

Translate this page into:
SAR studies of quinoline and derivatives as potential treatments for Alzheimer’s disease
⁎Corresponding authors. SunYaj@jlu.edu.cn (Ya-Juan Sun), tancheng@jlu.edu.cn (Cheng Tan)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Quinoline analogs are an important class of N-based heterocyclic compounds, which have received extensive attention because of their use in medicinal chemistry and organic synthesis. Over the past few decades, several new scaffold-based functionalization synthesis strategies have been reported for quinolines. Quinoline derivatives have a wide range of biological activities, including anti-Alzheimer’s disease activity. Herein, we review research on quinoline and related analogs as anti-Alzheimer’s disease agents from 2001 to 2022 and particularly highlight the structure–activity relationships and molecular binding modes. This review provides information for the rational design of more effective and target-specific drugs for Alzheimer's disease.
Keywords
Quinoline
Anti-Alzheimer
Multitarget-directed ligand
Acetylcholinesterase
Structure–activity relationship
Neuroprotective activity

- AD
-
Alzheimer's disease
- Aβ
-
beta-amyloid
- ACh
-
acetylcholine
- SAR
-
structure–activity relationship
- AChE
-
acetylcholinesterase
- BuChE
-
butyrylcholinesterase
- PAS
-
peripheral anionic site
- PD
-
Parkinson's Disease
- MAO
-
monoamine oxidase
- APP
-
precursor protein
- hChE
-
human cholinesterase
- hAChE
-
human acetylcholinesterase
- hBChE
-
human butyrylcholinesterase
- eeAChE
-
esterase acetylcholinesterase
Abbreviations
1 Introduction
Alzheimer's disease (AD) is a highly complex neurodegenerative disease, which mainly affects people over the age of sixty, and much research has been devoted to the occurrence and development of its pathology (Iqbal and Grundke-Iqbal, 2000, De-Paula, et al., 2012, Anand, et al., 2014, Kim, et al., 2021, Long and Holtzman, 2019, Gerring, et al., 2021). >46 million people worldwide are estimated to be affected by AD, and by 2050, 131.5 million people will suffer from this neurodegenerative disease (Prince, et al., 2015, Lemes, et al., 2016, Obulesu and Jhansilakshmi, 2014, Lee, et al., 2020, Kim, et al., 2020, Pan, et al., 2020). The pathogenesis of AD is not fully understood; however, many factors, including beta-amyloid (Aβ) deposition, oxidative stress, tau protein aggregation, reduced acetylcholine (ACh) levels, neuro-inflammation, and bimetallic imbalance, are believed to be involved in the pathogenesis of AD (Jakob-Roetne and Jacobsen, 2009, Shaik, et al., 2016, Sharma and Kumar, 2009, Bondi, et al., 2017, Hampel et al., 2018, Solis et al., 2020).
Quinoline (also known as 1-azanaphthalene and benzopyridine) is a heterocyclic aromatic organic compound (Nainwal, et al., 2019, Kaur and Kumar, 2021, Corio, et al., 2021, Patil, et al., 2021, Di Mola, et al., 2019, Marasco, et al., 2021, Kankanala, et al., 2016). Coal tar remains the main commercial source, although many reactions have been developed to synthesize quinoline. Quinoline and quinoline derivatives exist widely in nature (Baccile, et al., 2016, Rivo, et al., 2020, Petruczynik, et al., 2019, Felicetti, et al., 2020, Fabiano-Tixier, et al., 2011, Rao, et al., 2009, Nugraha et al., 2020). Because the quinoline moiety provides an easy-to-use scaffold for the design and synthesis of drugs, quinoline is recognized as a classic privileged structure. The quinoline pharmacophore is an important functionality, which has the potential for a wide range of biological and pharmacological activities, including anti-Alzheimer activity (Chu, et al., 2019, Musiol, 2017, Nqoro, et al., 2017, Rüb, et al., 2017, Shen, et al., 2016, Rani, et al., 2016, Boyd, et al., 2007, Cherny, et al., 2001)(Fig. 1).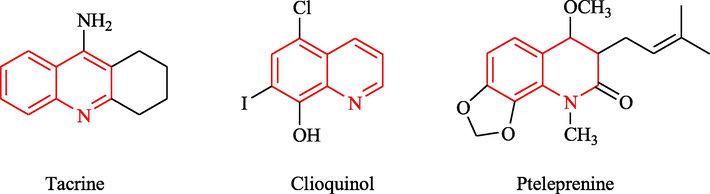
There approved anti-Alzheimer containing quinoline moiety.
Over the past several decades, many quinolone analogs have been designed, synthesized, and screened for pharmacological effects and adverse reactions in vitro and in vivo, and some compounds possessed considerable activity. This review highlights the recent progress in quinoline derivatives as potential anti-Alzheimer agents. The structure–activity relationship (SAR) and molecular docking studies were reviewed to provide information toward the further design of useful quinoline derivatives.
2 Anti-Alzheimer activity of quinoline and their analogs
Quinoline scaffolds are present in many natural and synthetic compounds with pharmacological activity. Recent studies have demonstrated that some quinoline and quinoline derivatives also have potent anti-acetylcholinesterase (AChE) and anti-butyrylcholinesterase (BuChE) effects. Molecular docking studies have indicated that the quinoline moiety can bind to the peripheral anionic site (PAS) of AChE via π-π stacking interactions (Sang, et al., 2017, Farina, et al., 2015, Shang, et al., 2018). To design drugs for AD, Okamura et al. (Okamura, et al., 2005) have synthesized a quinolone moiety with a phenyl ring substituted at the 2-position (compounds 1 and 2), which showed higher binding affinity to tau fibrils and lower binding affinity to Aβ fibrils (Fig. 2). The brain uptake after intravenous injection of [11C] 1 was 11.3 %, 5.0 %, 3.1 %, and 2.1 % ID/g at 2, 10, 30, and 60 min, respectively, suggesting that the quinolone moiety is potentially useful in anti-Alzheimer agents.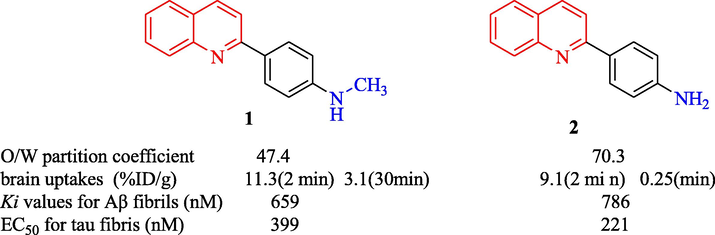
Structural and anti-Alzheimer of quinoline compounds 1,2.
In AD pathology, iron promotes the accumulation of Aβ peptides and increases the toxicity of Aβ peptides, and iron chelation has been shown to be a feasible neuroprotective strategy. Increased levels of iron in the process of aging and AD will lead to iron-dependent oxidative stress neurodegeneration (Youdim, et al., 2004a). Clioquinol (3) promotes metal uptake to target Aβ-responsive synaptic Zn and Cu. Clioquinol (3) and the 8-hydroxyquinoline analog (4) are shown in Fig. 3a, The Adlard group (Adlard, et al., 2008) has also found that 3 caused metal-induced Aβ-aggregation as an Zn/Cu ionophore and displayed greater blood–brain barrier permeability, which indicated it had potential as a disease-modifying drug for AD. Shachar et al. (Shachar, et al., 2004) have found that 4 could inhibit Fe/ascorbate- and substrate-induced mitochondrial membrane lipid peroxidation (IC50 = 12.7 µM) and 4 (1 µg) could also prevent 6-hydroxydopamine-induced striatal dopaminergic damage. In addition, this study showed that 4 exerted neuroprotective effects by acting as a brain-penetrating iron chelator (Fig. 3a). Youdim et al. (Youdim, et al., 2004a) and (Ryan, et al. 2015) have also reported that compounds 4 and 5 displayed activity against the neurodegenerative diseases Parkinson's Disease (PD) and AD (Fig. 3a). In addition, Youdim et al. (Youdim et al., 2005b; Youdim et al., 2006c) have reported that compound 6 was a brain permeable, iron chelating, brain-selective monoamine oxidase (MAO) inhibitor, with a propargyl alcohol neuroprotective effect. Compound 6 showed a reduction in amyloid precursor protein (APP) expression and Aβ peptide secretion, with simultaneous activation of α-secretase and release of the neuroprotective neurotrophic solution APP alpha; therefore, 6 is a potential agent for the treatment of AD (Fig. 3a)(Gal, et al., 2005; Prati, et al., 2016). The above studies confirmed that 8-hydroxyquinoline is a strong iron chelator with antioxidant, anti-PD and anti-AD properties. In addition, they also found that a piperazine moiety can enhance the biological property, penetration propargyl moiety is an APP or protein regulation and processing to soluble APP alpha.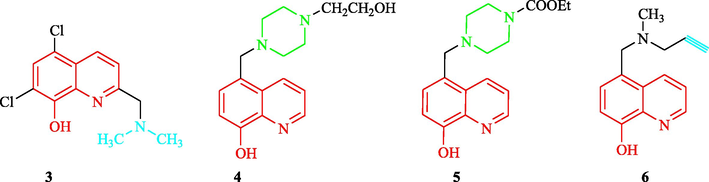
Structural and anti-Alzheimer of 8-hydroxyquinoline analogs 3–6.
To search for and discover compounds with strong anti-Alzheimer activity, Prati et al. (Prati, et al., 2016) have synthesized two classes of 8-hydroxyquinoline derivatives and investigated their inhibition of human cholinesterase (hChE). The results showed that the two series of compounds were not active, or weaker inhibitors, than hAChE (inhibition rate: 9.0 %–63.8 % for 5-chloro-8-hydroxyquinoline derivatives), but showed a dramatic inhibitory effect on human butyrylcholinesterase (hBChE) (49.2 %–89.1 % for 8-hydroxyquinoline compounds) at 40 μM. A comparison of compounds 7a and 7b indicated that the Cl atom on the benzene ring had a deleterious effect, removal of the Cl atom resulted in a fourfold increase in the inhibitory activity (with IC50 values from 23.3 to 5.71 μM). The compound with the highest anti-hBChE activity was 7a. Compound 7c with a bromine substitution also showed good BChE inhibition. Compound 7b had anti-BChE, anti-aggregation, and Cu2+ and Zn2+complexing properties in vitro, and may be worthy of further study (Fig. 3b). The study found that BChE inhibition was influenced by the presence, position of a substituent on the benzyl moiety, with the most potent compounds bearing a substituent at position 2. Yang et al. (Yang, et al., 2018) have synthesized two groups of analogs, 8 and 9, with an 8-hydroxyquinoline containing a piperazine ring and evaluated their anti-AD activity. Eight compounds in series 8 and four compounds in series 9 had Aβ1-42 aggregation inhibition activities from 32.46 % to 73.41 %, which were better than that of clioquinol (25.72 %) at 10 μM. Compound 8a with a 2,4-difluorobenzene ring (IC50 = 5.05 μM) showed the strongest Aβ1-42 aggregation inhibition activity. Compounds 8b and 9a (IC50 = 7.35 and 5.64 μM, respectively) with a 2-chlorobenzene ring displayed better inhibitory effects against Aβ1-42 aggregation than resveratrol (IC50 = 12.43 μM). In addition, an F or Cl substituent at the 2-position of the aromatic ring for in series 8 compounds was beneficial for the inhibitory effect. A methoxyl substituent was detrimental for the inhibition. Compound 9a could chelate biometals and inhibit Cu2+/Zn2+-induced Aβ1-42 aggregation (Fig. 3b).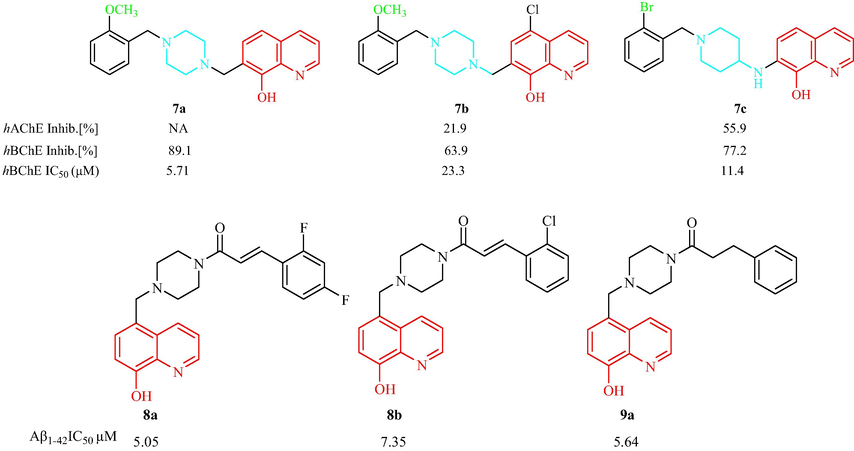
Structural of 8-hydroxyquinoline analogs including with piperazine, piperidine ring 7–9.
