

Translate this page into:
Structure design and mechanism study of Sc@P10C12M2 catalysts for methanol dehydrogenation to methyl formate
⁎Corresponding authors. renruipeng@tyut.edu.cn (Rui-Peng Ren), lykang@tyut.edu.cn (Yong-Kang Lv)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The Sc@P10C12M2 catalysts were first constructed and utilized for methanol conversion reaction. In this work, DFT-D3 calculations were employed to investigate the conversion of CH3OH to CHOOCH3 on four different types of the Sc@P10C12M2 catalysts (M = Fe, Co, Ni, and Cu). The results indicate that the selectivity and formation activity of CHOOCH3 are influenced by the choice of metals. Specifically, the Sc@P10C12Ni2 catalyst demonstrates superior catalytic activity with an activation free energy and activity of 0.64 eV and 1.97 lgk/s−1 for the rate-limiting step at 298 K.
This research reveals a structure–activity relationship for Sc@P10C12M2 catalysts, highlighting that catalytic performance is correlated with metal types, spin states, and charges. These insights provide theoretical guidance for the rational design of efficient methanol conversion catalysts.
Keywords
Methanol
Methyl formate
Sc@P10C12M2
DFT
Low temperatures

1 Introduction
Methanol, as a clean energy source, has a wide range of sources and can be obtained from coal, oil, and natural gas. The output of methanol is increasing year by year, and the methanol capacity is in surplus (Sen et al., 2022). The best approach is to develop a diversified downstream technology roadmap for methanol. Methanol can be utilized to produce aldehydes, ethers, alcohols, esters, olefins and other chemical products. It also serves as a raw material for important industrial reactions such as methanol steam reforming (Sá et al., 2010). Among the various downstream products of methanol, methyl formate can be used as a refrigerant, foaming agent, and fuel additive, playing an essential role in C1 chemistry (Jenner, 1995). The production of methyl formate involves methanol conversion technology which mainly includes esterification of methanol with formic acid (Indu et al., 1993), methanol carbonylation (Girolamo et al., 1996), direct dehydrogenation of methanol (Huang et al., 2013), selective oxidative dehydrogenation of methanol (Liu and Iglesia, 2005), and carbon dioxide hydrogenation-condensation with methanol (Evans and Newell, 1978). Among these methods, the direct dehydrogenation of methanol to methyl formate is considered an efficient and economical method due to its single raw material and simple operation, which is worth further development and utilization (Huang et al., 2013; Sato et al., 1997).
The core and key of methanol conversion technology lies in the development and design of efficient catalysts. The direct dehydrogenation of methanol to methyl formate commonly utilizes transition metal catalysts (Iwasa et al., 1995; Wang et al., 2021; Yang et al., 2018) such as Cu, Ni, Pt, Pd, etc., with reaction temperatures typically ranging from 200 to 300 ℃. Recently, there has been significant interest in Cu-based catalysts due to their unique activities, including CuZn/SBA-15 (Wang et al., 2021), Cu@mSiO2 (Yang et al., 2018) and Cu(1 1 1) (Wu et al., 2022); all of which have demonstrated excellent catalytic performance. However, the key challenges in the methanol to methyl formate reaction include copper agglomeration, sintering and high temperature deposition (Quan et al., 2022). Non-metal atoms like P can be used as dopants to regulate charge transfer between elements and inhibit sintering while affecting catalytic activity (Zhang et al., 2022). Additionally, methyl formate is prone to pyrolysis into CO at high temperatures leading to reduced selectivity (Zhang et al., 2002). In recent years, research focus has shifted towards catalytic reactions under mild conditions (Torimoto et al., 2019; Sun et al., 2021), aiming to avoid issues related to metal agglomeration, sintering and carbon deposition while maintaining selectivity for methyl formate. The unique structure of phosphorus-doped catalysts combined with low temperature conditions holds promise for addressing the issue of catalytic stability in the methanol conversion process.
Encapsulated catalysts, as unique structures, have demonstrated excellent activity and stability in various reactions (Chen et al., 2022; Chen et al., 2015; Kramer et al., 2017), garnering increasing attention from researchers. Shen et al. (Shen et al., 2023) utilized Co@NPC as catalysts for the hydrogenation of cinnamaldehyde, and found that P doping improved catalytic selectivity while N doping increased catalytic activity. Kramer et al. (Kramer et al., 2017) selected M@NC as catalysts for the preparation of allyl group aromatics from allyl group benzene, and observed that the type and quantity of metals had a significant impact on yield. These “armored” catalysts effectively enhance the activity and stability of the metal, with electron transfer between metals and carbon atoms positively affecting catalytic performance (Deng et al., 2013; Yu et al., 2020). Fullerene-like structures (B12N12 (Esrafili and Nurazar, 2014), Al12N12 (Zhang et al., 2018) were employed for CH3OH cracking, indicating that 4- and 6-membered rings fullerene-like cages were conducive to methanol activation. Building upon the exceptional stability and activity of carbon-based encapsulated catalysts, carbon cages with 4- and 6-membered rings have been designed to potentially possess remarkable catalytic properties. X12C12 (Schulman et al., 1987; Krylova et al., 2016; Hitler et al., 2023; Edet et al., 2022) cage nanostructures have garnered significant attention due to their unique chemical and physical properties in other fields such as C24, Si12C12, N12C12 and P12C12; these can serve as shell structures for “armored” catalysts. Metal-modified X12C12 cages are anticipated to enable efficient preparation of methyl formate from methanol at low temperatures while ensuring material stability and effective methanol activation.
Density functional theory (DFT) is a widely utilized method for investigating the electronic structure of multi-electron systems, commonly employed in computational materials science and computational chemistry in condensed matter physics (Kohn and Sham, 1965). In this study, four types of metal-modified P12C12 catalysts were innovatively designed based on DFT, and their catalytic properties were systematically studied at 298 K. The carbon resistance and thermal stability of the catalysts were analyzed, while the mechanism of CH3OH to CHOOCH3 was explored on the Sc@P10C12M2 (M = Fe, Co, Ni, Cu) catalysts. Additionally, the formation of CO by-products was investigated. The study involved calculations of reaction free energies, activation free energies, rate constants and activity. It also aimed to clarify the relationship between different types of metals and catalytic activity while explaining in detail the influence of electronic factors on catalytic performance. The structures proposed can offer theoretical guidance for similar reactions and catalytic systems.
2 Computational details
2.1 Calculation methods
All DFT calculations were conducted using the Vienna Ab initio Simulation Package (VASP) (Kresse and Hafner, 1993; Kresse and Furthmüller, 1996; Kresse and Furthmüller, 1996). DFT calculations with a dispersion correction method (DFT-D3) (Grimme et al., 2010) were employed to consider van der Waals interactions of CH3OH adsorption and conversion on the Sc@P10C12M2 catalysts. The interaction between ionic cores and electrons was described by projector augmented wave (PAW) pseudopotentials (Kresse and Joubert, 1999). The kinetic cutoff energy of the plane-wave basis was set at 400 eV. A 1 × 1 × 1 k-point sampling grid with a width of 0.05 eV of smearing was used in this calculation. The transition state (TS) was determined by the climbing-image nudged elastic band (CI-NEB) method (Henkelman et al., 2000). Through the vibrational analysis, the TSs were identified with one imaginary frequency. The atomic structure of the catalyst has been sufficiently optimized until the force on each atom is less than 0.06 eV/Å. The ab initio molecular dynamics(AIMD) (Fan et al., 2020) simulation was performed at 298 K with 600 steps and 5 fs time steps in NVT ensemble. The implicit solvation model of VASPsol was used to calculate the solvation effect (Mathew et al., 2014).
