

Translate this page into:
Structure and activity of new degraded products of limonoid from root bark of Dictamnus dasycarpus, and insights from broadened NMR spectra into self-aggregation of hydroxy acids
⁎Corresponding authors. wangyanan@imm.ac.cn (Yanan Wang), sujuanwang@imm.ac.cn (Sujuan Wang)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
New degraded products of limonoid were isolated from the root bark of Dictamnus dasycarpus. The A/B ring degraded biogenetic pathway of limonoid was firstly hypothesized. Broadened NMR spectra and self-aggregation of hydroxyl acids were studied. Compounds 1–3 significantly inhibited OA-induced lipid accumulation in HepG2 cells.
Abstract
Five degraded products of limonoid, including two previously undescribed compounds (1, 3) and one pair of new natural products (4i, 4ii), were isolated from the root bark of Dictamnus dasycarpus Turcz. Their structures were elucidated by extensively spectroscopic methods (NMR, MS, ECD, etc), as well as quantum chemical calculations. The broadening NMR lines, along with the relaxation time and DOSY experiment revealed the self-aggregation of 1 in chloroform through H-bond between –COOH and –OH, which provided additional evidence to distinguish acid and ester/lactone in structure elucidation. Compounds 1–3 attenuated oleic acid-induced lipid accumulation in HepG2 cells significantly, with inhibition rates of 19.8%, 23.6%, and 13.5% at 10 μM, much stronger than fenofibrate, affording a novel scaffold for drug discovery against fatty liver disease.
Keywords
Dictamnus dasycarpus Turcz
Degraded limonoid
Self-aggregation
Fatty liver disease

1 Introduction
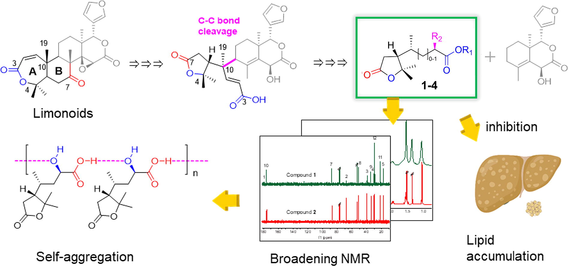
Root bark of Dictamnus dasycarpus Turcz., called Baixianpi in China, has long been used in traditional Chinese medicine for the treatment of skin inflammation, eczema, dermatoses, psoriasis, pruritus, scabies, and other diseases (Lv et al., 2015). Previous biological studies have revealed its widely biological activities, such as anticancer, anti-inflammation, and neuroprotection (Lv et al., 2015). In phytochemistry, fraxinellone-type and obacunone-type limonoids are considered to be characteristic components of Dictamnus species (Tan et al., 2011; Lv et al., 2015; Luo et al., 2022). Although fraxinellone derivatives were speculated to be degraded from obacunone by losing the A/B ring. The A/B-ring degraded products and the possible degraded biogenetic pathway have not yet been reported. As part of our ongoing research to find more bioactive constituents from D. dasycarpus Turcz. (Tian et al., 2021; Zhang et al., 2022), this paper described the discovery of four A/B-ring degradated products of limonoid (1, 3, 4i, 4ii in Fig. 1), their chemical structures, as well as their putative degraded biogenetic pathway.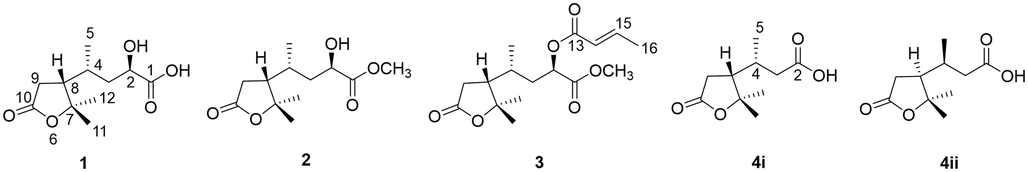
The chemical structures of compounds 1–4ii.
Interestingly, compound 1 exhibited broadening 1H NMR lines, along with almost disappearing 1-COOH and 2-CHOH carbon lines, while its methyl ester, compound 2, did not. The relaxation time in different concentrations and solvents revealed the self-aggregation due to H-bond formation between intermolecular 1-COOH and 2-OH in chloroform, which was confirmed by the diffusion ordered spectroscopy (DOSY) experiment. Once the carboxylic acid was esterified, the self-aggregation disappeared. This intermolecular self-aggregation could be widely observed in other hydroxy acid derivatives, providing more evidence to distinguish acid and ester/lactone in structure elucidation, besides the MS technique.
In addition, compounds 1–3 exhibited significant inhibitory activities against oleic acid (OA)-induced lipid accumulation in HepG2 cells at 10 μM, more outperforming than positive fenofibrate at 20 μM. Given no approved drug available for the treatment of fatty liver disease (FLD), these compounds afford a promising scaffold for anti-FLD drug development.
2 Materials and methods
2.1 General experimental procedures
Optical rotations were measured on a P-2000 automatic digital polarimeter (JASCO, Tokyo, Japan). UV and ECD spectra were collected on a J-815 spectropolarimeter (JASCO, Tokyo, Japan). IR spectra were recorded on a Nicolet IS 50 FTIR ATR microspectrometer (Thermo, MA, USA). NMR spectra were acquired using Avance III HD 400, 600, or Avance NEO 700 spectrometers (Bruker, Rheinstetten, Germany). Chemical shifts were given in δ (ppm) and are referenced to TMS. HRESIMS data were recorded on a Q Exactive Focus orbitrap mass spectrometer (Thermo, MA, USA) or an Agilent 6520 Accurate-Mass Q-TOF LC/MS mass spectrometer. Open column chromatography was performed using silica gel (60–100 mesh or TLC grade, Qingdao Marine Chemical Co., Qingdao, China), MCI gel (CHP20P, Mitsubishi Chemical Industries Ltd., Japan) or Sephadex LH-20 (GE, Uppsala, Sweden) as the stationary phase. Flash columns packed with C18 (20–35 μm, Agela, Tianjin, China) were used for separation. Preparative HPLC was performed on a FLEXA HP50 instrument (Agela, Tianjin, China) with an RID detector 2300 (Knauer, Germany), using HPLC columns: Capcell Pak MG-II C18/PFP (10 × 250 mm, 5 µm, Shiseido, Tokyo, Japan) or YMC-Pack ODS-A (20 × 250 mm, 5 µm, YMC, Japan). TLC analyses were carried out on silica gel GF254. The spots were visualized under UV light or by spraying with 10% H2SO4 in 95% EtOH followed by heating. All solvents were analytical grade or chromatographic grade (Tongguang Chemical Plant, Beijing, China).