Wang et al. (Wang, et al., 2014a) have reported seven 8-hydroxyquinoline analogs containing piperidine ring as cholinesterase (ChE) inhibitors. Two compounds 10a (n = 3 in the linker) and 10b (n = 2 in the linker) (10a: esterase acetylcholinesterase (eeAChE), IC50 = 2.7 μM and eqBuChE, IC50 = 4.5 μM) and (10b: eeAChE, IC50 = 1.8 μM and eqBuChE, IC50 = 1.6 μM) displayed better inhibition activity. However, 10b exhibited the best activity profile of all the compounds investigated. The SAR analysis showed (1) a trend was detected with α-aminonitrile series, linker length, α-aminonitriles compounds proved to be more potent than amines at inhibiting both enzymes; (2) bearing no methyl in linker connecting N-benzyl-piperidin-4′-yl residue to the N-propargyl core was inactive; (3) regarding tertiaryamines effect at inhibiting both ChE increased from n = 1 to n = 3. Molecular modeling of eeAChE and eqBuChE with 10b R- and S-enantiomers indicated —CN group formed a hydrogen bond of side chain of Asn83 for R-enantiomer, this bond was bifurcated in the case of S- enantiomer; the hydrophobic interactions with the catalytic triad residues of Ser198 and His438 were found in this orientation. In mode B, only one hydrogen bond was observed for R-enantiomer, it was formed between —CN and —OH group of Thr120, S-enantiomer —OH group of quinoline ring established a hydrogen bond with the catalytic triad residue His438 (Fig. 3c). In 2015, Knez et al. (Knez, et al., 2015) reported the preparation and ChE inhibitory activity and anti-aggregation effects of a family of nitroxoline-based analogs with an 8-hydroxyquinoline scaffold. Six analogs displayed hBChE inhibitory activity and compounds 11a and 11b exhibited good inhibitory effects with the IC50 values of 2.14 and 0.215 μM, respectively. The SAR analysis was shown all the synthesized derivatives had weaker hBChE inhibitory activity compared with the lead compound. The derivatives with benzoxazole or benzothiazole skeletons were active when the alkalinity of the piperidine nitrogen was retained, and when the compounds had a methylene group linkage between the aromatic bicyclic and piperidine structures. In addition, 11b had promising selective Cu2+-chelation properties. In the crystal structure, the 5-NO2-quinolin-8-ol moiety of 11b was located in the acyl-binding pocket of the hBChE active site. Molecular modeling of 11b BuChE indicated the hydroxyl group of the nitroxoline was hydrogen bonded to the His438 ε-imidazole nitrogen, and the OH group on the quinoline nitrogen formed a hydrogen bond with Ser198. The NO2 group interacted with the carbonyl groups of the residues Ser287 and Phe285 (Fig. 3c).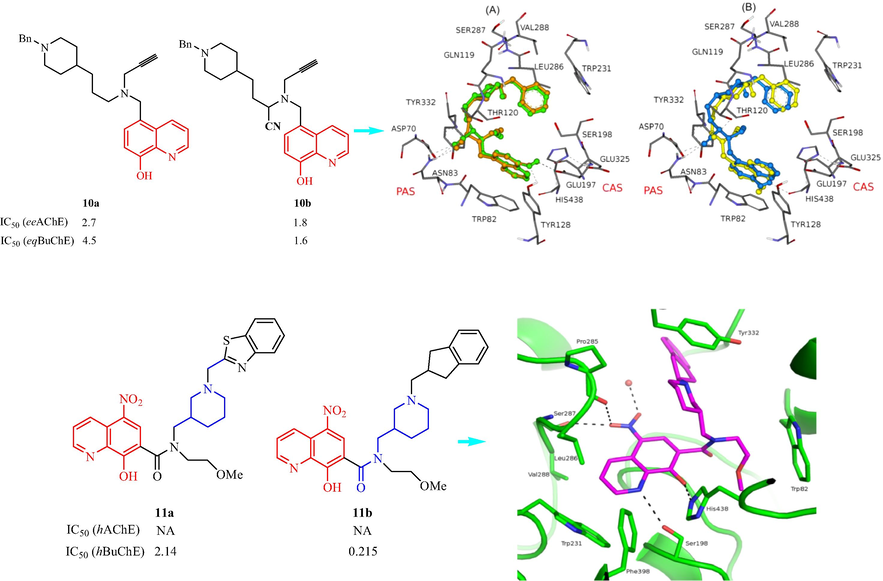
Structural of 8-hydroxyquinoline analogs (10,11) containing piperidine ring.
In 2018, Wang et al. (Wang, et al., 2018b) synthesized a family of quinoline-indole analogs as multi-target-directed ligands for AD therapy. Four classes of analogs were synthesized and compounds 12a (X = H, R1 = OH) (inhibition, 51.2 %) and 12b (X = Cl, R1 = OH) (inhibition, 66.8 %) exhibited better activity than clioquinol (1.9 %) and melatonin (19.3 %) (Fig. 3d). Compound 12b, a metal chelator, which had a neuroprotective effect with antioxidant activity in cells with an median effective concentration (EC50) value of 0.1 μM, a 71.6 % and 85.8 % reduction in of self- or Cu2+-induced Aβ-aggregation, and 72.7 % and 83.3 % disintegration of prefabricated self- or Cu2+-related Aβ1−42 aggregate fibrils, displayed the best anti-AD activity of all the tested compounds. The SAR analysis of the inhibitory effect on Aβ 1-42 self-induced aggregation for these analogs showed: (1) in presence of a Cl-atom on oxine moiety of quinoline ring played a pivotal role in the inhibitory activity; (2) methylation of the hydroxyl group or cyclization with methoxy group seemed unfavorable for the effects on quinoline ring; (3) it demonstrated that phenolic hydroxyl group on indole moiety was preferable for inhibitory activity. In 2020, Fernández-Bachiller et al. (Fernández-Bachiller, et al., 2010) have synthesized a class of tacrine-8-hydroxyquinoline analogs as multi-target-directed ligands for AD therapy (Fig. 3d). Compounds 13a, 13b, and 13c inhibited hAChE with IC50 values of 0.5–5.5 nM and inhibited hBuChE with IC50 values of 6.5–55 nM. Compound 13b displayed the best hAChE inhibition of the tested compounds with 700 times higher activity compared with tacrine, and 13a also showed good antioxidant properties, being 3.3 times more potent than VE in oxygen-radical absorbance capacity. In the propidium displacement assay, 13a, 13b, and 13c could reduce Aβ-aggregation through AChE by 22 %, 19 %, and 27 %, respectively, at a concentration of 0.3 μM. The three compounds could selectively complex Cu2+ and penetrate the central nervous system (CNS). The results displayed Clloquinol for its metal-chelating, neuroprotective, antioxidant properties. Molecules containing an unsubstituted 8-hydroxy quinoline fragment displayed the best AChE inhibitory effects. Tacrine for the inhibition of ChE through its binding to the CAS.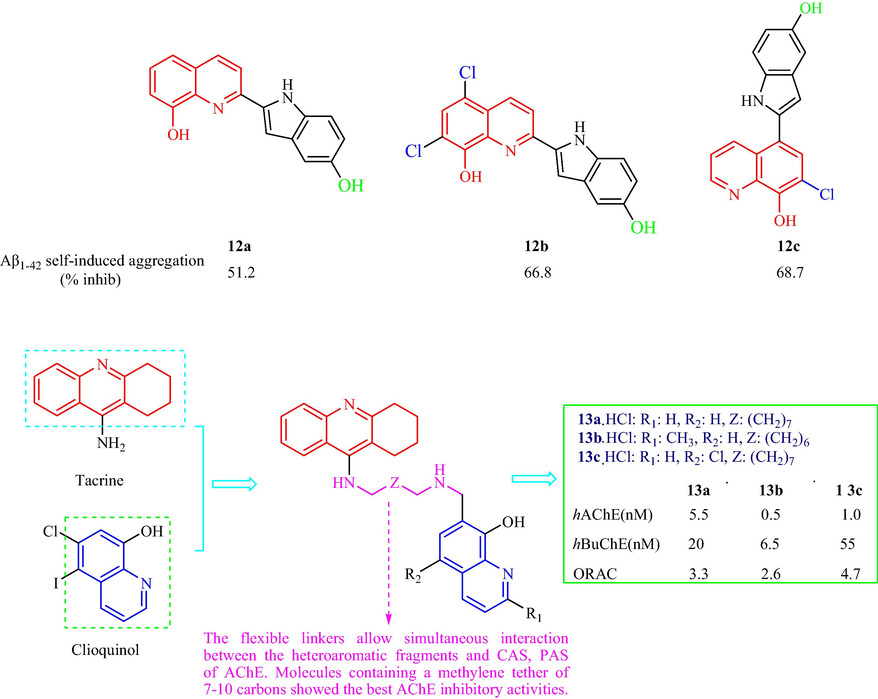
Structural of 8-hydroxyquinoline analogs (12,13) containing indole and tacrine ring.
Fiorito et al. (Fiorito, et al., 2013) have synthesized a family of quinoline analogs and assessed the phosphodiesterase 5 (PDE5) inhibition. The compounds maintained a CN group at the C7-position, a –CH2OH group at the C3-position, and the benzylamino moiety of the quinoline ring, because these factors have been shown to be important for PDE5 activity and selectivity (Fig. 4) (Bi, et al., 2004). In the biological analysis, six compounds with substituents at the C8-position showed PDE5 inhibition with IC50 values from 0.27 to 15.0 nM, the effect of the substituents was in the order of cyclopropyl > -N(CH3)2 > morpholinyl > –NH-cyclopropyl > –NH(CH2)2N(CH3)2 > –NH(CH2)2. Among these compounds, compounds 14a, 14b, and 14c showed good phosphodiesterase inhibition (PDEI) activity. In addition, 14a and 14b also showed selectivity toward all 11 PDEs. The selectivity of 14a and 14b toward PDE6 and PDE11 was higher than that of sildenafil, vardenafil, ortadalafil. The results indicated that 14a elicited effects by which impairments in synaptic plasticity and memory were rescued by elevated levels of Aβ42. Therefore, 14a is a potential PDE5I for the treatment of AD. In 2013, Silva et al. (Silva, et al., 2013) prepared a class of quinolinodonepezil analogs and assessed the AChE and BuChE inhibition. Two quinolinodonepezil compounds, 15b and 15c, were more potent inhibitors of eeAChE than hAChEas shown in Fig. 4. 15a did not have inhibitory activity. 15b and 15c were inactive against eqBuChE and were more potent inhibitors of hBuChE than eqBuChE. The N-benzylpiperidine unit was thought to be important for eeAChE inhibition as it was the main element involved in the binding to AChE. Pudlo et al. (Pudlo, et al., 2014) have designed and synthesized two families of 1,2-dihydroquinoline-3-carboxamides linked to benzylpiperidines as AChE inhibitor for AD (Fig. 4). In the first series, when R2 was hydroxyl, the order of activity for the anti-AChE inhibitory effect was nBu > Me > H (n = 0 or 1) and when R2 was hydrogen, the order of activity was Me (n = 1) > Me (n = 0) > Ph; an NH2 group (16b: R2 = NH2, R1 = CH3) had an effect similar to the OH group (16a: R2 = OH, R1 = CH3). In the second series, compounds had 6,7-di-OCH3or 6,7-di-OH groups on the quinolone ring, and 16c (R1 = H, R3 = OMe) exhibited the most inhibition of AChE with an IC50 value of 0.11 μM, but even at 60 μM there was no observed inhibition of BuChE. However, 16d (R1 = H, R3 = OH), the O-demethylated derivative, inhibited AChE with an IC50 value of 0.48 μM. No BuChE inhibition was displayed at 10 μM. 16c and 16d also showed hAChE inhibition with IC50 values of 0.23 and 0.98 μM, respectively. The SAR analysis displayed: (1) with a methylene linkage (n = 1) between piperidine ring and N-amide present a higher effect than compounds without spacer (n = 0); (2) AChE inhibition increases when N-quinoline is substituted; (3) an amino group has similar activity than a hydroxyl group at 4 position on the quinoline ring. The molecular modeling of 16d hAChE indicate piperidinium is snaking along gorge, making cation π interactions with aromatic rings in Tyr337 and even less in Phe338. The quinolone moiety is linked to peripheral anionic site by π-stacking interactions with Trp286, an H-bond between hydroxyl group in position 4 of quinolone and a hydroxyl of Tyr124 and another one between carbonyl in position 2 of quinolone and a NH of Phe295.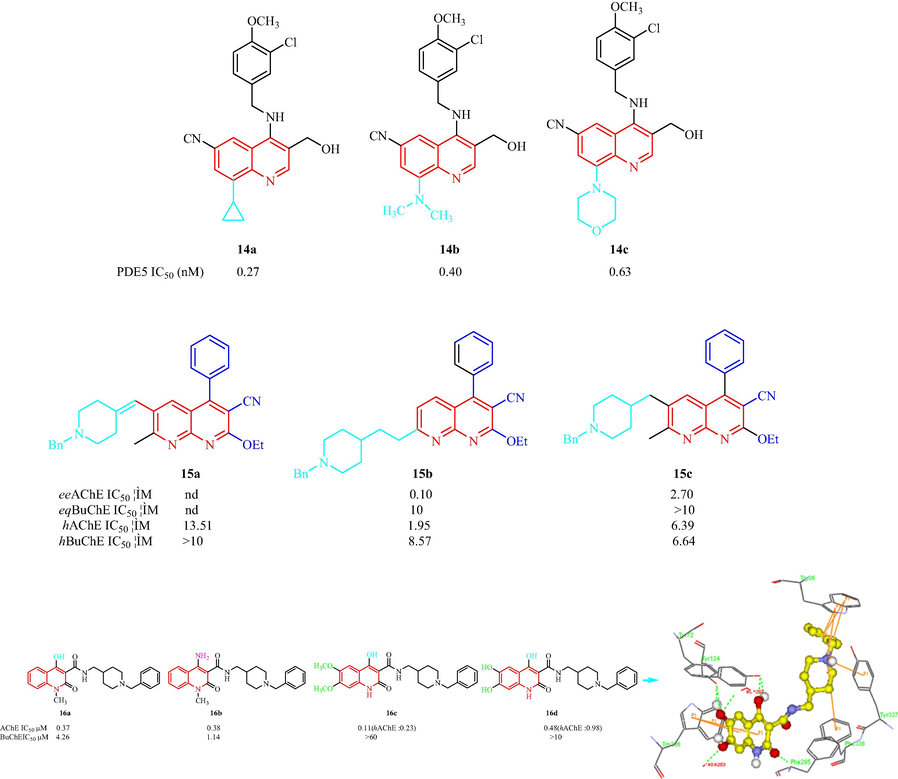
Structural and anti-Alzheimer of quinoline and related analogs 14–16.