The binding energies (Eb) is calculated according to the following formula:
The adsorption free energies (Gads) (Shao et al., 2023; Lan et al., 2023) is defined as follows:
The reaction free energies (ΔG) (Shao et al., 2023; Lan et al., 2023) and activation free energies (Ga) (Shao et al., 2023; Lan et al., 2023) are calculated according by the following equations:
Where EIS, ETS and EFS correspond to the total energies of initial state (IS), TS and final state (FS), respectively; GIS, GTS and GFS are the corresponding free energy corrections at 298 K. When the structure of IS, TS and FS is determined, various thermodynamic data such as enthalpy (H), entropy (S) and free energy (G) can be obtained by a vibrational analysis calculation. In this work, the values of Gadsorbate/sub, Gadsorbate, Gsub GIS, GTS, and GFS at a specified temperature can be obtained in the VASPKIT package (Wang et al., 2021).
Based on the transition state theory (Zha et al., 2018), the formula for calculating the reaction rate constant is as follows:
In the formula, Ga is activation free energy, h is Planck's constant, kB is Boltzmann's constant, T is reaction temperature (298 K).
2.2 Calculation models
The stable configuration of Sc@P10C12M2 (M = Fe, Co, Ni, Cu) catalysts models designed in this work are depicted in Fig. 1. The binding energies of Sc@P10C12Fe2, Sc@P10C12Co2, Sc@P10C12Ni2, and Sc@P10C12Cu2 are −5.01, −5.04, −4.99 and −4.78 eV, respectively. The negative binding energies indicate the relative stability of the catalysts to a certain extent. Taking Sc@P10C12Fe2 as an example, it can be seen from Fig. S1(a) that the binding energies increases with the increase of iron atomic distance. Additionally, Fig. S1(b) demonstrates that the binding energies of Sc supported by P10C12M2 are higher than those of Sc encapsulated byP10C12M2, indicating that Sc is stably encapsulated. Overall, the results suggest that these catalyst models exhibit a degree of stability and provide valuable insights into their potential applications in catalysis research.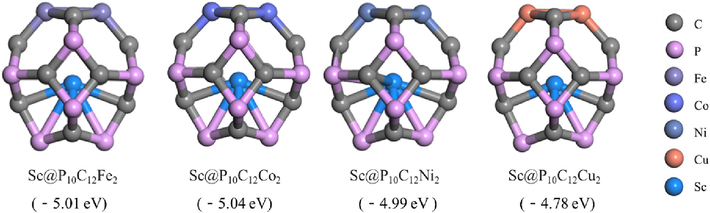
The stable configuration of Sc@P10C12M2.
3 Results and discussion
3.1 Molecular dynamic simulation
The thermodynamic stability of Sc@P10C12M2 catalysts was verified by performing molecular dynamics simulations. A simulation with a duration of 3 ps was conducted at a temperature of 298 K with 600 steps and 5 fs time steps. As depicted in Fig. 2, the energy fluctuated within a very narrow range during the simulation, and no significant structural changes were observed. These results indicate that Sc@P10C12M2 are thermodynamically stable at 298 K.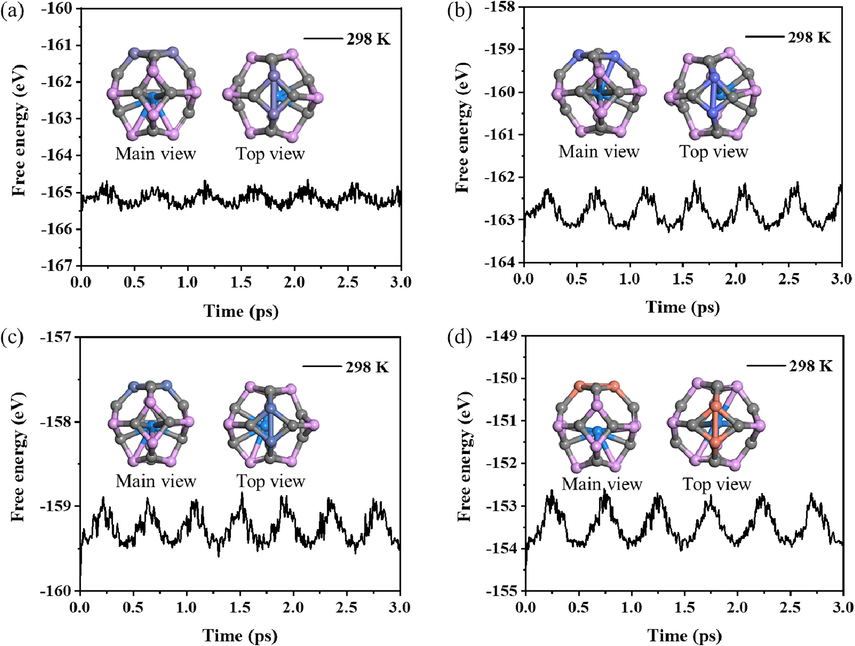
Total energy fluctuations of Sc@P10C12M2 during molecular dynamics simulations and geometry at the end of the simulation.
3.2 Adsorption behavior of CH3OH and related species on the Sc@P10C12M2 catalysts
To assess the stability of various adsorption species, the most stable adsorption sites of CH3OH and related species were calculated. The adsorption free energies of CH3OH and related species on the Sc@P10C12M2 catalysts are given in Table 1, and the corresponding configuration diagrams are shown in Fig. S2. The adsorption free energies of CH3OH range from −0.19 to 0.14 eV on the Sc@P10C12M2 catalysts.
Gads(eV)
Sc@P10C12Fe2
Sc@P10C12Co2
Sc@P10C12Ni2
Sc@P10C12Cu2
CH3OH
−0.19
0.08
0.14
0.06
CH3O
−2.79
−2.09
−1.12
−0.75
CH2O
−1.04
−0.59
−0.33
−0.72
CHO
−2.79
−2.55
−2.67
−2.70
CO
−1.14
−0.99
−1.55
−1.79
CHOOCH3
0.02
0.32
0.58
0.42
In addition, the presence of water or water vapor is virtually unavoidable in the actual methanol conversion system, which necessitates a study into its impact on methanol conversion (Zhang et al., 2016). Fig. S3 illustrates the influence of water solvation on adsorption energies and utilizes the implicit solvation model of VASPsol to calculate solvation effect. As depicted in Fig. S3; the implicit solvation model has minimal impact on the calculated energy, with an average change of only 0.02 eV compared to the gas phase on the Sc@P10C12Ni2 catalyst. The effect of implicit water solvation on adsorption energy is negligible, consistent with findings reported in the literature (Meia and Deskins, 2021).
3.3 Conversion mechanism of CH3OH on the Sc@P10C12M2 catalysts
3.3.1 On the Sc@P10C12Fe2 catalyst
The CH3OH cracking on the Sc@P10C12Fe2 catalyst initiates with the adsorbed CH3OH, leading to the reaction (CH3OH → CH3O + H). The dehydrogenation step through TS1-1 requires overcoming an activation free energy of 0.68 eV with an exothermicity of −0.67 eV. The formation of CH2OH via C–H bond scission from CH3OH has an activation free energy of 1.05 eV, which is higher than that of the CH3O formation. Another possible reaction pathway for CH3OH cracking via C–O bond cleavage to produce CH3 and OH is also investigated, where the step through TS1-3 needs to overcome an activation free energy of 1.46 eV. These results indicate that the formation of CH3O from CH3OH is more favorable. The CH2O and H are generated during the CH3O dissociation through TS1-4, the reaction (CH3O → CH2O + H) has an activation free energy of 1.19 eV with exothermicity of −0.26 eV. The formation of CH3 via C–O bond scission from CH3O has an activation free energy of 1.89 eV. After TS1-6, CHO and H are produced, and the reaction is exothermicity of −0.89 eV with an activation free energy of 0.65 eV. Another way is CH2O to generate CH2 through C–O bond cleavage, the activation free energy of this step is 0.92 eV, indicating that CH2 is difficult to generate. Subsequently, the dehydrogenation of CHO to CO via TS1-8 is an endothermic by 0.27 eV and requires overcoming an activation free energy of 0.78 eV. Additionally, it is noted that breaking the C–O bond can generate CH with an activation free energy of 1.56 eV.
