

Translate this page into:
Structure-based strategies for synthesis, lead optimization and biological evaluation of N-substituted anthra[1,2-c][1,2,5]thiadiazole-6,11-dione derivatives as potential multi-target anticancer agents
⁎Corresponding author at: Graduate Institute of Cancer Biology and Drug Discovery, College of Medical Science and Technology, Taipei Medical University, Taipei 11031, Taiwan huanghs99@tmu.edu.tw (Hsu-Shan Huang)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
As part of our research on developing multi-target small molecule anticancer agents, we designed, synthesized, and biologically evaluated a series of novel diversified analogues based on our thiadiazole-fused anthraquinone lead compound NSC745885. We initially screened our compounds based on their cytotoxicities against two prostate cancer cell lines (PC-3 and DU-145). Cytotoxicities of the selected compounds (3, 5, 6, 10, 11, 14, 15, 17, 18) were then evaluated using the single-dose testing against a panel of 60 cancer cell lines. Compounds which exceeded the threshold inhibition criteria (3, 6, 10, 11, 14, 17) were further evaluated using the five-dose cytotoxicity experiments against the panel of 60 cancer cell lines. Our compounds exhibited potent antiproliferative effects against the tested cancer cell lines with 50% growth inhibition (GI50) values in the sub-micro molar range. Furthermore, 3 and 6 showed high selectivity towards the leukemia subpanel, whereas 6 showed high selectivity towards the prostate subpanel. Our potent compound 11 (RV59, NSC763967) showed broad-spectrum cytotoxicity against different types of cancer cells, while being less cytotoxic than doxorubicin towards different normal cells (SV-HUC-1, WMPY-1, and RWPE-1). COMPARE analysis of the cytotoxicity data indicated that 11 is similar to the apoptosis-based anticancer drugs. We confirmed the apoptotic effects of 11 by microscopy, Western blotting, and flow cytometry of treated cancer cells, and found that it caused cells to exhibit apoptotic morphology, inhibited cyclin D1 and COX-2 in a dose-dependent manner, and accumulated cells at the G0/G1 phase with reduction of cells in the S and G2/M phases of cell cycle. Moreover, we tested the inhibition capabilities of our compounds towards Topoisomerases (TOP) using computational modeling and found that they are specific inhibitors to TOP1. Our data presented here presents our compounds as potential multi-target anticancer drugs.
Keywords
Apoptosis
Cell cycle
Cyclin D1
COX-2
Topoisomerase
Thiadiazole-fused anthraquinone derivative

1 Introduction
Multi-target small molecule inhibitors have attracted great attention because of their advantages in treating complex cancers by simultaneously targeting multiple targets and possibly leading to synergistic effects (Raevsky et al., 2018; Yuan et al., 2020). Many years of intensive research have been devoted to discovering heterocyclic compounds, which are very common as naturally occurring substances and are essential for living cells. The majority of heterocyclic compounds and fragments present in most pharmaceuticals currently marketed have consistently attracted substantial attention due to their broad biological and therapeutic applications, in addition to their intrinsic versatility and unique physicochemical properties (Abdella et al., 2020; Jampilek, 2019).
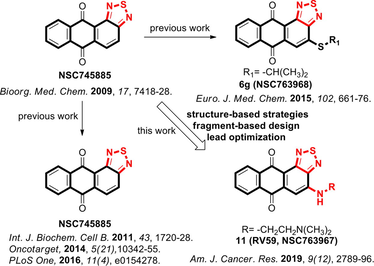
As is well known, the heterocyclic anthracycline antibiotics and their anthraquinone derivatives remain evergreen drugs with broad clinical indications, however, their pharmacological indices should be improved (Lee et al., 2015; Minotti et al., 2004; Monneret, 2001; Wu et al., 2017). From our previous studies involving the privileged anthraquinone scaffold and its associated biological activities, we found that the thiadiazole moiety imparted unprecedented broad-spectrum bioactivities to the anthraquinone scaffold (Ali et al., 2016b; Chang et al., 2011; Huang et al., 2009; Tang et al., 2014). Prompted by the varied biological activities of such compounds, we envisioned our approach towards the synthesis of novel heterocyclic small molecules that bear the anthraquinone scaffold fused with the thiadiazole moiety. Accordingly, our group have developed a series of 6,6,6,5-tetraheterocyclic derivatives using the fragment-based drug design (FBDD) approach, structure-based design, and ring fusion strategies, in which different side chain moieties were conjugated to the thiadiazole-fused anthraquinone scaffold. Thorough biological analysis of such compounds showed that the thiadiazole-fused anthraquinones exhibit broad-spectrum activities against several types of cancer (Ali et al., 2016b; Chang et al., 2011; Lee et al., 2015; 2013b; Tang et al., 2014). Moreover, addition of sulfur to other compounds similar bearing the tricyclic, tetracyclic, tetraheterocyclic or pentaheterocyclic ring systems typically lead to analogs with improved bioactivities because the sulfur atom imparts substantially increased binding to the intracellular targets as well as increased liposolubility (Fig. 1) (Tripathy et al., 2007). Among our library of small molecule inhibitors, we found that the thiadiazole-containing anthraquinone-like compound 1 (NSC745885) exhibits unique and promising pharmacological activities. Accordingly, we considered it as an advanced chemical lead for further optimization (Chang et al., 2011; Shen et al., 2019; Tang et al., 2014).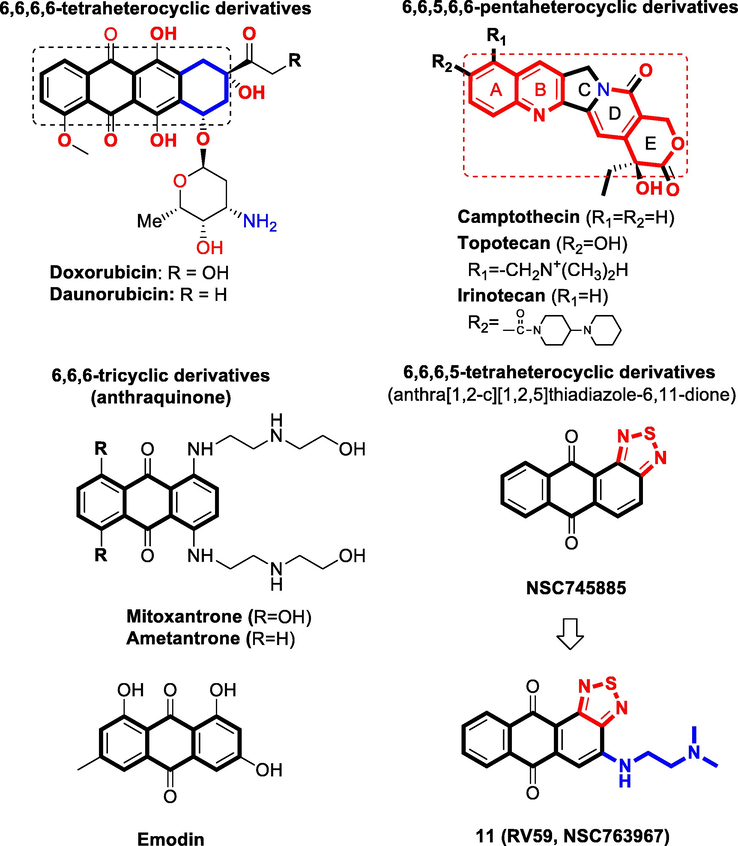
Drugs related to our series of novel compounds with the core structure of 6,6,6-tricyclic, 6,6,6,6-tetraheterocyclic, 6,6,6,5-tetrahetrocyclic, 6,6,5,6,6-pentaheterocyclic scaffolds and representative investigational structural sets used in the related pharmacophore studies.
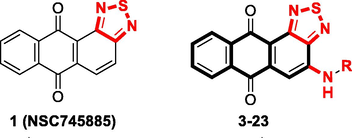
Here, we designed and synthesized a novel series of compounds based on the anthra[1,2-c][1,2,5]thiadiazole-6,11-dione scaffold using different N-substitutions on the 4-position as depicted in Scheme 1. We screened the synthesized compounds based on their cytotoxicity against two human prostate cancer cell lines (PC-3 and DU-145) using sulforhodamine B (SRB) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays (Table 1). Moreover, compounds 3 (NSC763958), 5 (NSC763965), 6 (NSC763954), 10 (NSC764639), 11 (NSC763967), 14 (NSC764640), 15 (NSC763966), 17 (NSC757967), and 18 (NSC763952) were tested by the National Cancer Institute (NCI), USA using single-dose cytotoxicity experiments against a panel of 60 human cancer cell lines (Table 2). Out of the tested compounds, 3 (NSC763958), 6 (NSC763954), 10 (NSC764639), 11 (NSC763967), 14 (NSC764640), and 17 (NSC757967) satisfied the pre-determined threshold of the growth inhibition criteria of the NCI and accordingly were evaluated using the five-dose cytotoxicity experiments against the panel of 60 cancer cell lines (Table 3). We found that many of our compounds exerted potent multi-log differential patterns of activity, with 50% growth inhibition (GI50) values in the sub-micro molar range against many cancer cell lines. To learn more about the toxicity of our compounds towards normal cells, we tested the cytotoxicity of compound 11 (RV59, NSC763967) against three normal cell lines (SV-HUC-1, WMPY-1, and RWPE-1) and found that it was less toxic to cells compared to doxorubicin. To gain insight into the targets and mechanisms of actions of our compounds, we selected our potent compound 11 (RV59, NSC763967) for further investigation. We performed COMPARE analysis experiments (Ali et al., 2016b) using comparative correlations of the cytotoxic activities of the included drugs in the NCI database to identify drugs that have similar targets and mechanisms of action to our test compound, and hence get insight into the possible targets and mechanisms of our drug. We observed that the cytotoxic profile of compound 11 (RV59, NSC763967) is highly correlated to those of the drugs that induce apoptosis and affect the cell cycle. Consequently, we aimed to confirm these findings for compound 11 (RV59, NSC763967), so we investigated its effect on the cell cycle of DU-145 cells as well as its apoptosis induction capabilities using the same cell line. We found that compound 11 (RV59, NSC763967) caused dose-dependent apoptotic cellular morphological changes to DU-145 cells which were clear under phase-contrast microscope, and arrested 86.04% of the DU-145 cells at the Sub-G1 phase of cell cycle. Moreover, it inhibited dose-dependently the expression of cyclin D1 DU-145 cells and cyclooxygenase (COX)-2 in PC-3 cells. Collectively, these data make it clear that compound 11 (RV59, NSC763967) causes cell cycle arrest and apoptosis to cancer cells. Furthermore, we found that some of our compounds, including compound 11 (RV59, NSC763967), are good topoisomerase (TOP1) inhibitors according to our molecular modeling experiments. Our data as presented in this manuscript present a novel series of potential multi-target anticancer compounds that represent appealing members for further drug development. GI50, concentration required for 50% inhibition of cell growth; TGI, total growth inhibition; LC50, 50% lethal concentration; CNS, central nervous system.![Synthesis routes of the N-substituted anthra[1,2-c][1,2,5]thiadiazole-6,11-dione derivatives. Reagents and conditions: (a) SOCl2, THF, r.t., 24 h; (b) KClO3, acetic acid, HCl, 90 °C, 5 h; (c) series aniline derivatives, ethylene glycol, 160 °C, 30 min; (d) series amine derivatives, copper acetate, dimethylformamide (DMF), 75 °C, 2 h.](/content/184/2021/14/2/img/10.1016_j.arabjc.2020.10.031-fig3.png)
Synthesis routes of the N-substituted anthra[1,2-c][1,2,5]thiadiazole-6,11-dione derivatives. Reagents and conditions: (a) SOCl2, THF, r.t., 24 h; (b) KClO3, acetic acid, HCl, 90 °C, 5 h; (c) series aniline derivatives, ethylene glycol, 160 °C, 30 min; (d) series amine derivatives, copper acetate, dimethylformamide (DMF), 75 °C, 2 h.