In 2015, Wang et al. (Wang, et al., 2015c) have reported the synthesis of a class of 2-arylethenylquinoline analogs as multi-target drugs for AD. Most of the analogs displayed Aβ1-42 aggregation inhibition from 23.6 % to 83.9 % at 20 μM. Of these compounds, 17a, 17b, and 17c possessing piperazine or piperidine ring at 4-position on quinoline ring displayed the most potent inhibitory effect on Aβ1-42 aggregation with inhibitory rates of 81.7 %, 83.9 %, and 81.8 %, respectively, and IC50 values of 10.8, 9.7, and 10.3 μM, respectively, which was greater inhibitory activity than resveratrol (IC50 = 11.4 μM) (Fig. 5). In addition, 17b (IC50 = 64.0 ± 0.1 μM for AChE and IC50 = 0.2 ± 0.1 μM for BuChE), and 17c (IC50 = 68.3 ± 0.1 μM for AChE and IC50 = 1.0 ± 0.1 μM for BuChE) displayed a good ChE inhibitory effect, with high selectivity toward BuChE. Furthermore, 17b was a selective metal chelator for Cu2+ and Fe2+. He et al. (He et al., 2017) and Safarizadeh and Garkani-Nejad (Safarizadeh and Garkani-Nejad, 2019) have also studied this series of analogs and their results supported the above conclusions. The SAR indicated flexible amino at 4-postion of quinoline scaffold contributed to the increased activity, substituent group with 4-dimethylamino or 4-diethylamino on benzene ring is favorable for inhibitory activity of Aβ1-42 aggregation. In 2018, a family of flexible amino-2-arylethenylquinoline analogs was prepared and investigated as multi-target agents for the treatment of AD by Wang et al. (Wang, et al., 2018d). All the derivatives displayed strong inhibition of Aβ1-42 self-induced aggregation (>77 % at 20 μM), which was similar to resveratrol (77.2 % at 20 μM). Compounds 18a, 18b, and 18c (Fig. 5) showed good inhibitory effects with inhibitory rates of 95.3 %, 92.1 %, and 90.2 %. 18b could effectively inhibit Cu2+-induced Aβ1-42 aggregation, had a protective effect on SH-SY5Y (human neuroblastoma cell) cytotoxicity induced by Aβ1-42 without evident toxicity, and reversed the memory impairment of mice in a model of AD. Xia et al. (Xia, et al., 2017) have designed and synthesized 27 2-arylethenyl-N-methylquinolinium analogs and evaluated the Aβ-aggregation inhibition, ChE inhibition, and antioxidant effect. The results showed that 19 compounds displayed Aβ-aggregation inhibitory activity with Aβ1-42 aggregation inhibition from 50.1 % to 98.7 %. Compounds 19a, 19b, and 19c showed the greatest inhibitory activity (91.3 %, 95.1 %, and 98.7 %, respectively, at 20 μM) of all the compounds (Fig. 5). In addition, most compounds displayed similar or weaker inhibitory activity toward hAChE than tacrine and donepezil. 19a, 19b, and 19c displayed better hAChE inhibitory activity, compared with tacrine and donepezil, with IC50 values of 1.10, 1.30, and 1.20 μM, respectively. In summary, the SAR of this series of derivatives implies (1) the flexible amino at 4-postion of quinoline scaffold contributed to the increased activity; (2) the substituent group with 4-dimethylamino or 4-diethylamino on benzene ring is favorable for the inhibitory activity. Molecular modeling was performed to investigate the binding mode between 19a and hAChE displayed the N atom on quinoline ring interacted with Tyr337 and Tyr341 via π-cationic interactions. A hydrogen bond formed between the hydroxyl and acyl groups of phe295 in the phenyl ring 19a binding pocket (Fig. 5).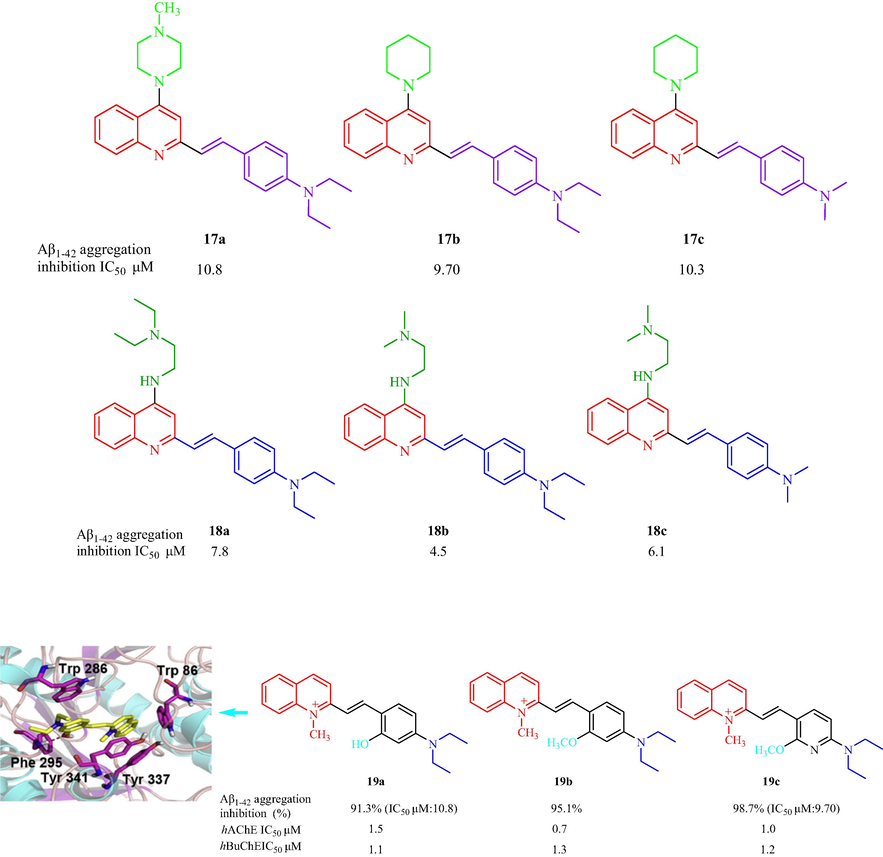
Structure of 2-arylethenylquinoline derivatives 17–19.
Rodríguez et al. (Rodríguez, et al., 2016) have synthesized a series of N-allyl/propargyl-4-substituted-1,2,3,4-tetrahydro-quinoline analogs and investigated the AChE and BChE inhibitory activity. The synthesized compounds had poor activity against AChE and BChE. The N-allyl-tetrahydroquinolines displayed inhibition of both AChE and BChE enzymes, with more inhibitory activity against BChE than AChE. The order of activity against BChE was 6-CH2CH3 > 6-Br > 6-CH3 > 6-OCH3 > 6-Cl > 6-F > 6-H; compounds 20a, 20b, and 20c (IC50 values of 31.66, 25.58, and 29.08 μM, respectively) had better inhibitory effects toward BChE than the other tested compounds. However, N-propargyl-tetrahydroquinolines exhibited weak inhibition against AChE with IC50values from 259.63 to 661.38 μM. A molecular docking study indicated that 20b had a π-π stacking interaction with residue Trp82 within the BChE active site (Fig. 6). In 2017, Sang et al. (Sang, et al., 2017) designed and synthesized 33 3,4-dihydro-2(1H)-quinoline-O-alkylamine derivatives and evaluated their multifunctional biological effects. All the derivatives displayed moderate to good inhibition of eeAChE with IC50 values from 0.56 to 23.5 μM, and showed similar potencies against eqBuChE with IC50 values from 0.87 to 39.6 μM. Three compounds 21a, 21b, and 21c showed good inhibition of AChE with IC50 values of 0.90, 0.76, and 0.56 μM, respectively. 21c exhibited the most potent activity against AChE and had an IC50 value of 2.3 μM against BuChE. SAR indicated piperazine substituent exhibited better inhibitory effects, AChE and BuChE inhibitory activity increased with an increase in methylene chain length. In addition, with isopropylpyrazine group and six methylene chain indicated the best AChE inhibitory activity. The molecular modeling studies displayed 4-benzylpiperidine of moiety of 21c bind to Try334, the long chain of methylene interacted with Phe330 and Tyr121 by hydrophobic interaction, carbonyl group and NH at quinoline nucleus bind to Trp84 and Ser124 intermolecular hydrogen bonds. In 2021, Bautista-Aguilera et al. (Bautista-Aguilera et al., 2021) have synthesized 19 quinolinones and 13 dihydroquinolinonesas potential multi-target drugs for the treatment of AD. None of the compounds inhibited human recombinant MAO. Three analogs displayed promising hAChE and hBuChE inhibition 22a (IC50 = 0.29 and 12.73 µM, respectively), 22b (IC50 = 0.96 and 6.70 µM, respectively), and 22c (IC50 = 1.58 and 16.73 µM, respectively) (Fig. 6). The SAR suggested isopropylpiperazine seemed also essential for the hrAChE activity with the butoxy group as the optimal linker between the core and basic centre. The molecular modeling of 22a hAChE indicated oxygen ether formed a hydrogen bond with Gly121 in the oxyanion hole, while Tyr124 formed a hydrogen bond with the hydrogen of one of the quaternized nitrogen of the piperazine moiety, carbon hydrogen interactions were observed between the ligand and Thr83, Tyr124, Ty337.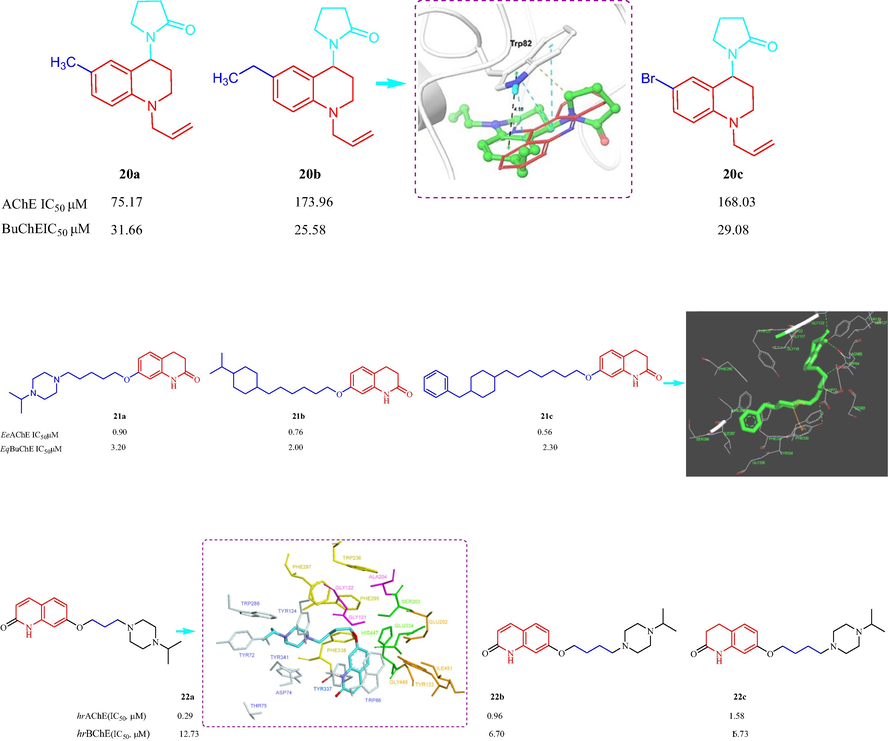
Structure of tetrahydro-quinoline and their derivatives 20–22.
In 2018, Umar et al. (Umar, et al., 2018) designed and synthesized seven 4-[(7-chloroquinolin-4-yl)oxy]-3-ethoxybenzaldehyde derivatives and evaluated the Aβ-aggregation inhibition, anti-oxidation, and metal chelation effects. Most of the compounds inhibited self-mediated Aβ1-42 aggregation ranging from 30 % to 53.72 % and compounds 23a and 23b (Fig. 7) displayed the most inhibition with values of 53.73 % and 53.63 %, respectively, at 50 μM. 23a and 23b also inhibited hAChE activity with values of 47.36 % and 58.26 %, respectively, which were higher than donepezil (23.66 %). The SAR suggested the quinolinone core looked more potent than the semiunsaturated dihydroquinolinone as hrAChE inhibitor, with electron donating groups like methyl, methoxy displayed higher potency while those with electron withdrawing groups like fluorine. The molecular modeling of 23a and 23b hAChE indicated an intermolecular hydrogen bond interaction was formed between O-atom for 23a attached to quinoline group and backbone NH2 group of Lys16. While an intermolecular hydrogen bonding interaction was observed for 23b between –NH group and carbonyl group of Leu17. In 2016, Mantoani et al. (Mantoani, et al., 2016) reported the preparation and assessment as hAChE and hBChE inhibitors of a class of tacrine-donepezil derivatives. The results are presented in Fig. 7, four compounds inhibited hAChE with IC50values from 109.4 to 146.6 µM. Among the tested compounds, compounds 24a and 24b showed the best inhibition against hAChE with IC50 values of 114.0 and 109.4 µM, respectively. The tested compounds did not display an inhibitory effect against hBChE. The binding mode for the docking of the inhibitor 24b with hAChE displayed π-π stacking interaction between the indanone ring of 24b with TRP286, and a second π-π stacking interaction between benzyl group with aromatic tyrosine residues Tyr124, 337 and 341.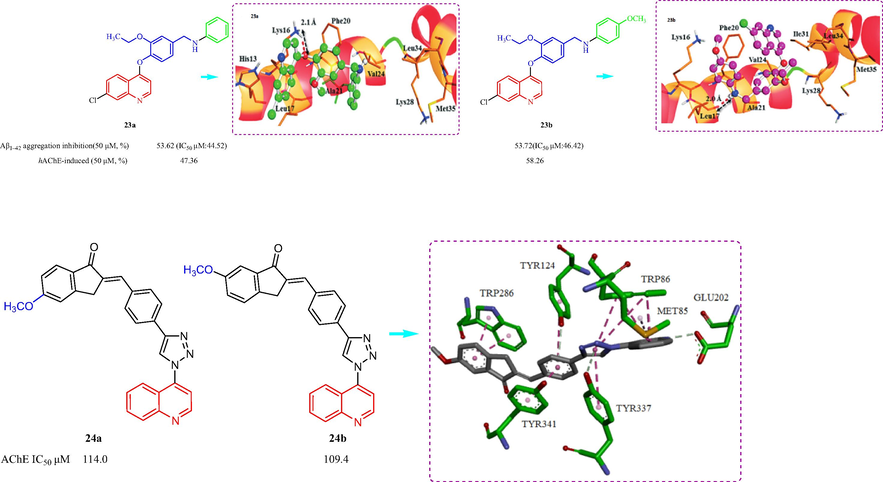
Structure of quinoline and their derivatives 23, 24.