2.2 Plant material
The root bark of Dictamnus dasycarpus Turcz. was collected from Mudanjiang City, Heilongjiang Province, China, in January 2019. A voucher specimen (ID-S-2933) was identified by Professor Lin Ma and deposited at the Institute of Materia Medica, Chinese Academy of Medical Science and Peking Union Medical College, China.
2.3 Extraction and isolation
The root bark of D. dasycarpus Turcz (50 kg) was extracted with boiling 95% EtOH (300 L × 3) and water (300 L × 3). The extract (14 kg) was dissolved in 40 L of water and partitioned in an EtOAc-H2O mixture 3 times.
The EtOAc extract (1.01 kg) was subjected to a silica gel column and eluted with CH2Cl2-MeOH (99:1–1:1) to obtain seven fractions (Fr. G1–7). Fr. G4 (320 g) was then subjected to a silica gel column and eluted with petroleum ether (PE)-acetone (50:1–5:1) to yield 9 fractions (Fr. B1–9).
Then, Fr. B2 (9.2 g) was subjected to RP-C18 flash column and eluted with MeOH-H2O (10–90%) to obtain 16 fractions (Fr. B2-1–16). Fr. B3 (8.2 g) was subjected to RP-C18 flash column and eluted with MeOH-H2O (50–90%) to obtain 17 fractions (Fr. B3-1–17).
After being merged, Fr. B2-3 and Fr. B3-1 were subjected to Sephadex LH-20 and eluted with CH2Cl2-MeOH (2:1) to yield two fractions (Fr. L1-1–2). Fr. L1-1 was subjected to RP-C18 flash column and then separated by preparative HPLC (YMC, 45% MeOH, 7 mL/min) to yield compound 1 (13.7 mg, 18.0 min). The 2nd fraction was then separated by semi-preparative HPLC (PFP, 45% MeOH, 3 mL/min) to yield compound 2 (117.0 mg, 30.0 min).
The H2O layer (10 kg) was loaded onto a microporous resin column and eluted with a stepwise gradient of EtOH-H2O (0–95%) to produce four fractions (Fr. D1–4). Fr. D2 (1.6 kg) of 4 fractions was then subjected to MCI gel column eluted with EtOH-H2O (0–85%) to yield four fractions (Fr. E1–4). Fr. D3 (248 g) was loaded onto MCI gel column with a gradient system of EtOH-H2O (0–100%) to gain eight fractions (Fr. M1–8).
Fr. M3-4, together with Fr. E3, were separated over silica gel column and eluted with CH2Cl2-MeOH (100:1–1:1) to afford Fr. H1–49. Fr. H6 was separated over silica gel with a gradient system of PE-acetone (5:1–2:1) to afford Fr. B1–5. The obtained Fr. B3 was subjected to a MG-II C18 HPLC column (50% MeOH-H2O, 3 mL/min) to afford 4 (3.2 mg, tR = 14.0 min), which was further resolved by CHIRALPAK IG column (4.6 × 250 mm, 5 μm) with n-hexane-i-PrOH (82:18) at 0.4 mL/min to afford 4i (0.2 mg, tR = 21.0 min) and 4ii (0.2 mg, tR = 25.0 min). Fr. H7 was separated over silica gel with a gradient system of PE-acetone (5:1–2:1) to afford Fr. C1–8. The obtained Fr. C3 was subjected to a Capcell Pak MG-II C18 HPLC column (65% MeOH-H2O, 3 mL/min) to afford 3 (12.0 mg, tR = 11.2 min).
2.3.1 (1) (2R,4R,8R)-dasycarpusacid
Colorless oil.
66.3 (c 0.08, MeOH); UV (MeOH) λmax (log ε) 206 (3.05) nm; ECD (MeOH) λmax (Δε) 210 (-1.7) nm; IR νmax 2978, 2934, 2877, 1742, 1422, 1393 cm−1; 1H NMR and 13C NMR data see Table 1; (+)-HRESIMS m/z 253.1042 [M + Na]+ (calcd for C11H18O5Na+, 253.1046). Measured at a600 or c700 MHz for 1H; b126 or d176 MHz for 13C. e,fOverlapped with each other. * The coupling constants were obtained from the 1H NMR spectrum at 0.5 mg/ml.
NO.
1
3
4
1Ha*
13Cb
1Hc
13Cd
1Hc
13Cd
1
178.3
170.8
2
4.32 dd (10.8, 2.8)
67.6
5.14 dd (11.3, 2.5)
69.3
177.3
3a
1.65 td (11.5, 2.9)
39.5
1.92 e
36.9
2.42 f
40.3
3b
1.59 m
1.61 ddd (13.6, 10.7, 2.6)
2.22 m
4
1.95 m
30.3
1.72 dqt (13.6, 6.7, 3.4)
30.6
2.11 m
31.2
5
1.08 d (6.3)
17.6
1.02 d (6.5)
17.9
1.10 d (6.2)
19.3
7
87.9
86.8
86.9
8
2.09 m
51.4
2.07 ddd (11.5, 9.5, 8.2)
51.3
2.16 m
50.8
9α
2.62 dd (17.4, 8.4)
34.7
2.65 dd (17.4, 8.2)
34.5
2.64 dd (17.2, 7.8)
34.6
9β
2.37 dd (17.4, 11.7)
2.35 dd (17.4, 11.6)
2.42 f
10
176.3
174.7
174.7
11
1.36 s
21.7
1.33 s
21.6
1.36 s
21.6
12
1.53 s
29.4
1.51 s
29.4
1.54 s
29.3
13
165.8
14
5.93 d (15.6)
146.9
15
7.06 dq (15.6, 6.9)
121.5
16
1.92 e
18.2
1-OCH3
3.75 s
52.6
2.3.2 (3) (2R,4R,8R)-2-O-crotonyldasycarpusester B
Colorless oil. 31.0 (c 0.20, MeOH); UV (MeOH) λmax (log ε) 209.5 (4.25) nm; ECD (MeOH) λmax (Δε) 217 (-5.6) nm; IR νmax 3501, 2976, 1761, 1724 cm−1. 1H NMR and 13C NMR data see Table 1. (+)-HRESIMS m/z 313.1641 [M + H]+ (calcd for C16H24O6H+, 313.1646).