Compound
R
SRB assay
MTT assay
PC-3 (µM)
DU-145 (µM)
1 (NSC745885)
2.50
7.41
3 (NSC763958)
0.21
0.16
4
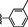
0.29
5.17
5 (NSC763965)

0.84
4.88
6 (NSC763954)
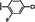
>15
9.15
7
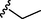
2.97
–
8
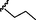
3.25
4.66
9
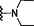
1.10
8.77
10 (NSC764639)

1.76
5.91
11 (NSC763967)
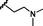
0.40
0.70
12
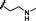
1.26
0.93
13
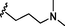
1.15
1.38
14 (NSC764640)
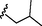
0.74
7.73
15 (NSC763966)
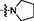
>15
9.56
16
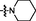
1.38
>15
17 (NSC757967)
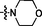
>15
>15
18 (NSC763952)
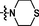
>15
11.89
19

1.20
2.28
20

9.43
7.36
22
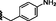
11.90
13.44
23
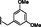
5.86
7.86
Doxorubicin (DXR)
0.25
0.29
Panel/Cell line
3 (NSC763958)
5 (NSC763965)
6 (NSC763954)
10 (NSC764639)
11 (NSC763967)
14 (NSC764640)
15 (NSC763966)
17 (NSC757967)
18 (NSC763952)
Growth percent (%)
Leukemia
CCRF-CEM
17.83
59.59
29.84
71.30
−12.55
43.73
86.62
48.31
43.60
HL-60 (TB)
1.11
46.13
8.17
26.51
−10.28
10.19
104.58
66.73
51.67
K562
16.97
33.45
35.29
29.82
−24.44
18.82
75.50
48.27
59.58
MOLT-4
−11.92
12.86
20.18
49.21
−41.28
21.97
92.84
47.64
42.86
RPMI-8226
16.20
21.59
40.73
39.61
−29.81
28.39
76.16
61.17
56.96
SR
−15.27
26.12
23.58
43.16
−49.98
11.00
84.23
68.92
55.02
Non-small-cell lung cancer
A549/ATCC
27.13
56.25
69.22
19.62
−60.09
9.87
63.51
100,58
62.74
EKVX
62.56
82.17
80.90
–
−25.35
–
75.67
84.52
86.63
HOP-62
40.78
65.10
79.67
49.76
−67.97
29.55
79.16
60.53
76.66
HOP-92
–
–
–
95.58
−25.30
74.44
–
90.59
–
NCI-H226
47.00
66.61
74.35
67.74
−69.78
51.61
78.66
87.42
78.10
NCI-H23
37.57
45.87
53.59
17.13
−43.43
6.55
59.74
39.65
33.45
NCI-H322M
64.54
94.85
91.66
60.29
−56.32
27.68
82.34
98.00
91.69
NCI-H460
11.85
21.80
81.55
14.22
−38.31
19.87
89.11
88.64
87.46
NCI-H552
12.41
53.06
47.67
40.20
−91.81
−0.77
83.41
79.03
38.70
Colon cancer
COLO 205
83.39
100.68
90.97
107.74
−99.38
77.77
82.13
104.19
107.31
HCC-2998
57.61
95.82
97.85
84.54
−86.18
25.32
104.58
107.48
107.83
HCT-116
24.23
31.32
52.51
35.18
−2.07
15.67
78.67
18.89
88.21
HCT-15
28.74
32.24
45.61
15.59
−60.44
7.06
61/0.25
55.97
61.93
HT29
52.26
96.29
89.33
94.39
−45.50
22.23
98.60
–
86.91
KM12
40.14
64.56
83.42
39.65
−40.00
17.75
95.18
83.91
83.12
SW-620
32.60
59.77
83.79
72.50
−21.66
32.17
95.28
72.70
103.06
CNS cancer
SF-268
39.57
52.27
69.25
59.16
−14.54
42.23
98.43
75.83
95.90
SF-295
34.71
75.76
86.04
75.88
−55.73
0.82
87.02
95.84
94.85
SF-539
47.90
70.48
75.99
79.80
−82.05
52.47
97.27
88.62
90.64
SNB-19
61.89
82.28
94.57
57.95
−77.31
41.44
90.45
84.82
100.15
SNB-75
43.18
80.91
83.19
–
−73.34
–
94.52
51.11
105.37
U251
19.18
34.17
43.08
47.53
−56.42
25.71
80.49
59.35
40.24
Melanoma
LOX IMVI
8.17
27.05
29.97
16.11
−90.91
6.15
69.55
13.57
30.16
MALME-3M
64.72
78.41
58.74
45.90
−85.22
11.56
70.73
61.99
48.56
M14
61.89
76.72
118.36
27.70
−96.08
10.89
94.61
75.06
117.70
MDA-MB-435 -
66.53
27.37
56.48
−58.82
−67.52
105.87
0.52
77.03
SK-MEL-2
32.64
93.85
72.52
76.32
−66.52
36.04
115.43
88.38
43.98
SK-MEL-28
53.13
74.97
87.69
72.51
−50.60
34.15
99.46
104.55
90.60
SK-MEL-5
8.03
24.13
49.49
−57.85
−98.79
−81.47
31.12
15.98
22.81
UACC-257
33.79
71.17
50.45
50.27
−88.80
18.82
84.14
79.36
35.78
UACC-62
57.68
67.41
85.80
0.33
−47.85
−44.11
96.64
62.43
83.96
Ovarian cancer
IGROV1
25.29
49.40
69.50
52.16
−28.40
34.57
75.83
54.88
75.28
OVCAR-3
22.37
57.92
10.25
41.79
−38.62
15.09
86.97
6.64
41.52
OVCAR-4
−5.96
31.44
−4.36
21.52
−71.27
16.50
77.50
28.69
0.28
OVCAR-5
79.06
109.85
114.60
75.16
−76.97
61.87
111.75
–
112.62
OVCAR-8
22.87
53.96
36.77
75.65
−28.91
53.06
82.12
43.29
41.72
NCI/ADR-RES 28.36
61.34
43.31
68.52
–23.20
40.95
86.36
43.58
54.16
SK-OV-3
–
–
–
96.39
–
64.43
–
104.65
–
Renal cancer
786-0
63.62
74.18
114.97
94.94
−14.06
61.88
96.67
109.16
120.26
A498
–
–
–
66.30
–
55.71
–
88.30
–
ACHN
38.07
57.60
74.11
67.09
–22.86
38.11
82.97
–
83.52
CAKI-1
27.55
50.33
72.65
48.83
6.88
24.81
70.65
61.73
76.06
RXF 393
45.36
71.05
66.49
80.98
−59.58
54.00
97.02
98.14
85.93
SN12C
46.94
74.58
86.43
68.21
−95.78
33.83
89.82
71.73
76.76
TK-10
63.75
95.15
110.93
106.78
−35.59
76.50
141.01
131.28
106.46
UO-31
17.74
56.97
61.81
38.63
−28.47
22.39
41.64
53.93
61.00
Prostate cancer
PC-3
35.60.
45.73
44.53
47.09
−18.35
36.84
71.01
56.59
56.99
DU145
47.51
68.26
74.73
27.01
−95.45
20.47
105.08
68.98
79.83
Breast cancer
MCF7
22.53
46.59
63.15
40.66
−49.41
27.68
59.71
55.12
49.73
MDA-MB-231/ATCC 31.45
73.83
35.85
53.53
−2.99
33.73
95.23
39.16
45.69
HS 578T
72.44
78.75
85.89
64.60
4.32
18.14
101.62
88.35
84.36
BT-549
75.04.
76.36
98.07
67.27
−99.43
35.83
86.83
93.78
104.74
T-47D
45.05
64.48
51.78
35.94
12.90
30.11
78.19
60.04
56.78
MDA-MB-468 12.85
58.18
14.40
4.48
−62.81
2.18
84.32
45.59
12.59
Mean
36.29
61.35
63.92
52.13
−49.52
25.82
85.69
67.97
70.31
Panel/Cell line (
M)
3 (NSC763958)
6 (NSC763954)
10 (NSC764639)
11 (NSC763967)
14 (NSC764640)
17 (NSC757967)
GI50
TGI
LC50
GI50
TGI
LC50
GI50
TGI
LC50
GI50
TGI
LC50
GI50
TGI
LC50
GI50
TGI
LC50
Leukemia
CCRF-CEM
0.19
>100
>100
1.92
4.72
>100
–
>100
>100
0.43
2.13
9.89
4.28
>100
>100
2.83
15.5
>100
HL-60(TB)
0.46
3.00
>100
1.91
4.89
>100
4.45
>100
>100
0.38
1.80
9.61
3.56
>100
>100
16.7
48.4
>100
K-562
0.08
31.2
>100
1.28
–
>100
4.43
>100
>100
0.26
1.14
>100
2.70
>100
>100
1.96
22.9
>100
MOLT-4
0.20
5.65
>100
1.67
–
>100
5.71
>100
>100
0.21
0.99
>100
2.85
18.5
>100
2.53
18.6
>100
RPMI-8226
0.66
21.7
>100
1.93
15.2
>100
3.08
>100
>100
0.48
2.85
>100
2.91
>100
>100
5.59
55.7
>100
SR
0.07
12.1
>100
1.47
–
>100
3.24
>100
>100
0.30
1.69
>100
2.68
>100
>100
3.81
27.2
>100
Non-small-cell lung cancer
A549/ATCC
1.64
23.5
94.2
25.5
>100
>100
5.61
55.3
>100
0.24
0.51
2.35
2.83
14.9
64.5
25.7
>100
>100
EKVX
–
–
–
–
–
–
–
–
–
–
–
–
–
–
–
>100
>100
>100
HOP-62
1.58
16.5
46.0
11.0
81.1
>100
5.03
>100
>100
0.32
1.33
8.81
3.23
22.5
>100
7.12
>100
>100
HOP-92
1.25
9.31
41.4
0.63
3.04
11.2
–
–
–
0.46
3.57
33.6
1.05
9.61
>100
8.63
36.9
>100
NCI-H226
2.97
30.2
>100
>100
>100
>100
5.31
>100
>100
1.26
2.57
5.23
3.30
>100
>100
26.5
>100
>100
NCI-H23
2.53
20.7
80.7
2.82
14.9
44.9
2.65
58.5
>100
0.32
2.23
18.8
1.42
12.3
>100
0.53
17.5
>100
NCU-H322M
19.8
>100
>100
>100
>100
>100
6.27
>100
>100
1.53
3.15
6.49
3.43
15.7
49.7
>100
>100
>100
NCI-H46-
0.40
13.0
87.5
5.79
23.7
74.5
4.29
>100
>100
0.43
1.92
7.02
3.04
16.9
56.7
16.3
75.0
>100
NCI-H522
0.69
12.5
61.0
11.9
32.7
89.6
8.21
57.5
>100
1.15
2.42
5.07
3.34
>100
>100
2.84
15.6
73.7
Colon cancer
COLON 205
9.19
>100
>100
>100
>100
>100
9.89
91.6
>100
1.62
2.98
5.46
2.58
7.44
28.1
48.8
>100
>100
HCC2998
8.25
>100
>100
54.8
>100
>100
3.65
>100
>100
0.89
2.35
5.75
4.77
>100
>100
71.0
>100
>100
HCT-116
0.32
11.2
43.3
1.60
3.12
6.10
4.41
>100
>100
0.20
0.59
12.4
2.82
55.1
>100
1.72
6.48
55.6
HCT-15
0.08
26.4
>100
2.62
>100
>100
1.82
>100
>100
0.19
0.43
0.99
0.86
>100
>100
5.38
>100
>100
HT-29
5.33
95.2
>100
81.1
>100
>100
5.84
>100
>100
0.54
2.75
16.0
3.99
>100
>100
37.2
>100
>100
KM12
0.81
24.0
>100
12.7
>100
>100
4.33
>100
>100
0.55
1.99
5.09
3.04
>100
>100
12.1
>100
>100
SW-620
0.59
15.8
50.7
4.20
19.7
69.2
–
>100
>100
0.26
1.15
4.82
3.34
>100
>100
4.61
31.5
>100
CNS cancer
SF-295
3.83
18.6
65.8
3.49
20.9
85.3
35.6
18.0
>100
0.20
0.45
1.01
2.59
7.27
30.0
31.2
>100
>100
SF-539
0.49
11.1
35.0
7.31
21.1
51.6
4.41
>100
>100
1.48
2.94
5.84
3.88
34.8
>100
4.90
80.8
>100
SNB-19
7.26
50.6
>100
>100
>100
>100
1.82
>100
>100
1.47
2.78
5.28
5.93
39.0
>100
4.43
>100
>100
SNB-75
0.32
10.3
35.2
–
–
–
5.84
48.4
>100
–
–
–
1.84
7.30
>100
4.35
81.9
>100
U251
0.61
13.5
45.9
5.54
18.9
45.1
4.33
>100
>100
0.44
2.34
15.2
2.24
46.8
>100
2.02
39.9
>100
Melanoma
LOX IMVI
0.19
11.3
37.6
1.54
3.72
9.01
2.80
>100
>100
0.15
0.33
50.8
2.43
>100
>100
0.37
11.3
>100
MALME-3M
2.91
10.7
38.7
6.03
20.1
50.2
2.61
7.98
>100
0.26
0.88
3.57
2.15
5.45
76.9
6.33
51.0
>100
M14
2.87
20.5
58.3
13.4
58.7
>100
7.81
>100
>100
0.16
0.31
0.58
3.91
16.0
50.8
7.30
44.5
>100
MDA-MB- 435
0.74
2.07
4.87
1.87
3.42
–
1.84
3.63
7.15
0.19
0.36
0.67
1.67
3.07
5.65
1.72
3.36
6.57
SK-MEL-2
3.91
20.8
78.2
10.1
23.5
54.5
10.0
>100
>100
0.33
1.32
5.04
3.61
23.5
>100
6.16
28.1
99.9
SK-MEL-28
5.14
19.7
44.7
26.2
98.4
>100
94.9
>100
>100
1.63
3.45
7.30
9.00
>100
>100
24.9
78.6
>100
SK-MEL-5
0.51
4.94
25.9
5.70
10.0
50.3
1.19
3.71
13.3
0.98
2.18
4.75
0.63
1.95
4.42
1.06
2.59
6.32
UACC-257
2.13
11.7
42.5
2.52
12.7
46.1
6.19
>100
>100
0.21
0.70
2.84
2.80
12.9
>100
4.23
31.8
>100
UACC-62
0.67
13.5
39.7
10.6
23.3
50.9
2.26
12.3
>100
0.17
0.30
0.56
1.54
5.07
60.8
2.32
11.4
86.0
Ovarian cancer
IGROV1
1.61
23.5
77.2
7.02
25.6
70.6
11.8
>100
>100
0.44
3.33
18.6
4.12
32.0
>100
6.63
35.1
>100
OVCAR-3
1.14
7.18
29.9
1.71
3.32
6.45
2.51
11.8
90.8
0.19
0.70
6.84
1.85
8.23
>100
1.49
4.71
24.4
OVCAR-4
1.14
2.35
4.84
2.33
7.86
29.5
1.13
17.7
84.7
0.32
1.44
3.97
1.47
27.4
>100
2.11
12.4
41.4
OVCAR-5
8.16
21.7
48.3
16.4
39.4
94.5
67.5
>100
>100
1.20
2.81
6.61
4.69
>100
>100
–
–
–
OVCAR-8
0.47
13.2
54.7
2.35
8.98
34.1
9.36
>100
>100
0.35
2.55
>100
3.94
>100
>100
2.47
24.5
>100
NCI/ADR-RES
0.82
12.5
>100
1.91
5.06
>100
8.16
80.8
>100
0.32
2.12
>100
3.79
>100
>100
1.46
28.5
>100
SK-OV-3
1.88
17.9
43.3
>100
>100
>100
23.6
>100
>100
1.29
3.20
7.91
6.75
55.3
>100
>100
>100
>100
Renal cancer
786-0
3.49
19.3
51.0
52.8
>100
>100
73.0
>100
>100
1.46
5.56
39.8
7.20
>100
>100
50.5
>100
>100
A489
10.3
62.9
>100
–
>100
>100
2.76
>100
>100
1.23
2.55
5.29
1.68
10.8
>100
51.2
>100
>100
ACHN
0.55
13.4
37.3
7.84
22.0
52.7
4.95
>100
>100
0.31
1.88
11.7
2.56
27.2
>100
–
–
–
CAKI-1
0.36
15.7
57.0
2.50
14.0
71.4
4.86
>100
>100
0.49
2.52
9.74
1.97
>100
>100
12.4
>100
>100
RXF 393
2.70
18.2
5.466
11.7
27.2
62.9
10.8
>100
>100
0.99
2.25
5.08
4.64
46.3
>100
16.2
>100
>100
SN12C
1.04
13.4
49.5
11.5
26.4
60.3
4.35
>100
>100
0.16
0.31
0.59
3.09
>100
>100
2.48
24.0
>100
TK-10
–
–
–
66.2
>100
>100
–
–
–
2.54
5.31
13.3
–
–
–
>100
>100
>100
UO-31
0.70
16.2
41.8
12.8
27.5
59.3
2.77
>100
>100
0.70
4.03
39.0
1.38
>100
>100
13.2
76.8
>100
Prostate cancer
PC-3
0.24
2.30
43.9
1.05
2.98
8.45
1.36
>100
>100
0.25
2.30
18.7
1.08
>100
>100
3.75
51.4
>100
DU-145
2.14
0.31
46.0
3.42
11.6
27.7
6.16
56.8
>100
0.17
0.31
0.57
4.99
18.3
46.2
10.1
84.0
>100
Breast cancer
MCF7
0.71
4.90
41.1
10.7
28.0
73.2
2.92
>100
>100
0.52
2.27
7.48
2.42
24.6
>100
3.12
19.9
59.9
MDA-MB-231/ATCC
0.53
6.55
44.0
2.15
11.2
46.1
5.08
>100
>100
0.25
1.58
7.67
2.81
38.2
>100
1.43
21.5
>100
HS 578-T
5.23
63.4
>100
3.61
47.0
>100
7.81
>100
>100
1.30
7.07
>100
3.18
61.2
>100
13.8
96.8
>100
BT-549
1.18
18.0
45.7
27.0
>100
>100
6.19
>100
>100
1.41
2.74
5.32
3.82
55.6
>100
34.3
>100
>100
T-47D
1.38
6.21
97.3
2.74
16.5
68.1
3.35
47.4
>100
1.20
3.11
8.04
1.95
15.5
>100
4.04
23.5
74.7
MDA-MB-468
1.17
3.21
8.79
2.37
6.76
31.1
2.07
9.66
>100
0.38
1.60
4.53
1.53
5.35
36.4
1.21
5.57
29.7
2 Experimental section
2.1 General experimental procedures
Melting points were determined by a melting point apparatus (Büchi® 545). All reactions were monitored by thin-layer chromatography (TLC; Silica Gel 60 F254). 1H nuclear magnetic resonance (NMR) spectra were recorded using GEMINI-300 MHz (Varian®) and AM-500 MHz (Bruker®) instruments. Chemical shift (δ) values were in ppm ranges relative to tetramethylsilane (TMS) as an internal standard. Fourier-transformed infrared (IR) spectra (KBr) were determined with a Perkin-Elmer 983G spectrometer. Mass spectra were obtained on Finnigan MAT 95 XL high-resolution mass spectroscopy (HRMS) and Finnigan/Thermo Quest MAT HRMS. Reagents and solvents were purchased from Merck and Aldrich and were used without further purification. Typical experiments illustrating the general procedures for the preparation of the anthraquinone derivatives are described below.
