

Translate this page into:
Synthesis, in silico and in vitro studies of novel quinazolinone derivatives as potential SARS-CoV-2 3CLpro inhibitors
⁎Corresponding author. o.akram@psau.edu.sa (Obaid Afzal),
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
A series of new quinazolinone derivatives (5a-l) were designed as 3CL protease inhibitors for SARS-CoV-2 infection. The designed derivatives were efficiently synthesized by S-alkylation/arylation of an intermediate, 6-fluoro-3-(4-fluorophenyl)-2-mercaptoquinazolin-4(3H)-one and their successful synthesis was established by analytical methods, viz. IR, 1H NMR, & 13C NMR spectroscopy. The in silico inhibitory potential against 3CLpro of SARS-CoV-2 were studied by means of docking and dynamics simulations, and compared with the co-crystallized ligand (VR4) of SARS-CoV-2 3CLpro. The compounds interacted strongly within the active catalytic dyad (Cys-His) site, thereby anticipated to obstruct the function of 3CLpro of SARS-CoV-2. Compounds 5b, 5c, 5i, 5j and 5l showed efficient binding with protease 3CLpro with XP Gscore of −7.4, −8.3, −7.8, −7.5 and −8.2 respectively. Furthermore, molecular dynamic simulation study of these compounds (5b, 5c, 5i, 5j and 5 l) showed stable interaction over 50 ns production run. Swiss ADME and pkCSM web tools showed favorable physicochemical and pharmacokinetic properties and fulfilled the criteria for drug-likeness of these selected studied compounds. The toxicity determination of these selected compounds predicted that some compounds were hepatotoxic, but were not AMES toxic. Compounds 5b, 5c, 5i, 5j and 5 l revealed their inhibitory potential against the SARS-CoV-2 3CLpro, and their IC50 values were attained at 1.58, 1.25, 1.97, 0.44 and 2.56 µM, respectively. In addition, these compounds were found to have devoid of any significant cytotoxicity even at a higher concentration of 20 µM against VeroE6 cells. These quinazolinone derivatives showed potent binding and inhibitory potential against SARS-CoV-2 3CLpro and may emerge as compounds that might act as prospective inhibitors.
Keywords
Quinazolinone
SARS-CoV-2
3CLpro
Protease inhibitors
Molecular docking
Molecular dynamics simulation

1 Introduction
The human SARS-CoV-2 (Severe Acute Respiratory Syndrome Corona Virus 2) instigated the worldwide pandemic COVID-19 infection in december 2019, and shares common genome (82 %) identical to SARS-CoV outbreak in 2002 (Abian et al., 2020). The virus consists of an enveloped single stranded positive RNA genome and is composed of both the nonstructural and structural proteins. The nonstructural protein (NSP) has open reading frame (ORF) from NSP1 to NSP16. The 5′-end of the SARS-CoV-2 large reading frame ORF1ab (accounts for two-third of the genome length) is translated to two viral precursor polyproteins (pp1a and pp1ab) that is cleaved by viral proteases, viz. papain-like protease (PLpro) and main protease (Mpro, also called as 3 Chymotrypsin-like cysteine protease, 3CLpro), to produce 16 mature non-structural polyproteins (NSP1-16) (Chen et al., 2022; Hartini et al., 2021; Liu et al., 2020). The NSPs are involved in transcription, replication and virus recombination in infections (Ferreira et al., 2022). The 3CLpro is one of the protease that is important in the lifecycle of the virus, with no homologous protein in humans. The 3CLpro cleaves polyprotein 1ab on at least eleven cleavage sites having common cleavage sequence, distinct from human protease and is a promising target for drug discovery (Chen et al., 2022). Thus, selective inhibitors against this protease will have no side-effects on humans (Abdel-Mohsen et al., 2023). The main protease (Mpro or 3CLpro) constitute extremely conserved genes with 96 % sequence similarity among SARS-CoV and SARS-CoV-2 (Abian et al., 2020). Since, the viral genome (positive-sense single stranded RNA; +ssRNA) after hijacking the host ribosomes produces large polypeptide (PP) chain after translation, and this PP chain after cleavage by 3CLpro (encoded by the viral genome) generates 11 NSPs (essentail for viral replication), 3CLpro is considered as a promising druggable target for anti-SARS-CoV-2 drug discovery (Hartini et al., 2021; Chen et al., 2022). The knowledge of the virus molecular structure, its life cycle and its interaction with the human cells led to the emergence of various vaccines and antiviral drugs for the treatment of SARS-CoV-2 infection. However, further research is needed to develop new antiviral drugs to overcome future drug resistance to avert the SARS-CoV-2 infections and to complement vaccines (Al Adem et al., 2023; Atzrodt et al., 2020).