2.3.3 (4) nordasycarpusacid
Colorless oil. UV (MeOH) λmax (log ε) 203 (3.56) nm; IR νmax 2979, 2937, 2878, 1743, 1418, 1394 cm−1; 1H NMR and 13C NMR data see Table 1; (+)-HRESIMS m/z 201.1126 [M + H]+ (calcd for C10H16O4H+, 201.1121). 4R,8R-nordasycarpusacid (4i): ECD (MeOH) λmax (Δε) 214 (‒0.2) nm; 4S,8S-nordasycarpusacid (4ii): ECD (MeOH) λmax (Δε) 213 (0.6) nm.
2.4 DOSY and relax time determination
The NMR spectra were recorded at 298 K with a Bruker Avance III HD 600 spectrometer operating at 600.15 MHz for 1H and 150.91 MHz for 13C, equipped with a 5 mm CPDCH He cooled gradient probehead (Bruker Biospin, Karlsruhe, Germany) optimized for 1H and 13C observation. All samples were stabilized at 298 K for 5 min before data collection. Spectra were processed with TopSpin 3.5pl7 Bruker’s software and were analyzed using the MestRenova 14.3.2 program (Mestrelab Research S. L., Spain).
DOSY spectra were acquired with the ledbpg2s pulse sequence, a spectral width of 13.7026 ppm, transmitter frequency offset of 3093.80 Hz, and matrices of 65,536 (F2) by 128 points (F1) were collected. The z-axis gradient strength varied linearly from 2% to 95% of its maximum value (0.66 T/m), the delay for gradient recovery was 0.5 ms, the diffusion time (big DELTA) was 60 ms, the eddy current delay was 5 ms, the relaxation delay (D1) was 1 s, 8 for number of scan, 0.695 ms for the length of the gradient pulse (little DELTA*0.5) optimized by ledbpgp2s1d NMR experiments.
The proton T1 NMR spectra were acquired with the t1ir pulse sequence, a spectral width of 20.0269 ppm, transmitter frequency offset of 4801.20 Hz and matrices of 16,384 (F2) by 15 points (F1) were collected. A list of 15 recovery delays (0.001 s ∼ 20 s) was used for the determination of T1. The relaxation delay (D1) was set to 21 s.
The proton T2 was measured with a CPMG sequence on the NMR device. There were 12 variable counters used for the determination of T2. The relaxation delay (D1) was set to 35 s, and the fixed echo time which allowed elimination of diffusion was set to 1 ms.
2.5 DFT calculation
Conformational search (<6 kcal/mol) was performed by CREST program (Pracht et al., 2020) using GFN2-xTB. Quantum chemistry was carried out by Gaussian 16C.01 (Gaussian Inc., Wallingford CT, USA) and ORCA 5.02 program (Neese et al., 2020) according to the multi-step conformer filtering procedure described previously (Li et al., 2023). The E0 was calculated by r2SCAN-3c. The ΔGcorr was generated by Shermo 2.3 program (Lu et al., 2021) after optimization and frequency calculation at B3LYP-D3(BJ)/TZVP level with IEFPCM solvent model. The E1 was calculated by ωB97M-V/def2-TZVP level with RIJCOSX approximation. The free energies ΔG1 were calculated using a Python script modified in-house. The NMR was calculated using Giao method at B972/pcSseg-2 level and weighted by a modified Python script (Willoughby et al., 2014). The DP4+ parameters [μ, σ, ν] were estimated as reported (Zanardi et al., 2021). The ECD was calculated using TDDFT method at B3LYP/def2-TZVP level with the overall theoretical UV and ECD curves Boltzmann-averaged by SpecDis 1.71 program (Bruhn et al., 2013).
2.6 ECD measurement of the molybdenum complex of compound 1
Compound 1 (0.5 mg) and Mo2(OAc)4 (1.0 mg) were dissolved in 1.0 mL anhydrous DMSO. The ECD spectrum of the obtained solution was measured immediately under wavelengths 250–500 nm. After 60 min, the stationary complex was formed, and the ECD spectrum of the solution was measured again and used to subtract the first ECD spectrum to obtain the Mo2(OAc)4 induced ECD spectrum of the compound.
2.7 Inhibitory assay against OA-induced lipid accumulation in HepG2 cells
After being exposed to different samples at 10 μM for 24 h, the cell viability was evaluated by MTT assay (Yan et al., 2009; Li et al., 2014) to ensure that they were not cytotoxic to HepG2 cells. To evaluate the effect of samples on lipid accumulation in HepG2 cells, the cells were incubated with compounds 1–3 or fenofibrate in the presence of 0.24 mM OA for 24 h. After being fixed with 4% Paraformaldehyde, the cells were stained with oil red O for 1 h in dark. Finally, the OD values were measured at the wavelength of 532 nm. The inhibition rates were calculated as (1 − ODsample/ODmodel) × 100%.
3 Results and discussion
3.1 Structure elucidation
Two previously undescribed compounds (1, 3) and one pair of new natural products (4i, 4ii), along with one known compound, dasycarpusester B (2) (Guo et al., 2012), were isolated from the root bark of D. dasycarpus Turcz (Fig. 1). Their structures were identified using extensively spectroscopic data and quantum chemical calculations.
Compound 1 was obtained as a colorless oil. Its molecular formula of C11H18O5 was established by a peak at m/z 253.1042 for the [M + Na]+ (calcd for C11H18O5Na+, 253.1046) in HRESIMS, indicating three degrees of unsaturation. The IR spectrum of 1 showed characteristic absorption bands for methyl (2978, 2934, 2877 cm−1) and carbonyl (1742 cm−1). The 1H NMR spectrum was broadened in CDCl3. As directed by the HSQC experiment, the 1D NMR data (Table 1) of 1 displayed three methyl signals at δH/δC 1.53/29.4 (CH3-12), 1.36/21.7 (CH3-11), and 1.08/17.6 (CH3-5), one oxygenated methine at δH/δC 4.32/67.6 (CH-2), two methylenes at δH 2.62/2.37 (H-9α/β), 1.65/1.59 (H-3) and δC 39.5 (C-3), 34.7 (C-9), as well as two methines at δH/δC 2.09/51.4 (CH-8), 1.95/30.3 (CH-4). Three quaternary carbons, including one hydroxy-substituted sp3 carbon at δC 87.9 (C-7) and two carbonyl carbon at δC 178.3 (C-1)/176.3 (C-10), were also observed in 13C NMR. Given that the NMR data in DMSO‑d6 (Fig. S1-4) were similar to those of dasycarpusacid (Guo et al., 2012) except for the H-2/3 and C-1, 1 was proposed as a stereoisomer of dasycarpusacid.
