

Translate this page into:
Stereoselective synthesis of diazaspiro[5.5]undecane derivatives via base promoted [5+1] double Michael addition of N,N-dimethylbarbituric acid to diaryliedene acetones
⁎Corresponding authors at: Department of Chemistry, College of Science, King Saud University, P.O. Box 2455, Riyadh 11451, Saudi Arabia (A. Barakat). Tel.: +966 114675884; fax: +966 114675992. mislam@ksu.edu.sa (Mohammad Shahidul Islam), ambarakat@ksu.edu.sa (Assem Barakat)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
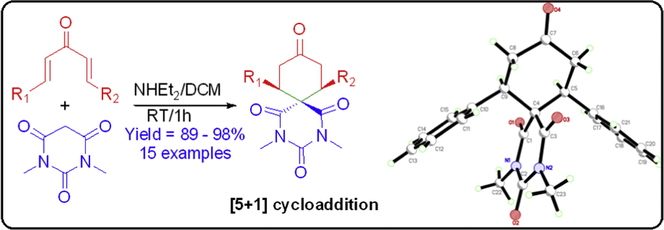
The nitrogen containing spiro-heterocycle is one of the privileged synthetic motif that constitutes various naturally occurring molecules and displays a broad range of pharmaceutical and biological activities. A new methodology was developed for the synthesis of 2,4-diazaspiro[5.5]undecane-1,3,5,9-tetraones spiro-heterocyclic derivatives via cascade cyclization of [5+1] double Michael addition reaction of N,N-dimethylbarbituric acid with the derivatives of diaryldivinylketones in the presence of diethylamine at ambient temperature. The developed protocol is highly capable of furnishing diazaspiro[5.5]undecane derivatives 3a–m in excellent yields (up to 98%), from easily accessible symmetric and non-symmetric divinylketones 2a–m, containing aryl and heteroaryl substituents. The diazaspiro-heterocyclic structure was mainly elucidated by NMR and X-ray crystallographic techniques. The single-crystal X-ray studies revealed that, the cyclohexanone unit of spirocycles often prefers a chair conformation rather than twisted conformation. The intermolecular hydrogen bonding and CAr—H⋯π, π–π stacking interactions driving forces are mainly responsible for the crystal packing.
Keywords
Cascade [5+1] cycloaddition
Double Michael reaction
Diethylamine catalysis
Diazaspiro[5.5]undecane
N,N-Dimethylbarbituric acid diaryliedene acetone

1 Introduction
Spiro-heterocycles scaffold is one of the eye-catching building blocks in organic synthesis due to their interesting conformational features as well as structural importance in biological systems (Pradhan et al., 2006). These spirocycles moiety by and large found in various biologically active natural products and clinical pharmaceuticals (Kotha et al., 2009; Bartoli et al., 2011). Beside the biological importance, these spirocycles have been extensively used for the synthesis of novel ligands, catalysts and some special organic optoelectronics synthetic materials (Saragi et al., 2007; Xie and Zhou, 2008; Ding et al., 2009). In addition, recent literature review suggests that the diazaspiro[5.5]undecane-1,3,5,9-tetraones motif has a wide range of therapeutic and biological properties such as CNS depressant (Behera et al., 2006), anticonvulsant (Rajopadhye and Popp, 1988), potent sedative-hypnotic (Kesharwani et al., 2009), antibacterial (Goel et al., 2005) and fungicidal (Behera et al., 2009). Moreover, these types of compounds can also be used as a yellow organic pigment and as a disperse dye with strong fluorescence property as it contains barbituric acid moiety (Theford et al., 2003; Karci, 2008; Wang and Kim, 2009). Due to their unique structural features, it has become one of the prominent research areas to the chemist. In order to access those versatile motifs, there is a need of synthetic strategy for the development of highly efficient methodology. So far, various literature has been documented for the synthesis of spirocycles compounds such as benzofuran spirocyclic (Li et al., 2013, 2014), Spiro-oxindole (Bencivenni et al., 2009; Chen et al., 2009; Wei and Gong, 2010; Jiang et al., 2010; Chen et al., 2011), spiro cyclohexanone rhodanines (Wu et al., 2012a,b) and spirocyclic azlactones (Weber et al., 2013). Over the last few decades, organocatalysis double Michael reaction of diversified donors with dienones has also been reported for the synthesis of several spirocyclic compounds (Xu et al., 2013; Weber et al., 2013; Fusco and Lattanzi, 2011; Wang et al., 2011; Li et al., 2011; Wu et al., 2011, 2012a,b). Moreover, double Michael addition reaction of diaryldienone with some active methylene compounds has been reported by Aggarwal et al under refluxing condition (Aggarwal et al., 2014). Michael addition reaction is one of the remarkable tool for C—C bond constructing process (Michael, 1887; Jung, 1991; Barakat et al., 2013; Islam et al., 2014; Al-Majid et al., 2014). Especially, intermolecular double-Michael reactions are the most powerful tool for the synthesis of spirocyclic product from the non-cyclic starting materials. Very few literature has been reported for the synthesis of diazaspiro[5.5]undecane derivatives, in spite of their enormous biological importance. Moreover, all the previously reported synthetic methodologies have some limitations, such as long reaction times, use of catalyst, substrate scope and high temperature. Therefore, a simple, convenient, facile and efficient methodology is required for constructing nitrogen containing spiro-heterocycles. Herein, we report, very useful, robust and environmentally benign methodologies for the synthesis of spiro-heterocyclic derivatives of diazaspiro[5.5]undecane-1,3,5,9-tetraones via double Michael reaction of 1,5-diaryl-1,4-pentadien-3-one derivatives with active methylene compound N,N-dimethylbarbituric acid in dichloromethane at room temperature, by simply using diethyl amine as base.
2 Results and discussion
In order to find out a simple, cost-effective and sustainable synthetic strategy, we initiated our research for the synthesis of diazaspiro compounds, containing N,N-dimethylbarbiturate scaffold with the help of double Michael addition reaction of diaryliedene acetones derivatives and active methylene compound N,N-dimethylbarbituric acid in dichloromethane at ambient temperature. Previously, Aggarwal group carried out this reaction using ethylene glycol as solvent at 100 °C, but it has some shortcoming, since it requires high temperature and high boiling solvent. To overcome these drawbacks, we decided to perform the reaction at room temperature using dichloromethane, which has reduced the effort for workup and purification process. Initially, we choosed dibenzylidene acetone 2a and N,N-dimethylbarbituric acid 1 as model substrate for the double Michael reaction using various solvent in basic medium. Aiming at screening the optimal parameters, the reaction was tested with the use of different mol% of diethylamine in several solvents such as dichloromethane (CH2Cl2), chloroform (CHCl3), tetrahydrofuran (THF), acetonitrile and toluene. The outcomes were summarized in Table 1. .