Quinazolinones has proven to have wide variety of activities including anti-HIV, antibacterial, anticancer, antihypertension and antifungal (Riadi et al., 2022). Quinazolines are reported to have potential antiviral activities against various RNA viruses such as hepatitis, influenza, and respiratory syncytial viruses. The effective antiviral quinazolinones derivative incorporating S-substituted coumarin moiety (1) was reported to inhibit chikungunya and hepatitis C virus (Zhang et al., 2020). A compound 7-chloro-2-((2,3,4-trifluorophenyl)amino)quinazolin-4(3H)-one (2) among the series of 2-aminoquinazolin-4-(3H)-one derivatives, was reported to be active against SARS-CoV-2 with IC50 2.6 µM and CC50 (cytotoxicity) more than 25 µM (Lee et al., 2021). To target the RNA genome (the causative agent of COVID-19 infection) within SARS-CoV-2, a small molecule having quinazoline moiety (3) was designed, that binds strongly with the attenuator hairpin of RNA genome with Kd of 11 nM. The compound (3) was futher ligated with a ribonuclease targeting chimera (RIBOTAC) to develop a compound (4) that specifically targets cellular ribonuclease to destroy the viral genome of SARS-CoV-2. This strategy was reported to improve the bioactivity of the compound (3) at least 10-fold. (Haniff et al., 2020). Two compounds (5 and 6) having core structure 11H-pyrido[2,1-b]quinazolin-11-one were recently identified as SARS-CoV-2 RNA-dependent RNA polymerase inhibitors and showed comparable in silico activity to standard drugs favipiravir and remdesivir (Manikanttha et al., 2023). The quinazolinone derivatives (7) was identified as potential viral protease inhibitors (3CLpro) after screening a large library using cell-based assay using GFP-split complementation (Rothan and Teoh, 2021). The methanolic extract of Strobilanthes cusia leaf reduced the virus yield in the cell infected with HCoV-NL63 with EC50 value of 0.64 µg/ml, and the most potent antiviral activity was exhibited by tryptanthrin (8) having indoloquinazoline moiety (EC50 1.52 µM) (Mani et al., 2020). Another plant containing quinazoline based alkaloids, vasicine (9), deoxyvasicine (10) and vasicinone (11) from Peganum harmala were found to be active in treating the symptoms of SARS-CoV-2 (Sharma et al., 2020). The structures of the above discussed quinazoline derivatives having antiviral potential against SARS-CoV-2 is represented in Fig. 1.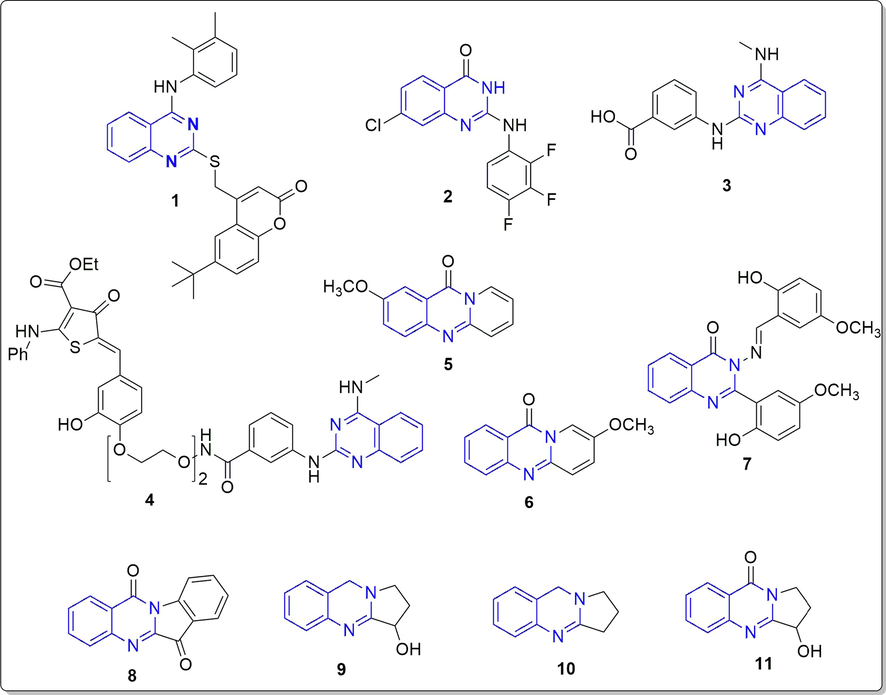
Reported quinazoline and quinazolinone derivatives as antiviral agents (1–11).
Based on the core structural framework of compound 2 (Fig. 1, 2-amino-quinazolin-4(3H)-one derivative), reported to have anti-SARS-CoV-2 potential with an IC50 of 2.6 µM, we designed some novel quinazolin-4(3H)-one derivatives as potential anti-SARS-CoV-2 main protease 3CLpro inhibitors. The strategy adopted in this present work is structure-based drug design i.e designing of inhibitors by the modifications of known hit compounds (Lee et al., 2021). Since, the aromatic ring substituted with halogen groups is important for antiviral acyivity, it was placed at position 3 of the quinazolinone ring in the designed compounds (Lee et al., 2021). The introduction of the flexible alkyl/aryl or heteroaryl groups are introduced with thioether linkage at position 2 of the quinazolinone ring in order to facilitate the binding within active catalytic dyad consisting Cys145 and His41 (Pillaiyar et al., 2022; Cannalire et al., 2020; Pillaiyar et al., 2016) (see Fig. 2). The derivatives were studied for their potential interaction inside the binding site of 3CLpro by molecular docking and dynamics simulation studies, and were also examined for their cytotoxicity and in vitro 3CLpro inhibitory potential.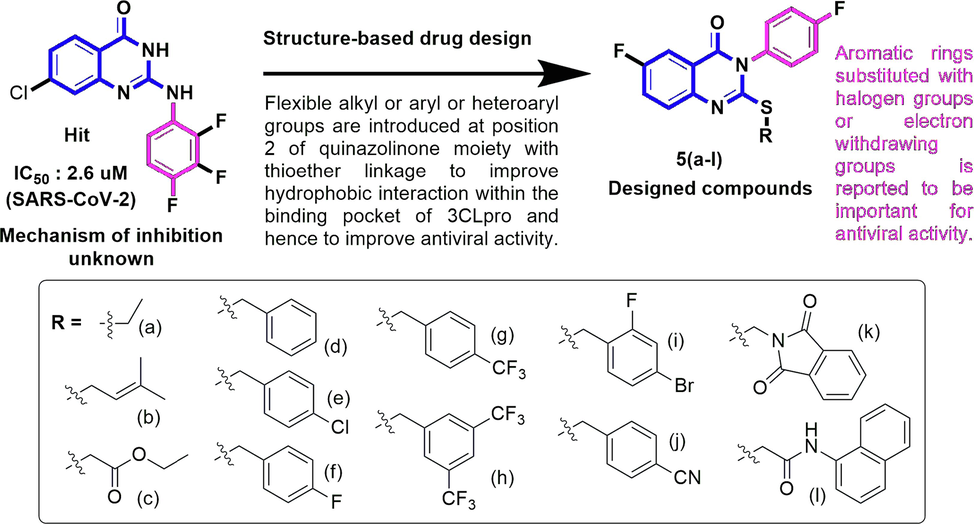
Designing of SARS-CoV-2 3CLpro Inhibitors.
2 Materials and methods
2.1 General
The advancement of the synthesis and pureness of the intermediates and final compounds were optimized with thin-layer chromatography (TLC) throughout the reaction time, by visualizing the spots of compounds in UV and Iodine vapor chambers. The melting points of the compounds were established by Dynalon Stuart apparatus. The IR spectra were documented by preparing KBr pellets of the synthesized compounds. The 1H and 13C NMR data were taken from BRUCKER-PLUS NMR spectrometer in solvent CDCl3 at 500 MHz and 125 MHz, correspondingly. Molecular mass (m/z) of the derivatives were obtained by UPLC-MS (Q-TOF-ESI) (Waters Corp., Milford, MA, USA).
2.1.1 6-Fluoro-3-(4-fluoro-phenyl)-2-mercapto-3H-quinazolin-4-one (3)
White solid; 81 % Yield; M.P.: 142–144 °C; IR (cm−1): 1544 (C⚌C str), 1689 (C⚌O str), 3055 (Ar C—H str); 1H NMR (500 MHz, CDCl3) δ (ppm): 7.42 (d, 2H, 2Hph), 7.52 (d, 1H, H8), 7.59 (d, 1H, H7), 7.98 (s, 1H, H5), 8.17 (d, 2H, 2Hph). 13C NMR (125 MHz, CDCl3) δ (ppm): 116.5 (CH), 126.9 (CH), 128.2 (CH), 128.7 (Cq), 129.0 (CH × 2), 129.3 (CH × 2), 137.8 (Cq), 138.7 (Cq), 138.9 (Cq), 144.5 (Cq), 155.4 (Cq), 159.4 (Cq); ESI-MS (m/z): 291.02 (M + 1)+, 292.03 (M + 2)+.