From the 1H-1H COSY spectrum (Fig. 2), an aliphatic chain of –CH(OH)CH2CH(CH3)CHCH2– could be established. The HMBC correlations (Fig. 2) from H3-11/H3-12 to C-7/C-8 confirmed the presence of two methyls at the quaternary C-7. The HMBC correlations from H-9 to C-7/C-8/C-10 and H-8 to C-7/C-9/C-10 proved the existence of 2,2-dimethylbutanolide. Additionally, the correlations from H3-5 to C-3/C-4/C-8 indicated that H3-5 is connected to C-4. The cross-peaks of H-2 to C-1/C-3/C-4 and H-3 to C-2/C-4 deduced the moiety of 2-hydroxy-4-methylbutyric acid. Finally, the correlations from H-8/H-9β to C-4 proved the linkage between C-4 and C-8.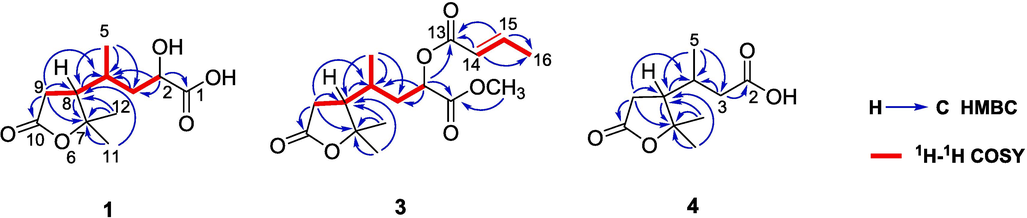
Key 1H–1H COSY and HMBC correlations of 1, 3, and 4.
Given the flexible moiety at C-8 and the unclearly described configuration of dasycarpusacid (Guo et al., 2012), the relative configuration of three stereocenters at C-1, C-4, and C-8 of 1 was further confirmed by comparing the experimental and theoretical NMR chemical shifts.
DFT calculations of NMR chemical shifts were performed using the reported multi-step conformer filtering protocol (Li et al., 2023) for four possible stereoisomers (1a-1d, Fig. 3). Correlation coefficient (R2), mean absolute error (MAE) and DP4+ probabilities were used to evaluate the theoretical chemical shifts of four candidates with experimental data (Table 2, Fig. 4A and 4B). Finally, 1b was identified as the correct structure with the highest R2, smallest MAE, and DP4+ possibility of > 70% for both 1H and 13C.
Possible stereoisomers of 1 and 4 for NMR calculation.
1a
1b
1c
1d
4a
4b
R2 (H)
0.9843
0.9953
0.9919
0.9729
0.9834
0.9450
R2 (C)
0.9991
0.9996
0.9996
0.9992
0.9992
0.9991
MAE (H)
0.10
0.06
0.07
0.11
0.06
0.06
MAE (C)
1.36
0.87
1.02
1.32
0.98
0.94
DP4+ (H)
2.23%
71.07%
26.53%
0.16%
98.75%
1.25%
DP4+ (C)
1.37%
71.26%
27.26%
0.01%
99.49%
0.51%
DP4+ (All data)
0.06%
87.45%
12.49%
0%
99.99%
0.01%
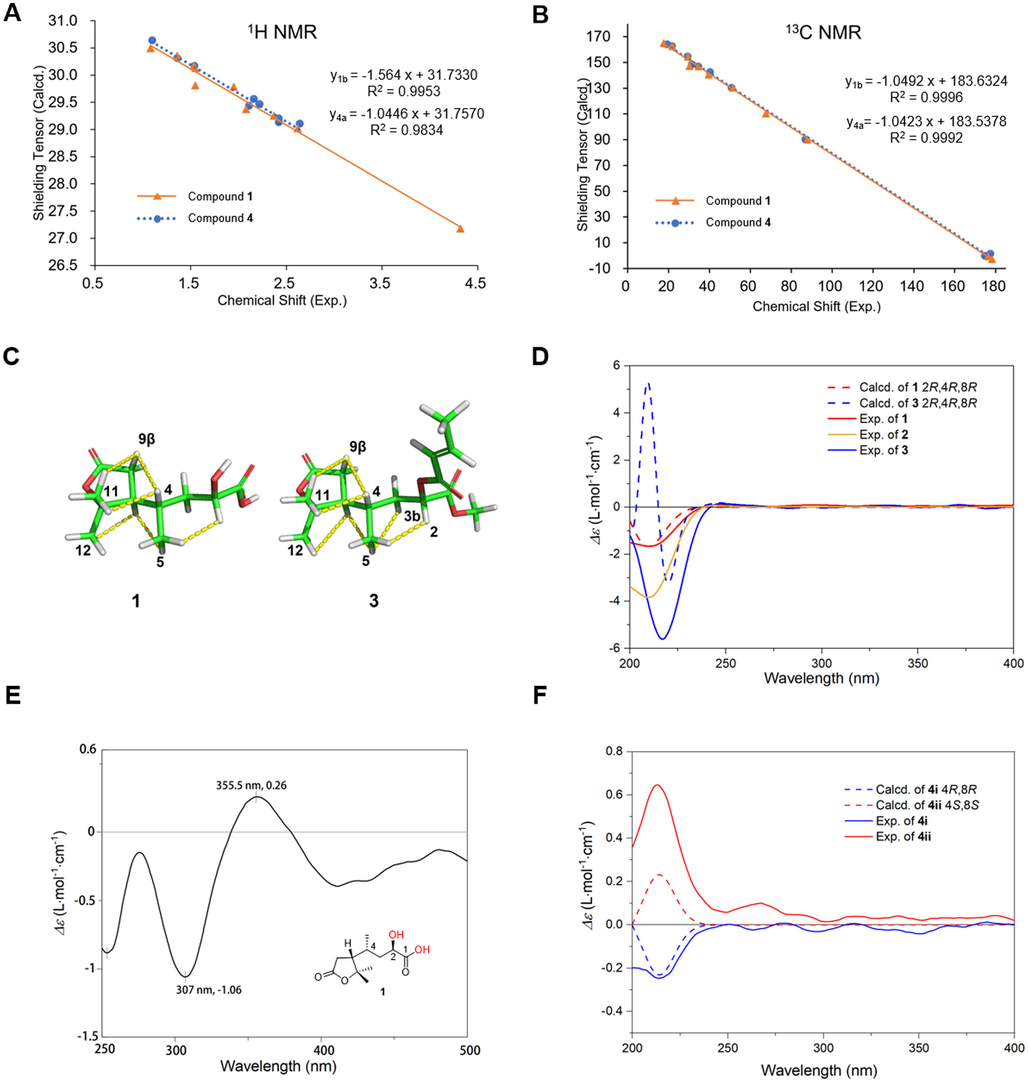
Relative and absolute configuration determination of 1, 3, 4. Linear correlation plots of experimental chemical shift versus calculated shielding tensor of 1b and 4a for 1H (A) and 13C (B). C: Key NOESY correlations of 1 and 3, illustrated by the most preferred conformer. D: Calculated ECD spectra of 1, 3 and experimental spectra of 1–3. E: The Mo2(OAc)4 induced ECD spectrum of 1. F: Calculated and experimental ECD spectra of 4i, 4ii.