As depicted in Scheme 1, CHOOCH3 can be synthesized through three pathways by intermediates (R10–13) (Yang et al., 2018; Wu et al., 2022; Quan et al., 2022; Zhang et al., 2022; Zhang et al., 2002; Torimoto et al., 2019; Sun et al., 2021; Chen et al., 2022; Chen et al., 2015; Kramer et al., 2017; Shen et al., 2023; Deng et al., 2013; Yu et al., 2020; Esrafili and Nurazar, 2014; Zhang et al., 2018; Schulman et al., 1987; Krylova et al., 2016; Hitler et al., 2023; Edet et al., 2022; Kohn and Sham, 1965; Kresse and Hafner, 1993; Kresse and Furthmüller, 1996; Kresse and Furthmüller, 1996; Grimme et al., 2010; Kresse and Joubert, 1999; Henkelman et al., 2000; Fan et al., 2020; Mathew et al., 2014; Shao et al., 2023; Lan et al., 2023; Wang et al., 2021; Zha et al., 2018; Zhang et al., 2016; Meia and Deskins, 2021; Beste and Overbury, 2017). From Scheme 1; these pathways include (1) dehydrogenation after coupling reaction of CH3O and CH2O, (2) direct coupling reaction by CHO and CH3O, and (3) dimerization reaction of CH2O. On the Sc@P10C12Fe2 catalyst, the groups formed by CH3OH cracking can be recombined to form CHOOCH3. For the CH2O + CH3O → CH2OOCH3, the step through TS1-10 needs to overcome activation free energy of 0.72 eV with endothermic of 0.50 eV. Next, the generated CH2OOCH3 is dehydrogenated to form CHOOCH3. This step has an activation free energy of 1.13 eV and is endothermic by 0.47 eV. For the CHO + CH3O → CHOOCH3, the reaction has an activation free energy of 2.53 eV and is endothermic by 1.41 eV. For the CH2O + CH2O → CHOOCH3, the reaction has an activation free energy of 2.94 eV and is endothermic by 1.28 eV. Among them, the indirect conversion of CH2O and CH3O to CHOOCH3 is the easiest.
Mechanism of CHOOCH3 and CO formation via CH3OH direct dehydrogenation.
The reaction pathway for CH3OH to CO is CH3OH → CH3O → CH2O → CHO → CO, and the dehydrogenation pathway is consistent with most catalysts (Lu et al., 2016; Ding et al., 2016; Damte et al., 2018); and the activation free energies are 0.68, 1.19, 0.65 and 0.78 eV, respectively. The main pathway for CH3OH to CHOOCH3 is CH3OH → CH3O → CH2O, CH2O + CH3O → CH2OOCH3, CH2OOCH3 → CHOOCH3 + H, and the activation free energies are 0.68, 1.19, 0.72 and 1.13 eV, respectively. The activation free energy of the rate-limiting step of CH3OH to CHOOCH3 and CO is 1.19 eV, indicating that CHOOCH3 and CO are formed simultaneously on the Sc@P10C12Fe2 catalyst. The corresponding IS, TS and FS are shown in Fig. S4, and the corresponding reaction free energies and activation free energies are shown at 298 K in Table 2.
Reactions
Sc@P10C12Fe2
Sc@P10C12Co2
Sc@P10C12Ni2
Sc@P10C12Cu2
ΔG(eV)
Ga(eV)
ΔG(eV)
Ga(eV)
ΔG(eV)
Ga(eV)
ΔG(eV)
Ga(eV)
R1
−0.67
0.68
−0.50
0.33
−0.45
0.32
−0.33
0.34
R2
−0.05
1.05
−0.45
1.40
−0.99
1.39
−1.78
2.21
R3
−1.36
1.46
−0.93
2.15
−0.45
2.07
−0.65
2.41
R4
−0.26
1.19
−0.60
0.94
−1.49
0.58
−2.04
0.68
R5
−0.57
1.89
0.05
2.78
−0.21
2.81
0.22
3.28
R6
−0.89
0.65
−1.12
0.50
−1.45
0.68
−1.81
0.53
R7
−0.59
0.92
−0.01
1.44
0.64
1.90
1.36
2.19
R8
0.27
0.78
0.18
0.85
−0.71
0.67
−1.60
1.05
R9
0.64
1.56
1.41
2.18
2.39
3.09
2.90
3.00
R10
0.50
0.72
0.55
0.83
0.45
0.64
0.15
0.52
R11
0.47
1.13
−0.35
0.53
−1.39
0.28
−1.73
0.25
R12
1.41
2.53
0.98
2.63
0.56
1.70
0.61
1.56
R13
1.28
2.94
0.75
3.10
0.60
3.24
0.62
3.30
3.3.2 On the Sc@P10C12Co2 catalyst
For the CH3OH cracking on the Sc@P10C12Co2 catalyst, the reaction initiates by the dehydrogenation of the adsorbed CH3OH to generate CH3O and H through TS2-1, this reaction is exothermic of −0.50 eV with an activation free energy of 0.33 eV. It can be seen that the formation of CH2OH needs to overcome an activation free energy of 1.40 eV. Another possible reaction pathway of CH3OH cracking via the C–O bond cleavage to produce CH3 and OH is also investigated, the step through TS2-3 needs to overcome an activation free energy of 2.15 eV. Subsequently, the formed CH3O further dehydrogenate to CH2O via TS2-4, which has an activation free energy of 0.94 eV with exothermicity of −0.60 eV. It can be seen that the formation of CH3 needs to overcome a higher activation free energy of 2.78 eV. The next step is the CHO formation through TS2-6, the step needs to overcome an activation free energy of 0.50 eV. Another possible reaction pathway of CH2O cracking via the C–O bond cleavage to produce CH2 and O is also investigated, the step through TS2-7 needs to overcome an activation free energy of 1.44 eV. Finally, the dehydrogenation of CHO to CO through TS2-8 is endothermic by 0.18 eV and needs to overcome an activation free energy of 0.85 eV. CH can also be generated by breaking the C–O bond, with an activation free energy of 2.18 eV.
As depicted in Scheme 1, for the CH2O + CH3O → CH2OOCH3, the reaction has an activation free energy of 0.83 eV and is endothermic by 0.55 eV. Next, the generated CH2OOCH3 is dehydrogenated to form CHOOCH3. This step has an activation free energy of 0.53 eV and is exothermic by −0.35 eV. For the CHO + CH3O → CHOOCH3, the step through TS1-12 needs to overcome an activation free energy of 2.63 eV with endothermicity of 0.98 eV. For the CH2O + CH2O → CHOOCH3, the reaction has an activation free energy of 3.10 eV and is endothermic by 0.75 eV. Among them, the indirect conversion of CH2O and CH3O to CHOOCH3 is the easiest.