Bosak et al. (Bosak, et al., 2019) also studied eight 4-aminoquinoline derivatives with different substituents, and investigated the hAChE and hBChE inhibition. All the tested compounds exhibited slight selectivity toward hAChE (with Ki constants from 0.46 to 11 μM) over hBChE, and compounds 25a, 25b, and 25c showed the highest affinity toward hAChE with Ki values of 0.46, 0.61, and 0.77 μM, respectively (Fig. 8). The SAR analysis showed that the replacement of chlorine and trifluoromethyl group on C(7) displayed strong AChE inhibition activity. It represented with long alkyl chain or adamantyl group on C(4)-amino group, is important for achieving high inhibition potency. The molecular modeling of 25a hAChE indicated to stabilize with residues Tyr124, Trp286, and Tyr341 in AChE, and protonated amino group was stabilized with Trp86 and Glu202 in AChE. In 2019, to simplify the structure of the hybrid product, Mo et al. (Mo, et al., 2019) designed and synthesized a series of quinoline-ferulic acid hybrids and evaluated the ChE inhibition. Most compounds displayed good inhibitory effects toward both AChE and BChE. Among them, 26a was found to be the most potent inhibitor against AChE (IC50 = 0.62 ± 0.17 μM) and 26b was the most potent inhibitor against BChE (IC50 = 0.10 ± 0.01 μM, Fig. 8). 26a and 26c, as representative compounds, were shown to be competitive inhibitors of AChE and BChE. SAR indicated incorporating a three-carbon chain or a four-carbon chain exhibited to enhance AChE and BChE inhibitory effects; the quinoline-cinnamic acid hybrids compared to quinoline-ferulic acid hybrids showed similar effects and target selectivity; compound with 2-methyl substitued on quinoline ring showed to enhance AChE and BChE inhibitory effects. The molecular modeling of 26a AChE indicated N-atom on quinoline ring interacts with Ser125 by a hydrogen bond, benzyloxyl moiety interacts with Leu289 through a π-alkyl contact, ethylene diamine linker shows van der Waals interactions with Tyr341, Asp72. For 26b in the active site of BChE displayed N-atom interacted with Asp70 by a salt bridge, carbonyl group of the cinnamic acid moiety forms a hydrogen bond with His438. A series of 4-N-phenylaminoquinolines derivatives were prepared and the inhibitory effects on AChE and BChE were assessed by Zhu et al. (Fig. 8) (Zhu, et al., 2019a). All the synthesized compounds inhibited AChE (IC50 values of 0.65–11.84 μM). The two analogs 27a and 27b inhibited AChE and BChE with IC50 values of 0.86 and 2.65 μM, respectively (27a) and 2.65 and 2.78 μM, respectively (27b). 27a displayed the best effect against both AChE and BChE. The SAR analysis indicated the order of inhibitory potency against AChE: meta ≥ ortho > para on the substituted position in the 4-N-phenyl ring, while para-substituted compounds were clearly less potent against BChE than meta-substituted or ortho-substituted ones. The molecular modeling of 27a AChE indicated a π-alkyl interaction between the 4-N-phenyl ring and sec-butyl moiety of Ile287, N-methylbenzylamine fragment interacted with CAS by π-π-stacked interaction with Trp84, quinoline moiety made a carbon hydrogen bond with Tyr70.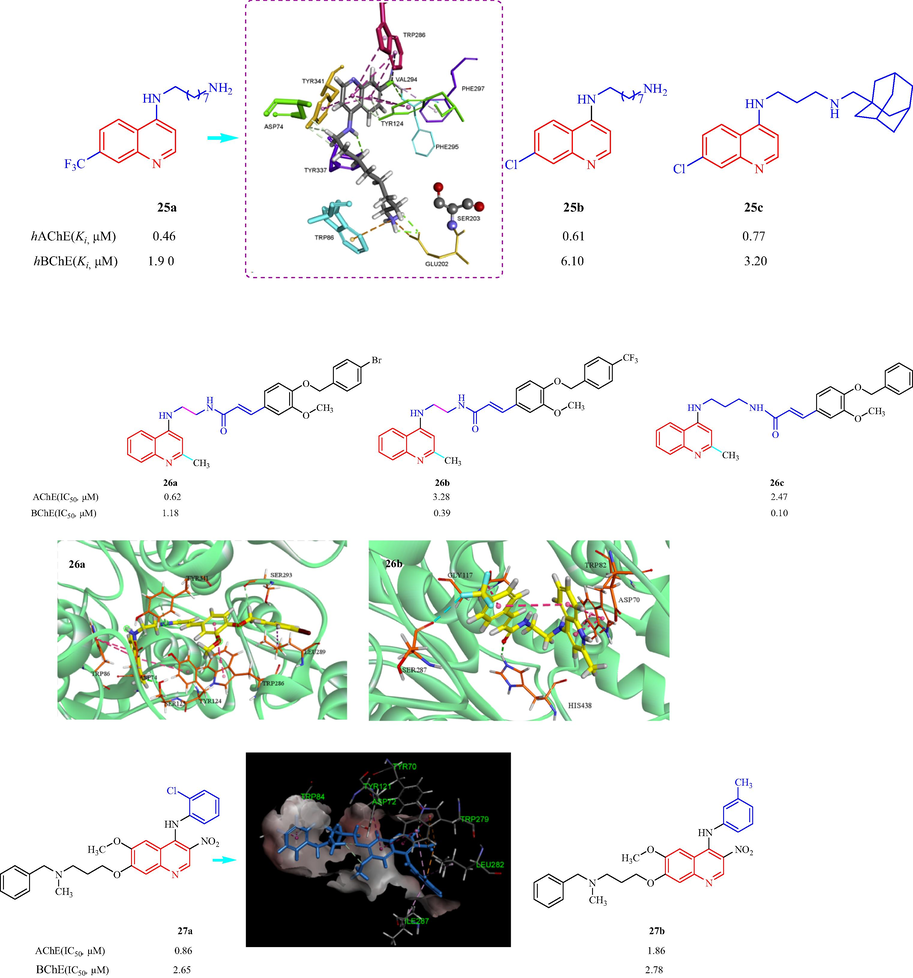
Structure of 4-aminoquinoline quinoline, 4-N-phenylaminoquinoline derivative 25–27.
In 2020, Fu et al. (Fu, et al., 2020) reported a family of quinolinone analogs bearing a dithiocarbamate as multifunctional AChEIs to treat AD. Most compounds displayed strong selective inhibition of eeAChE, and good selectivity for eeAChE over eqBuChE. Compound 28a, which had a four carbon linker and a piperidine at the terminal position showed the most effect against eeAChE and hAChE (IC50 = 0.22 and 0.16 μM, respectively). 28b with di-CH3 groups on the coumarin ring also inhibited eeAChE and hAChE (IC50 = 0.34 and 0.39 μM, respectively) (Fig. 9). The SAR analysis indicated including a four-carbon atom linker showed better inhibitory effect; the replacement of piperidine group using other cyclic amines were not beneficial for AChE inhibition. The molecular modeling of 28a for AChE indicated piperidine group established a ‘arene-H’ interaction with Trp 86, the polymethylene chain was folded in a conformation to make it interact with mid-gorge site by a ‘arene-H’ interaction with Tyr 341. In 2021, Zaib et al. (Zaib, et al., 2021) reported a class of quinoline-thiosemicarbazone derivatives as inhibitors of ChE (Fig. 9). All the synthesized compounds were fully selective for AChE activity, with IC50 values ranging from 0.12 to 60.9 μM. Compound 29a exhibited the greatest inhibitory effect (IC50 = 0.12 ± 0.02 μM), which was five times stronger than the positive control drug galantamine (IC50 = 0.62 ± 0.01 μM). The SAR analysis indicated that an ethylmorpholine group at the R3 position on the thioamide unit resulted in better inhibition than a benzene ring; compounds with an ethylmorpholine were determined to be lead candidates. Moving –OCH3 group from the six-position to the seven-position on the quinoline ring led to a reduction in the inhibition. The substitution of ethyl morpholine with a benzene ring was detrimental for the inhibition. All compounds showed < 50 % inhibition against BChE. The molecular docking of 29a and 29b analysis suggested the active pocket like π-π T-shaped by quinoline ring and π-sulfur by sulfur atom in thiosemicarbazone moiety with Trp86. the hydrogen bonding was noticed between oxygen atom and Gly122, nitrogen atom of morpholine ring exhibited interactions with Glu452 for 29a. While for 29b, nitrogen atom of morpholine ring was involved in formation of salt bridge and π-cation, interactions with Glu452 and Tyr449, respectively. Munir et al. (Munir, et al., 2021) have prepared a library of piperidine-containing quinolinyl thiosemicarbazones and investigated the AChE and BChE inhibition (Fig. 9). First, the effect of methyl substitution at the 6-(R1) and 8-(R2) positions of quinoline was studied while keeping the thiosemicarbazide chain unchanged. Five compounds displayed IC50 values < 20 µM (including 30a, IC50 = 19.85 μM and 30c, IC50 = 13.85 μM, against AChE). Compound 30b showed the strongest inhibitory effect toward AChE and BChE (IC50 = 9.68 and 11.59 μM, respectively), when the 8-position of the quinoline was substituted with a methyl group, the anti-cholinesterase activity of the meta-substituted chlorine and fluorine derivatives was the greatest. Molecular docking studies of 30b for AChE indicated two hydrogen bonds with sulfur and NH of carbothioamide by Arg296, a hydrogen bond with sulfur of thiocarbonyl by Ser293 (Fig. 9).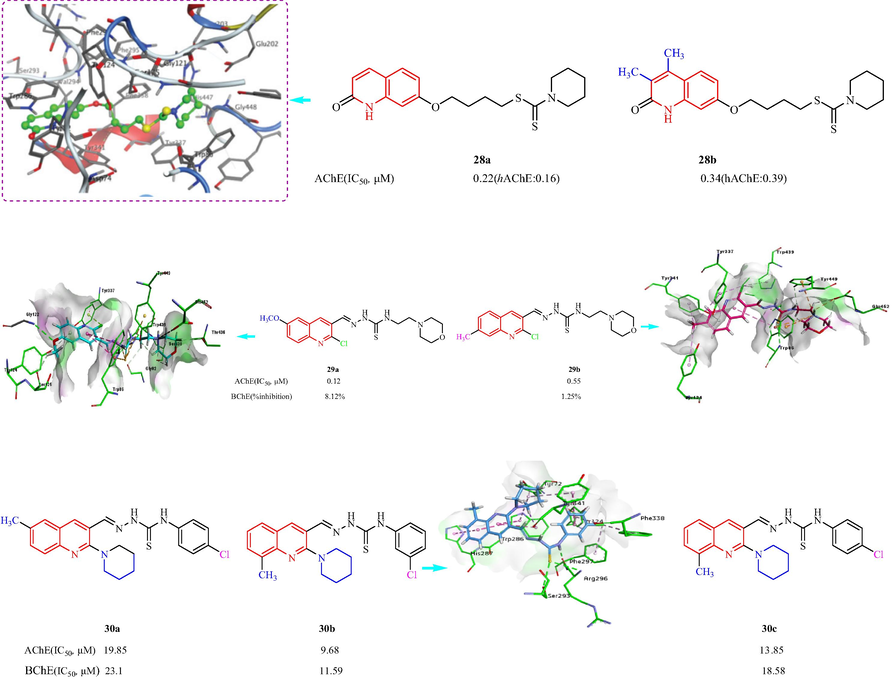
Structure of quinolinone-dithiocarbamate and quinoline-thiosemicarbazone 28–30.
Maalej et al. (Maalej et al., 2012a) have designed and prepared a family of racemic tacrine analogs and studied the inhibition of hAChE and hBuChE (Maalej et al., 2011b). Compounds with electron-donating groups, such as 3-OCH3-4-OH, 2,4-di-OCH3, 4-OH, 2-OCH3, or 4-CH3 displayed good inhibitory effects with IC50 values from 0.33 to 0.40 μM. Analysis of the SAR indicated that R group substitutions on the benzene ring increased the inhibition in the order: H > 4-OH-3-OCH3 > 2,4-di-OCH3 > 4-CH3 > 3,4,5-tri-OCH3 > 2-OCH3 > 4-OH. Compounds 31a (IC50 = 0.30 ± 0.04 μM) and 31d (IC50 = 0.33 ± 0.04 μM) showed activity against hAChE (Fig. 10a). Compounds bearing the electron-withdrawing groups 4-NO2, 3,4-di-Cl, and 2,6-di-Cl were 13–19 times less potent than other compounds, the activity was in the following order: 2,6-di-Cl > 3,4-di-Cl > 4-NO2, but were still selective hAChE inhibitors. Molecular modeling of hAChE with 31 R- and S-enantiomers indicated benzochromene moiety interacts with Trp286 and tetrahydroquinoline moiety interacts with Tyr341 via π-π stacking. The hydrogen bond interaction between hydroxyl substituent in the phenyl ring and hydroxyl group of Thr75 for R-, while S-enantiomers, hydroxyl group of Thr75 establishes hydrogen bond interactions with two oxygen atoms of methoxy and hydroxyl groups. Eghtedari et al. (Eghtedari, et al., 2017) have designed and synthesized a series of multifunctional tacrine derivatives, 4H-pyrano[2,3-b]quinoline-3-carboxylates (Fig. 10a), as ChE inhibitors. The IC50 values against AChE indicated that all the derivatives showed marked inhibitory activity with IC50 values from 0.069 to 6.22 μM. The activity increased in the order of 3-Br > 2-Br > 2-Cl > 3-Cl > 2-F > 4-F > 4-NO2 > 3-F > 4-Cl > 2,4-Cl2 > 3-NO2 > 4-Br for the electron-withdrawing groups. The order of the activity was H > 4-OCH3 > 3-OCH3 > 4-CH3 for electron-donating groups. And the study found the electron-withdrawing groups were more effective than electron-donating groups for AChE inhibition. Among the tested compounds, 32d exhibited the most potent inhibition of AChE and BuChE with IC50values of 0.069 and 1.35 μM of, respectively. Inhibition assays showed that most derivatives exhibited potent activity against AChE and the potential for activity against BuChE. Analysis of the SAR concluded that Cl or Br substituents at the ortho- or meta- positions of the 4-benzene ring can increase the anti-cholinesterase effects. Mahdavi et al. (Mahdavi, et al., 2019a) have synthesized 17 benzochromeno-quinolinone compounds as tacrine analogs and evaluated the pharmacological action with the aim of developing anti-AD agents (Fig. 10a). Fifteen compounds showed AChE inhibition (IC50 = 0.86–27.52 μM). Two analogs 33a and 33b were discovered to be good anti-AChE and anti-BChE agents with IC50 = 0.86 and 0.89 μM and IC50 = 6.03 and 13.85 μM, respectively. Surprisingly, 33b had an inhibitory effect on β-secretase 1 (IC50 = 19.60 μM), as well as the ability to chelate Cu2+, Fe2+, and Zn2+. In addition, the effect of halogenations had no significant effect, but the position of the substituent on the aryl moiety was important for AChE inhibitory activity, the order of activity of 2-halo substituted compounds was 2-bromine > 2-chlorine > 2-fluorine. In contrast, the order of activity was 4-fluorine > 4-bromine > 4-chlorine. In the molecular docking assay of 33b, a hydrogen bond was formed between 33b and Tyr334. In 2001, Marco et al. (Marco et al., 2001) investigated the AChE and BChE inhibitory activity of a family of pyrano[2,3-b]quinolones. The tested analogs showed less activity than tacrine (IC50 = 1.3 × 10-7 M). The most active compound was 34a with IC50 = 8.86 ± 0.42 × 107 M, which was approximately-seven times less active compound (Fig. 10a). Analysis of the SAR of the compounds indicated: (1) the type of substituents on the aromatic ring at C4 appeared to have no effect on the inhibition; (2) the order of AChE inhibitory activity of the substituted derivatives was 4-OCH3 > H > 4-CH3 > 3-NO2 > 4-Cl > 4–CN; (3) electron-donating substituents increased the activity more than electron-withdrawing substituents; and (4) replacing the phenyl ring in tacrine with a pyran ring yielded an analog with better AChE inhibitory activity.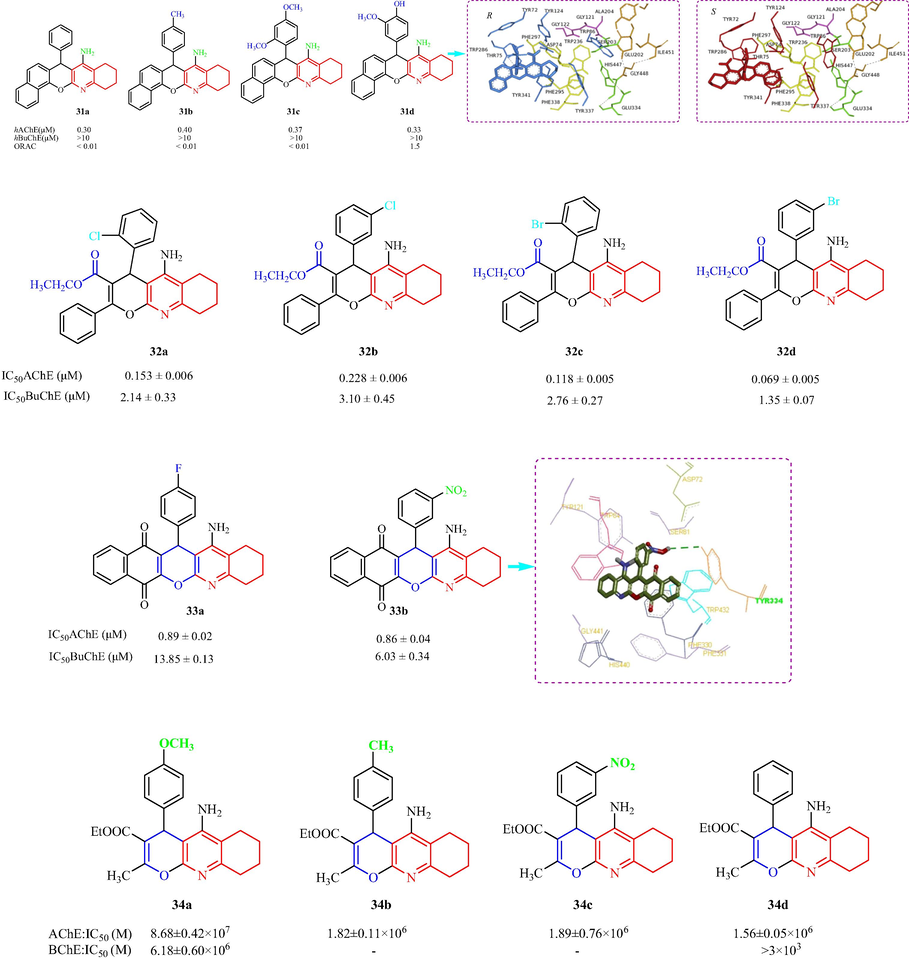
The structural of tacrine-quinoline and related analogs 31–34.