.
Entry
Solvent
Temperature (t)
Time (h)
Base
Base eqv.
Yieldd (%)
1b
CH2Cl2
rt
24
–
–
–
2
CH2Cl2
rt
24
NHEt2
0.1
–
3
CH2Cl2
rt
24
NHEt2
0.5
–
4c
CH2Cl2
rt
24
NHEt2
1
33
5c
CH2Cl2
rt
16
NHEt2
2
80
6
CH2Cl2
rt
2
NHEt2
2.5
98
7
CH2Cl2
40 °C
45 min
NHEt2
2.5
90
8
CHCl3
rt
2
NHEt2
2.5
94
9
ACN
rt
24
NHEt2
2.5
60
10
THF
rt
24
NHEt2
2.5
70
11b
Toluene
rt
24
NHEt2
2.5
–
12b
Toluene
80 °C
24
NHEt2
2.5
–
13c
CH2Cl2
rt
24
NEt3
2.5
65
14c
CH2Cl2
rt
24
Py
2.5
60
At the outset, the reaction was performed at room temperature in CH2Cl2 without using any base but the reaction remains unsuccessful. Then the reaction was performed in same solvent using different mol% of diethylamine and the results were shown in Table 1, entries 2–6. With the use of 10 and 50 mol% of diethylamine in DCM, no noticeable product formation was observed within 24 h (Table 1, entries 2 & 3), while with the use of 1 equiv. of diethylamine, 2,4-Dimethyl-7,11-diphenyl-2,4-diazaspiro[5.5]undecane-1,3,5,9-tetraone 3a (yield 33%) was obtained (Table 1, entries 2 & 3). In order to improve the yield of the reaction, diethylamine 2 equiv. and 2.5 equiv. were used successively for 16 h and 2 h at room temperature, which afforded good to excellent yield (80% and 98%) respectively (Table 1, entries 5c & 6). When the reaction was performed at slightly elevated temperature (40 °C), very good yield was obtained within 40–45 min reaction time (Table 1, entry 7). The reaction was further screened in different solvents such as chloroform, acetonitrile, tetrahydrofuran and toluene at room temperature. The results show that the reaction underwent in chloroform smoothly, producing excellent yield (94%) while in case of acetonitrile and tetrahydrofuran, only 70% and 75% yields were observed (Table 1, entries 8–10). However, in toluene the reaction remains unsuccessful even at 80 °C (Table 1, entries 11 & 12). Finally, the reaction was tested again, using triethylamine and pyridine as base in DCM at room temperature, yielded moderate yield (65% and 60%) respectively (Table 1, entries 13 & 14). It is obvious that the solvent CH2Cl2 remains the best choice for this double Michael reaction, as it is yielding diazaspiro compound 3a with excellent yield with the use of diethylamine at room temperature.
From the above discussion, we realized that, dichloromethane was the most practical choice for this double Michael reaction of diarylidene acetone 2a with N,N-dimethylbarbituric acid 1. In order to illustrate the generality of this of this reaction, the scope of substrates were extended up to various diarylidene acetone derivatives 2b–m under the optimized reaction parameters. Twelve examples were carried out and the results were depicted in Table 2.
#
Diarylidene 2b–m
R1/R2
3b–m
Yield (%)b
1
2b
p-CH3Ph
3b
96
2
2c
p-ClPh
3c
97
3
2d
2,6-Cl2Ph
3d
89
4
2e
2,4-Cl2Ph
3e
91
5
2f
p-BrPh
3f
93
6
2g
m-NO2Ph
3g
95
7
2h
p-CH3OPh
3h
97
8
2i
2-Naphthayl
3i
96
9
2j
2-Thiophene
3j
98
10
2k
2-Furan
3k
98
11
2l
m-BrPh
3l
95
12
2m
Ph & p-CH3Ph
3m
96
As it is evident from Table 2, that the substrates 2b–m were reacted well under the optimized parameters and the corresponding diazaspiro products 3b–m were obtained in excellent yield (89–98%) (Table 2, entries 1–12). Higher yields in case of all substrates signifying that the electron-withdrawing and electron-donating substituents at different positions on the aromatic ring do not have any remarkable influence on the reactivity of the reaction. However slightly lower yields were observed in case of substrates 2d and 2e, which could be attributed to the bulky environment of the substrate (Table 2, entries 3 & 4).
A plausible mechanism has been proposed for the double Michael addition reaction of diaryliedene acetones and N,N-dimethylbarbituric acid for the formation of diazaspiro derivatives (Fig. 1). The active methylene group of the compound 1 was activated by abstracting one of the methylene protons by diethylamine, which in turn enhance the formation of enolate form (1A). Then this enolate attacks to one of the double bonds of the diarylidene acetone 2a–m by intermolecular Michael addition reaction, leading to the formation of intermediate (3) which underwent rearrangement process and gave another intermediate (4). Again the second active methylene proton was activated by diethylamine intermediate (5) which involves second intramolecular Michael addition reaction to the another double bond in a best suitable fashion so the stable spiro products 3a–m were formed with aryl groups in the equatorial position.![A possible mechanistic pathway for [5+1] double Michael addition of N,N-dimethylbarbituric acid to diaryliedene acetones.](/content/184/2017/10/1/img/10.1016_j.arabjc.2015.03.007-fig2.png)
A possible mechanistic pathway for [5+1] double Michael addition of N,N-dimethylbarbituric acid to diaryliedene acetones.