The reaction pathway for CH3OH to CO is CH3OH → CH3O → CH2O → CHO → CO, and the activation free energies are 0.33, 0.94, 0.50 and 0.85 eV, respectively. The main pathway for CH3OH to CHOOCH3 is CH3OH → CH3O → CH2O, CH2O + CH3O → CH2OOCH3, CH2OOCH3 → CHOOCH3 + H, and the activation free energies are 0.33, 0.94, 0.83 and 0.53 eV, respectively. The activation free energy of the rate-limiting step of CH3OH to CHOOCH3 and CO is 0.94 eV, indicating that CHOOCH3 and CO are formed simultaneously on the Sc@P10C12Co2 catalyst. The corresponding IS, TS and FS are shown in Fig. S5, and the corresponding reaction free energies and activation free energies are shown at 298 K in Table 2.
3.3.3 On the Sc@P10C12Ni2 catalyst
For the CH3OH cracking on the Sc@P10C12Ni2 catalyst, the initial step of CH3OH → CH3O + H via TS3-1 has an activation free energy of 0.32 eV and the reaction free energy is −0.45 eV. As another possible pathway, the CH2OH formation is unlikely due to the high activation free energy of 1.39 eV. Similarly, the formation of CH3 is unlikely, with an activation free energy of 2.07 eV. The following step is the dehydrogenation of CH3O to CH2O through TS3-4 with an activation free energy of 0.58 eV, and the reaction free energy is −1.49 eV. It can be seen that the formation of CH3 needs to overcome a higher activation free energy of 2.81 eV. After the TS3-6, the CHO and H are generated. This step is exothermic by −1.45 eV with an activation free energy of 0.68 eV. As another possible pathway, the CH2 formation is unlikely due to the high activation free energy of 1.90 eV. Finally, the CO is formed from the CHO dehydrogenation through TS3-8, and the corresponding activation free energy and reaction free energy are 0.67 eV and −0.71 eV, respectively. As another possible pathway, the CH formation is unlikely due to the high activation free energy of 3.09 eV.
As depicted in Scheme 1, for the CH2O + CH3O → CH2OOCH3, the reaction has an activation free energy of 0.64 eV and is endothermic by 0.45 eV. Next, the generated CH2OOCH3 is dehydrogenated to form CHOOCH3. This step has a small activation free energy of 0.28 eV and is exothermic by −1.39 eV. For the CHO + CH3O → CHOOCH3, the step through TS1-12 needs to overcome an activation free energy of 1.70 eV with endothermicity of 0.56 eV. For the CH2O + CH2O → CHOOCH3, the reaction has an activation free energy of 3.24 eV and is endothermic by 0.60 eV. Among them, the indirect conversion of CH2O and CH3O to CHOOCH3 is the easiest.
The reaction pathway for CH3OH to CO is CH3OH → CH3O → CH2O → CHO → CO, and the activation free energies are 0.32, 0.58, 0.68 and 0.67 eV, respectively. The main pathway for CH3OH to CHOOCH3 is CH3OH → CH3O → CH2O, CH2O + CH3O → CH2OOCH3, CH2OOCH3 → CHOOCH3 + H, and the activation free energies are 0.32, 0.58, 0.64 and 0.28 eV, respectively. The activation free energies of the rate-limiting step of CH3OH to CHOOCH3 and CO are 0.64 and 0.68 eV, indicating that CHOOCH3 is easier to form on the Sc@P10C12Ni2 catalyst. The corresponding IS, TS and FS are shown in Fig. S6, and the corresponding reaction free energies and activation free energies are shown at 298 K in Table 2.
3.3.4 On the Sc@P10C12Cu2 catalyst
For the CH3OH cracking on the Sc@P10C12Cu2 catalyst, the initial step of CH3OH → CH3O + H via TS4-1 has an activation free energy of 0.34 eV, and the reaction free energy is −0.33 eV. The formation of CH2OH requires an activation free energy of 2.21 eV, which is higher than that of the CH3O formation. Another possible reaction pathway of CH3OH cracking via the C–O bond cleavage to produce CH3 and OH is also investigated, the step through TS4-3 needs to overcome an activation free energy of 2.41 eV. Therefore, the CH3O formation is easy to occur. Subsequently, the formed CH3O further dehydrogenate to CH2O through TS4-4. The activation free energy and reaction free energy are 0.68 and −2.04 eV, respectively. It can be seen that the formation of CH3 needs to overcome a higher activation free energy of 3.28 eV. The next step is CH2O dissociation through TS4-6. The reaction of the CHO and H production from CH2O species is calculated to be exothermic by −1.81 eV with an activation free energy of 0.53 eV. It can be seen that the formation of CH2 needs to overcome a higher activation free energy of 2.19 eV. Finally, the CO is formed from the CHO dehydrogenation through TS4-8, and the corresponding activation free energy and reaction free energy are 1.05 eV and −1.60 eV, respectively. As another possible pathway, the CH formation is unlikely due to the high activation free energy of 3.00 eV.
As depicted in Scheme 1, for the CH2O + CH3O → CH2OOCH3, the reaction has an activation free energy of 0.52 eV and is endothermic by 0.15 eV. Next, the generated CH2OOCH3 is dehydrogenated to form CHOOCH3. This step has activation free energy of 0.25 eV and is exothermic by −1.73 eV. For the CHO + CH3O → CHOOCH3, the step through TS1-12 needs to overcome an activation free energy of 1.56 eV with endothermicity of 0.61 eV. For the CH2O + CH2O → CHOOCH3, the reaction has an activation free energy of 3.30 eV and is endothermic by 0.62 eV. Among them, the indirect conversion of CH2O and CH3O to CHOOCH3 is the easiest.
The reaction pathway for CH3OH to CO is CH3OH → CH3O → CH2O → CHO → CO, and the activation free energies are 0.34, 0.68, 0.53 and 1.05 eV, respectively. The main pathway for CH3OH to CHOOCH3 is CH3OH → CH3O → CH2O, CH2O + CH3O → CH2OOCH3, CH2OOCH3 → CHOOCH3 + H, and the activation free energies are 0.34, 0.68, 0.52 and 0.25 eV, respectively. The activation free energies of the rate-limiting step of CH3OH to CHOOCH3 and CO are 0.68 and 1.05 eV, indicating that CHOOCH3 is easier to form on the Sc@P10C12Cu2 catalyst. The corresponding IS, TS and FS are shown in Fig. S7, and the corresponding reaction free energies and activation free energies are shown at 298 K in Table 2.
3.3.5 General discussion
For the CH3OH → CH3O + H, the activation free energies(eV) follow the order: Sc@P10C12Fe2(0.68)>Sc@P10C12Cu2(0.34)>Sc@P10C12Co2(0.33)>Sc@P10C12Ni2(0.32), which are lower than the energy barrier on the most catalyst surfaces (Lu et al., 2016; Ding et al., 2016; Damte et al., 2018; Zhang et al., 2018). For the CH3O → CH2O + H; the activation free energies(eV) follow the order: Sc@P10C12Fe2(1.19)>Sc@P10C12Co2(0.94)>Sc@P10C12Cu2 (0.68)>Sc@P10C12Ni2(0.58), which are close to the energy barrier on the most catalyst surfaces (Ding et al., 2016; Damte et al., 2018; Zhang et al., 2018; Hung et al., 2015). For the CH2O + CH3O → CH2OOCH3; the activation free energies(eV) follow the order: Sc@P10C12Co2(0.83)>Sc@P10C12Fe2(0.72)>Sc@P10C12Ni2 (0.64)>Sc@P10C12Cu2(0.52), and for the CH2OOCH3 → CHOOCH3 + H, the activation free energies(eV) follow the order: Sc@P10C12Fe2(1.13)>Sc@P10C12Co2(0.53)>Sc@P10C12Ni2 (0.28)>Sc@P10C12Cu 2(0.25), Sc@P10C12Fe2 (1.13 eV) is close to the energy barrier on the most catalyst surfaces, while the other three catalysts (0.53, 0.28, 0.25 eV) are lower than the energy barrier on the most catalyst surfaces (Wu et al., 2022; Lin et al., 2012). From the above results, it can be seen that the activation free energy of the rate-limiting step of CH3OH to CHOOCH3 is 0.64 eV on the Sc@P10C12Ni2 catalyst at 298 K, which is lower than that of Cu3Zn(1 1 1) (1.24 eV) (Wu et al., 2022), Cu(1 1 1) (1.43 eV) (Wu et al., 2022) and PdZn(1 1 1) (1.24 eV) (Lin et al., 2012) catalysts. In general, the activation free energy of CH3OH to CHOOCH3 is the lowest on the Sc@P10C12Ni2 catalyst.