The NOESY correlations (Fig. 4C) of H-9β/H-11, H-9β/H-4, and H-11/H-4 indicated that they are positioned nearby. Similarly, the cross-peaks of H-12/H-8, H-8/H-5, and H-5/H-2 indicated that they are spatially close to each other. Despite the flexible moiety, all above correlations could be illustrated by the most preferred conformer, with a population of > 40% (conf-1).
Different from dasycarpusacid (Guo et al., 2012), compound 1 exhibited a negative Cotton effect (CE) at 210 nm in the ECD spectrum. Therefore, the absolute configuration of 1 was determined to be 2R,4R,8R-dasycarpusacid by TDDFT calculation at B3LYP/def2-TZVP level, as shown in Fig. 4D. Considering that the 2-OH located on a flexible moiety, 2R configuration was further proved by Mo2(OAc)4 induced CD experiment (Fig. 4E), with positive CE at 356 nm followed by a negative one at 307 nm (Snatzke et al., 1981; Frelek et al., 2003).
Compound 3 gave a molecular formula of C16H24O6, deduced by a peak at m/z 313.1641 [M + H]+ (calcd for C16H24O6H+, 313.1646) in HRESIMS. The 1H and 13C NMR spectroscopic data were similar with those of 1, except for the existence of one methoxyl [δH 3.75 (3H, s, 1-OCH3) and δC 52.6 (1-OCH3)], one methyl [δH 1.92 (3H, dd, J = 7.0, 1.7 Hz, H3-16) and δC 18.2 (C-16)], one trans-double bond [δH 7.06 (1H, dq, J = 15.6, 6.9 Hz, H-14), δH 5.93 (1H, dq, J = 15.5, 1.9 Hz, H-15) and δC 146.9 (C-14), 121.5 (C-15)], and a carbonyl at δC 165.8 (C-13).
Besides of the aliphatic chain similar to 1, a –CH = CHCH3 moiety could be observed in 1H-1H COSY spectrum (Fig. 2). The HMBC cross-peaks of dasycarpusacid skeleton were identical to those of 1. Moreover, the correlations from olefinic protons H-14/H-15 to C-13/C-16 proved the existence of crotonic ester. The correlations from H-2 to C-13 indicated that the crotonyl moiety was linked to 2-OH. The correlations from the methoxyl at δH 3.75 to C-1 proved the existence of methyl ester. Finally, the planar structure of compound 3 was deduced as 2-O-crotonyldasycarpusester B (Fig. 1).
In NOESY spectrum (Fig. 4C), correlations between H-9β/H-11, H-9β/H-4, H-11/H-4, H-8/H-12, H-8/H-5, and H-5/H-2 were identical to those of 1, indicating that 3 had the same relative configuration. The ECD spectrum of 3 exhibited a negative CE at 217 nm, similar to that of 1. Therefore, 3 was characterized to be 2R,4R,8R, which were further supported by TDDFT calculation at B3LYP/def2-TZVP level (Fig. 4F).
Compound 4, named nordasycarpusacid, was isolated as a natural product for the first time since its synthesis in 1911 (Perkin, 1911). It had a molecular formula of C10H16O4, established by a peak at m/z 201.1126 [M + H]+ (calcd for C10H16O4H+, 201.1121) in HRESIMS. The 1H and 13C NMR spectroscopic data were similar with those of 1, except for the disappearance of one oxygenated methine. The HMBC cross-peaks of γ-lactone moiety were identical to those of 1. The correlations from H3-5 [δH 1.10 (1H, d, J = 6.2 Hz)] to C-3 (δC 40.3)/C-4 (δC 31.2)/C-8 (δC 50.8), H-8 [δH 2.16 (1H, m] to C-4 (δC 31.2), H-3a [δH 2.42 (1H, m)] to C-4 (δC 31.2) and H-3b [δH 2.22 (1H, m)] to C-2 (δ C 177.3) indicated that a 3-butanoic acid moiety was linked to C-8 (Fig. 1).
The relative configurations of C-4 and C-8 were determined by NMR calculations of two stereoisomers of 4 (Fig. 3). The results of R2 and DP4+ probabilities (Table 2, Fig. 4) supported that 4a was the correct structure. Being racemic, 4 was chirally resolved to 4i and 4ii, which were determined to be 4R,8R and 4S,8S, respectively, by TDDFT calculation at B3LYP/def2-TZVP level (Fig. 4F).