The formation of diazaspiro product 3a was confirmed exclusively by 1H NMR as illustrated in Fig. 2. The 1H NMR spectrum of the diazaspiro compound 3a, shows a doublet of doublet at 2.59 & 2.63 with coupling values J = 15.36 Hz and 4.40 Hz, for the equatorial protons of C2 and C6 positions, while another doublet of doublet appeared at 3.99 and 4.02 with J = 14.68 Hz and 4.40 Hz due to the axial protons of C3 and C5 positions. On the other hand a triplet was appeared at 3.72 with J = 14.68 Hz for the axial protons of C2 and C6 positions (see Fig. 2).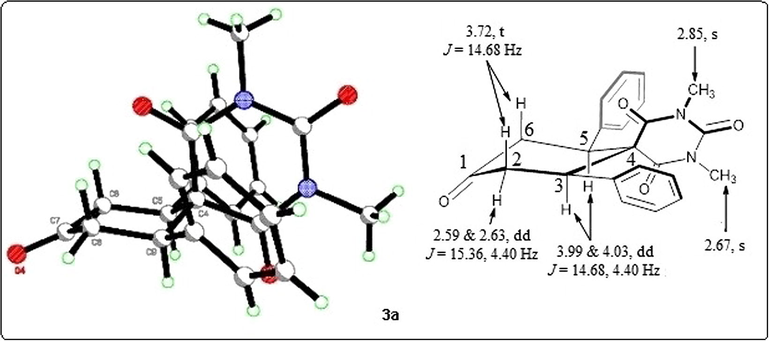
Schematic representation of the chair conformation of the spirocycles Cis-3a.
The compounds of 3a, 3h and 3i were obtained as crystals by slow diffusion of diethyl ether into the solution of pure compounds 3a, 3h and 3i in dichloromethane at room temperature and allowed to stand for 2 days. Data were collected on a Bruker APEX-II D8 Venture area diffractometer, equipped with graphite monochromatic Mo Kα radiation at 293 (2) K. Cell refinement and data reduction were carried out by Bruker SAINT. SHELXS-97 was used to solve structure (Sheldrick, 2008; Spek, 2009). The final refinement was carried out by full-matrix least-squares techniques with anisotropic thermal data for non-hydrogen atoms on F2. All the hydrogen atoms were placed in calculated positions (Table 3). The crystal structures for compounds 3a, 3h & 3i are shown in Fig. 3. a = 11.5470 (6) Å b = 14.5322 (7) Å c = 11.7043 (6) Å β = 99.3103 (15)° a = 37.494 (2) Å b = 7.8447 (4) Å c = 15.3990 (8) Å β = 95.955 (3)° a = 8.2784 (4) Å b = 12.7854 (7) Å c = 15.1885 (8) Å α = 108.445 (2)° β = 105.303 (2)° γ = 93.295 (2)°
Compound 3a
Compound 3h
Compound 3i
Empirical formula
C23H22N2O4
C25H25N2O6
C31H26N2O4·CHCl3
Formula weight
390.42
449.47
609.90
Temperature (K)
100
100
100
Wavelength (Mo Kα radiation, λ)
0.71073 Å
0.71073 Å
0.71073 Å
Crystal system
Monoclinic
Monoclinic
Triclinic
Space group
P21/n
C2/c
P1
Unit cell dimensions
Volume
1938.15(17) Å3
4504.9 (4) Å3
1453.37 (13) Å3
Z
4
8
2
Density (calculated)
1.338 Mg m−3
1.325 Mg m−3
1.394 Mg m−3
Absorption coefficient
0.09 mm−1
0.10 mm−1
μ = 0.36 mm−1
F(0 0 0)
824
1896
632
Crystal size
0.41 × 0.29 × 0.20 mm
0.59 × 0.41 × 0.35 mm
0.35 × 0.23 × 0.321 mm
Theta range for data collection
θmax = 30.6°, θmin = 2.3°
θmax = 30.6°, θmin = 2.7°
θmax = 30.6°, θmin = 2.6°
Index ranges
h = −16/16
h = −53/53
h = −11/11
k = −20/20
k = −11/11
k = −18/18
l = −16/16
l = −22/22
l = −21/21
Reflections collected/unique
107,047/5461
98,664/4506
36,460/6087
Completeness to theta = 30.57°
0.999
0.998
0.999
Absorption correction
Multi-scan
Multi-scan
Multi-scan
Goodness-of-fit on F2
1.06
1.08
1.12
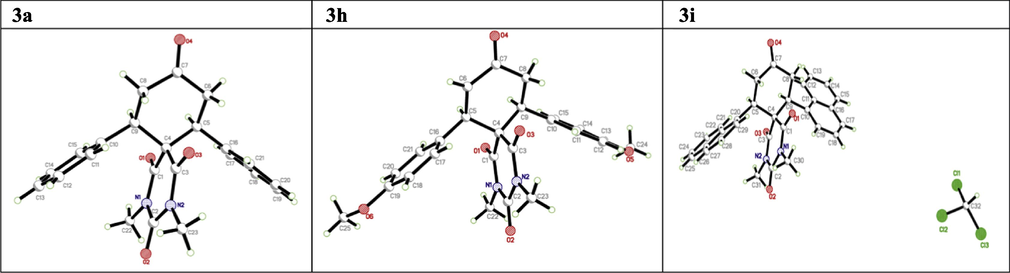
The ORTEP diagram of the final X-ray model of compound 3a, 3h and 3i with displacement ellipsoids drawn at 30% probability level. H-Atoms were placed and not included in refinement.
The structures of 3a, 3h and 3i were confirmed by X-ray crystal structure analysis (Bruker AXS GmbH). CCDC-1007513; CCDC-1004326 and CCDC-1004327 contains the supplementary crystallographic data for this compound. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
3 Conclusion
In conclusion, we developed a simple, highly efficient, cost economical process for the synthesis of diazaspiro compounds through cascade [5+1] double Michael addition reaction using diethylamine as a powerful base at room temperature. The products were isolated in its pure form with excellent yields (up to 98%). Further exploration of the substrate scope for this reaction would be investigated in our laboratory and very shortly, we will report the biological evaluations of the obtained spiro compounds, which are in progress. This methodology provides substantial improvements in the reaction time, yields as well as make ease in the workup process. We also examined the crystal structure and packing of diazaspiro compound. Further studies on expanding the application of this method and the biological evaluation of these spiro-heterocycles derivatives are in progress.
4 Experimental
4.1 General remarks
All the glassware were oven-dried before use and the reactions were conducted under an inert atmosphere. The progress of the reaction was monitored by TLC (Merck Silica Gel 60 F–254 thin layer plates). The chemicals were purchased from Aldrich, and Fluka, etc., and were used without further purification, unless otherwise stated. Petroleum ether (PE), hexane and ethyl acetate were distilled prior to use especially for column chromatography. All the major solvents were dried by using slandered drying techniques mentioned in the literature. Melting points were measured on a Gallen-kamp melting point apparatus in open glass capillaries and are uncorrected. IR Spectra were measured as KBr pellets on a Nicolet 6700 FT-IR spectrophotometer. The NMR spectra were recorded on a Jeol-400 NMR spectrometer. 1H NMR (400 MHz), and 13C NMR (100 MHz) were run in deuterated chloroform (CDCl3). Chemical shifts (δ) are referred in terms of ppm and J -coupling constants are given in Hz. Mass spectrometric analysis was conducted by using ESI mode on AGILENT Technologies 6410–triple quad LC/MS instrument. Elemental analysis was carried out on Elmer 2400 Elemental Analyzer, CHN mode.