2.1.2 2-Ethylsulfanyl-3-(4-fluoro-phenyl)-6-fluoro-3H-quinazolin-4-one (5a)
White solid; 76 % Yield; M.P.: 146–148 °C; IR (cm−1): 1556 (C⚌C str), 1672 (C⚌O str), 3034 (Ar C—H str); 1H NMR (CDCl3, 500 MHz,) δ (ppm): 1.34 (t, 3H, CH3), 3.15 (dd, 2H, CH2), 7.21 (d, 2H, Hph), 7.28 (d, 2H, Hph), 7.52 (d, 2H, H7,8), 8.00 (s, 1H, H5). 13C NMR (CDCl3,125 MHz) δ (ppm): 13.99 (CH3), 21.41 (CH2), 116.8 (CH), 117.0 (CH), 119.6 (CH), 126.2 (CH), 126.7 (CH), 131.3 (Cq), 132.1 (CH), 136.0 (CH), 136.2 (Cq), 146.1 (Cq), 156.3 (Cq), 162.1 (Cq), 162.3 (Cq), 164.3 (Cq); ESI-MS (m/z): 319.05 (M + 1)+, 320.06 (M + 2)+.
2.1.3 6-Fluoro-3-(4-fluoro-phenyl)-2-(3-methyl-but-2-enylsulfanyl)–3H-quinazolin-4-one (5b)
White solid; 69 % Yield; M.P.: 147–149 °C; IR (cm−1): 1541 (C⚌C str), 1665 (C⚌O str), 3028 (Ar C—H str); 1H NMR (CDCl3, 500 MHz) δ (ppm): 1.78 (s, 6H, 2 × CH3), 3.87 (dd, 2H, CH2), 5.33 (dd, 1H, CH), 7.28 (m, 2H, 2Hph), 7.35 (m, 2H, Hph), 7.55 (dd, 1H, H8), 7.85 (dd, 1H, H7), 8.39 (s, 1H, H5). 13C NMR (CDCl3, 125 MHz) δ (ppm): 17.8 (CH3), 25.6 (CH3), 31.2 (CH2), 116.6 (CH), 116.8 (CH), 117.0 (CH), 118.7 (CH), 120.8 (CH), 127.9 (CH), 129.4 (CH), 130.8 (CH), 130.9 (Cq), 131.3 (Cq), 137.6 (Cq), 138.1 (Cq), 146.5 (Cq), 158.2 (Cq), 160.6 (Cq), 162.1 (Cq); ESI-MS (m/z): 359.09 (M + 1)+, 360.10 (M + 2)+.
2.1.4 6-Fluoro-3-(4-fluoro-phenyl)-4-oxo-3,4-dihydro-quinazolin-2-ylsulfanyl]-acetic acid ethyl ester (5c)
White solid; 74 % Yield; M.P.: 152–154 °C; IR (cm−1): 1542 (C⚌C str), 1672 (C⚌O str), 3018 (Ar C—H str); 1H NMR (CDCl3, 500 MHz) δ (ppm): 1.28 (s, 3H, CH3), 3.87 (s, 2H, CH2), 4.19 (dd, 2H, CH2), 7.20 (m, 2H, Hph), 7.24 (m, 2H, Hph), 7.42 (dd, 1H, H8), 7.51 (dd, 1H, H7), 7.97 (s, 1H, H5). 13C NMR (CDCl3, 125 MHz) δ (ppm): 14.2 (CH3), 21.2 (CH3), 34.9 (CH2), 116.8 (CH), 117.0 (CH), 119.4 (CH), 126.1 (CH), 126.6 (CH), 131.2 (CH), 131.5 (CH), 136.2 (Cq), 136.3 (Cq), 145.5 (Cq), 154.7 (Cq), 161.7 (Cq), 162.3 (Cq), 164.3 (Cq), 168.6 (Cq); ESI-MS (m/z): 377.07 (M + 1)+, 378.08 (M + 2)+.
2.1.5 2-Benzylsulfanyl-3-(4-fluoro-phenyl)-6-fluoro-3H-quinazolin-4-one (5d)
White solid; 83 % Yield; M.P.: 149–151 °C; IR (cm−1): 1571 (C⚌C str), 1682 (C⚌O str), 3046 (Ar C—H str); 1H NMR (DMSO, 500 MHz,) δ (ppm): 4.43 (s, 2H, SCH2), 7.27–7.34 (m, 3H, Hph), 7.40–7.47 (m, 4H, Hph), 7.55–7.59 (d, 2H, Hph), 7.68 (d, 1H, H8), 8.02 (m, 1H, H7), 8.16 (d, 1H, H5). 13C NMR (DMSO, 125 MHz) δ (ppm): 36.4 (CH2), 116.9 (CH), 117.1 (CH), 118.5 (CH), 121.8 (CH), 127.8 (CH), 128.9 (Cq), 129.0 (CH × 3), 129.8 (CH × 3), 132.1 (Cq), 132.2 (CH), 137.00 (Cq), 138.2 (Cq), 146.6 (Cq), 158.4 (Cq), 160.2 (Cq), 161.9 (Cq); ESI-MS (m/z): 381.08 (M + 1)+, 382.09 (M + 2)+.
2.1.6 2-(4-Chlorobenzylsulfanyl)-3-(4-fluoro-phenyl)-6-fluoro-3H-quinazolin-4-one (5e)
White solid; 87 % Yield; M.P.: 153–155 °C; IR (cm−1): 1566 (C⚌C str), 1678 (C⚌O str), 3055 (Ar C—H str); 1H NMR (DMSO, 500 MHz) δ (ppm): 4.38 (t, 2H, SCH2), 7.32–7.35 (m, 2H, Hph), 7.37–7.41 (m, 2H, Hph), 7.45–7.48 (m, 2H, Hph), 7.52–7.54 (m, 2H, Hph), 7.66 (m, 1H, H8), 8.00 (m, 1H, H7), 8.12 (d, 1H, H5). 13C NMR (DMSO, 125 MHz) δ (ppm): 35.5 (CH2), 116.9 (CH), 117.1 (CH), 118.5 (CH), 121.8 (CH), 127.8 (CH), 128.9 (CH), 129.0 (CH × 2), 131.7 (CH × 2), 132.2 (Cq), 132.3 (CH), 132.4 (Cq), 136.5 (Cq), 138.2 (Cq), 146.6 (Cq), 149.6 (Cq), 158.2 (Cq), 160.2 (Cq), 164.0 (Cq); ESI-MS (m/z): 415.02 (M + 1)+, 417.01 (M + 3)+.