Table 3 lists the rate constants of the main pathway of CH3OH to CHOOCH3 on the Sc@P10C12M2 catalysts at 298 K. The activity of the main pathway of CH3OH to CHOOCH3 on the Sc@P10C12M2 catalysts at 298 K are shown in Fig. 3. For the CH3OH → CH3O + H, the activity(lgk/s−1) follows the order: Sc@P10C12Ni2(7.38)>Sc@P10C12Co2(7.21)>Sc@P10C12Cu2 (7.04) >Sc@P10C12Fe2(1.29). For the CH3O → CH2O + H, the activity(lgk/s−1) follows the order: Sc@P10C12Ni2(2.98)>Sc@P10C12Cu2(1.29)>Sc@P10C12Co2 (−3.11)>Sc@P10C12Fe2(−7.33). For the CH2O + CH3O → CH2OOCH3, the activity(lgk/s−1) follows the order: Sc@P10C12Cu2(4.00)>Sc@P10C12Ni2(1.97)>Sc@P10C12Fe2 (0.62)>Sc@P10C12Co2(−1.25). For the CH2OOCH3 → CHOOCH3 + H, the activity(lgk/s−1) follows the order: Sc@P10C12Cu2(8.57)>Sc@P10C12Ni2(8.06)>Sc@P10C12Co2 (3.83)>Sc@P10C12Fe2(−6.32). In general, the activity of CH3OH to CHOOCH3 is the highest on the Sc@P10C12Ni2 catalyst. In addition, the CH2O → CHO + H, as a competitive reaction to the formation of CHOOCH3, has an activity of 1.29 lgk/s−1, which is lower than that of CHOOCH3 (1.97 lgk/s−1), indicating that CHOOCH3 is formed as a preferred product and has high selectivity on the Sc@P10C12Ni2 catalyst. Moreover, the reaction mechanism of CH3OH to CHOOCH3 in this study is consistent with that of other catalysts (Yang et al., 2018; Wu et al., 2022).
k(s−1)
Sc@P10C12Fe2
Sc@P10C12Co2
Sc@P10C12Ni2
Sc@P10C12Cu2
R1
1.96 × 101
1.63 × 107
2.40 × 107
1.10 × 107
R4
4.64 × 10-8
7.85 × 10-4
9.63 × 102
1.96 × 101
R10
4.13 × 100
5.69 × 10-2
9.31 × 101
9.96 × 103
R11
4.80 × 10-7
6.75 × 103
1.14 × 108
3.67 × 108
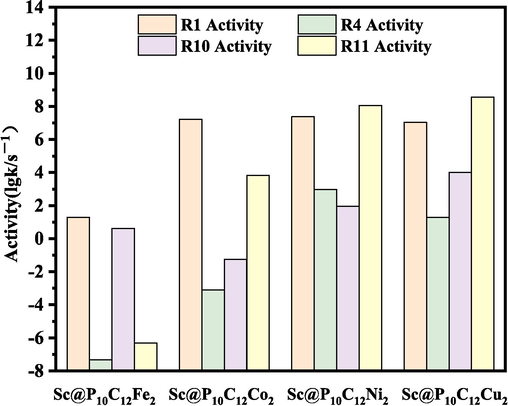
The activity (lgk/s−1) of the main pathway of CH3OH to CHOOCH3 on the Sc@P10C12M2 catalysts.
3.4 Electronic structure analysis
3.4.1 DOS and magnetic moment analysis
Fig. 4 (a), (b), (c), and (d) show the density of states of Sc@P10C12Fe2, Sc@P10C12Co2, Sc@P10C12Ni2 and Sc@P10C12Cu2, respectively. The zero point is set at the fermi level. It can be observed from Fig. 4 that the Sc@P10C12M2 systems exhibit semiconductor characteristics with asymmetric spin up and spin down states, as well as magnetism. In addition, as illustrated in Fig. 4 (e), the magnetic moments of 2Fe, 2Co, 2Ni and 2Cu are calculated. It can be noted that the relationship between the magnetic moment and the activation free energy of the rate-limiting step of CH3OH to CHOOCH3.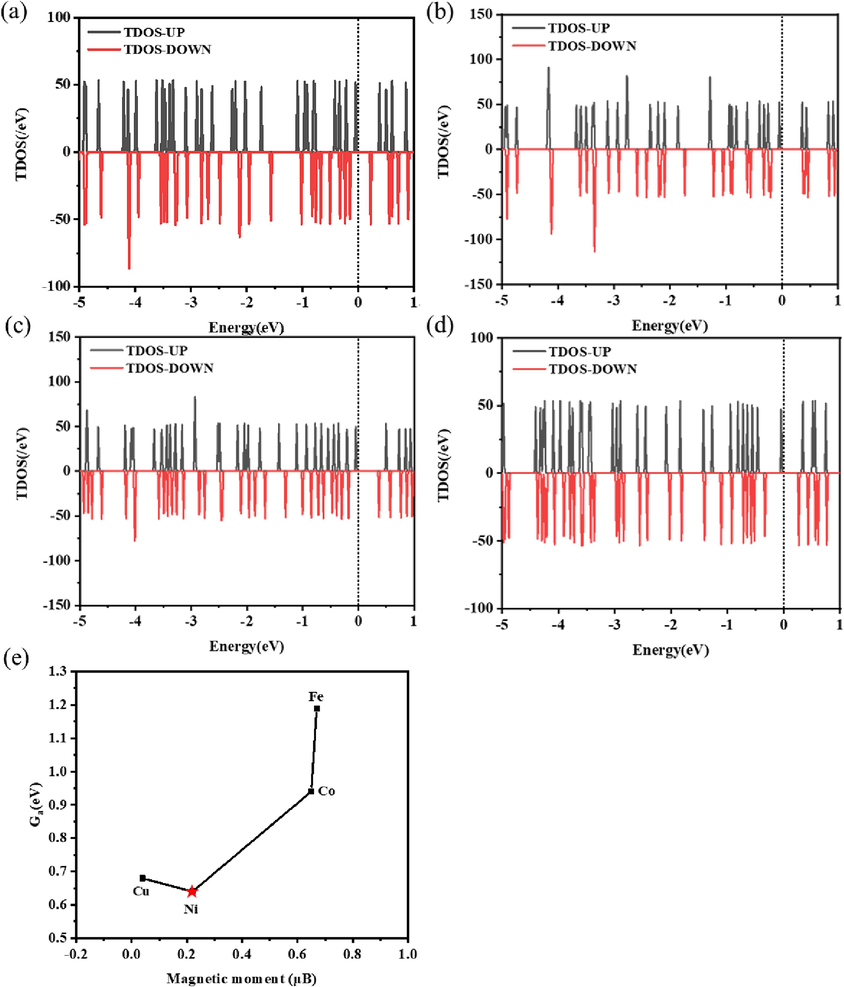
The density of states of (a) Sc@P10C12Fe2, (b) Sc@P10C12Co2, (c) Sc@P10C12Ni2 and (d) Sc@P10C12Cu2. (e) The relationship between the magnetic moment of 2Fe, 2Co, 2Ni, 2Cu and the activation free energy of the rate-limiting step of CH3OH to CHOOCH3.