4.2 General procedure of double Michael addition reaction for the synthesis of spiro-compounds 3a–m (GP1)
A solution of N,N-dimethylbarbituric acid (1) (2 mmol) and diarylidene acetone derivatives (2b–m) (2 mmol) in 10 mL of dry CH2Cl2 was charged into a 50 mL round bottom flask under inert atmosphere. The Et2NH (2.5 mmol) was then added to the reaction mixture and stirred at room temperature for up to 1.5–2 h, until TLC showed complete consumption of both the reactants. After the completion of reaction, the crud product directly subjected to column chromatography using 100–200 mesh silica gel and ethyl acetate/n-hexane (2:8, v/v) as an eluent to afford the pure products 3a–m. The solid products were further crystallized from a mixture of CHCl3/n-heptane.
4.2.1 2,4-Dimethyl-7,11-diphenyl-2,4-diazaspiro[5.5]undecane-1,3,5,9-tetraone (3a) (Aggarwal et al., 2014)
Diarylidene acetone 2a (468.2 mg, 2 mmol) reacted with compound 1 (312.1 mg, 2 mmol) according to GP1 yielded white solid spiro-product 3a (765 mg, 1.96 mmol, 98%); m.p. 125–127 (150–152) °C; 1H NMR (400 MHz, CDCl3) δ: 2.59 & 2.63 (dd, 2H, J = 15.36 Hz, 4.40 Hz, CH2(e)), 2.85 (s, 3H, —NCH3), 3.01 (s, 3H, —NCH3), 3.72 (t, 2H, J = 14.68 Hz, CH2(a)), 3.99 & 4.03 (dd, 2H, J = 14.68 Hz, 4.40 Hz, CH), 7.06–7.08 (m, 4H, Ar—H), 7.21–7.26 (m, 6H, Ar—H); 13C NMR (100 MHz, CDCl3) δ: 27.98, 28.39, 42.99, 50.55, 60.95, 127.56, 128.69, 128.94, 137.17, 149.70, 169.04, 170.71, 208.29; IR (KBr, cm−1) νmax = 2959, 2925, 1716, 1675, 1484, 1422, 1381, 1125, 755, 706; [Anal. Calcd. for C23H22N2O4: C, 70.75; H, 5.68; N, 7.17; Found: C, 70.69; H, 5.65; N, 7.01]; LC/MS (ESI, m/z): [M+], found 390.21, C23H22N2O4 requires 390.16.
The structure of 3a was unambiguously deduced by single-crystal X-ray diffraction structure analysis (Bruker AXS GmbH). CCDC-1007513 contains the supplementary crystallographic data for this compound. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. A colorless crystal suitable for X-ray analysis was obtained from recrystallization of the compound from DCM/Et2O at room temperature after 2 days.
4.2.2 2,4-Dimethyl-7,11-di-p-tolyl-2,4-diazaspiro[5.5]undecane-1,3,5,9-tetraone (3b) (Aggarwal et al., 2014)
Diarylidene acetone 2b (524.3 mg, 2 mmol) reacted with compound 1 (312.1 mg, 2 mmol) according to GP1 yielded white solid spiro-product 3b (803 mg, 1.92 mmol, 96%); m.p. 122–124 (159–160) °C; 1H NMR (400 MHz, CDCl3) δ: 2.25 (s, 3H, CH3), 2.55 & 2.58 (dd, 2H, J = 14.68 Hz, 4.40 Hz, CH2(e)), 2.87 (s, 3H, —NCH3), 3.01 (s, 3H, —NCH3), 3.68 (t, 2H, J = 14.68 Hz, CH2(a)), 3.94 & 3.98 (dd, 2H, J = 13.92 Hz, 4.40 Hz, CH), 6.93 (d, 4H, J = 8.04 Hz, Ar—H), 7.01 (d, 4H, J = 8.04 Hz, Ar—H); 13C NMR (100 MHz, CDCl3) δ: 21.13 27.94, 28.41, 43.15, 50.19, 61.08, 127.39, 129.58, 134.19, 138.39, 149.65, 169.29, 170.88, 208.63; IR (KBr, cm−1) νmax = 3019, 2970, 1740, 1678, 1441, 1370, 1221, 902, 672, 520; [Anal. Calcd. for C25H26N2O4: C, 71.75; H, 6.26; N, 6.69; Found: C, 71.58; H, 6.37; N, 6.81]; LC/MS (ESI, m/z): [M+], found 418.3, C25H26N2O4 requires 418.19.
4.2.3 7,11-Bis(4-chlorophenyl)-2,4-dimethyl-2,4-diazaspiro[5.5]undecane-1,3,5,9-tetraone (3c) (Ramachary et al., 2006)
Diarylidene acetone 2c (604.1 mg, 2 mmol) reacted with compound 1 (312.1 mg, 2 mmol) according to GP1 yielded white solid spiro-product 3c (889 mg, 1.92 mmol, 97%); m.p. 211–213 (224–226) °C; 1H NMR (400 MHz, CDCl3) δ: 2.55 & 2.58 (dd, 2H, J = 14.68 Hz, 4.40 Hz, CH2(e)), 2.89 (s, 3H, —NCH3), 3.04 (s, 3H, —NCH3), 3.64 (t, 2H, J = 14.68 Hz, CH2(a)), 3.95 & 3.98 (dd, 2H, J = 14.68 Hz, 4.40 Hz, CH), 6.99 (d, 4H, J = 8.80 Hz, Ar—H), 7.22 (d, 4H, J = 8.80 Hz, Ar—H); 13C NMR (100 MHz, CDCl3) δ: 28.49, 28.68, 42.94, 49.87, 60.58, 128.86, 129.01, 135.25, 135.52, 149.41, 168.75, 170.44, 207.20; IR (KBr, cm−1) νmax = 3015, 2970, 1740, 1678, 1437, 1369, 1218, 904, 672, 521; [Anal. Calcd. for C23H20Cl2N2O4: C, 60.14; H, 4.39; N, 6.10; Found: C, 59.97; H, 4.46; N, 6.19]; LC/MS (ESI, m/z): [M+], found 458.1, C23H20Cl2N2O4 requires 458.08.