2.2 General procedure A: Preparation of compounds (3 ∼ 23)
A mixture of 1 (300 mg, 1 mmole) in 20 ml of ethylene glycol and 3 mmole of serial organoamine was heated to 160 °C for 30 min. After its complete conversion (as determined by TLC), the solution was cooled to 80 °C and diluted with hot water. Precipitates were filtered off, washed with water, recrystallized from ethanol, and dried to produce 3 ∼ 6. The other amino derivatives, 7 ∼ 23, were similarly obtained.
2.3 General procedure B: Preparation of compounds (3 ∼ 23)
A mixture of 2 (1 mmol) in 20 ml of dry dimethylformamide (DMF) and copper acetate was heated to 75 °C, and then serial organoamine compounds (2 mmol) in the same solvent were added. Reaction mixtures were refluxed for 2 h at 75 °C. Then, the crude products were purified by ethanol to afford the desired compounds.
2.3.1 4-(o-Tolylamino)anthra[1,2-c][1,2,5]thiadiazole-6,11-dione (3)
The pure compound was obtained as a purplish-red powder (yield 28%). Mp: 177–178 °C (EtOH). FT-IR (KBr, νmax cm−1): 1541.0 (CO), 1670.2 (C⚌N). 1H NMR (300 MHz, CDCl3): δ ppm 2.37 (s, —CH3, 3H), 7.29 (d, J = 7.2 Hz, 1H), 7.34–7.40 (2H, m), 7.49 (s, 1H),7.52 (d, J = 7.2 Hz, 1H), 7.72 (td, J = 7.8 Hz, J = 1.5 Hz, 1H), 7.80 (td, J = 7.5 Hz, J = 1.5 Hz,1H), 8.19 (dd, J = 7.8 Hz, J = 1.2 Hz, 1H), 8.36 (d, J = 7.8 Hz, 1H). 13C NMR (75 MHz, CDCl3): δ ppm 17.85, 100.55, 115.84, 125.01, 126.82, 126.93, 127.30, 127.71, 131.95, 132.71, 133.25, 133.39, 134.35, 134.67, 136.57, 139.74, 142.68, 150.15, 153.25, 180.57, 184.59. HRMS (EI) m/z:calcd [M]+, 371.0728 (C21H13N3O2S +); found, 371.0730.
2.3.2 4-(m-Tolylamino)anthra[1,2-c][1,2,5]thiadiazole-6,11-dione (4)
The pure compound was obtained as a purplish-red powder (yield 23%). Mp: 175–176 °C (EtOH). FT-IR (KBr, νmax cm−1): 1504.4 (C = C), 1544.9 (CO), 1674.1 (C⚌N). 1H NMR (300 MHz, CDCl3): δ ppm 2.43 (s, 3H), 7.09 (d, J = 7.2 Hz, 1H), 7.35–7.36 (m, 2H), 7.36–7.38 (m, 1H), 7.72 (t, J = 7.2 Hz, 1H), 7.80 (t, J = 7.8 Hz, 1H), 7.88 (s, 1H), 8.20 (d, J = 7.2 Hz, 1H), 8.35 (d, J = 7.5 Hz, 1H). 13C NMR (75 MHz, CDCl3): δ ppm 21.52, 100.81, 116.08, 119.70, 123.37, 126.88, 126.95, 127.31, 130.01, 132.71, 133.27, 134.45, 134.67, 138.50, 139.68, 140.34, 141.77, 150.28, 153.16, 180.48, 184.48. HRMS (EI) m/z:calcd [M]+, 371.0728 (C21H13N3O2S +); found, 371.0727.
2.3.3 4-(p-Tolylamino)anthra[1,2-c][1,2,5]thiadiazole-6,11-dione (5)
The pure compound was obtained as a purplish-red powder (yield 25%). Mp: 197–198 °C (EtOH). FT-IR (KBr, νmax cm−1): 1544.9 (CO), 1587.3 (CO), 1668.3 (C⚌N). 1H NMR (300 MHz, CDCl3): δ ppm 2.42 (m, 3H), 7.27–7.36 (m, 4H), 7.72 (t, J = 7.5 Hz, 1H), 7.79–7.82 (m, 2H), 8.19 (d, J = 7.5 Hz, 1H), 8.34 (d, J = 7.5 Hz, 1H). 13C NMR (75 MHz, CDCl3): δ ppm 21.07, 100.46, 115.83, 123.02, 126.78, 126.93, 127.29, 127.58, 130.77, 132.68, 133.23, 134.44, 134.67, 135.83, 136.13, 139.68, 142.11, 150.19, 153.16, 180.45, 184.51. HRMS (EI) m/z: calcd [M]+, 371.0728 (C21H13N3O2S +); found, 371.0722.
2.3.4 4-(4-Chloro-2-fluorophenylamino)anthra[1,2-c][1,2,5]thiadiazole-6,11-dione (6)
The pure compound was obtained as a purplish-red powder (yield 61%). Mp: 212–213 °C (EtOH). FT-IR (KBr, νmax cm−1): 1542.9 (CO), 1568.0 (CO), 1670.2 (C⚌N). 1H NMR (300 MHz, CDCl3): δ ppm 7.29–7.33 (m, 2H), 7.63–7.71 (m, 1H), 7.76–7.79 (m, 2H), 7.84 (d, J = 7.5 Hz, 1H), 8.23 (d, J = 7.5H, 1H), 8.37 (d, J = 7.2 Hz, 1H). 13C NMR (75 MHz, CDCl3): δ ppm 101.08, 117.39, 117.62, 123.94, 125.27, 125.38, 125.42, 126.85, 127.17, 132.18, 133.37, 133.85, 134.69, 138.89, 140.15, 152.5, 180.46, 183.94. HRMS (EI) m/z: calcd [M]+, 409.0088 (C20H9ClFN3O2S +); found, 409.0083.
2.3.5 4-(Ethylamino)anthra[1,2-c][1,2,5]thiadiazole-6,11-dione (7)
The pure compound was obtained as a purplish-red powder (yield 88%). Mp: 162–163 °C (EtOH). FT-IR (KBr, νmax cm−1): 1500.5 (C⚌C), 1562.2 (CO), 1674.1 (C⚌N). 1H NMR (300 MHz, CDCl3): δ ppm 1.48 (t, J = 7.2 Hz, 3H), 3.59–3.65 (m, 2H), 6.13 (s, 1H), 7.24 (s, 1H), 7.72 (t, J = 7.5 Hz, 1H), 7.80 (t, J = 7.5 Hz, 1H), 8.23 (d, J = 7.2 Hz, 1H), 8.35 (d, J = 7.2 Hz, 1H).13C NMR (75 MHz, CDCl3): δ ppm 14.31, 38.29, 98.14, 114.47, 126.88, 127.27, 132.78, 133.04, 134.63, 135.37, 139.90, 144.98, 149.85, 153.25, 180.44, 184.92. HRMS (EI) m/z: calcd [M]+, 309.0572 (C16H11N3O2S +); found, 309.0573.
2.3.6 4-(Propylamino)anthra[1,2-c][1,2,5]thiadiazole-6,11-dione (8)
The pure compound was obtained as a purplish-red powder (yield 74%). Mp: 186–187 °C (EtOH). FT-IR (KBr, νmax cm−1): 1500.5 (C⚌C), 1556.4 (CO), 1583.4 (CO), 1668.3 (C⚌N). 1H NMR (300 MHz, CDCl3): 1.11 (t, J = 7.5 Hz, 3H), 1.85 (q, J = 7.5 Hz, 2H), 3.41 (t, J = 6.6 Hz, 2H), 7.23 (s, 1H), 7.74 (t, J = 7.2 Hz, 1H), 7.79 (t, J = 7.2 Hz, 1H), 8.21 (d, J = 6.9 Hz, 1H), 8.34 (d, J = 7.8 Hz, 1H). 13C NMR (75 MHz, CDCl3): δ ppm 11.52, 22.31, 45.21, 98.03, 114.10, 126.76, 127.15, 132.58, 132.97, 134.44, 134.58, 139.73, 145.03, 149.49, 153.08, 180.25, 184.80. HRMS (EI) m/z: calcd [M]+, 323.0728 (C17H13N3O2S+); found, 323.0728.
2.3.7 4-(Diethylamino)anthra[1,2-c][1,2,5]thiadiazole-6,11-dione (9)
The pure compound was obtained as a purplish-red powder (yield 80%). Mp: 204–205 °C (EtOH). FT-IR (KBr, νmax cm−1): 1552.6 (CO), 1674.1 (C⚌N). 1H NMR (300 MHz, CDCl3): δ ppm 1.38 (t, J = 6.9 Hz, 6H), 4.00 (q, J = 6.9 Hz, 4H), 7.23 (s, 1H), 7.65 (t, J = 7.2 Hz, 1H), 7.74 (t, J = 7.2 Hz, 1H), 8.15 (d, J = 7.5 Hz, 1H), 8.31 (d, J = 7.8 Hz, 1H). 13C NMR (75 MHz, CDCl3): δ ppm 13.15, 47.40, 102.08, 112.84, 126.71, 127.27, 132.72, 132.81, 134.59, 135.12, 138.60, 145.78, 151.20, 155.44, 179.69, 185.22. HRMS (EI) m/z: calcd [M]+, 337.0885 (C18H15N3O2S +); found, 337.0888.
2.3.8 4-(Butylamino)anthra[1,2-c][1,2,5]thiadiazole-6,11-dione (10)
The pure compound was obtained as a purplish-red powder (yield 80%). Mp: 188–189 °C (EtOH). FT-IR (KBr, νmax cm−1): 1500.5 (C⚌C), 1550.7 (CO), 1670.2 (C⚌N). 1H NMR (300 MHz, CDCl3): δ ppm 1.04 (t, J = 7.2 Hz, 3H), 1.51–1.58 (m, 2H), 1.83 (t, J = 7.5 Hz, 2H), 3.57 (q, J = 6.6 Hz, 2H), 6.17 (s, 1H), 7.27 (s, 1H), 7.73 (t, J = 7.5 Hz), 7.81 (t, J = 7.5 Hz, 1H), 8.24 (d, J = 7.5 Hz, 1H), 8.36 (d, J = 7.5 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ ppm 13.78, 20.22, 30.95, 43.12, 97.83, 113.78, 126.59, 126.95, 132.28, 132.78, 134.14, 134.40, 139.45, 144.74, 149.17, 152.79, 180.01, 184.56. HRMS (EI) m/z: calcd [M]+, 337.0885 (C18H15N3O2S +); found, 337.0893.
2.3.9 4-((2-(Dimethylamino)ethyl)amino)anthra[1,2-c][1,2,5]thiadiazole-6,11-dione (11)
The pure compound was obtained as a purplish-red powder (yield 79%). Mp: 256–257 °C (EtOH). FT-IR (KBr, νmax cm−1): 1552.6 (CO), 1666.4 (C⚌N). 1H NMR (300 MHz, CDCl3): δ ppm 2.37 (s, 6H), 2.76 (t, J = 6.0 Hz, 2H), 3.55–3.61 (m, 2H), 6.88 (bd, 1H), 7.18 (s, 1H), 7.70 (td, J = 1.2 Hz, J = 7.5 Hz, 1H), 7.79 (td, J = 7.5 Hz, J = 1.2 Hz, 1H), 8.20 (dd, J = 7.5 Hz, J = 1.2 Hz, 1H), 8.34 (dd, J = 7.5 Hz, J = 1.2 Hz, 1H). 13C NMR (75 MHz, CDCl3): δ ppm 40.32, 45.18, 57.12, 98.26, 114.24, 126.83, 127.24, 132.54, 133.00, 134.55, 134.65, 139.75, 145.05, 149.61, 153.25, 180.38, 184.96. HRMS (EI) m/z: calcd [M]+, 352.0994 (C18H16N4O2S +); found, 352.0994.