Khoobi et al. (Khoobi, et al., 2015) have designed and prepared a series of 2,4,6,7,8,9-hexahydro- pyrazolo[4′,3′:5,6]pyrano[2,3-b]quinolin-5-amine analogs, and evaluated the anti-cholinesterase activity in vitro. The IC50 values indicatedthat the synthesized analogs, with the exception of the 2-OCH3-phenyl substituents, showed AChE inhibition (IC50 = 0.19–3.27 μM). The most active analog was compound 35b with 3,4-diOCH3-phenyl substituents with an IC50 value of 0.19 μM (Fig. 10b), which was more active than tacrine. However, the 4-OCH3-phenyl and 1-naphthyl compounds were almost as effective against AChE as tacrine. For electron-donating groups, the order of AChE inhibitory activity was 3,4-diOCH3 > 4-OCH3 > 2,5-diOCH3 > 3,4,5-triOCH3 > 2-OCH3. The molecular modeling of AChE with 35 R- and S-enantiomers indicated R-enantiomer, dimethoxyphenyl moiety oriented toward the CAS, 3,4-diOCH3-phenyl moiety was a T-shape π-π stacking with Phe330 and hydrogen binding with His440. S- toward PAS, T-shape π-π stacking of 3,4-diOCH3-phenyl with Trp279 and a hydrogen bond between pyrazole ring and Ser286. Pourabdi et al. (Pourabdi et al., 2016) have designed and prepared a class of tacrine-based pyrazolo [4′,3′:5,6]pyrano[2,3-b]quinolines derivatives as inhibitors of AChE and BuChE for anti-AD activity. The majority of the derivatives inhibited AChE (IC50 < 2 μM), in particular, the 3-propyl compound 36a, 3-phenyl derivatives 36b and 36c, and the 3-(furan-2-yl) analog 36d showed superior activity compared with tacrine (Fig. 10b). 36c (IC50 = 0.034 μM) was six times more active than tacrine. In addition, all the derivatives inhibited BuChE (IC50 values ≤ 0.570 μM), except for the tri-phenyl (R1 = R2 = R3 = Ph) derivative, and most compounds inhibited BuChE to a greater extent than AChE. The putative binding mode of 36c in the AChE binding site displayed the pyrazole fragment was involved in hydrogen bonding to Glu199, the cyclohexyl moiety was installed in the hydrophobic pocket by Trp84, Phe330, Trp432 and Ile439. The interactions between 36c and BuChE displayed binding site Ser198 and Trp82 were inclued in binding with ligand through hydrogen bond and T-shape π-π interaction, respectively, another hydrogen bonding was exhibted between pyrazole ring and Asp70, aliphatic cyclohexyl fragment is directed into the lipophilic pocket formed by residues Leu286, Val288 and Trp231. In 2017, Mahdavi et al. (Mahdavi, et al., 2017b) prepared a family of pyrazolo[4′,3′:5,6]pyrano [2,3-b]quinolin-5-amines, and assessed AChE and BuChE inhibition in vitro. Eleven derivatives showed potent AChE inhibition (IC50 = 2.77–29.57 μM), compound 37a with a phenyl substituent at the 4-position displayed the highest inhibitory activity (IC50 = 2.77 μM). Eight compounds had good inhibitory activity against BuChE (IC50 values from 0.06 to 10.0 μM), and the compound with a 2,4-diOCH3-phenyl group displayed the best BuChE inhibitory activity (IC50 = 0.06 μM). The SAR analysis is shown in Fig. 10b. Overall, derivatives 37a and 37b showed good activity against AChE and BChE.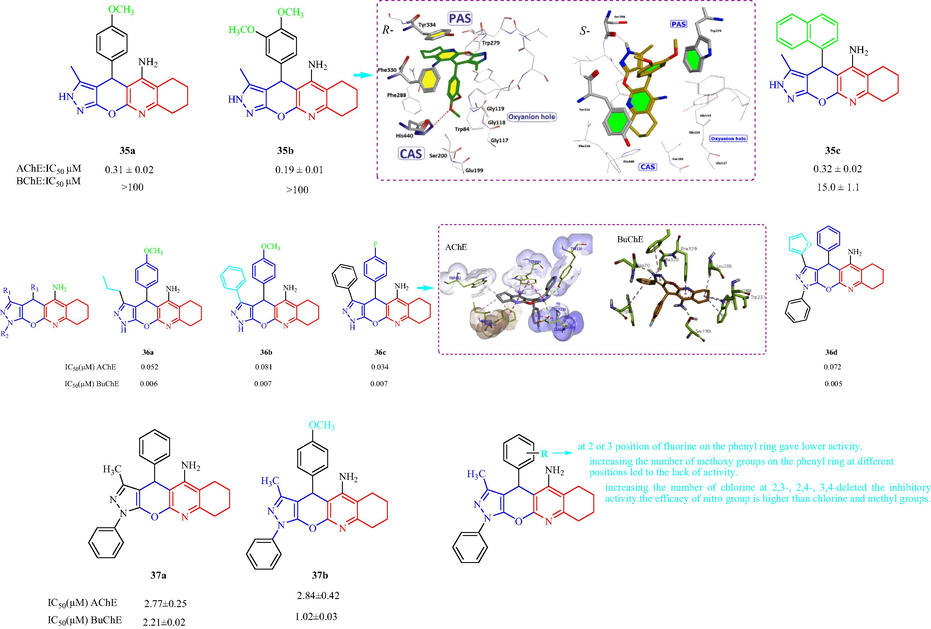
The anti-Alzheimer of structural tacrine-quinoline and related analogs 35–37.
In 2013, Khoobi et al. (Khoobi, et al., 2013) reported a class of 8-amino-tetrahydrochromeno [3′,4′:5,6] pyrano[2, 3-b]quinolin-6(7H)-one analogs and evaluated the AChE and BuChE inhibition. Most of the prepared derivatives displayed selective activity against AChE and three compounds 38a, 38b, and 38c showed better activity against eeAChE with IC50 values of 5, 14, and 16 nM, respectively, compared with the other compounds (Fig. 10c). 38a (with a 4-F-phenyl substituent) displayed the best activity against eeAChE. The compounds with methyl, ethyl, and 3-fluorophenyl groups at the 7-positioncan be thought of as dual inhibitors for both enzymes. In the analysis of the SAR of this series of compounds, the order of AChE inhibitory activity was n-butyl > ethyl > methyl for alkyl substituents at position 7; the activity for heteroaryl substituents had the following order: benzodioxole-5-yl > 4-pyridyl > 2-thienyl; and the order of activity for the substituents on the phenyl ring at position 7 was 4-F > 2-Cl > 2,4-Cl2 > 3-F > 4-Cl > 3-NO2 > 3-Cl for electron-withdrawing groups and 4-CH3 > 3,4,5-tri-OCH3 for electron-donating groups. Molecular modeling of AChE with 38a indicated a hydrogen bonding of coumarin carbonyl moiety with hydroxyl of Tyr121. The phenyl ring at position 7 was oriented toward the hydrophobic pocket of the binding cavity composed of Phe330, Tyr334 and Phe331. Hariri et al. (Hariri, et al., 2016) have designed and synthesized a family of pyrano[3′,4′:5,6] pyrano[2,3-b]quinolinone derivatives and assessed the tested compounds for AChE and BChE inhibition n vitro. The synthesized compounds exhibited good AChE inhibition with IC50 values from 0.37 to 5.62 μM. However, most of the tested derivatives did not display BChE inhibition. Among the tested compounds, compound 39a with 2,3-dichlorine atoms on the aromatic ring displayed the greatest AChE inhibition (IC50 = 0.37 μM) (Fig. 10c). Furthermore, derivatives with 2-NO2-5-Cl, 2-Cl, and 4-Cl substituents displayed better anti-AChE effects with IC50 values of 0.72, 0.80, and 0.82 μM, respectively. The electronic features of the substituents and their positions on the 12-aryl group played important roles in the AChE inhibitory activity. In analysis of the SAR, the order of AChE inhibitory activity for the electron-withdrawing groups was 2,3-diCl > 2-NO2-5-Cl > 4-Cl > 2-Cl > 2-NO2 > 3-NO2 > 2-F > 4-F; and for electron-donating groups was 4-Me > 3-Me > H > 2-Me ≈ 4-MeO > 3,4,5-triMeO. The molecular modeling of AChE with 39 R- and S-enantiomers indicated R-enantiomer 2,3-diCl moiety oriented toward Leu282 and hydrophobic interaction oriented toTyr70, Tyr121, Tyr334,Trp279 residues of PAS, for S-enantiomers 2,3-diCl moiety has oriented toward Asp72 of the PAS. In 2011, Bartolini et al. (Bartolini, et al., 2011) reported the synthesis and the evaluation of the AChE inhibition and Aβ1–42 self-aggregation in vitro of (R/S)-p-methoxytacripyrine (40) (Fig. 10c). 40 could have inhibitory activity against both ChEs by modulation of the chirality at the stereocenter C4. Specifically, enantiomer (S)-40 (Aβ1–42 self-aggregation inhibition: 88.6 %) had approximately 10-fold greater inhibitory activity than (R)-40 (Aβ1–42 self-aggregation inhibition: 84.1 %). Molecular docking simulation AChE with 40 R- and S-enantiomers suggested R-40 hAChE placed in the binding pocket residues involved in catalysis, Ser203, Glu334, His447, indole ring of Trp286 forms a π-π interaction with p-OCH3-phenyl ring, a hydrogen bond between amino group of S-40 and carboxylate group of Asp74.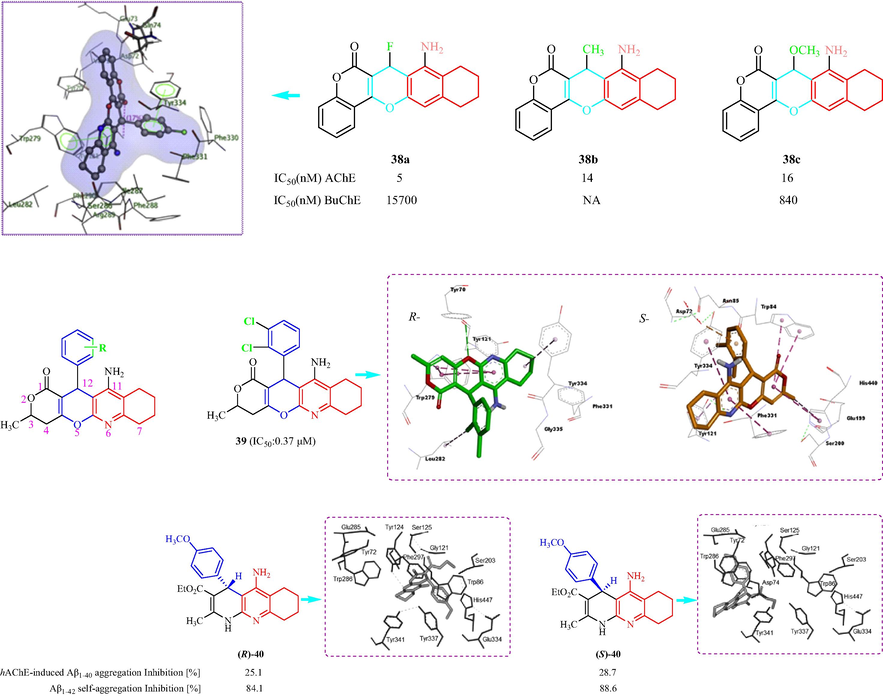
The anti-Alzheimer of structural tacrine-quinoline and related analogs 38–40.