2.1.7 2-(4-Fluorobenzylsulfanyl)-3-(4-fluoro-phenyl)-6-fluoro-3H-quinazolin-4-one (5f)
White solid; 84 % Yield; M.P.: 158–160 °C; IR (cm−1): 1554 (C⚌C str), 1671 (C⚌O str), 3062 (Ar C—H str); 1H NMR (DMSO, 500 MHz) δ (ppm): 4.66 (t, 2H, SCH2), 7.24–7.29 (m, 2H, Hph), 7.47–7.53 (m, 2H, Hph), 7.54–7.99 (m, 2H, Hph), 7.62–7.67 (m, 2H, Hph), 7.81–7.85 (m, 1H, H8), 8.00–8.14 (m, 1H, H7), 8.62–8.64 (d, 1H, H5). 13C NMR (DMSO, 125 MHz) δ (ppm): 36.0 (CH2), 116.8 (CH), 117.0 (CH), 119.2 (CH), 121.0 (CH × 2), 127.9 (CH), 129.6 (CH), 130.6 (CH × 3), 130.8 (CH), 130.9 (Cq), 134.9 (Cq), 137.9 (Cq), 146.3 (Cq), 148.4 (Cq), 157.1 (Cq), 160.5 (Cq), 162.1 (Cq), 164.2 (Cq); ESI-MS (m/z): 399.05 (M + 1)+, 400.04 (M + 2)+.
2.1.8 6-Fluoro-3-(4-fluorophenyl)-2-(4-trifluoromethylbenzylsulfanyl)–3H-quinazolin-4-one (5g)
White solid; 77 % Yield; M.P.: 164–166 °C; IR (cm−1): 1546 (C⚌C str), 1682 (C⚌O str), 3044 (Ar C—H str); 1H NMR (CDCl3, 500 MHz) δ (ppm): 4.34 (d, 2H, SCH2), 7.10–7.11 (d, 2H, Hph), 7.17–7.18 (d, 2H, Hph), 7.20–7.25 (m, 2H, Hph), 7.36–7.38 (m, 2H, Hph), 7.50–7.52 (m, 1H, H8), 7.79–7.82 (m, 1H, H7), 8.31–8.32 (d, 1H, H5). 13C NMR (CDCl3, 125 MHz) δ (ppm): 36.2 (CH2), 101.5 (CF3), 116.9 (CH), 117.1 (CH), 119.3 (CH), 121.1 (CH × 2), 121.2 (CH), 128.0 (CH), 129.8 (CH), 130.8 (CH × 2), 131.0 (CH), 131.1 (Cq), 135.0 (Cq), 138.0 (Cq), 146.5 (Cq), 148.6 (Cq), 157.3 (Cq), 160.7 (Cq), 162.2 (Cq), 164.4 (Cq); ESI-MS (m/z): 449.25 (M + 1)+, 450.02 (M + 2)+.
2.1.9 2-(3,5-Ditrifluoromethylbenzylsulfanyl)-6-Fluoro-3-(4-fluorophenyl)–3H-quinazolin-4-one (5h)
White solid; 72 % Yield; M.P.: 172–174 °C; IR (cm−1): 1552 (C⚌C str), 1661 (C⚌O str), 3048 (Ar C—H str); 1H NMR (CDCl3, 500 MHz) δ (ppm): 4.37 (d, 2H, SCH2), 7.22–7.30 (d, 6H, Hph), 7.40 (s, 1H, Hph), 7.54–7.57 (m, 1H, H8), 7.83–7.86 (m, 1H, H7), 8.34–8.35 (d, 1H, H5). 13C NMR (CDCl3, 125 MHz) δ (ppm): 36.2 (CH2), 101.5 (CF3 × 2), 116.9 (CH), 117.1 (CH), 119.3 (CH), 121.1 (CH), 121.0 (CH), 128.1 (CH), 129.1 (CH), 130.0 (CH × 2), 131.0 (CH), 131.4 (Cq), 135.0 (Cq), 138.5 (Cq), 146.5 (Cq), 148.6 (Cq), 157.2 (Cq), 160.4 (Cq), 162.6 (Cq), 164.4 (Cq); ESI-MS (m/z): 517.01 (M + 1)+, 518.02 (M + 2)+.
2.1.10 2-(4-Bromo-2-fluorobenzylsulfanyl)-6-Fluoro-3-(4-fluorophenyl)–3H-quinazolin-4-one (5i)
White solid; 75 % Yield; M.P.: 169–171 °C; IR (cm−1): 1586 (C⚌C str), 1689 (C⚌O str), 3068 (Ar C—H str); 1H NMR (CDCl3, 500 MHz) δ (ppm): 4.38 (s, 2H, SCH2), 7.22–7.30 (d, 6H, Hph), 7.37–7.42 (m, 1H, Hph), 7.54–7.57 (m, 1H, H8), 7.83–7.86 (m, 1H, H7), 8.34–8.35 (d, 1H, H5). 13C NMR (CDCl3, 125 MHz) δ (ppm): 29.7 (CH2), 117.0 (CH), 117.1 (CH), 119.1 (CH), 119.3 (CH), 119.4 (CH), 121.2 (CH), 121.6 (CH), 123.0 (CH), 123.1 (CH), 127.5 (CH), 128.1 (Cq), 129.8 (Cq × 2), 131.0 (Cq), 131.1 (Cq), 132.4 (Cq), 138.0 (Cq), 146.4 (Cq), 157.1 (Cq), 160.6 (Cq); ESI-MS (m/z): 476.58 (M + 1)+, 478.42 (M + 3)+.
2.1.11 2-(4-Cyanobenzylsulfanyl)-3-(4-fluoro-phenyl)-6-fluoro-3H-quinazolin-4-one (5j)
White solid; 73 % Yield; M.P.: 157–159 °C; IR (cm−1): 1554 (C⚌C str), 1671 (C⚌O str), 2257 (CN str), 3065 (Ar C—H str); 1H NMR (DMSO, 500 MHz,) δ (ppm): 4.40 (t, 2H, SCH2), 7.25–7.31 (m, 2H, Hph), 7.53–7.61 (m, 5H, Hph), 7.73–7.74 (m, 1H, H8), 7.94–7.96 (m, 1H, H7), 8.01–8.03 (d, 1H, H5). 13C NMR (DMSO, 125 MHz,) δ (ppm): 35.6 (CH2), 119.7 (CH), 121.3 (CH), 122.2 (CH), 124.3 (CH), 125.8 (CH), 126.8 (CH), 129.2 (CH × 2), 129.8 (CH × 3), 130.2 (Cq), 131.4 (Cq), 131.6 (Cq), 135.6 (Cq), 136.3 (Cq), 136.5 (Cq), 140.1 (Cq), 145.6 (Cq), 154.7 (Cq), 161.7 (Cq); ESI-MS (m/z): 406.02 (M + 1)+, 407.01 (M + 2)+.