2.3.10 4-((2-(Methylamino)ethyl)amino)anthra[1,2-c][1,2,5]thiadiazole-6,11-dione (12)
The pure compound was obtained as a purplish-red powder (yield 34%). Mp: 169–170 °C (EtOH). FT-IR (KBr, νmax cm−1): 1508.2 (C⚌C), 1558.4 (CO), 1670.2 (C⚌N). 1H NMR (300 MHz, CDCl3): δ ppm 2.54 (s, 3H) 3.07 (t, J = 5.4 Hz, 2H), 3.63–3.64 (m, 2H), 7.21 (s, 1H), 7.68–7.73 (m, 1H), 7.77–7.79 (m, 1H), 8.19 (d, J = 7.2 Hz, 1H), 8.33 (d, J = 7.8 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ ppm 35.76, 41.81, 49.34, 97.97, 113.65, 126.58, 126.92, 132.19, 132.78, 134.08, 134.42, 139.30, 144.75, 149.10, 152.70, 179.99, 184.42. HRMS (EI) m/z: calcd [M]+, 338.0837 (C17H14N4O2S +); found, 338.0839.
2.3.11 4-((3-(Dimethylamino)propyl)amino)anthra[1,2-c][1,2,5]thiadiazole-6,11-dione (13)
The pure compound was obtained as a purplish-red powder (yield 42%). Mp: 118–119 °C (EtOH). FT-IR (KBr, νmax cm−1): 1508.2 (C⚌C), 1558.4 (CO), 1670.2 (C⚌N). 1H NMR (300 MHz, CDCl3): δ ppm 1.94–1.96 (m, 2H), 2.35 (s, 6H) 2.55–2.57 (m, 2H), 3.64–3.66 (m, 2H), 7.18 (s, 1H), 7.70 (t, J = 7.2 Hz, 1H), 7.76 (t, J = 7.2 Hz, 1H), 8.21 (d, J = 7.5 Hz, 1H), 8.34 (d, J = 7.8 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ ppm 25.15, 43.51, 45.35, 58.35, 97.51, 113.24, 126.56, 126.95, 132.38, 132.65, 134.34, 134.37, 139.58, 145.46, 149.44, 153.07, 179.98, 184.86. HRMS (EI) m/z: calcd [M]+, 366.1150 (C19H18N4O2S +); found, 366.1139.
2.3.12 4-(Isobutylamino)anthra[1,2-c][1,2,5]thiadiazole-6,11-dione (14)
The pure compound was obtained as a purplish-red powder (yield 86%). Mp: 202–203 °C (EtOH). FT-IR (KBr, νmax cm−1): 1502.4 (C⚌C), 1556.4 (CO), 1666.4 (C⚌N). 1H NMR (300 MHz, CDCl3): δ ppm 1.10 (t, J = 6.6 Hz, 6H), 2.73 (t, J = 6.6 Hz, 1H), 3.53 (t, J = 6.6 Hz, 2H), 6.22 (s, 1H) 7.19 (s, 1H), 7.70 (t, J = 7.2 Hz, 1H), 7.78 (t, J = 7.5 Hz, 1H), 8.19 (d, J = 7.5 Hz, 1H), 8.32 (d, J = 7.8 Hz, 1H). 13C NMR (75 MHz, CDCl3): δ ppm 20.38, 28.33, 51.11, 98.14, 114.25, 126.84, 127.21, 132.72, 132.81, 132.99, 134.59, 139.84, 145.27, 149.00, 153.37, 180.36, 184.57. HRMS (EI) m/z: calcd [M]+, 337.0885 (C18H15N3O2S +); found, 337.0889.
2.3.13 4-(Pyrrolidin-1-yl)anthra[1,2-c][1,2,5]thiadiazole-6,11-dione (15)
The pure compound was obtained as a purplish-red powder (yield 83%). Mp: 256–257 °C (EtOH). FT-IR (KBr, νmax cm−1): 1552.6 (CO), 1674.1 (C⚌N). 1H NMR (300 MHz, CDCl3): δ ppm 1.61–1.62 (m, 2H), 2.11–2,16 (m, 6H), 7.07 (s, 1H), 7.70 (td, J = 7.5 Hz, J = 1.5 Hz, 1H), 7.77 (td, J = 7.8 Hz, J = 1.5 Hz, 1H), 8.18 (dd, J = 7.5 Hz, J = 1.2 Hz, 1H), 8.37 (dd, J = 7.8 Hz, J = 0.9 Hz, 1H). 13C NMR (75 MHz, CDCl3): δ ppm 28.54, 51.49, 94.31, 101.67, 112.40, 126.73, 127.20, 132.63, 132.89, 134.51, 138.64, 135.09, 185.09. HRMS (EI) m/z: calcd [M]+, 335.0728 (C18H13N3O2S+); found, 335.0724.
2.3.14 4-(Piperidin-1-yl)anthra[1,2-c][1,2,5]thiadiazole-6,11-dione (16)
The pure compound was obtained as a purplish-red powder (yield 85%). Mp: 207–208 °C (EtOH). FT-IR (KBr, νmax cm−1): 1527.5 (C⚌C), 1556.4 (CO), 1666.4 (C⚌N). 1H NMR (300 MHz, CDCl3): δ ppm 1.82–1.83 (m, 6H), 4.06–4.08 (m, 4H), 2.73 (s, 4H), 4.12 (s, 4H), 7.52 (s, 1H), 7.72 (td, J = 7.5 Hz, J = 1.5 Hz, 1H), 7.80 (td, J = 7.5 Hz, J = 1.5 Hz, 1H), 8.23 (d, J = 7.2 Hz, 1H), 8.36 (dd, J = 7.8 Hz, J = 0.9 Hz, 1H). 13C NMR (75 MHz, CDCl3): δ ppm 24.60, 26.19, 51.413, 104.88, 114.78, 126.86, 127.34, 132.83, 132.89, 133.00, 134.67, 138.42, 147.81, 185.05. HRMS (EI) m/z: calcd [M]+, 349.0885 (C19H15N3O2S +); found, 349.0889.
2.3.15 4-Morpholinoanthra[1,2-c][1,2,5]thiadiazole-6,11-dione (17)
The pure compound was obtained as a dark-brown powder (yield 70%). Mp: 259–260 °C (EtOH). FT-IR (KBr, νmax cm−1): 1525.6 (C⚌C), 1556.4 (CO), 1668.3 (C⚌N). 1H NMR (300 MHz, CDCl3): δ ppm 3.98–4.03 (m, 4H), 4.05–4.06 (m, 4H), 7.51 (s, 1H), 7.76 (t, J = 7.2 Hz, 1H), 7.81 (t, J = 7.2 Hz, 1H), 8.23 (d, J = 7.2 Hz, 1H), 8.35 (d, J = 7.2 Hz, 1H). 13C NMR (75 MHz, CDCl3): δ ppm 49.85, 66.89, 105.28, 116.32, 126.97, 127.40, 132.65, 133.31, 134.64, 134.78, 138.12, 147.50, 150.79, 154.60, 180.50, 184.60. HRMS (EI) m/z: calcd [M]+, 351.0678 (C18H13N3O3S +); found, 351.0682.
2.3.16 4-Thiomorpholinoanthra[1,2-c][1,2,5]thiadiazole-6,11-dione (18)
The pure compound was obtained as a light-brown powder (yield 62%). Mp: 219–220 °C (EtOH). FT-IR (KBr, νmax cm−1): 1521.7 (C⚌C), 1556.4 (CO), 1662.5 (C⚌N). 1H NMR (300 MHz, CDCl3): δ ppm 2.85–2.88 (m, 4H), 4.34–4.40 (m, 4H), 7.36 (s, 1H), 7.69 (t, J = 7.5 Hz, 1H), 7.76 (t, J = 7.5 Hz, 1H), 8.15 (d, J = 7.2 Hz, 1H), 8.35–8.38 (d, J = 7.5 Hz, 1H). 13C NMR (75 MHz, CDCl3): δ ppm 27.28, 52.49, 105.21, 115.39, 126.79, 127.24, 132.50, 134.51, 134.66, 138.03, 146.57, 150.45, 154.59, 180.02, 184.93. HRMS (EI) m/z: calcd [M]+, 367.0449 (C18H13N3O2S2+); found, 367.0444.
2.3.17 4-(4-Methylpiperazin-1-yl)anthra[1,2-c][1,2,5]thiadiazole-6,11-dione (19)
The pure compound was obtained as a purplish-red powder (yield 60%). Mp: 200–201 °C (EtOH). FT-IR (KBr, νmax cm−1): 1506.3 (C⚌C), 1558.4 (CO), 1670.2 (C⚌N). 1H NMR (300 MHz, CDCl3): δ ppm 2.40 (s, 3H), 2.69–2.71 (m, 4H), 4.09–4.11 (m, 4H), 7.53 (s, 1H), 7.73 (t, J = 7.8 Hz, 1H), 7.80 (t, J = 7.8 Hz, 1H), 8.23 (d, J = 7.5 Hz, 1H), 8.35 (d, J = 7.5 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ ppm 46.05, 49.30, 54.8, 105.18, 115.38, 126.67, 127.11, 132.26, 133.02, 134.24, 134.55, 137.81, 147.21, 150.35, 154.29, 180.20, 184.48. HRMS (EI) m/z: calcd [M]+, 364.0994 (C19H16N4O2S+); found, 364.0986.
2.3.18 4-(4-Ethylpiperazin-1-yl)anthra[1,2-c][1,2,5]thiadiazole-6,11-dione (20)
The pure compound was obtained as a purplish-red powder (yield 85%). Mp: 256–257 °C (EtOH). FT-IR (KBr, νmax cm−1): 1556.4 (CO), 1666.4 (C⚌N). 1H NMR (300 MHz, CDCl3): δ ppm 1.17 (t, J = 7.2 Hz, 3H), 2.53 (q, J = 7.2 Hz, 2H), 2.71–2.73 (m, 4H), 4.10–4.12 (m, 4H), 7.52 (s, 1H), 7.73 (t, J = 7.5 Hz, 1H), 7.80 (t, J = 7.5 Hz, 1H), 8.23 (d, J = 7.5 Hz, 1H), 8.35 (d, J = 7.5 Hz, 1H). 13C NMR (75 MHz, CDCl3): δ ppm 11.99, 49.58, 52.42, 52.83, 105.25, 113.92, 126.92, 127.38, 132.67, 133.20, 134.68, 134.76, 138.22, 147.53, 150.72, 153.81 180.46, 184.86. HRMS (EI) m/z: calcd [M]+, 378.1150 (C20H18N4O2S +); found, 378.1151.
2.3.19 4-(4-(2-Chlorophenyl)piperazin-1-yl)anthra[1,2-c][1,2,5]thiadiazole-6,11-dione (21)
The pure compound was obtained as a purplish-red powder (yield 29%). Mp: 251–252 °C (EtOH). FT-IR (KBr, νmax cm−1): 1508.2 (C⚌C), 1558.4 (CO), 1670.2 (C⚌N). 1H NMR (300 MHz, CDCl3): δ ppm 3.33–3.36 (m, 4H), 4.23–4.25 (m, 4H), 7.24–7.32 (m, 1H), 7.42 (dd, J = 7.8 Hz, J = 1.5 Hz, 1H), 7.56 (s, 1H), 7.76 (t, J = 7.8 Hz, 1H), 7.81 (t, J = 7.5 Hz, 1H), 8.22 (d, J = 7.5 Hz, 1H), 8.35 (d, J = 7.8 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ ppm 49.68, 51.16, 105.38, 115.58, 120.48, 124.33, 126.69, 127.10, 127.33, 127.61, 127.75, 128.95, 130.81, 132.24, 133.04, 134.15, 134.55, 137.74, 147.31, 148.55, 180.20, 184.32. HRMS (EI) m/z: calcd [M]+, 460.0761 (C24H17ClN4O2S +); found, 460.0761.
2.3.20 4-((4-Aminobenzyl)amino)anthra[1,2-c][1,2,5]thiadiazole-6,11-dione (22)
The pure compound was obtained as a purplish-red powder (yield 56%). Mp: 187–188 °C (EtOH). FT-IR (KBr, νmax cm−1): 1556.4 (CO), 1664.4 (C⚌N). 1H NMR (300 MHz, CDCl3): δ ppm 4.60 (d, J = 5.1 Hz, 2H), 6.71 (d, J = 8.1 Hz, 2H), 7.23 (d, J = 8.1 Hz, 2H), 7.35 (s, 1H), 7.72 (t, J = 7.2 Hz, 1H), 7.78 (t, J = 7.2 Hz, 1H), 8.22 (d, J = 7.2 Hz, 1H), 8.35 (d, J = 7.2 Hz, 1H). 13C NMR (75 MHz, CDCl3): δ ppm 47.54, 98.55, 114.59, 115.68, 126.34, 126.88, 127.27, 129.50, 132.70, 133.09, 134.52, 134.66, 139.84, 144.74, 146.81, 149.66, 153.18, 180.50, 184.89. HRMS (EI) m/z: calcd [M]+, 386.0837 (C21H14N4O2S +); found, 386.0843.
2.3.21 4-((3,5-Dimethoxybenzyl)amino)anthra[1,2-c][1,2,5]thiadiazole-6,11-dione (23)
The pure compound was obtained as a purplish-red powder (yield 62%). Mp: 220–221 °C (EtOH). FT-IR (KBr, νmax cm−1): 1550.7 (CO), 1662.5 (C⚌N). 1H NMR (300 MHz, CDCl3): δ ppm 3.90 (d, J = 3.9 Hz, 6H), 4.66 (d, J = 5.4 Hz, 2H), 6.89 (d, J = 8.1 Hz, 1H), 6.99 (t, J = 8.1 Hz, 2H), 7.35 (s, 1H), 7.75 (t, J = 7.8 Hz, 1H), 7.80 (t, J = 7.8 Hz, 1H), 8.22 (d, J = 7.8 Hz, 1H), 8.35 (d, J = 7.8 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ ppm 47.56, 55.96, 55.99, 98.43, 111.09, 111.44, 114.39, 120.31, 126.65, 127.01, 128.58, 128.78, 132.28, 132.94, 134.09, 134.49, 139.41, 144.30, 148.98, 149.742, 152.79, 180.24, 184.45. HRMS (EI) m/z: calcd [M]+, 431.0940. (C23H17N3O4S +); found, 431.0948.