In 2017, Chen et al. (Chen, et al., 2017) have prepared a class of tacrine-ferulic acid analogs as multi-target ChE inhibitors and 36 analogs had inhibitory activity against AChE with IC50 values < 100 nM. Among two compounds 41a and 41b displayed the best inhibitory effects. 41a exhibited hAChE inhibition (IC50 = 60.6 ± 5.6 nM), and hBuChE inhibition (IC50 = 86.1 ± 15.5 nM) and 41b showed hAChE inhibition (IC50 = 55.1 ± 4.9 nM) and hBuChEI inhibition (IC50 = 55.9 ± 3.3 nM) (Fig. 10d). The compounds efficiently inhibited human ChEs. The SAR analysis indicated that methoxy and methyl groups increase inhibitory activity on AChE, which was para- > meta- > ortho-. When substituted by Cl, the activity on AChE was para- > meta- > ortho-, different halogen atoms, the activity was –Cl ≈–Br > –F. The molecular docking of hAChE with 41a indicated tetrahydroacridin moiety formed multiple π–π stacking contacts with the aromatic side chains of Trp86 and Tyr124, while for 41b formed π–π stacking Trp286 and Tyr341. Hepnarova et al. (Hepnarova, et al., 2018) have designed and synthesized 21 tacrine-benzyl quinolone carboxylic acid analogs and evaluated the compounds as inhibitors of hAChE and hBChE in vitro. Three different series were synthesized with different tacrine cores: unsubstituted tacrine; 6-chlorotacrine; and 7-methoxytacrine. The analogs were inhibitors of ChEs. Compounds 42a, 42b, and 42c showed better AChE inhibition with IC50 values of 1.53, 0.129, and 0.0419 µM, respectively (Fig. 10d), compared with the other compounds. In addition, all the analogues demonstrated micromolar to nanomolar hBChE inhibition. The SAR analysis displayed linker between two basic pharmacophores played a crucial role in contacting both anionic sites, which found the best effect was two or three methylene units tethered hybrids. In addition, the observation can be made between series with descending hAChE affinity from 6-OCH3-tacrine ≥ unsubsituted tacrine ≥ 7-Cl-tacrine, whcih 7-Cl-tacrine was found to be the most active hAChE inhibitor. Docking simulations 42c with hAChE suggested 1,4-dihydroquinoline provide favorable stacking to Tyr341, Tyr124 is engaged in anchoring 4-methoxybenzyl moiety (Fig. 10d). In 2018, Zhu et al. (Zhu, et al., 2018b) designed and prepared a family of tacrine-ferulic acid hybrids modified with a benzyl group to investigate ChE inhibitors asmulti-target ligands. All the tested analogues displayed AChE and BuChE inhibition with IC50 values from 37 to 284.1 nM, and 52.7 to 256.7 nM, respectively. Among the tested compounds, 43a (R = 4-Br), 43b (R = 3,4-diCH3), and 43c (R = 4-CF3) showed the highest activity against AChE (with IC50 values of 49.5, 37.0, and 128.6 nM, respectively), and against BuChE (IC50 values of 69.4, 101.4, and 52.7 nM, respectively) (Fig. 10d). The SAR analysis displayed dimethyl substitution at benzyloxyl showed to enhance effects against AChE. The electron-withdrawing groups: –CN and-CF3 showed reduced AChE inhibitory effect while they were favorable for BuChE inhibition. In addition, Docking simulations 43b with AChE displayed 1,2,3,4-tetrahydroacridin core was located at CAS by a π-π stacking interaction with Trp86, phenyl core of the ferulic acid moiety formed p-p stacking interactions with Trp286 and Tyr341(Fig. 10d). In 2018, Galdeano et al. (Galdeano, et al., 2018) reported the synthesis of polar tacrine derivatives, including 6-chlorotacrine, and huprine Y, and investigated the inhibition of hAChE and hBChE. All the derivatives showed hAChE and hBChE inhibitory effects with IC50 values from 0.01 to 12 µM, and 0.05 to 22.3 µM, respectively. Compounds 44a, 44b, and 44c displayed the most potent and selective hAChE inhibitory effects of all the derivatives, with IC50 values of 0.01, 0.06 and 0.20 µM, respectively, and were selective for hAChE and hBChE inhibition (Fig. 10d). The SAR analysis showed triazole-containing side chain is terminated with a polyphenol-like aromatic ring, resulting compounds are slightly more potent hAChE and hBChE inhibitors than the parent tacrine and 6-chlorotacrine. Docking simulations 44a with hAChE indicated triazole ring is engaged in two perpendicular π-stacking interactions with Tyr334, Phe300, and in H-bonds with Tyr121 and Asp72. While 44b showed huprine plane was shifted away from catalytic His440, and upwards towards Phe330. Hamulakova et al. (Hamulakova, et al., 2021) have prepared two classes of tacrine-indole ligands and evaluated the hAChE and hBChE inhibitory effects. The tested derivatives showed hChE inhibitory activity with IC50 values from 160 to 25 nM and 160 to 39 nM for hAChE and hBChE, respectively. Compounds 45a and 45b showed the best inhibition for AChE with IC50 values of 25 and 70 nM, respectively (Fig. 10d). The analysis of the SAR suggested four-methylene spacer appeared to best hAChE inhibition, six-methylene bridge conferred the best inhibition to hBChE, methoxy group on indole nitrogen enhanced inhibitory activity for hBChE with a chain length of n = 4–6. In 2022, the Przybyłowska group designed and synthesized a family of 14 N- and O-phosphorylatedtacrine analogs and investigated the inhibitory effects against hAChE and hBChE as latent anti-AD agents (Przybyłowska, et al., 2022). The tested analogs displayed activity against AChE with IC50 values from 6.11 to 676.7 nM, respectively, and against BuChE (IC50 = 1.969–186.2 nM). The compounds with the most inhibitory activity against AChE were 46a (pIC50 = 1.513 nM) and 46b (pIC50 = 0.786 nM); and 46c and 46d exhibited the best inhibition against BuChE with pIC50 values of 0.913and 0.998 nM, respectively. These synthesized compounds also inhibited BChE. In addition, the molecular docking of 46b was investigated and 46b exhibited low docking energies of –12.5 and –10.7 kcal/mol for AChE and BChE, respectively.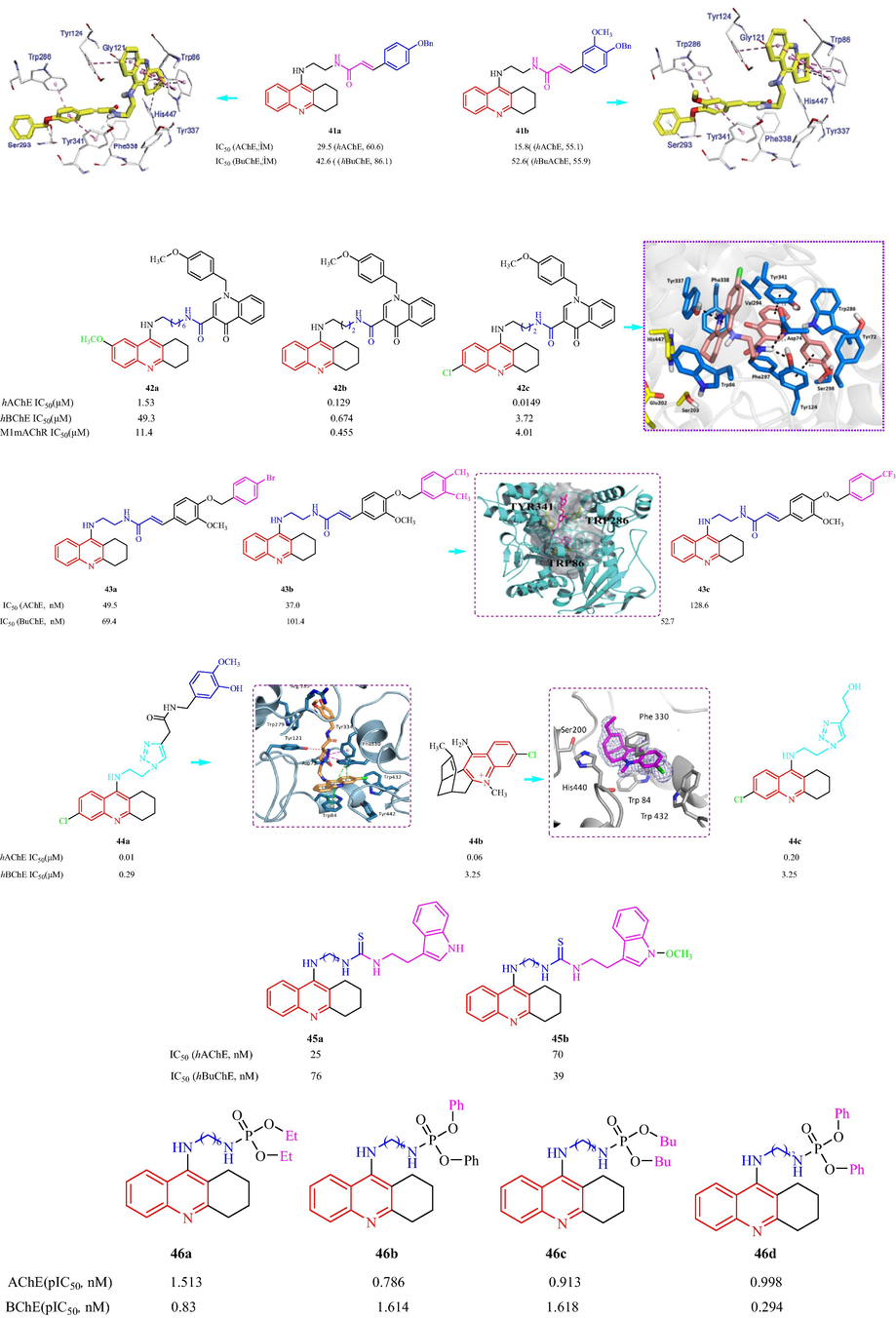
Structural formula tacrine-quinoline analogs 41–46.
In 2016, Grychowska et al. (Grychowska, et al., 2016) have also reported a class of arylsulfonyl analogues based on N-4-(pyrrolidin-3-yl-amino)-1H-pyrrolo[3,2-c]quinoline as 5-HT6R antagonists. The 18 synthesized compounds showed medium to high affinity for the 5-HT6R with Ki values from 3 to 101 nM. Among them, compounds 47a and 47b exhibited the most affinity for the 5-HT6R [with Ki values of 7 ± 1 (S-enantiomer) and 12 ± 3 (R-enantiomer) and 3 ± 1 (S-enantiomer) and 7 ± 2 (R-enantiomer), respectively] (Fig. 11a). However, it was suggested that the S isomers were the best antagonists and displayed a better antagonistic action on the 5-HT6 sites. The SAR analysis suggested tert-butyl or isopropyl groups in para position significantly decreased affinity, suggesting that bulky substituents at this position were unfavorable for binding to the 5-HT6R. The electron-withdrawing substituent in meta position of phenyl ring with electron-donating methoxy or methyl groups maintained the high affinity for 5-HT6R. Najafi et al. (Najafi, et al., 2016) have designed and synthesized a family of acridine-chromenone and quinoline-chromenone derivatives and assessed the AChE and BuChE inhibition. Twelve compounds displayed anti-AChE effects (IC50 = 16.17–83.10 μM). Most of the analogs did not show any effects against BChE. Among the tested compounds, 48a with CH3 and Cl substituents displayed the best inhibitory activity (IC50 = 16.17 μM). Substitution of the five-membered ring (in 48a) with a 6-membered ring, resulted in the formation of a tetrahydroacridine scaffold (48b), which decreased the inhibitory activity against AChE (IC50 = 18.03 μM). 48c with a tetrahydroacridine moiety and no other substituents had an IC50 value of 19.07 μM. The SAR analysis displayed five or six-membered ring induce more activity, seven-membered ring fused to quinoline moiety which showed lower activity comparing with its counterpart. Docking simulations 48a with hAChE showed π-π interactions between quinoline moiety and Trp84 and Phe330, hydrophobic interaction of Cl with Ile439, Met436, Tyr442 and Trp432, phenoxy group occupied to make π-anion interaction with Asp72 and π-π interaction with Phe330, 4-methylchromenone moiety established π-π and hydrophobic interaction with Trp279 (Fig. 11a). Czarnecka et al. (Czarnecka, et al., 2017a) have reported a series of 2,3-dihydro-1H-cyclopenta[b]quinoline derivatives using 5,6-dichloronicotinic acid. All the synthesized derivatives were AChE inhibitors with IC50 values ranging from 0.052 to 0.744 μM. Among the reported derivatives, 49a (IC50 = 0.052 μM) and 49b (IC50 = 0.053 μM) showed greater inhibition of AChE, which was three and two times that of tacrine and donepezil, respectively. All the derivatives also showed good BuChE inhibitionwithIC50 values ranging from 0.071 to 1.863 μM. The most active compound toward BuChE, 49c, had an IC50 value of 0.071 μM (Fig. 11a). In addition, in 2019, Czarnecka et al. (Czarnecka et al., 2019b) reported eight cyclopentaquinoline derivatives with a tetrahydroacridine structure and evaluated the EeAChE and EqBuChE inhibition in vitro. Four compounds exhibited IC50 values below 100 nM for EeAChE inhibition, which suggested the best inhibition was obtained with alkyl linkers with 6 to 9 carbons in the chain. Eight compounds also displayed inhibitory activity against BuChE with IC50 values ranging from 42 to 662 nM. Among these compounds, compounds 50a and 50b displayed the most activity against EeAChE and EqBuChE with IC50 values of 0.067 and 0.073 μM, respectively and 0.153 and 0.042 μM, respectively. The molecular docking of hAChE with 50a indicated an amide nitrogen atom formed a hydrogen bond with hydroxyl group of Tyr121 toward AChE. While for 50b with BuChE, a hydrogen bond was with the main chain of Tyr332 in anionic site (Fig. 11a). Makhaeva et al. (Makhaeva, et al., 2020a) have reported the synthesis of two series of derivatives of 4-amino-2,3-polymethylenequinoline. All the derivatives inhibited AChE and BChE, displaying selectivity toward BChE (IC50 = 0.084–4.27 µM), and with IC50 values ranging from 1.90 to 45.0 µM against AChE. Among these compounds, 51a, 51b, and 