Melander (Melander et al., 2014) et al. demonstrated that the reactivity of metal surfaces is dependent on their magnetic state. Bhattacharjee (Bhattacharjee et al., 2016) points out that in order to understand trends in the catalytic activity of transition metals, spin polarization of metals should be considered. The literature above indicates that spin polarization is a significant factor influencing the catalytic activity of metals, with spin-polarized surfaces showing lower activity than non-spin-polarized surfaces. In other words, a low degree of spin polarization corresponds to higher activity levels.
In this work, the catalytic activity is correlated with the magnetic state and spin polarization of metals (Fe, Co, Ni, Cu). It is observed that the spin polarization of Ni and Cu is low while their activity is high, whereas the spin polarization of Fe and Co is high but their activity is low. This overall trend aligns with previous literature (Melander et al., 2014). The tiny magnetic moment of 2Ni and 2Cu may contribute to Sc@P10C12Ni2 and Sc@P10C12Cu2 exhibiting high activity from the point of view of magnetic states and spin polarization. However, slight variations in the activity of Ni and Cu may be attributed to differences in the number of metal transfer electrons as well as active center charge. Charge analysis was subsequently conducted to further investigate these differences.
3.4.2 The charge density difference analysis
The formula for calculating the charge density difference is as follows: Δρ = ρAB − ρA – ρB (Ammar et al., 2020). Here, ρAB represents the structural charge density after interface optimization, while ρA and ρB represent the charge densities of A and B, respectively. According to formula, the charge density change after A and B form AB can be analyzed. The charge density difference of the CH3OH molecule adsorbed on the catalysts of Sc@P10C12M2 is shown in Fig. 5, which is obtained by subtracting the charge densities of isolated CH3OH molecule and Sc@P10C12M2 from that of the Sc@P10C12M2 with the CH3OH adsorbed. Blue and yellow represent charge loss and accumulation, respectively. It can be observed from Fig. 5 that there is a noticeable charge transfer between the methanol molecule and the Sc@P10C12M2 catalysts. Specifically, CH3OH transferred 0.066, 0.067, 0.043 and 0.040 electrons to the Sc@P10C12Fe2, Sc@P10C12Co2, Sc@P10C12Ni2, and Sc@P10C12Cu2 catalysts, respectively.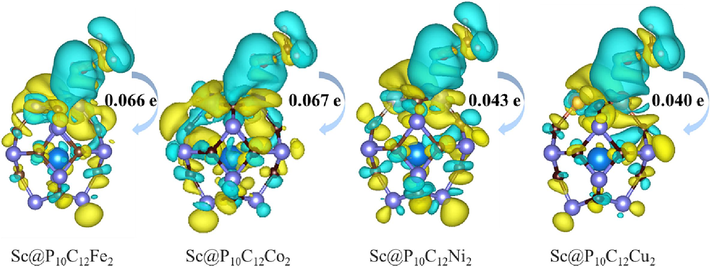
The charge density difference for the most stable configuration of CH3OH adsorbed on the Sc@P10C12M2 catalysts, the isosurface value is set to 0.0002 e/bohr3.
3.4.3 Bader charge analysis
Since the adsorption and activation sites involved in methanol conversion are limited to the C-M−C−M four-membered ring, the charges on the four-membered ring of the four catalysts are calculated. It was determined that the entire four-membered ring gains electrons, with electron counts of 0.37, 0.71, 0.89 and 0.98 e, respectively. These findings indicate that when there is a moderate number of electrons (0.89 e) in the reaction center of the catalyst, efficient conversion of methanol to methyl formate can occur on the Sc@P10C12Ni2 catalyst as shown in Fig. 6.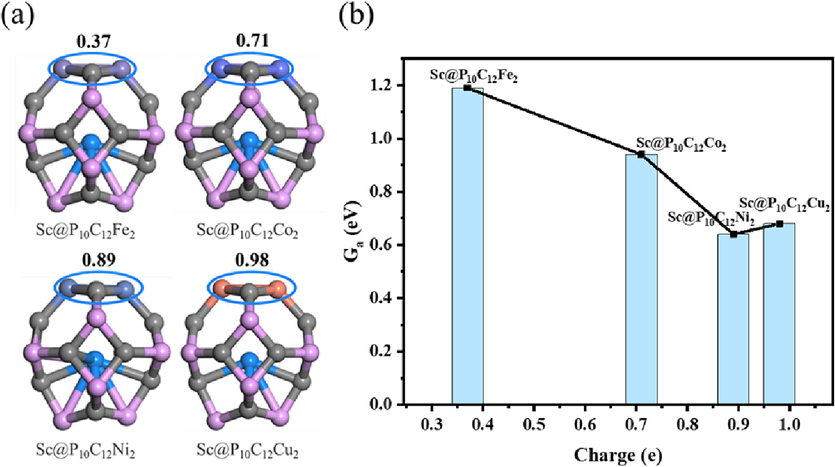
Charge (e) in the four-membered ring and its relation to the activation free energy of the rate-limiting step on the Sc@P10C12M2 catalysts.