2.4 Initial in vitro cytotoxicity screening of compounds
In an initial attempt to investigate the cytotoxic activities of the newly synthesized analogues 3 ∼ 23, the 50% inhibitory concentration (IC50) values of each compound were measured by SRB and MTT assays against two prostate cancer cell lines (PC-3 and DU-145) obtained from the American Type Culture Collection (ATCC). Both cell lines were maintained in RPMI-1640 culture medium containing 5% heat-inactivated fetal calf serum in an atmosphere of 5% CO2 in a humidified incubator. The tetrazolium reagent (MTT; 3-(4, 5-di-methylthiazol)-2,5-diphenyltetrazolium bromide, USB) was designed to yield a colored formazan complex upon metabolic reduction by viable cells (Denizot and Lang, 1986; Mosmann, 1983). Approximately 2 × 103 cells were plated into each well of a 96-well plate and incubated in an 5% CO2 atmosphere at 37 °C for 24 h. To assess the in vitro cytotoxicity, each compound was dissolved in DMSO, prepared immediately before the experiments, and diluted into complete medium before addition to cell cultures. Test compounds and the vehicle control (0.1% DMSO) were then added to the culture medium at various designated concentrations. After 72 h, an amount of 100 μL of an MTT solution was added to each well, and samples were incubated at 37 °C for 4 h. A 100 μL solution of DMSO was added to each well and incubated at 37 °C for another 3 h. The absorbance at 570 nm was measured using an enzyme-linked immunosorbent assay (ELISA) reader.
2.5 One-dose and Five-dose assays of the NCI-60 human tumor cell lines Screen program
The growth inhibition capabilities of our selected compounds were tested against a panel of 60 human cancer cell lines under the NCI Drug Screen Program. Compounds were tested initially at a single high dose (10 μM) concentration, then those who showed significant growth inhibition capabilities and fulfilled the pre-determined threshold inhibition criteria of the NCI proceeded to the Five-dose assay. Details of the methodology are described in our previous publications (Ali et al., 2016b; Huang et al., 2012, 2009, 2010; Lee et al., 2013a, 2012a, 2012b, 2015).
2.6 Selectivities of the compounds tested by the Five-dose NCI-60 assays towards different types of cancer
The panel of NCI-60 cell lines used to evaluate our compounds by the Five-dose assays includes nine tumor subpanels; leukemia, melanoma, lung, colon, kidneys, ovaries, breasts, prostate, and central nervous system (Stinson et al., 1992). In order to investigate the selectivity of our compounds towards each cancer type, we calculated their selectivity ratios by dividing the average GI50values of each particular subpanel by the average GI50value of all the tested cell lines (mean graph midpoint (MID)) (Ali et al., 2016b).
2.7 Identification of the drugs with similar profile to our compounds using COMPARE analysis
Results from the NCI five-dose cytotoxicity experiments of our compounds were used as “seed” in the COMPARE algorithms to correlate to those in the NCI databases using the Pearson’s correlation coefficient calculations according to our previous publication (Ali et al., 2016b).
2.8 Apoptotic morphological changes in prostate cancer cells
DU-145 cells were plated at a density of 5 × 105 cells per well in a 10-cm dish and then treated with various concentrations of compound 11 for 48 h. Cells were directly examined and photographed under a phase-contrast microscope (Cheng et al., 2012).
2.9 Western blot assay
Treated cells were collected and washed with PBS. After centrifugation, cells were lysed in lysis buffer. The lysates were incubated on ice for 30 min and centrifuged at 12 × 104 g for 15 min. Supernatants were collected, and protein concentrations were determined using the Bradford assay. Protein samples were electrophoresed on 12% sodium dodecylsulfate (SDS)-polyacrylamide gels and transferred to a polyvinylidene difluoride membrane (Ali et al., 2016a; Ali et al., 2020). Immunoreactivity was detected using a Western blot chemiluminescence regent system (Huang et al., 2005) β-actin was used as a loading control.
2.10 Flow cytometry
DU-145 cells (105 cells/well) were treated with various concentrations of compound 11 (0.25, 0.5, or 1.5 µM) or with DMSO for 48 h. Cells were washed with phosphate-buffered saline (PBS) and removed using trypsin-EDTA (Invitrogen, Carlsbad, CA) (Ali et al., 2020). Cells were then washed with PBS, fixed, and permeabilized in 3 ml of 70% ice-cold ethanol cooled at −20 °C. After an overnight incubation, cells were washed again in PBS then stained for 30 min at room temperature with 50 μg/mL propidium iodide (PI), and 50 μg/mL DNase-free ribonuclease A in PBS for 30 min at room temperature. Cells were analyzed using a FACScan flow cytometer (Riccardi and Nicoletti, 2006).
2.11 Molecular modelling
We used the crystal structure of the DNA topoisomerase I (TOP1) from the RCSB protein databank; code: 1K4T (Staker et al., 2002), and that of the DNA topoisomerase III (TOP3) from the RCSB protein databank; code: 4CGY (Bocquet et al., 2014). We established hypothetical binding models of our test compounds to the obtained X-ray crystal structures using molecular docking and calculated the docking score for each compound.
3 Results and discussion
3.1 Chemistry
According to our previous series of studies (Chang et al., 2011; 2013a, 2013b; Huang et al., 2009; Lee et al., 2015; 2013b), the structural complexity of NSC745885, arising from the presence of a tetraheterocyclic ring restricted most structure–activity relationship (SAR) modifications to derivatization of the parent lead structure rather than de novo chemical synthesis. We planned our strategy of synthesizing N-substituted anthra[1,2-c][1,2,5]thiadiazole-6,11-diones as outlined in Scheme 1. Compound 1 was prepared in good yield by the reaction of 1,2-diaminoanthraquinone with thionyl chloride and trimethylamine. In an attempt to prepare the final products (3 ∼ 23) in the highest yields, two different methods of synthesis were utilized as follows: (i) an aniline series was obtained by treating 1 with copper acetate as a catalyst in dimethylformamide (DMF) for several hours to yield the corresponding N-substituted side chains; and (ii) 1 was reacted by nucleophilic substitution of chlorine atoms to obtain 2 and then with a series of nitrogen derivatives in ethylene glycol to produce diverse N-substituted anthra[1,2-c][1,2,5]thiadiazole-6,11-dione derivatives (3 ∼ 23).
3.2 Initial in vitro cytotoxicity screening of compounds and their structure activity relationship (SAR) observations
To initially screen the active members of our library of compounds, we evaluated their cytotoxicities against two androgen-independent human prostate cancer cell lines namely PC-3 and DU-145 (Table 1). Generally, most of the tested compounds demonstrated potent cytotoxic activities in both cell lines. From our structure activity relationship (SAR) observations, we found that compounds with the alkyl amine-side chain substitutions (alkyl nitrogen series) exhibited greater antitumor activities (IC50 values of 0.40 to > 15 µM) than those with the aminated benzene group substitutions (phenyl nitrogen series). In addition, results of the alkyl nitrogen series indicated that the length of the carbon side chain of the anthra[1,2-c][1,2,5]thiadiazole-6,11-dione scaffold played an important role in enhancing the cytotoxic activities of our synthesized compounds. For the phenyl nitrogen and benzyl nitrogen series, increasing the length between the core scaffold and the benzene side chain by adding a CH3 group (22 and 23) may have increased the inhibitory activity against the prostate cancer cell lines. Compounds 3 (NSC763958) and 11 (NSC763967) were the most active derivatives with IC50 values of 0.21 and 0.40 µM against PC-3 cells and 0.16 and 0.70 µM against DU-145 cells, respectively. We further investigated the effect of compound 11 on the viability of DU-145 cells and found that it exerts potent cytotoxic effect on cells in a time-dependent and dose-dependent manner (Fig. 2 (a - b)).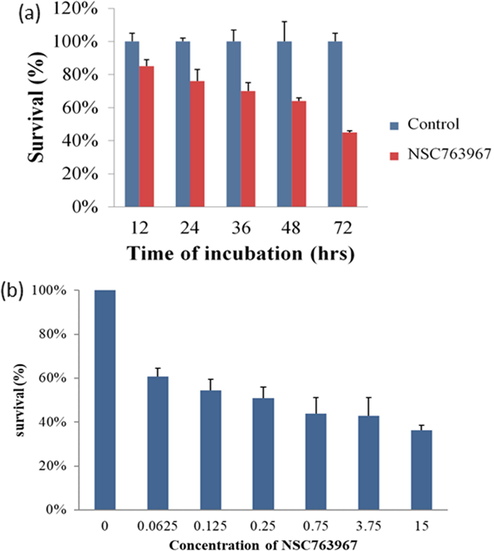
Time- and concentration-dependent cytotoxic effects of compound 11 on the human prostate cancer cell line DU-145 determined by the MTT assay after exposure of cells to the IC50 dose of compound 11 for 12, 24, 36, 48, and 72 h (a) or to 0.0625, 0.125, 0.25, 0.75, 3.75, 15 μM of compound 11 for 72 h (b).
3.3 Evaluating the cytotoxic activities of our compounds by the One-dose and Five-dose assays of the NCI-60 human tumor cell lines Screen program
As shown in Table 2, compounds 3 (NSC763958), 5 (NSC763965), 6 (NSC763954), 10 (NSC764639), 11 (NSC763967), 14 (NSC764640), 15 (NSC763966), 17 (NSC757967), and 18 (NSC763952) were selected for evaluation by the One-dose assays of the NCI. Results for each compound were reported as the percentage of growth of cells treated with a concentration of 10 μM of the compound compared to the percentage of growth of the untreated control cells. Among the tested compounds, 11 was the most potent one which reduced the growth of all tested cell lines by a mean of −49.52%. The most inhibited cell lines were NCI-H552, COLO 205, LOX IMVI, M14, SK-MEL-5, SN12C, DU145, and BT-549 which showed percentages of growth of −91.81%, −99.38%, −90.91%, −96.08%, −98.79%, −95.78%, −95.45%, and −99.43, respectively, after treatment with compound 11. Compound 10 achieved inhibitory effects against the SK-MEL-5 melanoma cell line and reduced its growth percentage to −57.85%. Compound 14 was effective against the melanoma cell lines MDA-MB-435, SK-MEL-5, and UACC-62 and reduced their growth percentages to −67.52%, −81.47%, and −44.11%, respectively.
Tested compounds that met the predetermined growth inhibition criteria of the NCI were selected to proceed to the Five-dose assays of the NCI-60 Human Tumor Cell Lines Screen program. Compounds 3 (NSC763958), 6 (NSC763954), 10 (NSC764639), 11 (NSC763967), 14 (NSC764640), and 17 (NSC757967) were selected for the Five-dose assays. Dose response curves were plotted for every compound using the percentages of growth of every tested cell line after the treatment with different concentrations of every compound (Fig. 3), and the 50% growth inhibitory concentration (GI50), total growth inhibition (TGI), and 50% lethal concentration (LC50) were calculated for every tested compound, and their values are presented in Table 3. Interestingly, compound 11 exhibited remarkable potent cytotoxic activity against most of the tested cell lines of the nine different cancer subpanels with GI50 values in the range of 0.145 ∼ 2.54 μM with a sub-micromolar GI50 mean value of 0.63 μM. Compound 11 showed high activity against leukemia (GI50 range is 0.21 ∼ 0.43 μM), non-small-cell lung cancer (GI50 range is 0.24 ∼ 1.53 μM), colon cancer (GI50 range is 0.20 ∼ 1.62 μM), central nervous system (CNS) cancer (GI50 range is 0.20 ∼ 1.48 μM), melanoma (GI50 range is 0.15 ∼ 1.63 μM), ovarian cancer (GI50 range is 0.19 ∼ 1.29 μM), renal cancer (GI50 range is 0.16 ∼ 1.46 μM), prostate cancer (GI50 range is 0.17 ∼ 0.25 μM), and breast cancer (GI50 range is 0.25 ∼ 1.41 μM). Such results indicate the exceptional high cytotoxicity of compound 11 toward the NCI-60 human tumor cell lines, extend our initial screening observations, and emphasize the potential of this compound as a future anticancer drug. Accordingly, we aimed to characterize further and understand more the biological activities of compound 11 in this study. Along the same line, we found that compound 3 exhibited potent cytotoxic capabilities with GI50 values in the range of 0.0668 ∼ 40.6 μM with a mean of 2.23 μM. The leukemia subpanel was quite sensitive to compound 3, while the non-small cell lung cancer, colon, CNS, melanoma, ovarian, renal, prostate, and breast panels were less sensitive. For compound 6, the GI50 values were in the range of 0.625 μM (non-small cell lung cancer: HOP-92) to > 100 μM. Compound 10 showed GI50 values in the range of 1.19 μM (melanoma: SK-MEL-5) to 94.9 μM (melanoma: SK-MEL-28). Compound 14 showed GI50 values in the range of 0.627 μM (melanoma: SK-MEL-5) to 9.00 μM (melanoma: SK-MEL-28). Compound 17 showed GI50 values in the range of 0.373 μM (melanoma: LOXIMVI) to >100 μM (non-small cell lung cancer: EKVX and NCI-H322M; ovarian cancer: SK-OV-3; and renal carcinoma: TK-10).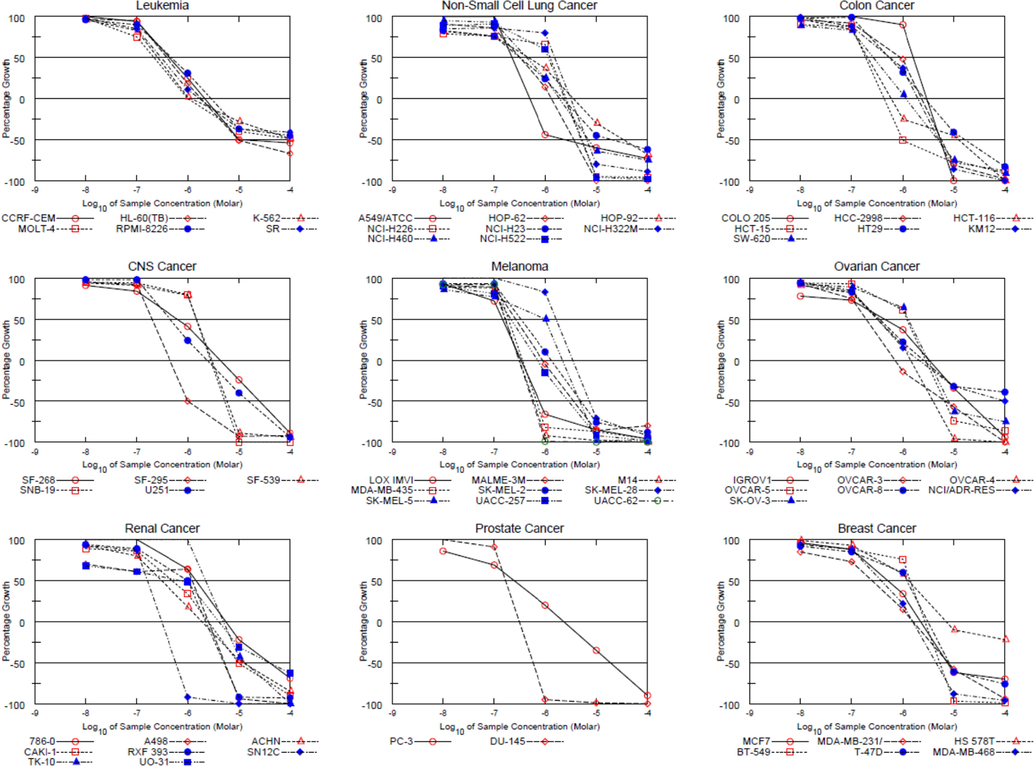
Dose response curves of the NCI-60 human cancer cell lines after exposure to compound 11 (RV59, NSC763967) obtained from the Five-dose assays.