2.1.12 2-[6-Fluoro-3-(4-fluoro-phenyl)-4-oxo-3,4-dihydro-quinazolin-2-ylsulfanyl]-N-naphthalen-1-yl-Acetamide (5l)
White solid; 65 % Yield; M.P.: 168–170 °C; IR (cm−1): 1546 (C⚌C str), 1665 (C⚌O str), 3066 (Ar C—H str), 3388 (NH str); 1H NMR (DMSO, 500 MHz) δ (ppm): 4.06 (s, 2H, SCH2), 7.21–7.32 (m, 3H, Hph), 7.43–7.46 (m, 2H, Hph), 7.58–7.66 (m, 2H, Hph), 7.70–7.73 (m, 1H, Hph), 7.80–7.82 (m, 1H, Hph), 7.83–7.85 (m, 1H, Hph), 7.83–7.85 (m, 1H, H8), 7.95–7.97 (m, 2H, Hph), 7.98–7.99 (m, 1H, H7), 8.36–8.38 (d, 1H, H5), 9.31 (s, 1H, NH); ESI-MS (m/z): 474.06 (M + 1)+, 475.05 (M + 2)+.
2.2 Extra-precision (XP) docking
The protein of SARS-CoV-2 3CLpro co-crystallized with a deuterated GC376 alpha-ketoamide analog (VR4) having 1.65 Å resolution was acquired from PDB (protein data bank) in pdb format (PDB ID: 7K0F) (Dampalla et al., 2021). The protein structure was refined with protein preparation wizard for extra-precision (XP) docking using Maestro Schrodinger version 9.4. The procedure includes the removal of the water and optimization of the hydrogen bonds. The energy of the 3D structure was minimized by FF (force field) OPLS_2005 to a RMSD (root mean square deviation) of 0.3 Å. The grid box of dimension 20 × 20 × 20 Å was generated by choosing the co-crystallized ligand (VR4). The energy minimization of the ligands including tautomeric and protonation states of the compounds (5a-l) at 7.0 ± 2.0 pH, were generated by using option LigPrep with Epik and OPLS_2005 FF. The ligands structures including the reference compound (VR4) were docked to 3CL protease using glide tool of glide extra-precision (XP) docking (Friesner et al., 2006).
2.3 ADMET analysis
The absorption, distribution, metabolism, excretion, and toxicity (ADMET) of the 5 potent compounds nominated on the basis of the docking scores, were determined by the web tools SwissADME and pkCSM (Daina et al., 2017; Pires et al., 2015).
2.4 MD simulation
The five selected compounds were evaluated for its stability and interactions inside the binding area of the protein by MD simulations studies, executed for 50 ns MD run. The ligands topology was generated using the package GROMACS2018.1 (OPLS-AA FFs) and the server SwissParam, as per previously explained protocol (Van Der Spoel et al., 2005; Zoete et al., 2011; Alamri, 2020). The simulation was executed at constant 300 K of temperature (isothermal) and 1 atm of pressure (isobaric). The trajectories were generated each 2 fs and were saved each 2 ps. In brief, the complex generated between the ligand and the proteins was within the space of at least 1 nm space hydrated with water molecules (TIP3P model) in a triclinic dimension. The solvated complex was then neutralized Na+Cl- counter ions. The complexes were energy minimized by steepest decent algorithm with 1000 kJ/mol/nm, and within 0.01 nm of maximum step size. The constraint of the bond length and the electrostatic calculations were run by Linear Constraint Solver algorithm (LINCS) and PME method. After the energy minimization for 100 ps, each complex was equilibrated using canonical ensembles NVT followed by NPT. The GROMACS analysis tool was used to calculate backbone and ligand RMSD, RMSF and the hydrogen bonds fluctuations.
2.5 Cellular viability assay
The cellular viability assay was executed by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5- diphenyltetrazolium bromide) assay. Concisely, VeroE6 cells (2 × 105 cells/well) were planted in 24-well plates. The varying concentrations (2.5, 5, 10 and 20 µM) of compounds 5b, 5c, 5i, 5j and 5l were then incubated in a CO2 incubator for 24 h at 37 °C. The control does not receive any compound treatment. After 24 h, wells were supplemented with MTT at a concentration of 2 mg/mL (HiMedia) at 37 °C and further incubated for 4 h. The formazan crystals obtained were liquefied in DMSO (100 μL) at 37 °C for 15 min. The OD (optical density) was estimated at 570 nm using a microplate reader (Islamuddin et al., 2021). Cell viability was calculated by the formula provided in Eq. (1).
2.6 3CLpro inhibition assay
The SARS-CoV-2 3CLpro assay kit, available commercially (BPS Bioscience, CA, USA) was employed in this study (in vitro) to investigate the inhibition of 3CLpro of SARS-CoV-2 by the compounds 5b, 5c, 5i, 5j and 5l. The experiments were performed according to the manufacturer’s instructions, and all the samples were assayed in triplicate. Briefly, recombinant 3CLpro (150 ng) of SARS-CoV-2 was incubated with the tested compounds 5b, 5c, 5i, 5j and 5l at varying concentrations of 10, 5, 2.5, 1.25 and 0.625 µM. Test samples (enzyme + test compound + substrate), DMEM as negative control (enzyme + DMEM + substrate), and a protease inhibitor GC376 as a positive control (enzyme + GC376 + substrate), were investigated during this study. The supplied substrate was used and the reaction mixture was incubated for 4 h at RT. A Synergy H1TM Hybrid Multi-Mode Microplate Reader (BioTek Instruments, Inc., VT, USA) was used to measure the fluorescence intensity at the wavelengths of 360 nm (excitation) and 460 nm (emission) (Ngwe Tun et al., 2022).
2.7 Statistical analysis
The data acquired after three autonomous experiments (in triplicates) were examined by one-way ANOVA (GraphPad Prism 8.0 software). Error bars indicates SEM (standard error of mean).
3 Results and discussion
3.1 Chemistry
The procedure adopted for the synthesize the novel thioquinazolin-4(3H)-one derivatives is described in our previous published article and represented in Scheme 1 (Riadi et al., 2022). The procedure started with the reaction of 4-fluorophenylisothiocyanate (1 mmol) with 5-flouroanthranilic acid (1 mmol) in the company of catalytic amount of triethylamine (Et3N; 1.1 mmol) and absolute ethanol (EtOH; 20 mL) as solvent. The reaction was refluxed for 2 h and optimized with the help of thin layer chromatography (TLC). The precipitate obtained after pouring the raw product in water was filtered, and then recrystallized with EtOH to attain the pure intermediates 3. The compound 3 (1 mmol) was reacted with alkyl/aryl halide (1 mmol) (4a-l) in the company of anhydrous K2CO3 (1.5 mmol) and refluxed under acetone (10 mL) for ten hours. The reaction product obtained was then filtered, and the solvent was evaporated in vacuo. The raw product was recrystallized from EtOH to attain the final products (5a-l).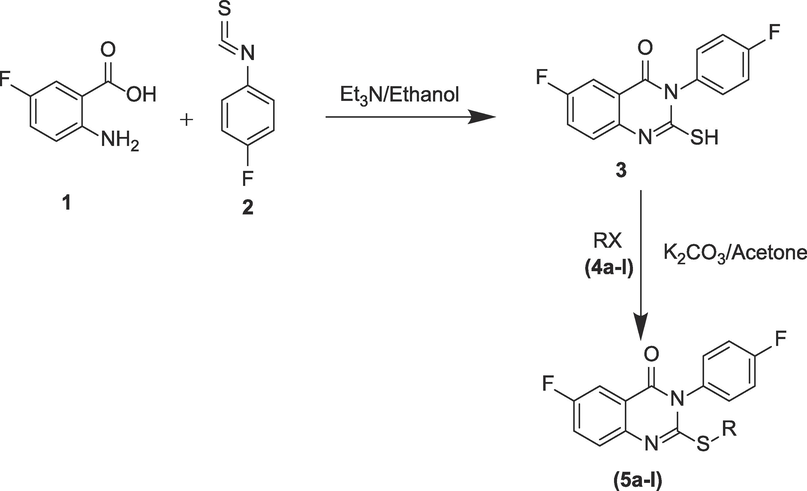
Route of synthesis for final target compounds (5a-l).