Fig. 7 illustrates the relationships between the electrons in the transition state of the primary reaction pathways from CH3OH to CHOOCH3 and the corresponding activation free energies on the Sc@P10C12M2 catalysts. For the R1 (CH3OH → CH3O + H) reaction, the number of electrons transferred by the Fe, Co, Ni and Cu on the catalysts are −0.849, −0.688, −0.628 and −0.683 e in the transition state, respectively. There is a strong linear relationship between the number of electrons transferred by metals and the activation free energies (R2 = 0.94). Similarly, for the R4 (CH3O → CH2O + H) and R11 (CH2OOCH3 → CHOOCH3 + H) reactions, there are significant correlations between the number of electrons transferred by metals and the activation free energies (R2 = 0.99 and R2 = 0.99). For the R10 (CH2O + CH3O → CH2OOCH3) reaction, the number of electrons transferred by the whole catalysts are −0.487, −0.460, −0.494 and −0.558 e in the transition state, respectively. A strong linear relationship exists between the number of electrons transferred by the catalysts and the activation free energies (R2 = 0.91). That is, for the dehydrogenation reactions (R1, R4 and R11), the electrons transferred by transition state metals (Fe, Co, Ni, Cu) in the transition state on the catalysts can serve as the activity descriptor, while for the R10 reaction, the electrons transferred by the entire catalysts can be utilized as the activity descriptor.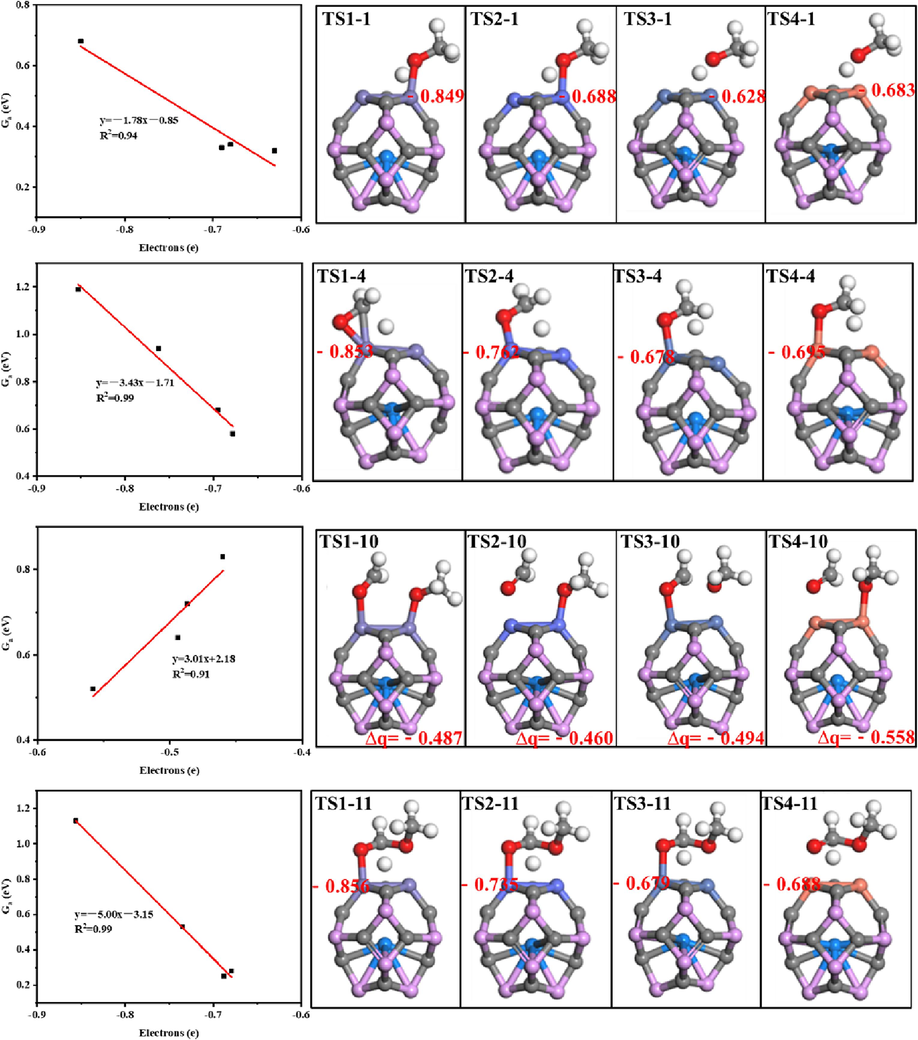
The relationships between the electrons (e) of the transition state and the corresponding activation free energies on the Sc@P10C12M2 catalysts.
Fig. S8 illustrates the electron distribution on the Sc@P10C12Ni2 catalyst. It is evident that the nickel and phosphorus atoms lose electrons, while carbon atoms gain electrons. Additionally, the scandium atoms within the catalyst lose electrons, exhibiting a similar pattern to the transfer of metal to outer electrons observed in other encapsulated catalysts (Kramer et al., 2017; Shen et al., 2023).
3.5 Carbon resistance
In this work, the carbon resistance can be assessed based on the deep dehydrogenation and deoxygenation of methanol. Specifically, the ease with which methanol forms C and/or CH substances on the catalyst directly correlates to its propensity for coking. On the one hand, the activation energy of CHXO is lower than that of CH, which greatly reduces the formation of CH. On the other hand, the activation free energy of CO to form elemental carbon on the Sc@P10C12Ni2 catalyst is 3.91 eV at 298 K, indicating a challenging reaction process. These factors collectively contribute to a substantial decrease in coking potential, ultimately demonstrating excellent carbon resistance of the catalyst.
4 Conclusions
The Sc@P10C12M2 catalysts were constructed for the first time to improve the catalytic performance of methanol conversion catalysts, and their catalytic properties were systematically studied by DFT at 298 K. The results indicate that (1) Sc@P10C12M2 catalysts exhibit certain stability at 298 K, (2) the selectivity and formation activity of CHOOCH3 are influenced by the types of metals (Fe, Co, Ni, Cu), and (3) the Sc@P10C12Ni2 demonstrates the highest catalytic activity, with an activation free energy and activity of 0.64 eV and 1.97 lgk/s−1 for the rate-limiting step at 298 K. The models and calculations presented in this study provide a valuable reference for the design and development of new nanocatalysts at low temperatures.
CRediT authorship contribution statement
Wannan Wang: Writing – original draft. Rui-Peng Ren: Writing – review & editing. Yong-Kang Lv: Resources.
Acknowledgement
The authors gratefully acknowledge the financial support of this study by the National Natural Science Foundation of China (22078226 and 22178244), Shanxi-Zheda Institute of Advanced Materials and Chemical Engineering (2021SX-FR001) and the Natural Science Foundation of Shanxi Province (20210302123118). The authors especially thank anonymous reviewers for their helpful suggestions.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Interaction and detection of formaldehyde on pristine and doped boron nitride nano-cage: DFT calculations. Mater. Today Commun.. 2020;25:101408
- [CrossRef] [Google Scholar]
- Methyl formate formation during methanol conversion over the (111) ceria surface. J. Phys. Chem. C. 2017;121(18):9920-9928.
- [CrossRef] [Google Scholar]
- An improved d-band model of the catalytic activity of magnetic transition metal surfaces. Sci. Rep.. 2016;6:35916.
- [CrossRef] [Google Scholar]
- Iron catalyst encapsulated in carbon nanotubes for CO hydrogenation to light olefins. Chinese J. Catal.. 2015;36(9):1631-1637.
- [CrossRef] [Google Scholar]
- Bimetallic alloys encapsulated in fullerenes as efficient oxygen reduction or oxygen evolution reaction catalysts: A density functional theory study. J. Alloys Compd.. 2022;894:162508
- [CrossRef] [Google Scholar]
- Methanol decomposition reactions over boron-doped graphene supported Ru-Pt catalyst. Phys. Chem. Chem. Phys.. 2018;20:9355-9363.
- [CrossRef] [Google Scholar]
- Iron encapsulated within pod-like carbon nanotubes for oxygen reduction reaction. Angew. Chem. Int. Ed.. 2013;125(1):389-393.
- [CrossRef] [Google Scholar]
- Insight into the reaction mechanisms of methanol on PtRu/Pt(111): A density functional study. Appl. Surf. Sci.. 2016;369:257-266.
- [CrossRef] [Google Scholar]
- Hydrogen storage capacity of C12X12 (X = N, P, and Si) Chem. Phys. Impact. 2022;5:100107
- [CrossRef] [Google Scholar]
- A density functional theory study on the adsorption and decomposition of methanol on B12N12 fullerene-like nanocage. Superlattices Microstruct.. 2014;67:54-60.
- [CrossRef] [Google Scholar]
- Conversion of CO2, H2, and alcohols into formate esters using anionic iron carbonyl hydrides. Inorg. Chim. Acta.. 1978;31:L387-L389.
- [CrossRef] [Google Scholar]
- Adsorption-induced liquid-to-solid phase transition of Cu clusters in catalytic dissociation of CO2. J. Phys. Chem. Lett.. 2020;11(19):7954-7959.
- [CrossRef] [Google Scholar]
- Methanol carbonylation to methyl formate catalyzed by strongly basic resins. Catal. Lett.. 1996;38:127-131.
- [CrossRef] [Google Scholar]
- A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys.. 2010;132:154104
- [CrossRef] [Google Scholar]
- A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys.. 2000;113:9901-9904.