4.2.4 7,11-Bis(2,6-dichlorophenyl)-2,4-dimethyl-2,4-diazaspiro[5.5]undecane-1,3,5,9-tetraone (3d)
Diarylidene acetone 2d (734 mg, 2 mmol) reacted with compound 1 (312.1 mg, 2 mmol) according to GP1 yielded white solid spiro-product 3d (936 mg, 1.78 mmol, 89%); m.p. 149–151 (3e_sh104) °C; 1H NMR (400 MHz, CDCl3) δ: 2.58 & 2.62 (dd, 2H, J = 16.12 Hz, 4.40 Hz, CH2(e)), 2.93 (s, 3H, —NCH3), 3.26 (s, 3H, —NCH3), 3.42 (t, 2H, J = 15.40 Hz, CH2(a)), 4.67 & 4.71 (dd, 2H, J = 13.92 Hz, 4.40 Hz, CH), 7.04 (d, 2H, J = 8.80 Hz, Ar—H), 7.15 & 7017 (dd, 2H, J = 8.76 Hz, 2.20 Hz, Ar—H), 7.38 (d, 2H, J = 2.20 Hz, Ar—H); 13C NMR (100 MHz, CDCl3) δ: 28.51, 29.10, 43.55, 45.20, 57.36, 128.28, 130.55, 133.99, 134.98, 149.55, 167.90, 169.91, 205.63; IR (KBr, cm−1) νmax = 3015, 2970, 2030, 1977, 1722, 1776, 1585, 1470, 1446, 1376, 1223, 1106, 1050, 528, 751, 521, 469; [Anal. Calcd. for C23H18Cl4N2O4: C, 52.30; H, 3.43; N, 5.30; Found: C, 52.41; H, 3.45; N, 5.37]; LC/MS (ESI, m/z): [M+], found 526.10.1, C23H18Cl4N2O4 requires 526.00.
4.2.5 7,11-Bis(2,4-dichlorophenyl)-2,4-dimethyl-2,4-diazaspiro[5.5]undecane-1,3,5,9-tetraone (3e)
Diarylidene acetone 2e (734 mg, 2 mmol) reacted with compound 1 (312.1 mg, 2 mmol) according to GP1 yielded white solid spiro-product 3e (957 mg, 1.82 mmol, 91%); m.p. 185–187 (3d_sh105) °C; 1H NMR (400 MHz, CDCl3) δ: 2.57 & 2.61 (dd, 2H, J = 16.12 Hz, 4.40 Hz, CH2(e)), 2.96 (s, 3H, —NCH3), 3.26 (s, 3H, —NCH3), 3.42 (t, 2H, J = 15.40 Hz, CH2(a)), 4.67 & 4.71 (dd, 2H, J = 15.40 Hz, 4.40 Hz, CH), 7.11–7.17 (m, 2H, Ar—H), 7.21 (s, 2H, Ar—H), 7.11–7.38 (m, 2H, Ar—H); 13C NMR (100 MHz, CDCl3) δ: 28.70, 29.14, 43.57, 45.24, 57.36, 127.75, 129.86, 130.50, 131.91, 133.02, 134.00, 149.55, 169.43, 169.91, 205.60; IR (KBr, cm−1) νmax = 2920, 1718, 1673, 1444, 1375, 1108, 1045, 828, 747, 465; [Anal. Calcd. for C23H18Cl4N2O4: C, 52.30; H, 3.43; N, 5.30; Found: C, 52.41; H, 3.45; N, 5.37]; LC/MS (ESI, m/z): [M+], found 526.10.1, C23H18Cl4N2O4 requires 526.00.
4.2.6 7,11-Bis(4-bromophenyl)-2,4-dimethyl-2,4-diazaspiro[5.5]undecane-1,3,5,9-tetraone (3f) (Aggarwal et al., 2014)
Diarylidene acetone 2f (780 mg, 2 mmol) reacted with compound 1 (312.1 mg, 2 mmol) according to GP1 yielded white solid spiro-product 3f (1.0 g, 1.86 mmol, 93%); m.p. 205–207 (153–154) °C; 1H NMR (400 MHz, CDCl3) δ: 2.55 & 2.58 (dd, 2H, J = 14.68 Hz, 4.40 Hz, CH2(e)), 2.90 (s, 3H, —NCH3), 3.02 (s, 3H, —NCH3), 3.59 (t, 2H, J = 14.68 Hz, CH2(a)), 3.94 & 3.97 (dd, 2H, J = 14.68 Hz, 4.40 Hz, CH), 6.92 (d, 4H, J = 8.80 Hz, Ar—H), 7.37(d, 4H, J = 8.80 Hz, Ar—H); 13C NMR (100 MHz, CDCl3) δ: 28.16, 28.59, 42.82, 49.95, 60.40, 122.90, 129.25, 129.86, 132.22, 132.42, 149.90, 169.53, 170.48, 207.15; IR (KBr, cm−1) νmax = 3020, 1712, 1675, 1640, 1415, 1376, 1284, 1177, 1067, 979, 806, 547, 443; [Anal. Calcd. for C23H20Br2N2O4: C, 50.39; H, 3.68; N, 5.11; Found: C, 50.51; H, 3.73; N, 5.17]; LC/MS (ESI, m/z): [M+], found 546.11, C23H20Br2N2O4 requires 545.98.
4.2.7 2,4-Dimethyl-7,11-bis(3-nitrophenyl)-2,4-diazaspiro[5.5]undecane-1,3,5,9-tetraone (3g)
Diarylidene acetone 2g (648 mg, 2 mmol) reacted with compound 1 (312.1 mg, 2 mmol) according to GP1 yielded white solid spiro-product 3g (912 mg, 1.9 mmol, 95%); m.p. 232–234 °C; 1H NMR (400 MHz, CDCl3) δ: 2.66 & 2.69 (dd, 2H, J = 15.40 Hz, 4.40 Hz, CH2(e)), 2.87 (s, 3H, —NCH3), 3.06 (s, 3H, —NCH3), 3.75 (t, 2H, J = 14.68 Hz, CH2(a)), 4.14 & 4.18 (dd, 2H, J = 13.92 Hz, 4.40 Hz, CH), 7.42–7.51 (m, 4H, Ar—H), 7.91(s, 2H, Ar—H), 8.13 (d, 2H, J = 8.04 Hz, Ar—H); 13C NMR (100 MHz, CDCl3) δ: 28.36, 28.71, 42.63, 49.98, 60.09, 122.40, 124.01, 130.28, 133.95, 138.95, 148.64, 168.26, 169.84, 205.46; IR (KBr, cm−1) νmax = 2953, 1715, 1673, 1527, 1420, 1381, 901, 808, 731, 681, 451; [Anal. Calcd. for C23H20N4O8: C, 57.50; H, 4.20; N, 11.66: Found: C, 57.56; H, 4.32; N, 11.43]; LC/MS (ESI, m/z): [M+], found 480.03, C23H20N4O8 requires 480.13.