3.2 Extra-precision (XP) docking
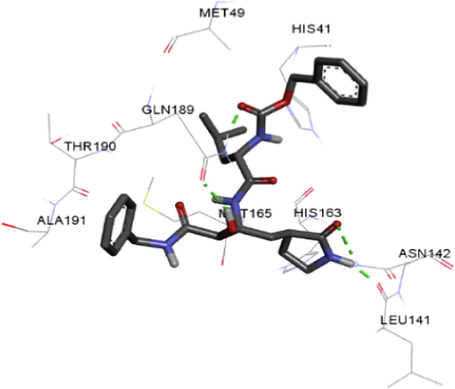
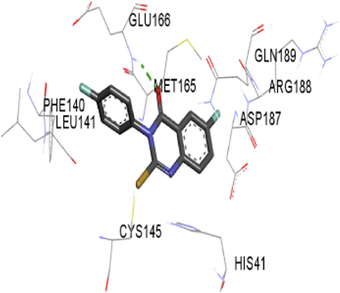
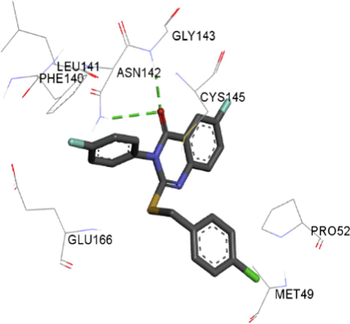
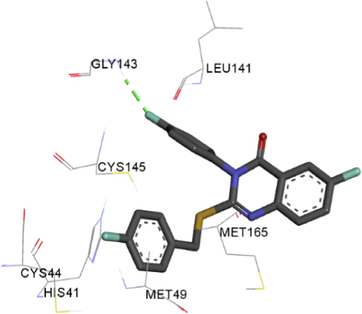
The synthesized compounds and the co-crystallized ligands (VR4) were docked in the binding site of SARS-CoV-2-3CLpro protein. The surface and ribbon representation of the protein with docked ligands are represented in Fig. 3. The ligands conformations of the five best bindings, compared with the docked co-crystallized ligands (VR4) are represented in Fig. 4. The extra-precision docking studies revealed that the co-crystallized ligand (VR4) binds effectively in the active site of 3CLpro of SARS-CoV-2, by four H-bonds with the amino acids, namely Leu141, Asn142 and Glu189. In addition, amide-pi stacked contact was witnessed between the distal phenyl ring and Thr190, along with several alkyl-alkyl and pi-alkyl hydrophobic contacts with His41, Met49, His163, Met165 and Ala191. Compounds 5b, 5c, 5i, 5j, and 5l were found to have good XP docking score and binding energy, among all the docked compounds. Among them, compound 5c was found to be the highest scoring compound, and it binds to the key amino acid residue Asn142 and Gln189 by two hydrogen bonds and Asp187 by one halogen bond. A pi-sulfur interaction was also observed between quinazolinone ring and Cys44. Thr25 is found to be involved in pi-sigma contact with the phenyl ring. Moreover, Cys145 and His163 were involved in alkyl-alkyl and pi-alkyl hydrophobic interactions. Compound 5l was found to interact with 2H-bonds with Gly143 and Glu166, and three H-bonds with Phe140, Glu166 and Arg188. One pi-pi T-shaped contact with His41, pi-sulfur with Cys145, and pi-alkyl contacts with Met49, Cys145, and Met165 were also observed. Compound 5i also interacted in similar fashion, having two H-bonds with Glu166 and Gln189, and three halogen bonds with Phe140, Glu166 and Gln189. One pi-pi T-shaped contact with His41, one pi-sulfur interaction with Cys145, and alkyl-alkyl and pi-alkyl contacts with Cys145, Met165 and Pro168 were also involved in binding. Compound 5j binding interaction revealed two hydrogen bonds (Cys44 with cyano & Gly143 with keto group), one halogen bond (His163), pi-pi stacked interaction with His41, and pi-alkyl interactions with Cys145. Similarly, compound 5b docking revealed one H-bond with Glu166, three halogens bonds with Phe140, Glu166, and Arg188, pi-sulfur contact with Cys145, pi-pi T-shaped contact with His41, and alkyl-alkyl and pi-alkyl hydrophobic contacts with Leu27, His41, Cys145, and Met165. The 3CLpro forms homodimer and each monomer is split into three domains (I-III). The active site of the cleft is between domain I (residue 10–99) and II (100–182) and has six sub sites corresponds to peptide substrate. The His41 and the Cys145 form the catalytic dyad of the active sites and is part of domain I and II (Ghosh et al., 2007). The His41 deprotonates the thiol of Cyst145 to act as nucleophile and form a thioester linkage with carbonyl carbon of Gln of peptide substrate. This thioester linkage formation is important step for the catalysis of 3CLpro and target antiviral compounds (Ferreira et al., 2022). Most of our compounds including 5a; 5b; 5d, 5e, 5h, 5i, 5k, and 5l showed pi-sulfur interaction with Cys145. The binding interaction of the compounds (5a-l) and the co-crystallized ligands are represented in tabular form (Table 1) and their 3D interaction are represented in Table 2.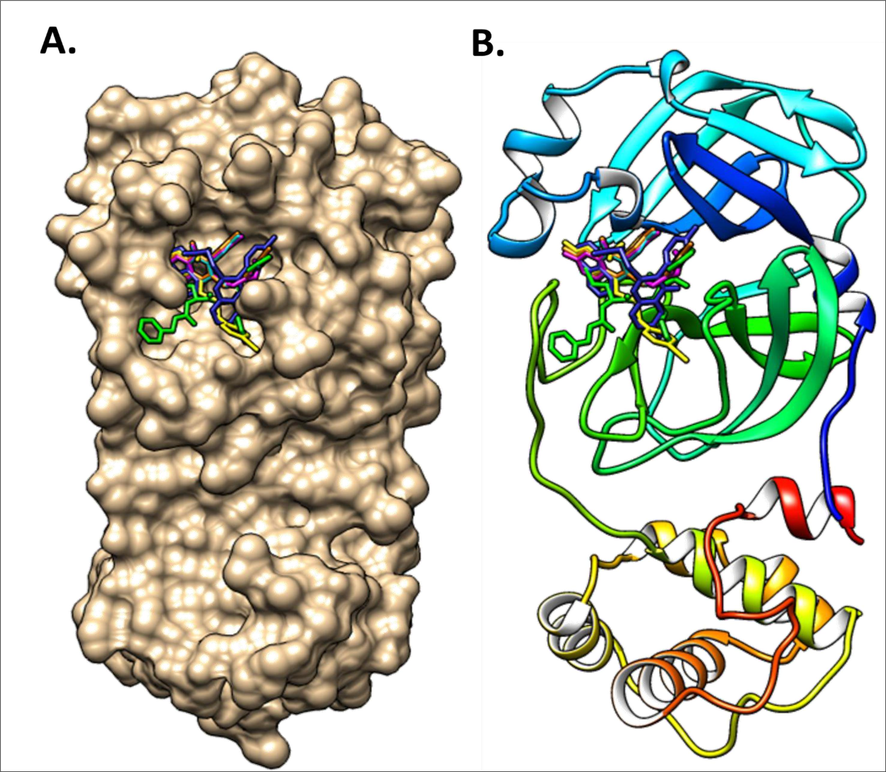
The docking conformation of the docked ligands in the binding pocket of main protease 3CLpro using molecular docking. A. Surface representation of all compounds binding mode in the active site of 3CLpro; B. Ribbon representation of all compounds binding mode in the active site of 3CLpro.