51c displayed better inhibition of hAChE and hBuChE with IC50 values of 4.03, 3.50 and 1.90 μM, respectively; and 0.419, 0.652 and 0.084 μM, respectively (Fig. 11a). Analysis of the SAR showed to increase of an aliphatic ring size of 4-amino-2,3-polymethylenequinoline fragment from C-5 to C-7 had no effect on anti-AChE activity, whereas its increase to C-8 reduced potency of AChE inhibition ten times, with a cycloheptaquinoline moiety (C-7) exhibited maximum activity toward BChE. In 2020, Makhaeva et al. (Makhaeva, et al., 2020b) also synthesized 11 derivatives of 4-amino-2,3-polymethyl-enequinoline and investigated the ChE inhibition. The tested derivatives displayed inhibitory effects toward AChE (with IC50 values from 0.131 to 9.01 µM) and BChE (IC50 values from 0.0431 to 0.924 µM), but showed selectivity toward BChE. The SAR results are shown in Fig. 11a. In 2018, Ekiz et al. (Ekiz, et al., 2018) reported the synthesis of bromoindenoquinolines and phenyl-substituted indenoquinoline amine analogs and investigated the inhibition of AChE and BChE (Fig. 11a). The monophenyl indenoquinolines inhibited AChE and BChE with IC50values ranging from 37 to 57 nM and 84 to 93 nM, respectively. Compounds 53a, 53b, 53c, and 53d displayed good inhibitory effects against AChE with IC50 values of 230, 37, 57, and 56 nM, respectively, and against BuChE with IC50 values of 2620, 93, 87 and 87 nM, respectively. The SAR analysis displayed the introduction of phenyl substituent on compounds led to increased inhibition effects against AChE and BChE. Six quinoline-piperonal analogs were prepared and the inhibitory activity against AChE and BChE was assessed by De Brum et al. (DeBrum et al., 2019). Five compounds showed EeAChE inhibition with IC50 values from 8.50 to 62.76 µM. Compound 54a showed selectivity for EeAChE over EqBChE; the inhibitory effect on EeAChE (IC50 = 8.50 ± 0.39 µM) was twice that of EqBChE (IC50 = 15.16 ± 0.75 µM). The docking studies of EeAChE with 54a indicated the lowest intermolecular energy and the residues Tyr124, Tyr133 and His447 (Fig. 11b).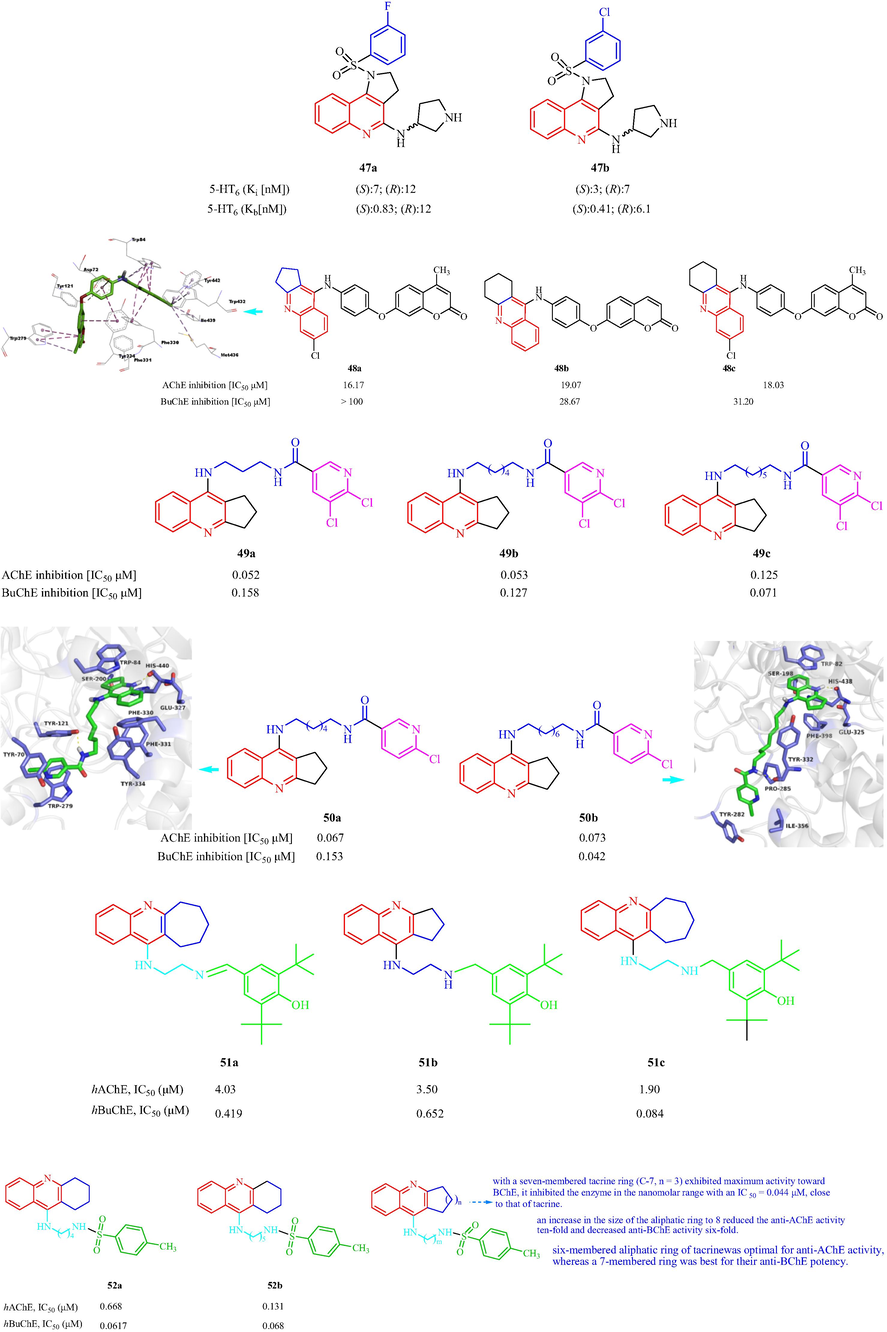
Structural formula tacrine-quinoline and related analogs 47–52.
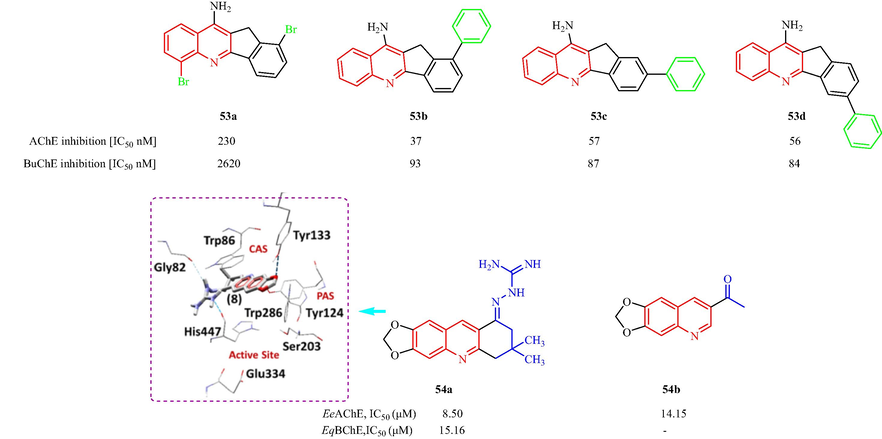
Structural formula tacrine-quinoline and related analogs 53, 54.
3 Conclusions
In summary, because the basic properties and extra-binding capacity of the quinoline scaffold play an important role in optimization the physicochemical properties in the drug design and development strategies. Therefore, in recent years, researchers have synthesized hybrid quinoline scaffolds with compounds containing other heterocyclic and evaluated various biological activities, also involving the inhibitory effects against anti-AD effects. The main part of this review focus on and highlights the synthesized quinolone and related analogs, including various heterocyclic such as indole, tacrine, triazole, pyrrolo, chromeno, and pyrano etc as anti-Alzheimer’s disease agents from 2001 to 2022, and also particularly highlight the structure–activity relationships and the molecular binding modes. Thus, to find the new characteristic structures or groups, discover the active lead compounds, further design of rationally anti-Alzheimer’s disease agents and drug molecules provide the basis.
Acknowledgements
This research was supported by the Province of Jilin Science and Technology Development Plan (No. 20210204115YY), and the Department of Education of Jilin Province (No. JJKH202111444KJ). We thanks Victoria Muir, PhD, from Liwen Bianji, (Edanz) (www.liwenbianji.cn/), edited the English text of a draft of this manuscript.
References
- Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Aβ. Neuron. 2008;59:43-55.
- [CrossRef] [Google Scholar]
- Therapeutics of Alzheimer’s disease: past, present and future. Neuropharmacol.. 2014;76:27-50.
- [CrossRef] [Google Scholar]
- Plant-like biosynthesis of isoquinoline alkaloids in Aspergillus fumigatus. Nat. Chem. Biol.. 2016;12:419-424.
- [CrossRef] [Google Scholar]
- Chemical and pharmacological studies on enantiomerically pure p-methoxy- tacripyrines, promising multi-target-directed ligands for the treatment of Alzheimer’s disease. ChemMedChem. 2011;6:1990-1997.
- [CrossRef] [Google Scholar]
- Quinolines as extremely potent and selective PDE5 inhibitors as potential agents for treatment of erectile dysfunction. Bioorg. Med. Chem. Lett.. 2004;14:1577-1580.
- [CrossRef] [Google Scholar]
- Alzheimer’s disease: Past, present, and future. J. Int. Neuropsychol. Soc.. 2017;23:818-831.
- [CrossRef] [Google Scholar]
- Structural aspects of 4-aminoquinolines as reversible inhibitors of human acetylcholinesterase and butyrylcholine-sterase. Chem. Biol. Interact.. 2019;308:101-109.
- [CrossRef] [Google Scholar]
- Synthesis, structure and stereochemistry of quinoline alkaloids from Choisya ternata. Org. Biomol. Chem.. 2007;5:2983-2991.
- [CrossRef] [Google Scholar]
- Design, synthesis, in vitro and in vivo evaluation of tacrine-cinnamic acid hybrids as multi-target acetyl-and butyrylcholinesterase inhibitors against Alzheimer’s disease. RSC Adv.. 2017;7:33851-33867.
- [CrossRef] [Google Scholar]
- Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron. 2001;30:665-676.
- [CrossRef] [Google Scholar]
- Quinoline and quinolone dimers and their biological activities: An overview. Eur. J. Med. Chem.. 2019;161:101-117.
- [CrossRef] [Google Scholar]
- Regioselective functionalization ofquinolines through C-Hactivation: A comprehensive review. Molecules. 2021;26:5467.
- [CrossRef] [Google Scholar]
- Discovery of new cyclopentaquinoline analogues as multifunctional agents for the treatment of Alzheimer’s disease. Int. J. Mol. Sci.. 2019;20:498-520.
- [CrossRef] [Google Scholar]
- New cyclopentaquinoline hybrids with multifunctional capacities for the treatment of Alzheimer’s disease. J. Enzyme Inhib. Med. Chem.. 2017;33:158-170.
- [CrossRef] [Google Scholar]
- Synthesis of new quinoline-piperonal hybrids as potential drugs against Alzheimer’s disease. Int. J. Mol. Sci.. 2019;20:3944-3959.
- [CrossRef] [Google Scholar]
- Metal-Free air oxidation in a convenient cascade approach for the access toisoquinoline-1,3,4(2H)-triones. Molecules. 2019;24:2177.
- [CrossRef] [Google Scholar]
- New tacrine-derived AChE/BuChE inhibitors: synthesis and biological evaluation of 5-amino-2-phenyl-4H-pyrano[2,3-b]quinoline-3-carboxylates. Eur. J. Med. Chem.. 2017;128:237-246.
- [CrossRef] [Google Scholar]
- Synthesis, characterization, and SAR of arylated indenoquinoline-based cholinesterase and carbonic anhydrase inhibitors. Arch. Pharm. Chem. Life Sci.. 2018;351:e1800167.
- [Google Scholar]
- Rapid and green analytical method for the determination ofquinolinealkaloids fromCinchona succirubrabased on microwave-integrated extraction and leaching (MIEL) prior to high performance liquid chromatography. Int. J. Mol. Sci.. 2011;12:7846-7860.
- [CrossRef] [Google Scholar]
- Structure-based des-ign and optimization of multitarget-directed 2H-chromen-2-one derivatives as potent inhibitors of monoamine oxidase B and cholinesterases. J. Med. Chem.. 2015;58:5561-5578.
- [CrossRef] [Google Scholar]
- C-2 phenyl replacements to obtain potentquinoline-basedStaphylococcus aureus Nor A inhibitors. J. Enzyme Inhib. Med. Chem.. 2020;35:584-597.
- [CrossRef] [Google Scholar]
- Novel tacrine-8-hydroxyquinoline hybrids as multifunctional agents for the treatment of Alzheimers disease, with neuroprotective, cholinergic, antioxidant, and copper-complexing properties. J. Med. Chem.. 2010;53:4927-4937.
- [CrossRef] [Google Scholar]
- Synthesis of quinoline derivatives: Discovery of a potent and selective phosphodies terase 5 inhibitor for the treatment of Alzheimer’s disease. Eur. J. Med. Chem.. 2013;60:285-294.
- [CrossRef] [Google Scholar]
- Design, synthesis and evaluation of quinolinone derivatives containing dithiocarbamate moiety as multifunctional AChE inhibitors for the treatment of Alzheimer’s disease. J. Enzyme. Inhib. Med. Chem.. 2020;35:118-128.
- [CrossRef] [Google Scholar]
- Novel multifunctional neuroprotective iron chelator-monoamine oxidase inhibitor drugs for neurodegenerative diseases. selective brain monoamine oxidase inhibition and prevention of MPTP-induced striatal dopamine depletion. J. Neurochem.. 2005;95:79-88.
- [CrossRef] [Google Scholar]
- Increasing polarity in tacrine and huprine derivatives: potent anticholinesterase agents for the treatment of myasthenia gravis. Molecules. 2018;23:634.
- [CrossRef] [Google Scholar]
- Integrative network-based analysis reveals gene networks and novel drug repositioning candidates for Alzheimer disease. Neurol. Genet.. 2021;7:e622.
- [Google Scholar]
- Novel 1H-pyrrolo[3,2-c]quinoline based 5-HT6 receptor antagonists with potential application for the treatment of cognitive disorders associated with Alzheimer’s disease. ACS Chem. Neurosci.. 2016;7:972-983.
- [CrossRef] [Google Scholar]
- The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain. 2018;141:1917-1933.
- [CrossRef] [Google Scholar]
- Design and synthesis of novel tacrine–indole hybrids as potential multitarget-directed ligands for the treatment ofAlzheimer’s disease. Future Med. Chem.. 2021;13:785-804.
- [CrossRef] [Google Scholar]
- Novel tacrine-based pyrano[3́,4́:5,6]pyrano[2,3-b]quinolinones: synthesis and cholinesterase inhibitory activity. Arch. Pharm. Chem. Life Sci.. 2016;349:1-10.
- [CrossRef] [Google Scholar]
- The protection of novel 2-arylethenylquinoline derivatives against impairment of associative learning memory induced by neural Aβ in C. elegans Alzheimer’s disease model. Neurochem. Res.. 2017;42:3061-3072.
- [CrossRef] [Google Scholar]
- The concept of hybrid molecules of tacrine and benzyl quinolone carboxylic acid (BQCA) as multifunctional agents for Alzheimer’s disease. Eur. J Med. Chem.. 2018;150:292-306.
- [CrossRef] [Google Scholar]
- Alzheimer disease is multifactorial and heterogeneous. Neurobiol. Agin.. 2000;21:901-902.
- [CrossRef] [Google Scholar]
- Alzheimer's disease: from pathology to therapeutic approaches. Angew. Chem. Int. Ed Engl.. 2009;48:3030-3059.