4.2.8 7,11-Bis(4-methoxyphenyl)-2,4-dimethyl-2,4-diazaspiro[5.5]undecane-1,3,5,9-tetraone (3h)
Diarylidene acetone 2h (588 mg, 2 mmol) reacted with compound 1 (312.1 mg, 2 mmol) according to GP1 yielded white solid spiro-product 3h (873 mg, 1.94 mmol, 97%); m.p. 131–133 °C; 1H NMR (400 MHz, CDCl3) δ: 2.53 & 2.56 (dd, 2H, J = 14.68 Hz, 4.40 Hz, CH2(e)), 2.88 (s, 3H, —NCH3), 3.01 (s, 3H, —NCH3), 3.68 (t, 2H, J = 14.68 Hz, CH2(a)), 3.72 (s, 3H, OCH3), 3.91 & 3.94 (dd, 2H, J = 13.92 Hz, 4.40 Hz, CH), 6.73 (d, 4H, J = 8.80 Hz, Ar—H), 6.96 (d, 4H, J = 8.80 Hz, Ar—H); 13C NMR (100 MHz, CDCl3) δ: 28.01, 28.45, 43.29, 49.73, 55.26, 61.39, 114.17, 128.64, 129.21, 149.98, 159.51, 169.25, 171.79, 208.54; IR (KBr, cm−1) νmax = 2957, 2838, 1713, 1670, 1609, 1510, 1449, 1420, 1248, 1031, 831, 729, 530, 452; [Anal. Calcd. for C25H26N2O6: C, 66.65; H, 5.82; N, 6.22; Found: C, 66.81; H, 5.71; N, 6.34]; LC/MS (ESI, m/z): [M+], found 450.15, C25H26N2O6 requires 450.18.
The structure of 3a was unambiguously deduced by single-crystal X-ray diffraction structure analysis (Bruker AXS GmbH). CCDC-1004326 contains the supplementary crystallographic data for this compound. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. A colorless crystal suitable for X-ray analysis was obtained from recrystallization of the compound from DCM/Et2O at room temperature after 2 days.
4.2.9 2,4-Dimethyl-7,11-di(naphthalen-1-yl)-2,4-diazaspiro[5.5]undecane-1,3,5,9-tetraone (3i)
Diarylidene acetone 2i (668 mg, 2 mmol) reacted with compound 1 (312.1 mg, 2 mmol) according to GP1 yielded white solid spiro-product 3i (941 mg, 1.92 mmol, 96%); m.p. 218–220 °C; 1H NMR (400 MHz, CDCl3) δ: 1.98 (s, 3H, —NCH3), 2.75 & 2.79 (dd, 2H, J = 15.4 Hz, 4.40 Hz, CH2(e)), 3.22 (s, 3H, —NCH3), 3.89 (t, 2H, J = 15.04 Hz, CH2(a)), 5.11 & 5.14 (dd, 2H, J = 13.92 Hz, 4.40 Hz, CH), 7.33–7.39 (m, 4H, Ar—H), 7.47 (t, 2H, J = 8.04 Hz, Ar—H), 7.57 (t, 2H, J = 8.04 Hz, Ar—H), 7.73 & 7.74 (dd, 2H, J = 7.56 Hz, 2.40 Hz, Ar—H), 7.80 (d, 2H, J = 8.08 Hz, Ar—H), 8.22 (d, 2H, J = 8.80 Hz, Ar—H); 13C NMR (100 MHz, CDCl3) δ: 28.03, 28.32, 44.37, 44.86, 59.34, 123.16, 124.20, 124.95, 126.19, 126.74, 128.94, 129.23, 130.91, 134.05, 134.22, 149.61, 169.47, 170.38, 208.33; IR (KBr, cm−1) νmax = 3049, 2919, 1711, 1666, 1421, 1374, 1266, 1241, 1018, 773, 467; [Anal. Calcd. for C31H26N2O4: C, 75.90; H, 5.34; N, 5.71; Found: C, 76.13; H, 5.41; N, 5.83]; LC/MS (ESI, m/z): [M+], found 490.23, C31H26N2O4 requires 490.19.
The structure of 3i was unambiguously deduced by single-crystal X-ray diffraction structure analysis (Bruker AXS GmbH). CCDC-1004327 contains the supplementary crystallographic data for this compound. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. A colorless crystal suitable for X-ray analysis was obtained from recrystallization of the compound from DCM/Et2O at room temperature after 2 days.
4.2.10 2,4-Dimethyl-7,11-di(thiophen-2-yl)-2,4-diazaspiro[5.5]undecane-1,3,5,9-tetraone (3j)
Diarylidene acetone 2j (520 mg, 2 mmol) reacted with compound 1 (312.1 mg, 2 mmol) according to GP1 yielded white solid spiro-product 3j (788 mg, 1.96 mmol, 98%); m.p. 136–138 °C; 1H NMR (400 MHz, CDCl3) δ: 2.80 & 2.83 (dd, 2H, J = 15.4 Hz, 4.40 Hz, CH2(e)), 3.11 (s, 3H, —NCH3), 3.14 (s, 3H, —NCH3), 3.66 (t, 2H, J = 14.64 Hz, CH2(a)), 4.34 & 4.37 (dd, 2H, J = 13.92 Hz, 4.40 Hz, CH), 6.84 (d, 2H, J = 3.72 Hz, Ar—H), 6.93–6.95 (m, 2H, Ar—H), 7.23 (d, 2H, J = 5.12 Hz, Ar—H); 13C NMR (100 MHz, CDCl3) δ: 28.19, 28.61, 44.16, 45.44, 61.50, 125.48, 125.87, 126.96, 139.84, 149.83, 168.60, 170.90, 205.77; IR (KBr, cm−1) νmax = 2959, 2921, 1715, 1668, 1420, 1373, 1260, 1036, 799, 702, 501, 444; [Anal. Calcd. for C19H18N2O4S2: C, 56.70; H, 4.51; N, 6.96; Found: C, 56.76; H, 4.43; N, 7.03]; LC/MS (ESI, m/z): [M+], found 402.11, C19H18N2O4S2 requires 402.07.