Dasycarpusacid derivatives were rare terpenes only found in Dictamnus genus. The biogenetic source of 1–4 is still unclear. Fortunately, a set of A,B-seco limonoids containing dasycarpusacid skeleton have been identified from Meliaceae (Qi et al., 2003; Qi et al., 2003; Cai et al., 2014; Lv et al., 2016; Sun et al., 2022). Therefore, we proposed that the dasycarpusacid was the degraded A/B ring of limonoid, as shown in Fig. 5.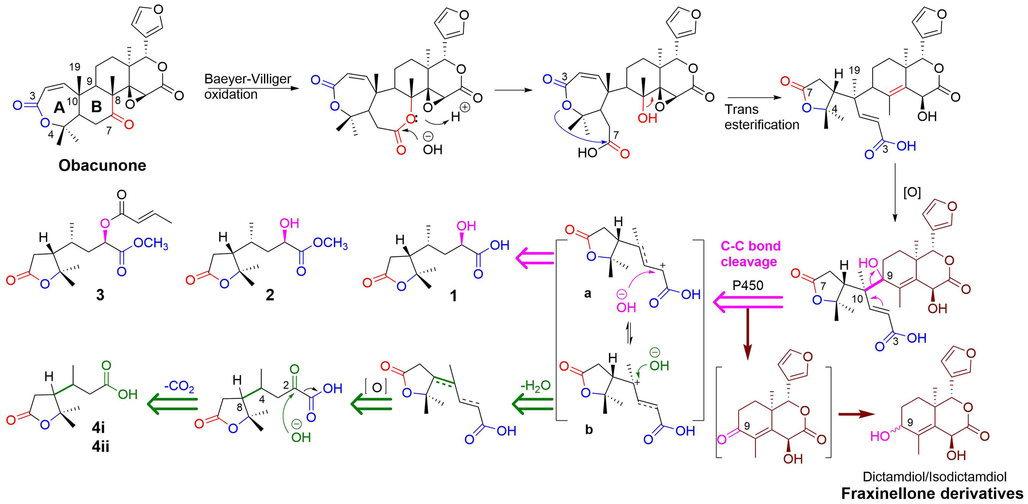
Plausible biogenetic pathway of A/B-ring degradation of limonoid generates rearranged terpenes 1–4.
Obacunone, the most abundant constituent of D. dasycarpus, acted as starting material of the degradation. The B ring was oxidized to lactone through a Baeyer-Villiger reaction. Following the hydrolysis of the B-ring lactone, the 7-COOH and the A-ring lactone were transesterified and rearranged to generate the dasycarpusacid skeleton. C–C bond cleavage occurred after oxidation at C-9, perhaps catalyzed by a P450 enzyme (Guengerich et al., 2018). Then the A/B rings were degraded to dasycarpusacid (1) via a carbenium intermediate (a). 1 could be esterified to generate 2 and 3. Alternatively, the carbenium intermediate (b) was hydroxylated, and an H2O was lost. The formation of the double bond led to the racemization of 4i/4ii at C-4 and C-8. After being oxidized to a carbonyl group, the C-2 was decarboxylated to form 4i/4ii. The residual obacunone finally formed to dictamdiol or isodictamdiol by a 9-carbonyl intermediate. For the first time, the biogenetic pathway of A/B-ring products of degraded products of limonoid was hypothesized to explain the generation of fraxinellone derivatives, based on the structures of dasycarpusacids and dictamdiol/isodictamdiol.
3.2 NMR broadening, relaxing time, and self-aggregation of compound 1
Broadened NMR brings many troubles to the structure elucidation of natural products. Misinterpretation of NMR data may lead to an incorrect structure. It is interesting to observe that all proton signals of 1 were enormously broadened in CDCl3. Carbon signals at C-8 moiety were dramatically reduced in intensity, as shown in Fig. 6A and 6B. Oppositely, the proton lines of methyl ester (2) were sharp, and the carbon intensity of C-8 moiety was normal.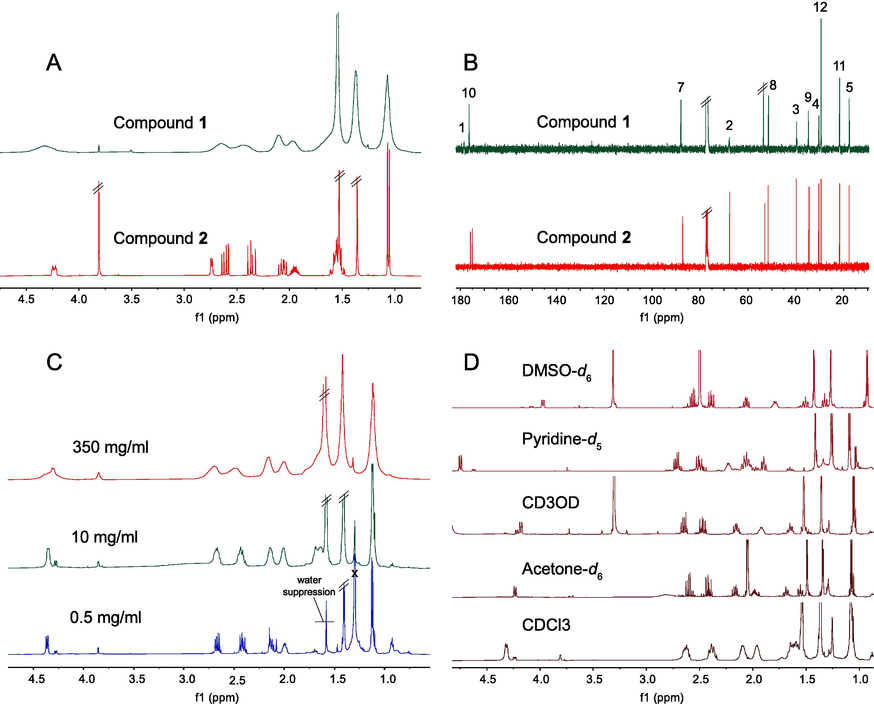
the NMR spectra of 1 and 2. A: 1H NMR spectra in CDCl3; B: 13C NMR spectra in CDCl3; C: 1H NMR spectra of 1 in CDCl3 at different concentrations; D: 1H NMR spectra of 1 in different solvents at 10 mg/mL.