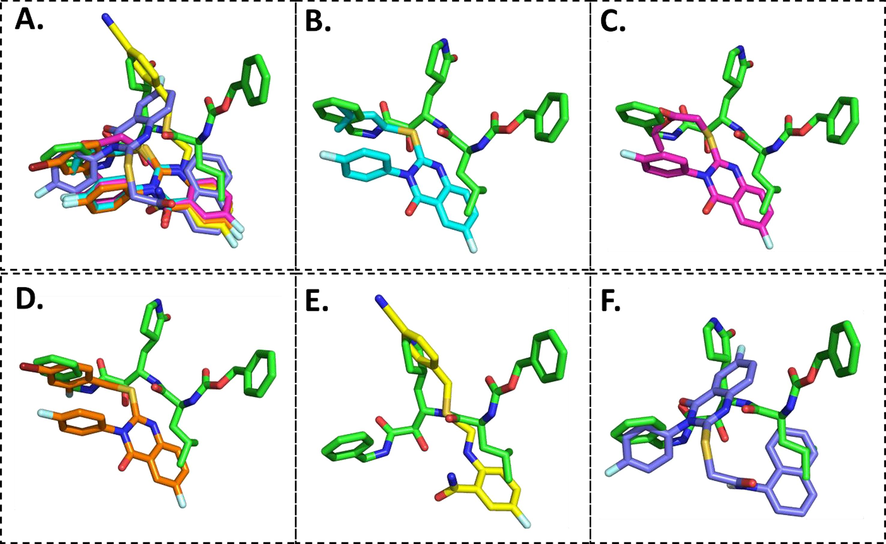
Overlay of the conformations of the docked ligands (green) vs. VR4 (co-crystallized), A. Top 5 high scoring quinazolinone derivatives, B. Compound 5b, C. Compound 5c, D. Compound 5i, E. Compound 5j, F. Compound 5 l in the active pocket of main protease 3CLpro.
Ligands
XP GScore
Glide emodel (kcal/mol)
H-bonds
Halogen bonds
pi-pi
pi-sulfur
Alkyl-alkyl & pi-alkyl
VR4
−9.4
−78.7
Four with Leu141, Asn142, Glu189
---
---
---
His41, Met49, His163, Met165, Ala191
3
−6.2
−44.5
One with Glu166
Four with Phe140, Glu166, Asp187, Arg188
His41
Cys145
Cys145, Met165
5a
−7.0
−68.4
One with Glu166
Four with Phe140, Glu166, Asp187, Arg188
His41
Cys145
Cys145, Met165
5b
−7.4
−54.3
One with Glu166
Three with Phe140, Arg1888
His41
Cys145
Leu27, His41, Cys145, Met165
5c
−8.3
−57.3
Two with Asn142, Gln189
One with Asp187
---
Cys44
Cys145, His163
5d
−6.5
−60.4
One with Glu166
Three with Phe140, Glu166, Arg188
His41
Cys145
Cys145, Met165
5e
−6.1
−59.8
Two with Asn142, Gly143
Three with Phe140, Leu141, Glu166
---
Cys145
Met49, Pro52
5f
−6.9
−53.6
One with Gly143
Two with His41,
Leu141---
Cys44
Met49, Cys145, Met165
5g
−7.3
−48.0
One with Thr190
Four with Thr26, Glu166, Gln189, Thr190
---
---
Met49, Cys145, Met165
5h
−7.2
−75.8
Two with Thr26,
Glu166Five with Thr24, Thr26, Phe140, Glu166, Arg188
His41
Cys145
Cys145, Met165
5i
−7.8
−64.58
Two with Glu166, Gln189
Three with Phe140, Glu166, Gln189
His41
Cys145
Cys145, Met165, Pro168
5j
−7.5
−57.5
Two with Cys44,
Gly143One with His163
His41
---
Cys145
5k
−7.0
−60.5
Two with His41,
Glu166Four with Phe140, Glu166, Asp187, Arg188
His41
Cys145
Met49, Cys145, Met165
5l
−8.2
−59.9
Two with Gly143, Glu166
Three with Phe140, Glu166, Arg188
His41
Cys145
Met49, Cys145, Met165
Ligands
R
3D interactions
VR4
---
3
H
5a
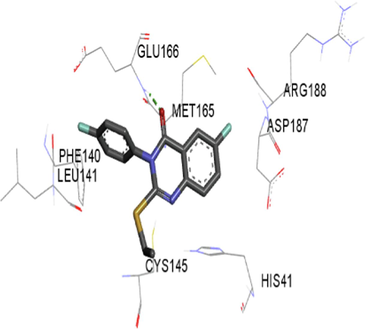
5b

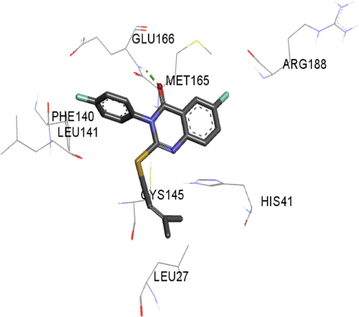
5c
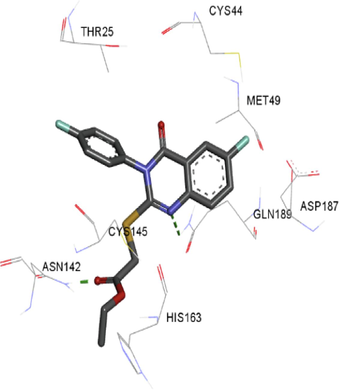
5d
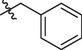
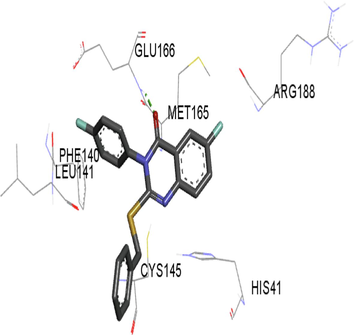
5e
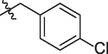
5f
5g
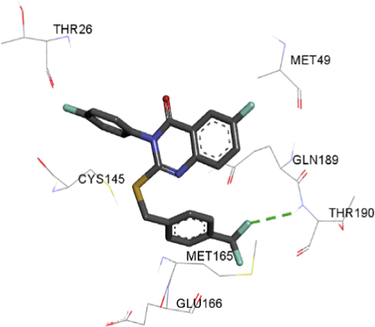
5h
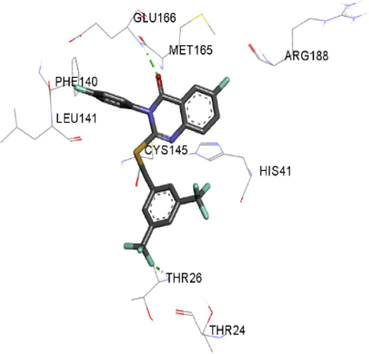
5i
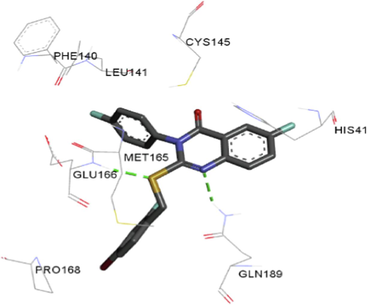
5j
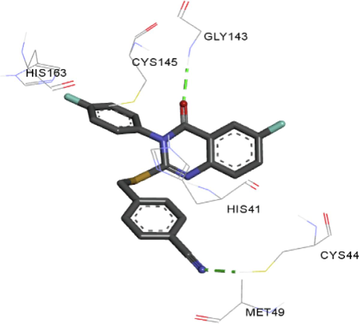
5k
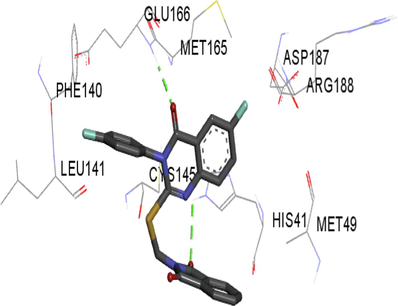
5l

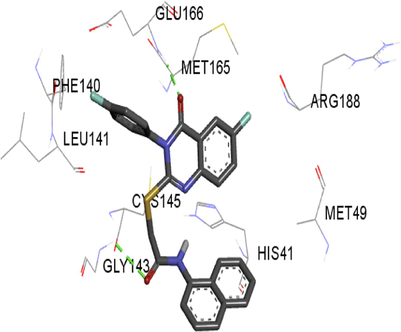
3.3 ADMET analysis
The compounds 5b, 5c, 5i, 5j and 5l having efficient bindings with the receptor SARS-CoV-2 3CLpro were selected for ADME and toxicity calculations. The ADME and the drug-likeness stuffs of the compounds are important factors for the development of new drugs. The absorption parameters, viz. water solubility (LogSw) and lipophilicity (LogPo/w) of the compounds shows whether the compounds are more lipophilic or hydrophilic. The lipophilicity of these compounds were in the range of 3.38 to 3.74 (recommended < 5), and the logSw values