4.2.11 7,11-Di(furan-2-yl)-2,4-dimethyl-2,4-diazaspiro[5.5]undecane-1,3,5,9-tetraone (3k)
Diarylidene acetone 2k (456 mg, 2 mmol) reacted with compound 1 (312.1 mg, 2 mmol) according to GP1 yielded white solid spiro-product 3k (725 mg, 1.96 mmol, 98%); m.p. 114–116 °C; 1H NMR (400 MHz, CDCl3) δ: 2.67 & 2.71 (dd, 2H, J = 15.40 Hz, 4.40 Hz, CH2(e)), 3.08 (s, 3H, —NCH3), 3.10 (s, 3H, —NCH3), 3.49 (t, 2H, J = 14.68 Hz, CH2(a)), 4.08 & 4.11 (dd, 2H, J = 13.96 Hz, 4.40 Hz, CH), 6.05 (d, 2H, J = 3.64 Hz, Ar—H), 6.25–6.26 (m, 2H, Ar—H), 7.23 (d, 2H, J = 1.40 Hz, Ar—H); 13C NMR (100 MHz, CDCl3) δ: 28.18, 28.80, 40.90, 43.32, 57.09, 107.39, 110.56, 142.56, 151.20, 151.32, 168.01, 170.75, 206.10; IR (KBr, cm−1) νmax = 3115, 1959, 1721, 1670, 1446, 1420, 1377, 1011, 922, 738, 465; [Anal. Calcd. for C19H18N2O6: C, 61.62; H, 4.90; N, 7.56; Found: C, 61.49; H, 5.11; N, 7.43]; LC/MS (ESI, m/z): [M+], found 370.18, C19H18N2O6 requires 370.12.
4.2.12 7,11-Bis(3-bromophenyl)-2,4-dimethyl-2,4-diazaspiro[5.5]undecane-1,3,5,9-tetraone (3l)
Diarylidene acetone 2l (780 mg, 2 mmol) reacted with compound 1 (312.1 mg, 2 mmol) according to GP1 yielded white solid spiro-product 3l (1037 mg, 1.90 mmol, 95%); m.p. 118–120 °C; 1H NMR (400 MHz, CDCl3) δ: 2.57 & 2.61 (dd, 2H, J = 14.68 Hz, 4.40 Hz, CH2(e)), 2.91 (s, 3H, —NCH3), 3.06 (s, 3H, —NCH3), 3.64 (t, 2H, J = 14.68 Hz, CH2(a)), 3.92 & 3.96 (dd, 2H, J = 14.68 Hz, 4.40 Hz, CH), 6.98 (d, 2H, J = 8.08 Hz, Ar—H), 7.11 (t, 2H, J = 8.08 Hz, Ar—H), 7.22, (s, 2H, Ar—H), 7.37 (d, 2H, J = 7.36 Hz, Ar—H); 13C NMR (100 MHz, CDCl3) δ: 28.09, 28.57, 42.67, 50.01, 60.52, 123.11, 126.25, 130.53, 130.69, 131.97, 139.22, 149.44, 168.66, 170.29, 206.89; IR (KBr, cm−1) νmax = 2959, 2921, 1710, 1667, 1423, 1349, 1285, 1256, 1070, 787, 749, 695, 443; [Anal. Calcd. for C23H20Br2N2O4: C, 50.39; H, 3.68; N, 5.11; Found: C, 50.51; H, 3.73; N, 5.17]; LC/MS (ESI, m/z): [M+], found 546.11, C23H20Br2N2O4 requires 545.98.
4.2.13 2,4-Dimethyl-7-phenyl-11-(p-tolyl)-2,4-diazaspiro[5.5]undecane-1,3,5,9-tetraone (3m)
Diarylidene acetone 2m (496 mg, 2 mmol) reacted with compound 1 (312 mg, 2 mmol) according to GP1 yielded white solid spiro-product 3m (776 mg, 1.92 mmol, 96%); m.p. 110–112 °C; 1H NMR (400 MHz, CDCl3) δ: 2.25 (s, 3H, CH3), 2.55 & 2.59 (dd, 2H, J = 14.68 Hz, 4.40 Hz, CH2(e)), 3.87 (s, 3H, —NCH3), 3.01 (s, 3H, —NCH3), 3.72 (t, 2H, J = 14.68 Hz, CH2(a)), 3.94 & 3.98 (dd, 2H, J = 14.68 Hz, 4.40 Hz, CH), 6.92 (d, 4H, J = 8.08 Hz, Ar—H), 7.01 (d, 4H, J = 8.08 Hz, Ar—H), 7.21–7.25 (m, 1H, Ar—H); 13C NMR (100 MHz, CDCl3) δ: 21.12, 27.97, 28.40, 44.17, 43.15, 50.19, 61.8, 127.39, 128.92, 129.57, 134.18, 138.39, 149.98, 168.18, 170.89, 208.66; IR (KBr, cm−1) νmax = 29.57, 2924, 1717, 1672, 1446, 1419, 1377, 1285, 814, 730, 560, 509; [Anal. Calcd. for C24H24N2O4: C, 71.27; H, 5.98; N, 6.93; Found: C, 71.36; H, 6.07; N, 7.01]; LC/MS (ESI, m/z): [M+], found 404.11, C24H24N2O4 requires 404.17.
Acknowledgment
The authors would like to extend their sincere appreciation to the Deanship of Scientific Research at King Saud University for its funding this Research group no. (RG -044-1435-1436).
References
- An efficient catalyst free synthesis of nitrogen containing spiro heterocycles via [5+1] double Michael addition reaction. RSC Adv.. 2014;4:13313-13321.
- [Google Scholar]
- Facile and promising method for Michael addition of indole and pyrrole to electron-deficient trans-β-nitroolefins catalyzed by a hydrogen bond donor catalyst Feist’s acid and preliminary study of antimicrobial activity. Sci. World J. 2014
- [CrossRef] [Google Scholar]
- Highly enantioselective FriedeleCrafts alkylation of indoles with α, β-unsaturated ketones with simple Cu(II)eoxazolineeimidazoline catalysts. Tetrahedron. 2013;69:5185-5192.