Broadened NMR may be caused by chemical exchange or self-aggregation, in addition to poor instrument performance. Given that all protons were broadening at different temperatures from 10 to 55 °C (Fig. S1-15), we speculated that the self-aggregation, rather than chemical exchange, caused the unexpected line-broadening of 1 in CDCl3. This suggestion was further supported by diffusion coefficients of 1 and 2, determined by the DOSY experiment (Fig. 7A, B) in CDCl3 and CD3OD.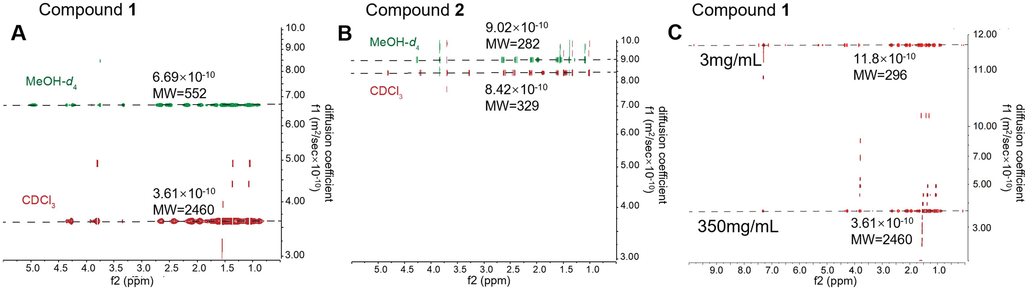
The DOSY spectra showed the diffusion coefficient (f1) of 1 and 2 in different solvents (A, B) or concentrations (C). DOSY of 1 (A) and 2 (B) in CDCl3 (red) and CD3OD (green). C: DOSY of 1 at 3 mg/ml and 350 mg/ml in CDCl3. The molecular weight (M.W.) was estimated from diffusion coefficient by MestReNova.
Concentration is a key factor affecting self-aggregation. Lower concentration of 1 resulted in the degradation of self-aggregation, which was indicated by a higher diffusion coefficient in the DOSY spectrum (Fig. 7C). Correspondingly, the proton of 1 at 350 mg/mL (Fig. 6C) was broadened due to its high concentration. But at 10 mg/mL, a common concentration for NMR analysis, the proton lines were still broadened. Until diluted to 0.5 mg/mL, with no self-aggregation, 1 exhibited sharp proton lines with good resolution in CDCl3. However, the concentration is too low for NMR determination.
In order to destroy the self-aggregation in NMR determination, we investigated the proton spectra of 1 in different solvents at routine concentration (10 mg/mL). Fortunately, the proton spectrum of 1 was only broadened in CDCl3. In CD3OD, acetone‑d6, DMSO‑d6, and pyridine‑d5, 1 exhibited sharp signals with slightly different chemical shifts. Compared with the DOSY data (Fig. 7A) in CD3OD, the smaller diffusion coefficient and the higher estimated M.W. of 1 in CDCl3, indicated that 1 was more prone to self-aggregation in CDCl3. In contrast, 2 has similar diffusion coefficient and estimated M.W. in both solvents.
NMR broadening is usually governed by T2 relaxation (Claridge, 2009). The T1 and T2 in CDCl3 are much shorter than those in the other four solvents (Table 3), further supporting the self-aggregation of 1 in CDCl3. The shorter T2 of C1-C4 (Table 4) and the near disappeared C-1 and C-2 (Fig. 6B) suggested that the self-aggregation of 1 in CDCl3 may be due to the hydrogen bond between 1-COOH and 2-OH (Fig. 8).
NO.
T1 (s)
CDCl3
Acetone‑d6
CD3OD
Pyridine‑d5
DMSO‑d6
2
0.12
2.88
2.10
0.71
0.89
3a
0.18
1.15
0.38
0.60
0.40
3b
0.42
1.17
0.61
0.41
0.16
4
0.19
8.12
0.47
0.52
0.65
5
0.30
1.07
0.48
0.52
0.20
8
0.21
2.61
0.63
0.60
0.86
9a
0.15
1.45
0.44
0.46
0.53
9b
0.17
1.43
0.42
0.45
0.53
11
0.26
1.01
0.45
0.54
0.32
12
0.42
1.37
0.61
0.70
0.61
NO.
T2 (s)
2
0.02
0.04
0.05
0.22
0.0021
3a
0.11
0.61
0.15
0.15
0.18
3b
0.24
0.73
0.59
0.19
0.16
4
0.04
7.88
0.08
0.19
0.044
5
0.10
0.73
0.24
0.29
0.20
8
0.05
0.15
0.35
0.15
0.11
9a
0.08
0.96
0.43
0.20
0.35
9b
0.08
0.91
0.44
0.26
0.43
11
0.15
1.00
0.44
0.44
0.32
12
0.24
1.35
0.59
0.64
0.48
No.
T1 (ms)
T2 (ms)
1
50
5
2
400
----
3
340
5
4
100
5
5
80
45.92
7
130
28.78
8
270
35.09
9
50
5
10
90
35.65
11
110
47.22
12
250
52.94
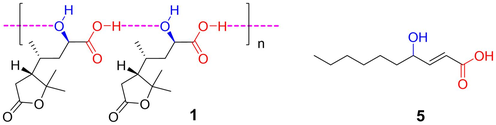
Proposed self-aggregation form of 1 in CDCl3 and another hydroxy acid (5) with broadened NMR spectra.
When CDCl3 was used as solvent, other hydroxyl acid compounds, such as compound 5 (Fig. S4-1), also exhibited broadened proton lines under variable temperatures of 0–55 °C (Fig. S4-6) and nearly disappeared carbon signals between COOH and C(OH). The low-intensity carbon signal of COOH is easy to ignore in structure elucidation, due to the lack of corresponding proton signal. In particular, with more significant fragment ion of losing water than quasi-molecular ion in mass spectra, it was easily elucidated as incorrect structure of lactone. Therefore, this discovery offers more proof to distinguish between acid and ester/lactone in structure elucidation.
3.3 Biological evaluation
At 10 μM, compounds 1–3 showed stronger inhibitory activity against OA-induced lipid accumulation in HepG2 cells, compared to the positive control fenofibrate at 20 μM (Table 5). Being mimic of lipid acid, the α-hydroxyl carboxylic chain of 1–3 may be the key group of competitively inhibiting the lipid accumulation in liver cells. This activity could be increased by methyl esterification on carboxyl (2) and be deduced by esterification on hydroxyl (3) with a big functional group.
Compounds
Concentration (μM)
OD
Inhibition rate (%)
blank
0.135 ± 0.009
–
modela
240
0.185 ± 0.004***
–
Fenofibrateb
20
0.153 ± 0.017#
12.2
1
10
0.148 ± 0.018##
19.8
2
10
0.141 ± 0.021##
23.6
3
10
0.16 ± 0.02#
13.6
FLD has emerged as one of the main causes of liver disease in the world. However, no drug has been approved for the therapy of FLD, and only twenties of natural products were bioactive for relieving FLD (Guo et al., 2022). Therefore, the promising activities of 1–3 against OA-induced lipid accumulation afford a novel scaffold for anti-FLD drug discovery and encourage us to discover more active compounds from D. dasycarpus.