- [CrossRef] [Google Scholar]
- Computational study of the interaction of C12P12 and C12N12 nanocages with alendronate drug molecule. ChemistrySelect. 2023;8(1):e202203607.
- [CrossRef] [Google Scholar]
- Catalytic activity of dehydrogenation of methanol to MF over Cu/SBA-15 and Cu-ZnO/SBA-15 prepared by grinding and impregnation. Adv. Mater. Res.. 2013;608–609:1476-1479.
- [CrossRef] [Google Scholar]
- Dependence on size of supported Rh nanoclusters in the decomposition of methanol. ACS Catal.. 2015;5(7):4276-4287.
- [CrossRef] [Google Scholar]
- Methanol-formic acid esterification equilibrium in sulfuric acid solutions: Influence of sodium salts. Ind. Eng. Chem. Res.. 1993;32(5):981-985.
- [CrossRef] [Google Scholar]
- Dehydrogenation of methanol to methyl formate over supported Ni, Pd and Pt catalysts. Anomalous catalytic functions of PdZn and PtZn alloys. React. Kinet. Catal. l. 1995;55:245-250.
- [CrossRef] [Google Scholar]
- Homogeneous catalytic reactions involving methyl formate. Appl. Catal. A-Gen.. 1995;121(1):25-44.
- [CrossRef] [Google Scholar]
- Self-consistent equations including exchange and correlation effects. Phys. Rev.. 1965;140(4A):A1133-A1138.
- [CrossRef] [Google Scholar]
- Nitrogen-doped carbon-encapsulated nickel/cobalt nanoparticle catalysts for olefin migration in allylarenes. ChemCatChem. 2017;9(15):2930-2934.
- [CrossRef] [Google Scholar]
- Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B. 1996;54(16):11169-11186.
- [CrossRef] [Google Scholar]
- Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci.. 1996;6(1):15-50.
- [CrossRef] [Google Scholar]
- Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B. 1993;48(17):13115-13118.
- [CrossRef] [Google Scholar]
- From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B. 1999;59:1758-1775.
- [CrossRef] [Google Scholar]
- Calculation of the structure of carbon clusters based on fullerene-like C24 and C48 molecules. Phys. Solid State. 2016;58:394-401.
- [CrossRef] [Google Scholar]
- C2H2 semi-hydrogenation over N-doped graphene supported diatomic metal catalysts: Unraveling the roles of metal type and its coordination environment in tuning catalytic performance. Appl. Surf. Sci.. 2023;641:158413
- [CrossRef] [Google Scholar]
- First-principles study of the methyl formate pathway of methanol steam reforming on PdZn(1 1 1) with comparison to Cu(1 1 1) J. Mol. Catal. A-Chem.. 2012;356:165-170.
- [CrossRef] [Google Scholar]
- Selective oxidation of methanol and ethanol on supported ruthenium oxide clusters at low temperatures. J. Phys. Chem. B. 2005;109(6):2155-2163.
- [CrossRef] [Google Scholar]
- Methanol oxidation on Pt3Sn(111) for direct methanol fuel cells: Methanol decomposition. ACS Appl. Mater. Interfaces. 2016;8(19):12194-12204.
- [CrossRef] [Google Scholar]
- Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys.. 2014;140:084106
- [CrossRef] [Google Scholar]
- An evaluation of solvent effects and ethanol oxidation. Phys. Chem. Chem. Phys.. 2021;23:16180-16192.
- [CrossRef] [Google Scholar]
- Effect of magnetic states on the reactivity of an FCC(111) iron surface. J. Phys. Chem. C. 2014;118(29):15863-15873.
- [CrossRef] [Google Scholar]
- Efficient CuZn/SiO2 lamellar catalysts for methanol dehydrogenation: New insights into the role of zinc. Appl. Catal. A-Gen.. 2022;637:118585
- [CrossRef] [Google Scholar]
- Catalysts for methanol steam reforming—A review. Appl. Catal. B: Environ.. 2010;99(1–2):43-57.
- [CrossRef] [Google Scholar]
- Vapor-phase dehydrocoupling of methanol to methyl formate over CuAl2O4. J Catal. 1997;169(2):447-454.
- [CrossRef] [Google Scholar]
- Symmetrical clusters of carbon atoms: The C24 and C60 molecules. Chem. Phys. Lett.. 1987;141:45-48.
- [CrossRef] [Google Scholar]
- Homogeneous hydrogenation of CO2 and CO to methanol: The renaissance of low-temperature catalysis in the context of the methanol economy. Angew. Chem. Int. Ed.. 2022;61(42):e202207278.
- [Google Scholar]
- C2H2 semi-hydrogenation over Pdn/TiO2 and PdnCO/TiO2 catalysts: Probing into the roles of Pd cluster size and pre-adsorbed CO in tuning catalytic performance. Fuel. 2023;358:130053
- [CrossRef] [Google Scholar]
- Geometric and electronic effects of Co@NPC catalyst in chemoselective hydrogenation: Tunable activity and selectivity via N, P Co-doping. J. Catal.. 2023;421:65-76.
- [CrossRef] [Google Scholar]
- Oxidative coupling of methanol with molecularly adsorbed oxygen on Au surface to methyl formate. J. Phys. Chem. Lett.. 2021;12(29):6941-6945.
- [CrossRef] [Google Scholar]
- Low-temperature heterogeneous catalytic reaction by surface protonics. B. Chem. Soc. Jpn.. 2019;92:1785-1792.
- [CrossRef] [Google Scholar]
- Highly active CuZn/SBA-15 catalyst for methanol dehydrogenation to methyl formate: Influence of ZnO promoter. Mol. Catal.. 2021;505:111514
- [CrossRef] [Google Scholar]
- VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun.. 2021;267:108033
- [CrossRef] [Google Scholar]
- Insights into the crucial role of a Zn promoter for methanol dehydrogenation to methyl formate over Cu(111) catalysts. Phys. Chem. Chem. Phys.. 2022;24(37):22661-22669.
- [CrossRef] [Google Scholar]
- A highly stable copper-based catalyst for clarifying the catalytic roles of Cu0 and Cu+ species in methanol dehydrogenation. Angew. Chem. Int. Ed.. 2018;57(7):1836-1840.
- [CrossRef] [Google Scholar]
- Chain mail for catalysts. Angew. Chem. Int. Ed.. 2020;59(36):15294-15297.
- [CrossRef] [Google Scholar]
- Identification of Pt-based catalysts for propane dehydrogenation via a probability analysis. Chem. Sci.. 2018;9(16):3925-3931.
- [CrossRef] [Google Scholar]
- Dissociation and oxidation mechanism of methanol on Al12N12 cage: A DFT study. Theor. Chem. Acc.. 2018;137:1-12.
- [CrossRef] [Google Scholar]
- Theoretical study on the influence of a secondary metal on the Cu(110) surface in the presence of H2O for methanol decomposition. RSC Adv.. 2016;6:15127-15136.
- [CrossRef] [Google Scholar]
- In situ FTIR studies of methanol adsorption and dehydrogenation over Cu/SiO2 catalyst. Fuel. 2002;81(11–12):1619-1624.
- [CrossRef] [Google Scholar]
- A comparative DFT study on the dehydrogenation of methanol on Rh(100) and Rh(110) Appl. Surf. Sci.. 2018;436:268-276.
- [CrossRef] [Google Scholar]
- Preparation of heteroatom-doped carbon materials and applications in selective hydrogenation. ChemistrySelect. 2022;7:e202102581.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2024.105806.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary Data 1
Supplementary Data 1