3.4 Selectivities of compounds tested by the Five-dose assays towards different types of cancer
We aimed to determine the selectivities of our compounds towards different types of cancer of the NCI-60 cell lines panel by calculating the selectivity ratios of the tested compounds using the obtained GI50 values (Ali et al., 2016b). Every tested compound was identified as “highly selective” to the cell line panel if its ratio is more than 6, identified as “moderately selective” if its ratio is between 3 and 6, and identified as “non-selective” if its ratio is less than 3. According to Table 4, compounds 3, 6, 10, 11, 14, and 17 exhibited broad spectrum growth inhibiting activities towards the tested cancer subpanels, except the following observations. Compounds 3, and 6 were highly selective for cell lines of the Leukemia subpanel with selectivity ratios of 7.96, and 7.88, respectively. Similarly, compound 6 was highly selective for cell lines of the Prostate Cancer subpanel with a selectivity ratio of 6.06. Compound 11 showed moderate selectivity towards cell lines of the Prostate Cancer subpanel with a selectivity ratio of 3.00. Compound 17 showed moderate selectivity towards cell lines of the Ovarian Cancer subpanel with a selectivity ratio of 4.05.
Compound Subpanel Cell line
3 (NSC763958)
6 (NSC763954)
10 (NSC764639)
11 (NSC763967)
14 (NSC764640)
17 (NSC757967)
Subpanel MIDb
Selectivity ratio
Subpanel MIDb
Selectivity ratio
Subpanel MIDb
Selectivity ratio
Subpanel MIDb
Selectivity ratio
Subpanel MIDb
Selectivity ratio
Subpanel MIDb
Selectivity ratio
Leukemia
0.28
7.96
1.70
7.88
4.18
2.13
0.34
1.85
3.16
0.98
5.57
2.06
Non-small cell lung cancer 3.86
0.58
20.0
0.67
5.34
1.67
0.71
0.89
2.71
1.15
12.52
0.91
Colon cancer
3.51
0.64
28.57
0.47
4.99
2.98
0.61
1.03
3.06
1.02
25.83
0.44
CNS cancer
2.50
0.89
5.44
2.46
10.4
0.86
0.90
0.70
3.30
0.94
9.38
1.22
Melanoma
2.12
1.05
25.60
0.52
14.65
0.61
0.45
1.40
3.08
1.01
6.04
1.90
Ovarian cancer
2.17
1.03
5.28
2.54
17.56
0.51
0.59
1.07
3.80
0.82
2.83
4.05
Renal cancer
2.73
0.82
23.62
0.57
14.78
0.60
0.99
0.64
3.22
0.97
24.33
0.47
Prostate Cancer
1.19
1.87
2.21
6.06
3.76
2.37
0.21
3.00
3.04
1.02
6.93
1.65
Breast cancer
1.70
1.31
8.10
1.65
4.57
1.95
0.84
0.63
2.62
1.19
9.65
1.19
MID a
2.23
13.39
8.91
0.63
3.11
11.45
3.5 Cytotoxicity of compound 11 towards normal human cells
Due to the exceptional potency of compound 11 and its promising role as an effective anticancer drug, we aimed to evaluate its cytotoxicity against three normal human cell lines and compare its results to those of the standard chemotherapeutic drug doxorubicin (DXR) to get insight into its safety profile. We observed that the survival of normal human urothelial cells (SV-HUC-1) was about 80% of the untreated population after 72 h of exposure to a concentration of 15 µM of compound 11, whereas their survival was about 42% for the same exposure time and concentration of DXR. For the normal human prostatic stromal myofibroblasts cells (WPMY-1), the survival percentages were about 50% and 19% of the untreated cells, after an exposure time of 72 h to a concentration of 15 µM of either compound 11 or DXR, respectively. Similarly, the survival percentages of normal human prostate epithelial cells (RWPE-1) were about 52% and 30% of the untreated cells, after exposure to 15 µM of either compound 11 or DXR, respectively, for 72 h. Collectively, these data show that compound 11 is a potent inhibitor of cancer cell growth while being less toxic to normal human cells compared to the standard chemotherapeutic drug doxorubicin.
3.6 Discovering drugs with similar profile to compound 11 using COMPARE analysis
Drugs exhibiting similar activity profiles in the NCI-60 cell lines experiments usually possess similar mechanisms of action and modes of drug resistance. A data mining strategy using the “COMPARE analysis” method was proved to be an effective and facile way to predict similar drugs to the test compound and the underlying molecular targets (Ali et al., 2016b). We used the fingerprint of GI50 values of compound 11 in the Five-dose assay of NCI-60 cell lines as a “seed” to correlate to drugs and compounds in the “Marketed Drugs”, “Investigational Drugs”, “Mechanistic Set”, “Standard Agents”, “Synthetic Compounds”, “BEC Referral Set”, and “Diversity Set” sets of the NCI database. Selected correlated drugs and compounds of importance to our study are displayed in Table 5. Similarities between our test compound and correlated drugs are presented as “Pearson’s Correlation Coefficient” values, where a value of 1 denotes perfect positive similarity (correlation), a value of zero denotes no similarity (correlation), and a value of −1 denotes negative similarity (correlation). We observed that compound 11 is positively correlated to Bafilomycin antibiotic, Nifuroxazide, Lapachol, Tryptanthrin, Austrocortirubin, Methotrexate, Aminopterin, Combretastatin A-4, Estramustine, Tipifarnib, Raltitrexed, Cyclopentenyl cytosine, Thioguanine, Cytosine arabinoside. These findings suggest further the multi-target nature of compound 11, however, the top similar drugs in our list exert their cytotoxic actions by hampering the cell cycle and causing apoptosis to cancer cells, suggesting strongly that compound 11 possess the same effects on the cell cycle and apoptosis of cancer cells. In order to verify these findings, we aimed to characterize further compound 11 by performing further experiments to discover the mechanisms of its underlying potent activity as follows in this manuscript.
Pearson’s Correlation Coefficient
Count of cell lines used in correlation
Drug/Compound name
Mechanisms of action and molecular targets of the drug/compound
0.56
47
Bafilomycin antibiotic
Induces the caspase-independent apoptosis and inhibits cell growth and autophagy (Yan et al., 2016; Yuan et al., 2015)
0.55
57
Nifuroxazide
Induces G2/M phase arrest and cell apoptosis (Zhu et al., 2016), and targets ALDH1 expressing cancer cells (Sarvi et al., 2018)
0.51
42
Lapachol
Inhibits glycolysis by targeting the pyruvate kinase M2 (PKM2) (Shankar Babu et al., 2018)
0.49
58
Tryptanthrin
Inhibits ERK1/2 and p38 pathways (Shankar et al., 2020)
0.49
59
Austrocortirubin
Induces DNA damage during the G0/G1, S, and G2 phases of cell cycle resulting in cessation of mitosis at the G2/M phase checkpoint (Wang et al., 2015)
0.49
59
Methotrexate
Inhibits dihydrofolate reductase (DHFR) (Hagner and Joerger, 2010)
0.47
59
Aminopterin
Inhibits dihydrofolate reductase (DHFR) (Mathews, 2012)
0.45
49
Combretastatin A-4
Induces apoptosis (activates caspase-3), induces G2-M phase arrest with sub-G1 formation (Shen et al., 2010), and inhibits angiogenesis and suppresses the vascular endothelial growth factor (VEGF)-induced proliferation, migration and capillary-like tube formation of cancer cells (Su et al., 2016)
0.45
59
Estramustine
Is an alkylating agent conjugated to estradiol that causes anaphase arrest to prostate cancer (Bilusic, 2016)
0.44
59
Tipifarnib
Inhibits the Farnesyl transferase (FTase) (Karp and Lancet, 2008)
0.43
50
Raltitrexed
Inhibits the thymidylate synthase (TS), thus hampering the formation of DNA and RNA of cells (Van Cutsem et al., 2002)
0.43
58
Cyclopentenyl cytosine
In cells, it is phosphorylated and transformed into cyclopentenyl cytosine 5′-triphosphate which inhibits the cytidine triphosphate (CTP) synthase), thus hampering the formation of DNA and RNA of cells (Schimmel et al., 2007)
0.42
59
Thioguanine
It is an analogue of guanine that disrupts the DNA by its incorporation into DNA in place of guanine (Nelson et al., 1975)
0.4
58
Cytosine arabinoside
Converted intracellularly into cytosine arabinoside triphosphate which incorporates into DNA and leads to cell death (Rein and Rizzieri, 2014)
3.7 Morphological changes and apoptosis caused by compound 11 to prostate cancer cells
To investigate whether compound 11 has any effect on the apoptosis of cells, we studied the cellular morphological changes due to the treatment by compound 11 on the prostate cancer cells DU-145. As shown in Fig. 4, there were significant morphological changes to DU-145 cells typical to apoptosis such as rounding and shrinkage of cells, membrane blebbing, presence of apoptotic bodies, and cell detachment from the culture wells. The observed morphological changes were dependent on the treatment concentration of compound 11, confirming the apoptotic capabilities of the compound.
Apoptotic morphological changes of DU-145 cells induced by compound 11 after exposure to concentrations of either 0.25, 1, or 2.5 µM of the compound for 48 h.
3.8 Detection of the change in expression of the apoptosis-related proteins after treatment with compound 11
To gain insight into the observed apoptotic activity of compound 11, we investigated the change in expression of the apoptosis-related proteins cyclin D1 (important regulator of cell proliferation and cell cycle; inhibitors of cyclin D1 induce apoptosis) and COX-2 (promotes cancer cell growth and carcinogenesis; inhibitors of COX-2 induce apoptosis) by Western blot analysis. We found that the expression levels of both cyclin D1 and COX-2 were inhibited in a concentration-dependent manner after exposure to compound 11 (Fig. 5 (a-b)). Inhibition of cyclin D1 by compound 11 suggests that it affects the cell cycle of cancer cells, accordingly, we aimed to investigate the effect of compound 11 on the cell cycle of DU-145 cells as presented in the following section. Inhibition of COX-2 by compound 11 may result in apoptosis that occurs through COX-2 independent pathways (Sobolewski et al., 2010), highlighting the multi-target properties of our compound.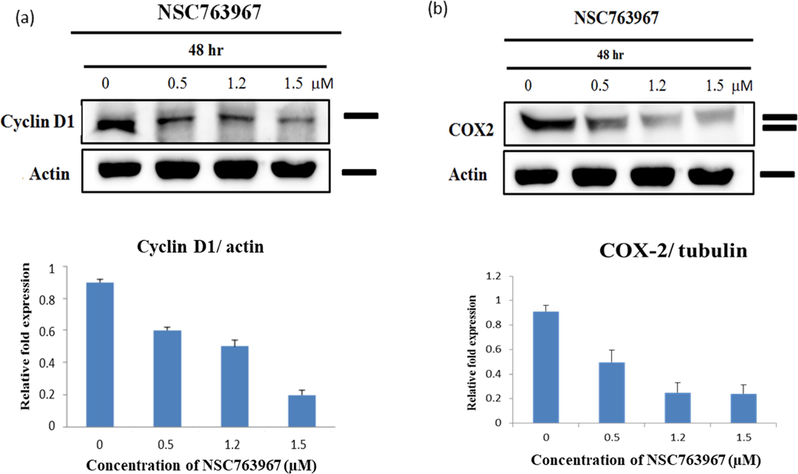
Western blot of whole-cell extracts of DU-145 cells after treatment with different concentrations of compound 11 and their corresponding relative expression quantification. (a) Compound 11 inhibited expression of cyclin D1 in a concentration-dependent manner. (b) Compound 11 inhibited expression of COX-2 in a concentration-dependent manner.
3.9 Effects of compound 11 on the cell cycle of prostate cancer cells
In order to investigate whether compound 11 exhibits its antiproliferative effect against the prostate cancer cells through induction of apoptosis, we performed flow cytometric experiments to analyze the cell cycle of treated DU-145 cells. As shown in Fig. 6, the effects of compound 11 on the cell cycle were concentration dependent. We observed an increase in the accumulation of cells in the G0/G1 phase of the cell cycle due to exposure to compound 11 with a significant reduction of cells in the S and G2/M phases. Also, compound 11 produced a high population of apoptotic cells (86.04%). Such findings support the previous ones regarding the mechanisms of cytotoxicity of compound 11. An other study involving 4-amine substituted derivatives of anthra[2,1-c][1,2,5]thiadiazole-6,11-dione showed that such compounds may modulate the cell cycle of cancer cells (Dong et al., 2017), supporting our current findings.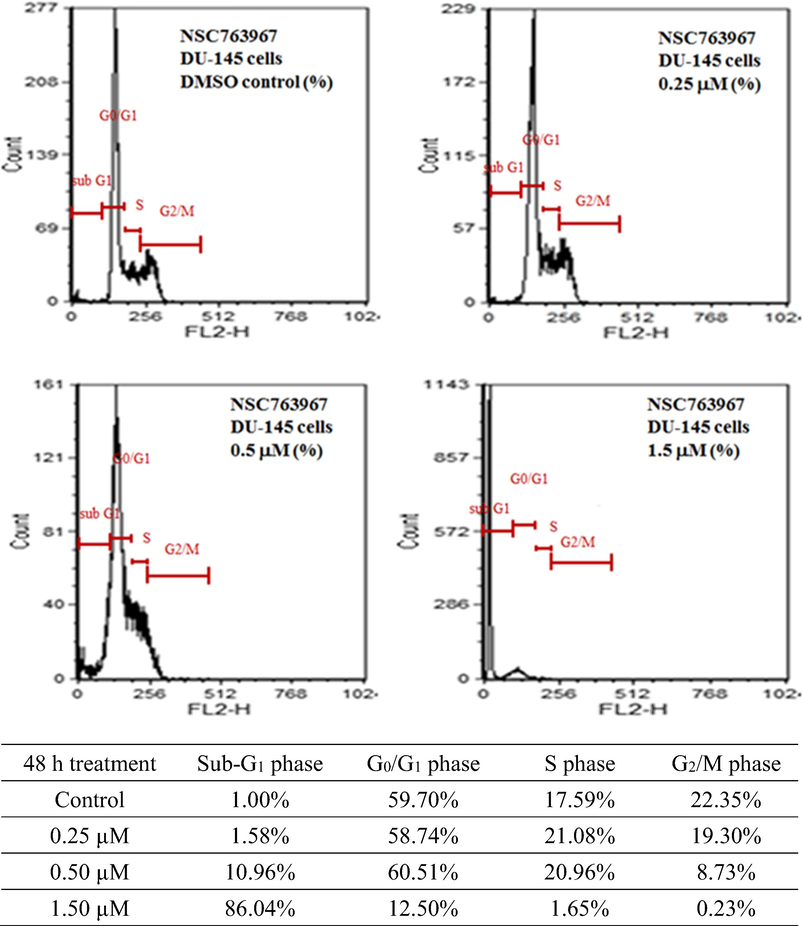
Effects of different concentrations of compound 11 on the cell cycle distribution of DU-145 cells determined by propidium iodide staining for DNA followed by flow cytometry.