Compounds 1–3 were determined for antidiabetic activity against α-glucosidase and PTP1B, as well as anti-inflammatory activity against LPS-induced NO production of macrophages, but none of them exhibited significant activity at 10 μM.
4 Conclusion
The present research work, led to the identification of five derivatives from the degradation of limonoids, including two previously undescribed compounds (1, 3) and one pair of new natural products (4i, 4ii) from Dictamnus dasycarpus Turcz. The self-aggregation of 1 in chloroform was observed through the broadened NMR spectra, which provided additional evidence to distinguish acid and ester/lactone in the structural elucidation. Given the structures of dasycarpusacids and dictamdiol/isodictamdiol, the biogenic pathway of A/B-ring products of degraded limonoid and fraxinellone derivatives was proposed for the first time. Finally, the isolated compounds 1–3 were tested for inhibitory activity against OA-induced lipid accumulation in HepG2 cells, with inhibition rates of 19.8%, 23.6%, and 13.5% at 10 μM, significantly stronger than fenofibrate, affording a promising scaffold for anti-FLD drug development.
Acknowledgments
This research was financially supported by the National Natural Science Foundation of China (NSFC 22177134) and the CAMS Innovation Fund for Medical Sciences (CIFMS 2021-I2M-1–028). DFT calculation was supported by Biomedical High Performance Computing Platform, Chinese Academy of Medical Sciences.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- SpecDis: quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality. 2013;25:243-249.
- [CrossRef] [Google Scholar]
- Limonoids from Aphanamixis polystachya and their antifeedant activity. J. Nat. Prod.. 2014;77:472-482.
- [CrossRef] [Google Scholar]
- Claridge, T.D.W., 2009. Chapter 2 Introducing high-resolution NMR. Tetrahedron Organic Chemistry Series. 27, 11-34. https://doi.org/10.1016/S1460-1567(08)10002-2.
- Dinuclear Transition Metal Complexes as Auxiliary Chromophores in Chiroptical Studies on Bioactive Compounds. Curr. Org. Chem.. 2003;7:1081-1104.
- [CrossRef] [Google Scholar]
- Formation and cleavage of C-C bonds by enzymatic oxidation–reduction reactions. Chem. Rev.. 2018;118:6573-6655.
- [CrossRef] [Google Scholar]
- Three new compounds from Dictamnus dasycarpus. J. Asian Nat. Prod. Res.. 2012;14:210-215.
- [CrossRef] [Google Scholar]
- Non-alcoholic fatty liver disease (NAFLD) pathogenesis and natural products for prevention and treatment. Int. J. Mol. Sci.. 2022;23:15489.
- [CrossRef] [Google Scholar]
- The antitumor effect of formosanin C on HepG2 cell as revealed by 1H-NMR based metabolic profiling. Chem. Biol. Interact.. 2014;220:193-199.
- [CrossRef] [Google Scholar]
- Aromatic diglycosides from Sophora tonkinensis and a multi-step conformer filtering procedure for TDDFT calculation of flexible glycoside. J. Asian Nat. Prod. Res.. 2023;25:411-421.
- [CrossRef] [Google Scholar]
- Shermo: A general code for calculating molecular thermochemistry properties. Comput Theor Chem.. 2021;1200:113249
- [CrossRef] [Google Scholar]
- Research progress of meliaceous limonoids from 2011 to 2021. Nat Prod Rep.. 2022;39:1325-1365.
- [CrossRef] [Google Scholar]
- Medicinal uses, phytochemistry and pharmacology of the genus Dictamnus (Rutaceae) J. Ethnopharmacol.. 2015;171:247-263.
- [CrossRef] [Google Scholar]
- Isolation and asymmetric total synthesis of perforanoid A. Angew. Chem. Int. Ed.. 2016;55:7539-7543.
- [CrossRef] [Google Scholar]
- The ORCA quantum chemistry program package. J. Chem. Phys.. 2020;152:224108
- [CrossRef] [Google Scholar]
- LXXXI.—Experiments on the synthesis of the terpenes. Part XIX. Synthesis of cis- and trans-Δ3-o-menthenol(8), Δ4-o-menthenol(8), and the corresponding menthadienes. J. Chem. Soc., Trans.. 1911;99:741-761.
- [CrossRef] [Google Scholar]
- Automated exploration of the low-energy chemical space with fast quantum chemical methods. Phys. Chem. Chem. Phys.. 2020;22:7169-7192.
- [CrossRef] [Google Scholar]
- Novel tetranortriterpenoid derivatives from Munronia henryi. Tetrahedron.. 2003;59:4193-4199.
- [CrossRef] [Google Scholar]
- Insect Antifeedants from Munronia henryi: Structure of Munroniamide. J. Agric. Food Chem.. 2003;51:6949-6952.
- [CrossRef] [Google Scholar]
- Circulardichroism—LXXV: Cottonogenic derivatives of chiral bidentate ligands with the complex [Mo2(O2CCH3)4] Tetrahedron.. 1981;37:349-361.
- [CrossRef] [Google Scholar]
- Diverse ring-seco limonoids from Munronia unifoliolata and their biological activities. Chin. J. Chem.. 2022;40:123-136.
- [CrossRef] [Google Scholar]
- Meliaceous limonoids: Chemistry and biological activities. Chemical Reviews.. 2011;111
- [Google Scholar]
- Antimicrobial alkaloids from the root bark of Dictamnus dasycarpus. J. Asian Nat. Prod. Res.. 2021;24:483-489.
- [CrossRef] [Google Scholar]
- A guide to small-molecule structure assignment through computation of (H and C) NMR chemical shifts. Nat. Protoc.. 2014;9:643-660.
- [CrossRef] [Google Scholar]
- In vitro and in vivo anticancer activity of steroid saponins of Paris polyphylla var. yunnanensis. Exp. Oncol.. 2009;31:27-32.
- [Google Scholar]
- Sensitivity analysis of DP4+ with the probability distribution terms: development of a universal and customizable method. J. Org. Chem.. 2021;86:8544-8548.
- [CrossRef] [Google Scholar]
- Undescribed protolimonoids from the root bark of Dictamnus dasycarpus Turcz. Fitoterapia.. 2022;163:105345
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.105517.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




