anti-HIV-1 activity of 2',5'-bis-O-(tert-butyldimethylsilyl)-β-D-ribofuranosyl-3'-spiro-5“-(4”-amino-1“,2”-oxathiole-2“,2-dioxide) purine and purine-modified nucleosides. J. Med. Chem.. 1992;35(16):2988-2995.
- [CrossRef] [Google Scholar]
- The alpha-carbonic anhydrase from the thermophilic bacterium Sulfurihydrogenibium yellowstonense YO3AOP1 is highly susceptible to inhibition by sulfonamides. Bioorg. Med. Chem.. 2013;21(6):1534-1538.
- [CrossRef] [Google Scholar]
- Transcriptomic analysis of responses to cytopathic bovine viral diarrhea virus-1 (BVDV-1) infection in MDBK cells. Mol. Immunol.. 2016;71:192-202.
- [CrossRef] [Google Scholar]
- Stereocontrolled synthesis of a C1–C10 building block (“southwestern moiety”) for the unnatural enantiomers of the polyene polyol antibiotics filipin III and pentamycin: A sultone-forming ring-closing metathesis for protection of homoallylic alcohols. Eur. J. Org. Chem.. 2014;2014(15):3210-3224.
- [CrossRef] [Google Scholar]
- δ-Sultone formation through Rh-Catalyzed C-H insertion. Org. Lett.. 2007;9(21):4363-4366.
- [CrossRef] [Google Scholar]
- Synthesis and anti-BVDV activity of novel delta-sultones in vitro: implications for HCV therapies. Bioorg. Med. Chem. Lett.. 2014;24(10):2388-2391.
- [CrossRef] [Google Scholar]
- Discovery of δ-sultone-fused pyrazoles for treating Alzheimer's disease: Design, synthesis, biological evaluation and SAR studies. Eur. J. Med. Chem.. 2019;181 111598
- [CrossRef] [Google Scholar]
- Hypervalent iodine reagent mediated oxidative heterocyclization of aldoximes with heterocyclic alkenes. J. Org. Chem.. 2017;82(22):11742-11751.
- [CrossRef] [Google Scholar]
- Visible-light initiated aerobic oxidations: a critical review. Green Chem.. 2018;20(21):4790-4833.
- [CrossRef] [Google Scholar]
- The structure-based optimization of δ-sultone-fused pyrazoles as selective BuChE inhibitors. Eur. J. Med. Chem.. 2020;201 112273
- [CrossRef] [Google Scholar]
- Arylnaphthalene lactone analogues: synthesis and development as excellent biological candidates for future drug discovery. RSC Adv.. 2018;8(17):9487-9502.
- [CrossRef] [Google Scholar]
- Pharmaceutical and medical significance of sulfur (SVI)-containing motifs for drug discovery: A critical review. Eur. J. Med. Chem.. 2019;162(15):679-734.
- [CrossRef] [Google Scholar]
- DL0410, A novel dual cholinesterase inhibitor, protects mouse brains against Abeta-induced neuronal damage via the Akt/JNK signaling pathway. Acta Pharmacol. Sin.. 2016;37(11):1401-1412.
- [CrossRef] [Google Scholar]