3.10 Molecular modeling studies for topoisomerase inhibition by our compounds
Drugs with similar structure to our compounds, such as topotecan, camptothecin, and doxorubicin proved to be potent inhibitors to different types of topoisomerase by interfering with the topoisomerase-DNA complexes, leading to DNA damage and death of cell (Bali et al., 2018; Foglesong et al., 1992; Li et al., 2017; Marinello et al., 2018). It is worth noting that other 4-thio/sulfonyl and 4-amine substituted derivatives of anthra[2,1-c][1,2,5]thiadiazole-6,11-dione showed strong topoisomerase I inhibition capabilities (Dong et al., 2017), suggesting that our compounds may have similar activities.
To investigate whether our series of compounds are potential topoisomerase inhibitors, we carried out molecular modeling experiments to study the interaction between our compounds and either of topoisomerase I (TOP1) or topoisomerase III (TOP3). We selected nine of our compounds with the lowest IC50 values against the PC-3 prostate cancer line (Table 1) for the molecular modeling experiments. We started with testing the binding of our compounds to the topotecan pocket of the crystal structure of human TOP1 (hTOP1)-DNA complex (Fig. 7 (a)). The docking poses of the nine compounds (Fig. 7 (b)) show that they bind to the DNA by intercalating into the G11:C12 and T10:A13 DNA base pairs, by the same mechanism as topotecan. The binding scores of our compounds (Fig. 7 (c)) revealed that they may bind to hTOP1 with high affinity. The highest binding score was 13.41 for compound 11 followed by compounds 3, 5, 9, 16, 14, 4, 10, and 1 with binding scores of 13.10, 13.01, 13.00, 12.76, 12.73, 12.32, 11.86, and 10.50, respectively. In general, the binding affinities were going along the same line with the observed cytotoxic activities and IC50 values (with few exceptions), suggesting strongly that the cytotoxic activities are related to binding of compounds to hTOP1.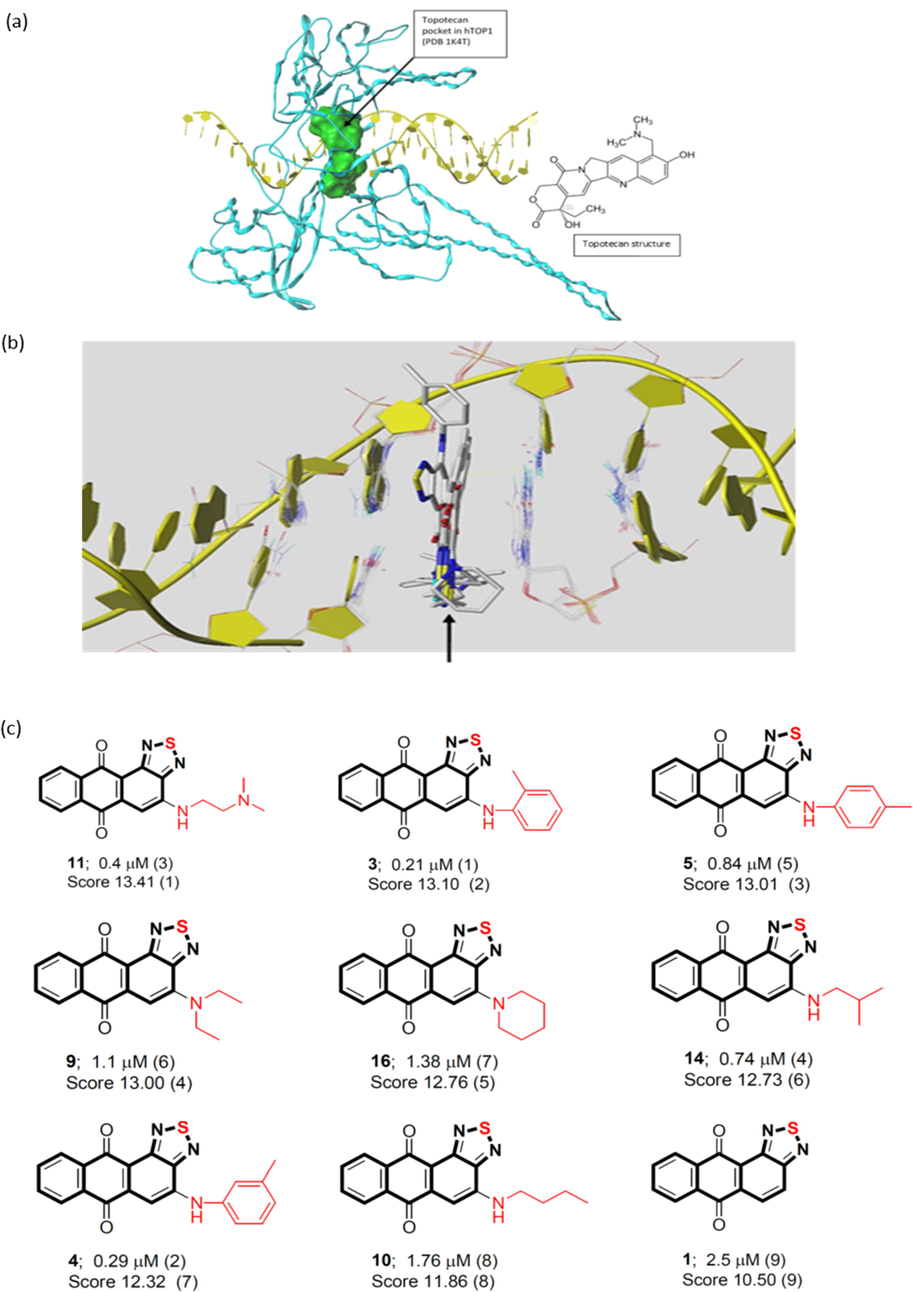
(a) Binding pocket of the human topoisomerase I (hTOP1) (PDB code 1K4T) defined by the ligand topotecan. The pocket was shown as molecular surface in green. (b) Binding poses of our compounds (marked by a black arrow) within DNA. (c) The binding scores of our compounds (1, 3, 4, 5, 9, 10, 11, 14, 16) to the hTOP1.
For the TOP3, we used the crystal structure of the hTOP3-RMI1 complex where a RMI1 loop (purple color) is inserted into the interface of the DNA binding site (Fig. 8 (a)). The catalytic tyrosine residue (Tyr362) and the metal ion Mg(II) are shown inside a drawn vertical box. Since no DNA is bound with the complex, the TOP3 is in its closed conformation, accordingly, we suggested that the binding of the TOP3 inhibitor to be at the RMI1 loop binding site at the hTOP3-RMI1 interface as highlighted in Fig. 8 (a). Three potential binding pockets for the inhibitor were identified at the RMI1-hTOP3 interface. Pocket 1 as shown in Fig. 8 (b), was defined by the RMI1 loop with the loop still bound. Pocket 2 as shown in Fig. 8 (c) was at the interface between TOP3 and RMI1 and was identified while the RMI1 loop was being removed, accordingly, ligands of pocket 2 should be competitive inhibitors of the RMI1 loop. Pocket 2 could be further classified as two pockets; 2n and 2c, as the interfaces of the N-terminal and C-terminal portions of the tail loops. Therefore, the three proposed pockets of hTOP3 inhibitors can be summarized as: 1) Pocket 1 – enclosed by the RMI1 loop and TOP3, stabilizing RMI1 binding; 2) Pocket 2n – defined by the N-terminal portion of the RMI1 loop, destabilizing RMI1 binding; 3) Pocket 2c – defined by the C-terminal portion of the RMI1 loop, destabilizing RMI1 binding. Docking scores (Fig. 8 (d)) showed that topotecan is a better binder for all three pockets (although with weaker binding affinity compared to TOP1) compared to compound 11. Compound 11 exhibited the highest binding affinity towards pocket 1 of hTOP3 with a docking score of 7.97 (its binding pose is presented in Fig. 8 (e)). Compound 11 fitted with the RMI1 loop very well and bound to RMI1 with two hydrogen bonds through Lys 105 and Thr111. Moreover, the hydrophilic binding surface includes some hydrophobic residues, Leu115, Pro98, Gln97 and Gln104, which may play a role in the binding at the interface. The binding of topotecan to pocket 2n (docking score = 7.70) is shown in Fig. 8 (f). Pocket 2n has many charged residues and hydrophobic clusters. Topotecan bound by hydrogen bonding with Arg255 and Arg338 at each end of the topotecan molecule. The binding of topotecan to pocket 2c (docking score = 7.44) is shown in Fig. 8 (g). Pocket 2c has both hydrophilic and hydrophobic features. topotecan bound to the pocket by hydrogen bonding with His288, Arg289 and Ser132 at one end and Glu265 at the other. Moreover, the hydrophobic residues Val93, Pro131, Leu210, Leu290, and Phe291 stabilized binding of the ligand. In summary, three different inhibition mechanisms are proposed in Fig. 8 (h). The ligand of Pocket 1 is stabilizing the RMI1 loop. The ligands of Pocket 2n and 2c inhibit the TOP3 activity by interfering with the RMI1 binding. Also, our compounds may have high binding affinity for TOP1 but not for TOP3, suggesting that they are TOP1 specific inhibitors.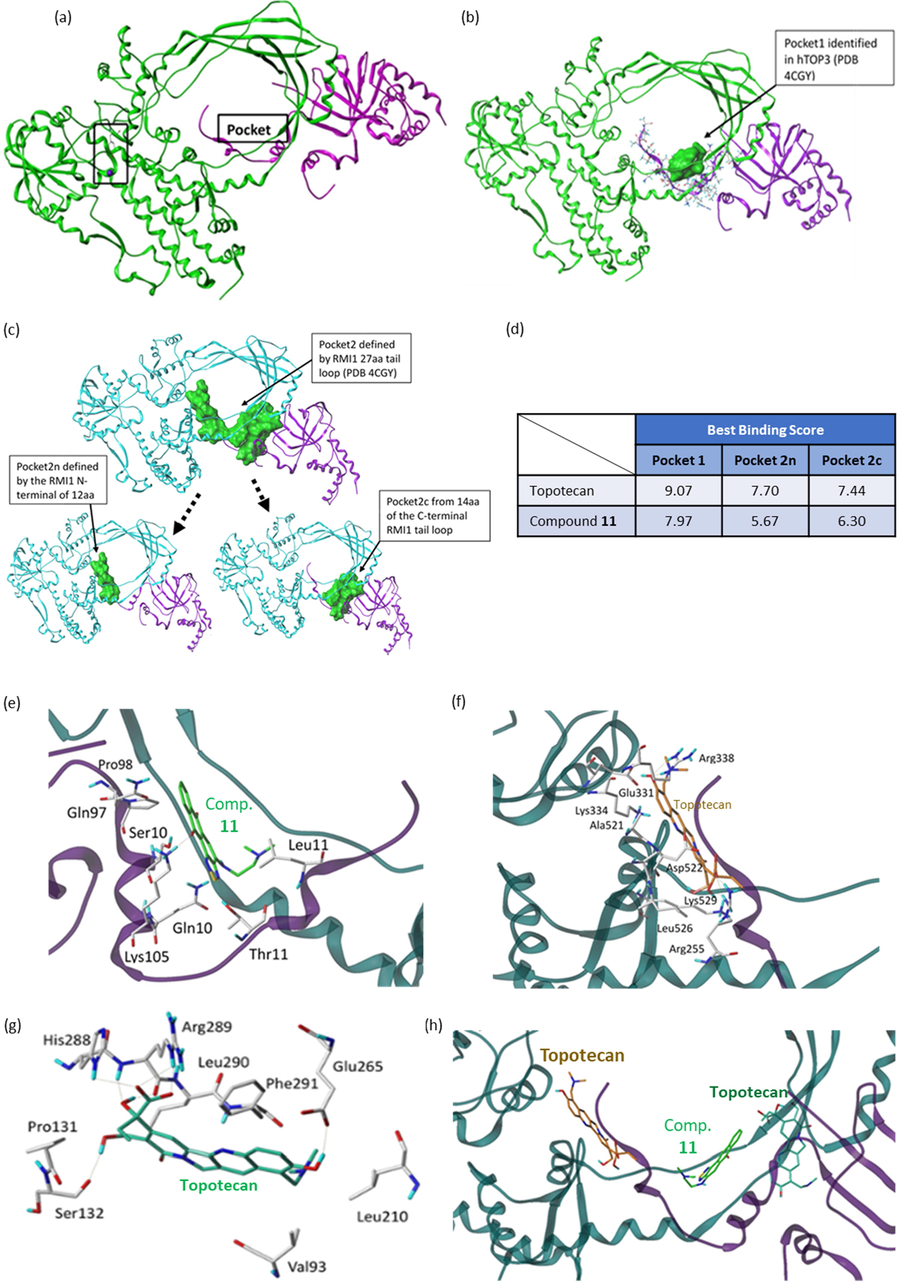
(a) Human topoisomerase III (hTOP3) (PDB code 4CGY) (green) in complex with RMI1 loop (purple). Tyr362 and Mg(II) are shown as stick and ball rendering. The proposed inhibitor binding pocket is also labeled. (b) The identified pocket 1 of hTOP3 presented in green molecular surface. (c) The identified pockets 2, 2n, and 2c of hTOP3 presented in green molecular surfaces. (d) The binding scores of topotecan and compound 11 to pockets 1, 2n, and 2c of hTOP3. (e) Docking pose of compound 11 in pocket 1 of hTOP3 (Score 7.97). (f) Docking pose of topotecan in pocket 2n of hTOP3 (Score 7.70). (g) Docking pose of topotecan in pocket 2c of hTOP3 (Score 7.44). (h) Docking poses of ligands in pockets 1, 2n and 2c of hTOP3 with consequent inhibition effects.