- [Google Scholar]
- Construction of spirolactones with concomitant formation of the fused quaternary centre – application to the synthesis of natural products. Nat. Prod. Rep.. 2011;28:763-782.
- [Google Scholar]
- Synth. Commun. Studies on spiroheterocycles Part-II: heterocyclisation of the spiro compounds containing cyclohexanone and thiobarbituric acid with different bidentate nucleophilic reagents.. 2006;36:3729-3742.
- [Google Scholar]
- Studies on spiroheterocycles part-III: synthesis of diazaspiroundecanetetraone derivatives containing biological active heterocycles. Phosphorus Sulfur Silicon Relat. Elem.. 2009;184:753-765.
- [Google Scholar]
- Targeting structural and stereochemical complexity by organocascade catalysis: construction of spirocyclic oxindoles having multiple stereocenters. Angew. Chem., Int. Ed.. 2009;2009(48):7200-7203.
- [Google Scholar]
- Organocatalytic direct asymmetric aldol reactions of 3-isothiocyanato oxindoles to ketones: stereocontrolled synthesis of spirooxindoles bearing highly congested contiguous tetrasubstituted stereocenters. Org. Lett.. 2011;13:2472-2475.
- [Google Scholar]
- Organocatalytic synthesis of spiro[pyrrolidin-3,3′-oxindoles] with high enantiopurity and structural diversity. J. Am. Chem. Soc.. 2009;131:13819-13825.
- [Google Scholar]
- Spiro skeletons: a class of privileged structure for chiral ligand design. Chem.-Asian J.. 2009;4:32-41.
- [Google Scholar]
- Quinine-catalysed double michael addition of malononitrile to 1,5-disubstituted pentadien-3-ones: a stereoselective route to cyclohexanones. Eur. J. Org. Chem. 2011:3728-3731.
- [Google Scholar]
- Synthesis and CNS depressant of newer spirobarbiturates. Indian J. Pharm. Sci.. 2005;67:194-199.
- [Google Scholar]
- Highly enantioselective Friedel-crafts alkylation of indole with electron deficient trans-b-nitroalkenes using Zn(II)–oxazoline–imidazoline catalysts. Tetrahedron: Asymmetry. 2014;25:245-251.
- [Google Scholar]
- A unique approach to the concise synthesis of highly optically active spirooxazolines and the discovery of a more potent oxindole-type phytoalexin analogue. J. Am. Chem. Soc.. 2010;132:15328-15333.
- [Google Scholar]
- Trost B.M., Fleming I., Semmelhack M.F., eds. Comprehensive Organic Synthesis. Vol vol. 4. Pergamon: Oxford; 1991. p. :1-68.
- The synthesis and solvatochromic properties of some novel heterocyclic disazo dyes derived from barbituric acid. Dyes Pigm.. 2008;77:451-456.
- [Google Scholar]
- Synthesis and biological evaluation of some new spiro derivatives of barbituric acid. Pharm. Chem. J.. 2009;43:315-319.
- [Google Scholar]
- Asymmetric organocatalytic double-conjugate addition of malononitrile to dienones: efficient synthesis of optically active cyclohexanones. Org. Lett.. 2011;13:374-377.
- [Google Scholar]
- Synthesis of optically enriched spirocyclic benzofuran-2-ones by bifunctional thiourea-base catalyzed double-Michael addition of benzofuran-2-ones to dienones. Chem. Asian J.. 2013;8:997-1003.
- [Google Scholar]
- Catalytic asymmetric synthesis of chiral benzofuranones. Adv. Synth. Catal.. 2014;356(6):1172-1198.
- [Google Scholar]
- Ueber die Addition von natriumacetessig- und natriummalonsäureäthern zu den aethern ungesättigter säuren. Prakt. Chem.. 1887;35:349-356.
- [Google Scholar]
- Potential anticonvulsants. 11. Synthesis and anticonvulsant activity of spiro[1,3-dioxolane-2,3′-indolin]-2′-ones and structural analogs. J. Med. Chem.. 1988;31:1001-1005.
- [Google Scholar]
- Development of drug intermediates by using direct organocatalytic multi-component reactions. Org. Biomol. Chem.. 2006;4:1641-1646.
- [Google Scholar]
- Synthesis and properties of some polycyclic barbiturate pigments. Dyes Pigm.. 2003;59:185-191.
- [Google Scholar]
- A highly organocatalytic stereoselective double Michael reaction: efficient construction of optically enriched spirocyclic oxindoles. Chem. Commun.. 2011;47:5593-5595.
- [Google Scholar]
- New solvatochromic merocyanine dyes based on Barbituric acid and Meldrum’s acid. Dyes Pigm.. 2009;80:314-320.
- [Google Scholar]
- Catalytic asymmetric synthesis of spirocyclic azlactones by a double Michael-addition approach. Chem. Eur. J.. 2013;19:8342-8351.
- [Google Scholar]
- Organocatalytic asymmetric formal [4 + 2] cycloaddition for the synthesis of spiro[4-cyclohexanone-1,3′-oxindoline] derivatives in high optical purity. Org. Lett.. 2010;12:1008-1011.
- [Google Scholar]
- Highly enantioselective synthesis of spiro[cyclohexanone-oxindoles] and spiro[cyclohexanone-pyrazolones] by asymmetric cascade [5+1] double Michael reactions. Eur. J. Org. Chem. 2012:1318-1327.
- [Google Scholar]
- Efficient synthesis of optically active 4-nitro-cyclohexanones via bifunctional thiourea-base catalyzed double-Michael addition of nitromethane to dienones. Chem. Commun.. 2011;47:3992-3994.
- [Google Scholar]
- Asymmetric construction of spirocyclohexanonerhodanines catalyzed by simple diamine derived from chiral tert-leucine. Chem. Commun.. 2012;48:9180-9182.
- [Google Scholar]
- Chiral diphosphine and monodentate phosphorus ligands on a spiro scaffold for transition-metal-catalyzed asymmetric reactions. Acc. Chem. Res.. 2008;41:581-593.
- [Google Scholar]
- A simple and highly efficient procedure for construction of quaternary carbons centers by tributylphosphine catalyzed bis-Michael addition. Tetrahedron. 2013;70:176-180.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.arabjc.2015.03.007.
Appendix A
Supplementary material
Supplementary data 1
Supplementary data 1