- [CrossRef] [Google Scholar]
- Isoquinoline-1,3-diones as selective inhibitors of tyrosyl DNA phosphodiesterase II (TDP2) J. Med. Chem.. 2016;59:2734-2746.
- [CrossRef] [Google Scholar]
- Synthetic and medicinal perspective ofquinolinesas antiviral agents. Eur. J. Med. Chem.. 2021;215:113220
- [CrossRef] [Google Scholar]
- Design, synthesis, docking study and biological evaluation of some novel tetrahydrochromeno[3́,4́:5,6]pyrano[2,3-b]quinolin-6(7H)-one derivatives against acetyl- and butyrylcholinesterase. Eur. J. Med. Chem.. 2013;68:291-300.
- [CrossRef] [Google Scholar]
- New tetracyclic tacrine analogs containing pyrano[2,3-c]pyrazole:efficient synthesis, biological assessment and docking simulation study. Eur. J. Med. Chem.. 2015;89:296-303.
- [CrossRef] [Google Scholar]
- Correction to: BMI1 is associated with CSF amyloid-β and rates of cognitive declinein Alzheimer’s disease. Alzheimers Res. Ther.. 2021;13:164.
- [CrossRef] [Google Scholar]
- A comprehensive evaluation of the process of copying a complex figure in early- and late-onsetAlzheimer disease: a quantitative analysis of digital pen data. J. Med. Internet. Res.. 2020;22:e18136.
- [Google Scholar]
- Structure-based development of nitroxoline derivatives as potential multifunctional anti-Alzheimer agents. Bioorg. Med. Chem.. 2015;23:4442-4452.
- [CrossRef] [Google Scholar]
- Magnetic resonance imaging texture predicts progression to dementia due toAlzheimer diseaseearlier than hippocampal volume. J. Psychiatry Neurosci.. 2020;45:7-14.
- [CrossRef] [Google Scholar]
- Cardanol-derived AChE inhibitors: towards the development of dual binding derivatives for Alzheimer’s disease. Eur. J. Med. Chem.. 2016;108:687-700.
- [CrossRef] [Google Scholar]
- Alzheimer disease: an update on pathobiology and treatment strategies. Cell. 2019;179:312-339.
- [CrossRef] [Google Scholar]
- Synthesis, biological assessment, and molecular modeling of racemic 7-aryl-9,10,11,12-tetrahydro-7H-benzo[7,8]chromeno[2,3-b]quinolin-8-amines as potential drugs for the treatment of Alzheimer’s disease. Eur. J. Med. Chem.. 2012;54:750-763.
- [CrossRef] [Google Scholar]
- Synthesis of novel tacrine analogs as acetylcholinesterase inhibitors. J. Heterocyclic Chem.. 2017;54:384-390.
- [CrossRef] [Google Scholar]
- Synthesis, biological assessment and molecular modeling of 14-aryl-10,11,12,14-tetrahydro-9H-benzo[5,6] chromeno[2,3-b]quinolin-13-amines. Bioorg. Med. Chem. Lett.. 2011;21:2384-2388.
- [CrossRef] [Google Scholar]
- Synthesis and biological activity of some benzochromenoquinolinones: tacrine analogs as potent anti-Alzheimer’s agents. Chem. Biodivers.. 2019;16:e1800488.
- [Google Scholar]
- New hybrids of 4-amino-2,3-polymethylene-quinoline and p-tolylsulfonamide as dual inhibitors of acetyl- and butyrylcholinesterase and potential multifunctional agents for Alzheimer’s disease treatment. Molecules. 2020;25:3915.
- [CrossRef] [Google Scholar]
- New multifunctional agents based on conjugates of 4-amino-2,3-polymethylenequinoline and butylated hydroxytoluene for Alzheimer’s disease treatment. Molecules. 2020;25:5891.
- [CrossRef] [Google Scholar]
- Novel triazole-quinoline derivatives as selective dual binding site acetylcholinesterase inhibitors. Molecules. 2016;21:193.
- [CrossRef] [Google Scholar]
- Plantisoquinolinealkaloids as potential neurodrugs: A comparative study of the effects of benzo[c]phenanthridine and berberine-based compounds on β-amyloid aggregation. Chem. Biol. Interact.. 2021;334:109300
- [CrossRef] [Google Scholar]
- Synthesis and acetylcholines-terase/butyrylcholinesterase inhibition activity of new tacrine-like analogues. Bioorg. Med. Chem.. 2001;9:727-732.
- [CrossRef] [Google Scholar]
- Design, synthesis, biological evaluation, and molecular modeling studies of quinoline-ferulic acid hybrids as cholinesterase inhibitors. Bioorg. Chem.. 2019;93
- [CrossRef] [Google Scholar]
- Microwave-assisted synthesis of (piperidin-1-yl)quinolin-3-yl) methylene)hydrazine-carbothioamides as potent Inhibitors of cholinesterases: a biochemical and in silico approach. Molecules. 2021;26:656-687.
- [CrossRef] [Google Scholar]
- An overview of quinoline as a privileged scaffold in cancer drug discovery. Expert. Opin. Drug Discov.. 2017;12:583-597.
- [CrossRef] [Google Scholar]
- Green recipes to quinoline: a review. Eur. J. Med. Chem.. 2019;164:121-170.
- [CrossRef] [Google Scholar]
- Design and synthesis of novel anti-Alzheimer’s agents: acridine-chromenone and quinoline-chromenone hybrids. Bioorg. Chem.. 2016;67:84-94.
- [CrossRef] [Google Scholar]
- Quinoline-based hybrid compounds with antimalarial activity. Molecules. 2017;22:2268-2290.
- [CrossRef] [Google Scholar]
- Antimalarialproperties ofisoquinolinederivative fromStreptomyces hygroscopicus subsp.hygroscopicus: an in silico approach. Biomed. Res. Int. 2020:6135696.
- [CrossRef] [Google Scholar]
- Neuroinflammation in Alzheimer’s disease: an understanding of physiology and pathology. Int. J. Neurosc.. 2014;124:227-235.
- [CrossRef] [Google Scholar]
- Investigation of causal effect of a trial fibrillation on Alzheimer dise-ase: A mendelian randomization study. J. Am. Heart Assoc.. 2020;9:e014889.
- [CrossRef] [Google Scholar]
- Advances in polymer based friedlander quinolinesynthesis. Turk. J. Chem.. 2021;45:1299-1326.
- [CrossRef] [Google Scholar]
- Comparison of anticancer activity and HPLC-DAD determination ofselectedisoquinoline alkaloids from Thalictrum foetidum, Ber-berissp. and Chelidonium majus extracts. Molecules. 2019;24:3417.
- [CrossRef] [Google Scholar]
- Synthesis and structure-activity relationship study of tacrine-based pyrano[2,3-c]pyrazoles targeting AChE/BuChE and 15-LOX. Eur. J. Med. Chem.. 2016;123:298-308.
- [CrossRef] [Google Scholar]
- Novel 8-hydroxyquinoline derivatives as multitarget compounds for the treatment of Alzheimer′s disease. ChemMedChem.. 2016;11:1284-1295.
- [CrossRef] [Google Scholar]
- Prince, M., Wimo, A., Guerchet, M., Ali, G.C., Wu, Y.T., Prina, M., 2015. World Alzheimer report. The global impact of dementia: ananalysis of prevalence, incidence, cost and trends. Alzheimer’s disease international. pp. 1-80. https://www.alz.co.uk/research/WorldAlzheimerReport.
- Design, synthesis and biological evaluation of novel N-phosphorylated and O-phosphorylated tacrine derivatives as potential drugs against Alzheimer’s disease. J. Enzyme Inhib. Med. Chem.. 2022;37:1012-1022.
- [CrossRef] [Google Scholar]
- Quinolone-benzylpiperidine derivatives as novel acetylcholinesterase inhibitor and antioxidant hybrids for Alzheimer disease. Bioorganic. Med. Chem.. 2014;22:2496-2507.
- [CrossRef] [Google Scholar]
- A review for the analysis of antidepressant, antiepileptic and quinolone type drugs in pharmaceuticals and environmental samples. Crit. Rev. Anal. Chem.. 2016;46:424-442.
- [CrossRef] [Google Scholar]
- In vitro activity of a new quinoline derivative, ER-2, against clinical isolates of Mycoplasma pneumoniae and Mycoplasma hominis. Antimicrob. Agents Chemother.. 2009;53:5317-5318.
- [CrossRef] [Google Scholar]
- Antimalarial properties of isoquinolinederivative from Streptomyces hygrosco- picussub sp. hygroscopicus: an in silico approach. Biomed. ResInt. 2020;6135696
- [CrossRef] [Google Scholar]
- Novel N-allyl/propargyl tetrahydroquinolines: synthesis via Three-component cationic Imino Diels-Alder reaction, binding prediction, and evaluation as cholinesterase inhibitors. Chem. Bio.l Drug Des.. 2016;88:498-510.
- [CrossRef] [Google Scholar]
- Alzheimer’s disease: characterization of the brain sites of the initial tau cytoskeletal pathology will improve the success of novel immunological anti-Tau treatment approaches. J. Alzheimer’s Dis.. 2017;57:683-696.
- [CrossRef] [Google Scholar]
- Stabilization of nontoxic Aβ-oligomers: Insights into the mechanism of action of hydroxyquinolines in alzheimer’s disease. J. Neurosci.. 2015;35:2871-2884.
- [CrossRef] [Google Scholar]
- Molecular docking, molecular dynamics simulations and QSAR studies on some of 2-arylethenylquinoline derivatives for inhibition of Alzheimer's amyloid-beta aggregation: Insight into mechanism of interactions and parameters for design of new inhibitors. J. Mol. Graph. Model.. 2019;87:129-143.
- [CrossRef] [Google Scholar]
- Design, synthesis and biological evaluation of 3,4-dihydro-2(1H)-quinoline-O-alkyl-amine derivatives as new multipotent cholinesterase/ monoamine oxidase inhibitors for the treatment ofAlzheimer’s disease. Bioorg. Med. Chem.. 2017;25:3006-3017.
- [CrossRef] [Google Scholar]
- Neuroprotection by a novel brain permeable iron chelator, VK-28, against 6-hydroxydopamine lession in rats. Neuropharmacol.. 2004;46:254-263.
- [CrossRef] [Google Scholar]
- Synthesis, pharmacological assessment, molecular modeling and in silico studies of fused tricyclic coumarin derivatives as a new family of multifunctional anti-Alzheimer agents. Eur. J. Med. Chem.. 2016;107:219-232.
- [CrossRef] [Google Scholar]
- Biologically activequinolineand quinazoline alkaloids part I. Med. Res. Rev.. 2018;38:775-828.
- [CrossRef] [Google Scholar]
- Nature’s derivative(s) as alternative anti-Alzheimer’s disease treatments. J. Alzheimers Dis. Rep.. 2009;3:279-297.
- [CrossRef] [Google Scholar]
- An update on potential therapeutic strategies for Parkinson’s disease based on pathogenic mechanisms. Expert Rev. Neurother.. 2016;16:711-722.
- [CrossRef] [Google Scholar]
- Synthesis, pharmacological assessment, and molecular modeling of acetylcholinesterase/butyrylcholinesterase Inhibitors: effect against amyloid-β-Induced neurotoxicity. ACS Chem. Neurosci.. 2013;4:547-565.
- [CrossRef] [Google Scholar]
- Alzheimer’s Disease: The link between Amyloid-β and neurovascular dysfunction. J. Alzheimers Dis.. 2020;76:1179-1198.
- [CrossRef] [Google Scholar]
- New amyloid beta-disaggregating agents: synthesis, pharmacological evaluation, crystal structure and molecular docking of N-(4-((7-chloroquinolin-4-yl)oxy)-3-ethoxybenzyl)amines. Med. Chem. Commun.. 2018;9:1891-1904.
- [CrossRef] [Google Scholar]
- Donepezil + propargylamine+8-hydroxyquinoline hybrids as new multifunctional metal-chelators, ChE and MAO inhibitors for the potential treatment of Alzheimer’s disease. Eur. J. Med. Chem.. 2014;80:543-561.
- [CrossRef] [Google Scholar]
- Design, synthesis, and biological evaluation of 2-arylethenylquinoline derivatives as multifunctional agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem.. 2015;89:349-361.
- [CrossRef] [Google Scholar]
- Design, synthesis, and evaluation of orally bioavailable quinoline-indole derivatives as innovative multitarget-directed ligands: promotion of cell proliferation in the adult murine hippocampus for the treatment of Alzheimer’s disease. J. Med. Chem.. 2018;61:1871-1894.
- [CrossRef] [Google Scholar]
- Preparation of 4-flexible amino-2-arylethenyl-quinoline derivatives as multi-target agents for the treatment of Alzheimer’s disease. Molecules. 2018;23:3100-3120.
- [CrossRef] [Google Scholar]
- Design, synthesis and evaluation of 2-arylethenyl-N-methylquinolinium derivatives as effective multifunctional agents for Alzheimer’s disease treatment. Eur. J. Med. Chem.. 2017;130:139-153.
- [CrossRef] [Google Scholar]
- Novel 8-hydroxyquinoline derivatives targeting β-amyloid aggregation, metal chelation and oxidative stress against Alzheimer’s disease. Bioorganic. Med. Chem.. 2018;26:3191-3201.
- [CrossRef] [Google Scholar]
- The path from anti Parkinson drug selegiline and rasagiline to multifunctional neuroprotective anti Alzheimer drugs ladostigil and m30. Curr. Alzheimer Res.. 2006;3:541-550.
- [CrossRef] [Google Scholar]
- Novel bifunctional drugs targeting monoamine oxidase inhibition and iron chelation as an approach to neuroprotection in Parkinson’s disease and other neurodegenerative diseases. J. Neural. Transm.. 2004;111:1455-1471.
- [CrossRef] [Google Scholar]
- Bifunctional drug derivatives of MAO-B inhibitor rasagiline and iron chelator VK-28 as a more effective approach to treatment of brain ageing and ageing neurodegenerative diseases. Mech. Ageing Dev.. 2005;126:317-326.
- [CrossRef] [Google Scholar]
- Hybrid quinoline-thiosemicarbazone therapeutics as a new treatment opportunity for Alzheimer’s disease-synthesis, in vitro cholinesterase inhibitory potential and computational modeling analysis. Molecules. 2021;26:6573.
- [CrossRef] [Google Scholar]
- Synthesis, pharmacology and molecular docking on multifunctional tacrine-ferulic acid hybrids as cholinesterase inhibitors against Alzheimer’s disease. J. Enzyme Inhib. Med. Chem.. 2018;33:496-506.
- [CrossRef] [Google Scholar]
- Design, synthesis, evaluation and molecular modeling study of 4-N-phenylaminoquinolines for Alzheimer disease treatment. Bioorg. Med. Chem. Lett.. 2019;29:1325-1329.
- [CrossRef] [Google Scholar]