4 Conclusions
In summary, a series of novel N-substituted anthra[2,1-c][1,2,5]thiadiazole-6,11-dione derivatives were synthesized as a part of our efforts to develop new anticancer agents. We investigated their cytotoxic and biochemical activities as multi-target small molecule inhibitors through a series of experiments involving two androgen-independent human prostate cancer cell lines (PC-3 and DU-145), normal human cell lines (SV-HUC-1, WPMY-1, and RWPE-1), and a panel of NCI-60 cancer cell lines. We aimed further to understand the mechanisms of action of our compounds by performing COMPARE analysis, microscopical, Western blot, flow cytometry, and molecular modeling experiments. We found compound 11 to exhibit potent cytotoxic activities against most of the tested cell lines with GI50 values in the sub-micromolar range, however, it showed less cytotoxicity to normal human cell lines compared to doxorubicin. Moreover, we found that compound 11 has similar cytotoxic activity profile to drugs that affect the cell cycle and apoptosis as well as other pathways and mechanisms. To confirm such findings, we performed further in vitro experiments and found that compound 11 caused morphological apoptotic changes to treated cells, inhibited expression of cyclin D1 and COX-2, and caused cells to accumulate in the G0/G1 phase of cell cycle, all observed results were dependent on the used concentration of compound 11. Also, molecular modeling experiments showed that our compounds are potent inhibitors to human topoisomerase I but not to topoisomerase III. Our results, as presented in this manuscript, suggest that the 6,6,6,5-tetraheterocyclic thiadiazole-fused anthraquinone scaffold is a chemotype for multi-target small molecule inhibitors and is worthy of further development.
Acknowledgments
The authors thank the NCI Developmental Therapeutics Program (DTP) for the 60-cancer-cell-line screening of selected compounds described in this paper, funded by the National Cancer Institute, National Institutes of Health (NIH-NCI). The present study was supported by grants (MOST108-2113-M-038-006 and MOST109-2113-M-038-003) from the Ministry of Science and Technology, Taiwan.
Declaration of competing interest
The authors declared that they have no conflicts of interest to this work. We declare that we do not have any commercial or associative interest that represents a conflict of interest in connection with the work submitted.
References
- Abdella, A.M., Abdelmoniem, A.M., Abdelhamid, I.A., Elwahy, A.H.M., 2020. Synthesis of heterocyclic compounds via Michael and Hantzsch reactions. 57, 4, 1476–1523. http://doi.org/10.1002/jhet.3883.
- Erlotinib-Conjugated Iron Oxide Nanoparticles as a Smart Cancer-Targeted Theranostic Probe for MRI. Sci. Rep.. 2016;6(1):36650.
- [CrossRef] [Google Scholar]
- Novel Anthra[1,2-c][1,2,5]Thiadiazole-6,11-Diones as Promising Anticancer Lead Compounds: Biological Evaluation, Characterization & Molecular Targets Determination. PLoS ONE. 2016;11(4):e0154278
- [CrossRef] [Google Scholar]
- Efficient Labeling Of Mesenchymal Stem Cells For High Sensitivity Long-Term MRI Monitoring In Live Mice Brains. Int. J. Nanomed.. 2020;15:97-114.
- [CrossRef] [Google Scholar]
- Activity of Topotecan toward the DNA/Topoisomerase I Complex: A Theoretical Rationalization. Biochemistry. 2018;57(9):1542-1551.
- [CrossRef] [Google Scholar]
- Chapter 54 - Castration Resistant Prostate Cancer: Role of Chemotherapy. In: Mydlo J.H., Godec C.J., eds. Prostate Cancer (Second Edition). San Diego: Academic Press; 2016. p. :509-514.
- [Google Scholar]
- Structural and mechanistic insight into Holliday-junction dissolution by Topoisomerase IIIα and RMI1. Nat. Struct. Mol. Biol.. 2014;21(3):261-268.
- [CrossRef] [Google Scholar]
- Different roles of p53 in the regulation of DNA damage caused by 1,2-heteroannelated anthraquinones and doxorubicin. Int. J. Biochem. Cell Biol.. 2011;43(12):1720-1728.
- [CrossRef] [Google Scholar]
- Structure-based design, synthesis and evaluation of novel anthra[1,2-d]imidazole-6,11-dione derivatives as telomerase inhibitors and potential for cancer polypharmacology. Eur. J. Med. Chem.. 2013;60:29-41.
- [CrossRef] [Google Scholar]
- Structure-based design, synthesis and biological evaluation of novel anthra[1,2-d]imidazole-6,11-dione homologues as potential antitumor agents. Eur. J. Med. Chem.. 2013;69:278-293.
- [CrossRef] [Google Scholar]
- B1, a novel topoisomerase II inhibitor, induces apoptosis and cell cycle G1 arrest in lung adenocarcinoma A549 cells. Anticancer Drugs. 2012;23(2):191-199.
- [CrossRef] [Google Scholar]
- Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods. 1986;89(2):271-277.
- [Google Scholar]
- Design, synthesis and evaluation of 4-substituted anthra[2,1-c][1,2,5]thiadiazole-6,11-dione derivatives as novel non-camptothecin topoisomerase I inhibitors. Bioorg. Med. Chem. Lett.. 2017;27(9):1929-1933.
- [CrossRef] [Google Scholar]
- Doxorubicin inhibits human DNA topoisomerase I. Cancer Chemother. Pharmacol.. 1992;30(2):123-125.
- [CrossRef] [Google Scholar]
- Cancer chemotherapy: targeting folic acid synthesis. Cancer Manage. Res.. 2010;2:293-301.
- [CrossRef] [Google Scholar]
- Synthesis, telomerase evaluation and anti-proliferative studies on various series of diaminoanthraquinone-linked aminoacyl residue derivatives. Arch. Pharm. (Weinheim). 2012;345(2):101-111.
- [CrossRef] [Google Scholar]
- Synthesis, cytotoxicity and human telomerase inhibition activities of a series of 1,2-heteroannelated anthraquinones and anthra[1,2-d]imidazole-6,11-dione homologues. Bioorg. Med. Chem.. 2009;17(21):7418-7428.
- [CrossRef] [Google Scholar]
- NSC746364, NSC746365, and NSC746366: the spectra of cytotoxicity and molecular correlates of response to telomerase activity. Anticancer Drugs. 2010;21(2):169-180.
- [CrossRef] [Google Scholar]
- YC-1 suppresses constitutive nuclear factor-kappaB activation and induces apoptosis in human prostate cancer cells. Mol. Cancer Ther.. 2005;4(10):1628-1635.
- [CrossRef] [Google Scholar]
- Heterocycles in Medicinal Chemistry. Molecules (Basel, Switzerland). 2019;24(21):3839.
- [CrossRef] [Google Scholar]
- Tipifarnib in the treatment of newly diagnosed acute myelogenous leukemia. Biologics: Targets Therapy. 2008;2(3):491-500.
- [CrossRef] [Google Scholar]
- Design, synthesis and antiproliferative evaluation of fluorenone analogs with DNA topoisomerase I inhibitory properties. Bioorg. Med. Chem.. 2013;21(22):7125-7133.
- [CrossRef] [Google Scholar]
- Design, synthesis and evaluation of telomerase inhibitory, hTERT repressing, and anti-proliferation activities of symmetrical 1,8-disubstituted amidoanthraquinones. Eur. J. Med. Chem.. 2012;50:102-112.
- [CrossRef] [Google Scholar]
- Synthesis, antiproliferative activities and telomerase inhibition evaluation of novel asymmetrical 1,2-disubstituted amidoanthraquinone derivatives. Eur. J. Med. Chem.. 2012;47(1):323-336.
- [CrossRef] [Google Scholar]
- Ring fusion strategy for synthesis and lead optimization of sulfur-substituted anthra[1,2-c][1,2,5]thiadiazole-6,11-dione derivatives as promising scaffold of antitumor agents. Eur. J. Med. Chem.. 2015;102:661-676.
- [CrossRef] [Google Scholar]
- New approaches of PARP-1 inhibitors in human lung cancer cells and cancer stem-like cells by some selected anthraquinone-derived small molecules. PLoS ONE. 2013;8(2):e56284
- [CrossRef] [Google Scholar]
- Camptothecin (CPT) and its derivatives are known to target topoisomerase I (Top1) as their mechanism of action: did we miss something in CPT analogue molecular targets for treating human disease such as cancer? Am. J. Cancer Res.. 2017;7(12):2350-2394.
- [Google Scholar]
- Anthracyclines as Topoisomerase II Poisons: From Early Studies to New Perspectives. Int. J. Mol. Sci.. 2018;19(11):3480.
- [CrossRef] [Google Scholar]
- DNA synthesis as a therapeutic target: the first 65 years. FASEB J.: Off. Publ. Feder. Am. Societ. Exper. Biol.. 2012;26(6):2231-2237.
- [CrossRef] [Google Scholar]
- Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev.. 2004;56(2):185-229.
- [CrossRef] [Google Scholar]
- Recent developments in the field of antitumour anthracyclines. Eur. J. Med. Chem.. 2001;36(6):483-493.
- [CrossRef] [Google Scholar]
- Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983;65(1–2):55-63.
- [Google Scholar]
- Nelson, J.A., Carpenter, J.W., Rose, L.M., Adamson, D.J., 1975. Mechanisms of Action of 6-Thioguanine, 6-Mercaptopurine, and 8-Azaguanine. 35, 10, 2872–2878.
- Applications of Multi-Target Computer-Aided Methodologies in Molecular Design of CNS Drugs. Curr. Med. Chem.. 2018;25(39):5293-5314.
- [CrossRef] [Google Scholar]
- Clinical potential of elacytarabine in patients with acute myeloid leukemia. Therapeutic Adv. Hematol.. 2014;5(6):211-220.
- [CrossRef] [Google Scholar]
- Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat. Protoc.. 2006;1(3):1458-1461.
- [CrossRef] [Google Scholar]
- ALDH1 Bio-activates Nifuroxazide to Eradicate ALDH(High) Melanoma-Initiating Cells. Cell Chem. Biol.. 2018;25(12):1456-1469.e1456.
- [CrossRef] [Google Scholar]
- Cyclopentenyl Cytosine (CPEC): An Overview of its in vitro and in vivo Activity. Curr. Cancer Drug Targets. 2007;7:504-509.
- [CrossRef] [Google Scholar]
- Lapachol inhibits glycolysis in cancer cells by targeting pyruvate kinase M2. PLoS ONE. 2018;13(2):e0191419
- [CrossRef] [Google Scholar]
- Pre-clinical evidences for the efficacy of tryptanthrin as a potent suppressor of skin cancer. Cell Prolif.. 2020;53(1) e12710–e12710
- [CrossRef] [Google Scholar]
- Combretastatin A-4 inhibits cell growth and metastasis in bladder cancer cells and retards tumour growth in a murine orthotopic bladder tumour model. Br. J. Pharmacol.. 2010;160(8):2008-2027.
- [CrossRef] [Google Scholar]
- RV-59 suppresses cytoplasmic Nrf2-mediated 5-fluorouracil resistance and tumor growth in colorectal cancer. Am. J. Cancer Res.. 2019;9(12):2789-2796.
- [Google Scholar]
- The role of cyclooxygenase-2 in cell proliferation and cell death in human malignancies. Int. J. Cell Biol.. 2010;2010 215158–215158
- [CrossRef] [Google Scholar]
- The mechanism of topoisomerase I poisoning by a camptothecin analog. J. Proc. Natl. Acad. Sci.. 2002;99(24):15387-15392.
- [CrossRef] [Google Scholar]
- Morphological and immunocytochemical characteristics of human tumor cell lines for use in a disease-oriented anticancer drug screen. Anticancer Res.. 1992;12(4):1035-1053.
- [Google Scholar]
- The anti-angiogenic effect and novel mechanisms of action of Combretastatin A-4. Sci. Rep.. 2016;6(1):28139.
- [CrossRef] [Google Scholar]
- Pharmacologic down-regulation of EZH2 suppresses bladder cancer in vitro and in vivo. Oncotarget. 2014;5(21):10342-10355.
- [CrossRef] [Google Scholar]
- 1,2,3-Thiadiazole substituted pyrazolones as potent KDR/VEGFR-2 kinase inhibitors. Bioorg. Med. Chem. Lett.. 2007;17(6):1793-1798.
- [CrossRef] [Google Scholar]
- Raltitrexed: current clinical status and future directions. Ann. Oncol.. 2002;13(4):513-522.
- [CrossRef] [Google Scholar]
- The fungal natural product (1S,3S)-austrocortirubin induces DNA damage in HCT116 cells via a mechanism unique from other DNA damaging agents. Bioorg. Med. Chem. Lett.. 2015;25(2):249-253.
- [CrossRef] [Google Scholar]
- Research Progress on the Antitumor Effects of Rhein: Literature Review. Anticancer Agents Med Chem. 2017;17(12):1624-1632.
- [CrossRef] [Google Scholar]
- Bafilomycin A1 induces caspase-independent cell death in hepatocellular carcinoma cells via targeting of autophagy and MAPK pathways. Sci. Rep.. 2016;6(1):37052.
- [CrossRef] [Google Scholar]
- Bafilomycin A1 targets both autophagy and apoptosis pathways in pediatric B-cell acute lymphoblastic leukemia. Haematologica. 2015;100(3):345-356.
- [CrossRef] [Google Scholar]
- LigBuilder V3: A Multi-Target de novo. Drug Design Approach.. 2020;8(142)
- [CrossRef] [Google Scholar]
- Nifuroxazide exerts potent anti-tumor and anti-metastasis activity in melanoma. Sci. Rep.. 2016;6(1):20253.
- [CrossRef] [Google Scholar]