were between −4.67 to −6.79, which indicated poor water solubility and more lipophilic nature of these compounds. The drug-likeness property predicts the molecules to be developed as orally bioavailable drug by comparing their physicochemical or structural characteristics. The Lipinski rule of 5 and the Veber methods predicts compounds about their pharmacological activity after oral administration (Lipinski et al., 2001; Veber et al., 2002). The five properties of Lipinski rule state oral drug should have no more than one violation that include molecular weight ≤ 500 g/mol; partition coefficient ≤ 5; number of H-bond accepters ≤ 10 and H-bond donors ≤ 5 and the number of rotatable bonds ≤ 10. The Veber rule includes two criteria that compounds should have ≤ 10 rotatable bonds and polar surface area should be less than 140 Å2. All the potent compounds passed the Lipinski and Veber parameters to be orally active (see Table 3). MW – molecular weight, logPo/w (iLOGP) – octanol/water partition coefficient; log Sw -aqueous solubility; Donor HB – number of groups with hydrogen bond donor (HBD); Acceptor HB – number of groups with hydrogen bond acceptor; TPSA – Topological Polar Surface Area; nrot – number of rotatable bonds; DL- Drug likeness; nLV – number of Lipinski rule violation; nVV- number of Veber’s rule violations.
Compd.
MW
logPo/w
logSw
HB donor
HB acceptor
TPSA (Å2)
nrot
DL
nLV
nVV
5b
358.4
3.49
−5.47
0
4
60.19
4
Yes
1
0
5c
376.3
3.38
−4.67
0
6
86.49
6
Yes
0
0
5i
477.3
3.74
−6.79
0
5
60.19
4
Yes
1
0
5j
405.4
3.42
−5.67
0
5
83.98
4
Yes
0
0
5l
459.4
3.57
−6.72
1
5
89.29
5
Yes
1
0
The absorption of the compounds is related to skin permeation (logKp) and Caco-2 permeability. The skin permeation of all the compounds were below −2.7, which is less than −2.5 means compound can penetrate the skin. The Caco-2 permeability was between 0.958 and 1.139, which is higher than the standard value. The larger the value of logarithm of permeability (log Papp > 8 × 10–6) the larger will be Caco-2 permeability (Sucharitha et al., 2021). The logKp (skin permeation) is linearly correlated with molecular size and lipophilicity. After absorption, the drug has to cross several membranes such as BBB, intestinal membrane etc. The BBB (blood brain barrier) permeability and HIA (human intestinal absorption) represent the absorption and the distribution of compounds. The data showed that except 5c all the tested drug can cross BBB. The predicted human intestinal absorption of the compounds was between 92.11 and 98.09 % indicating good absorption. The (fraction unbound in plasma) hFU is another parameter for estimating the pharmacokinetics of the drugs (volume of distribution) and portion of free drug in plasma that escape to surroundings (del Amo et al., 2013). The results showed it ranges from 0.190 to 0.298. The substrate permeability glycoprotein (P-gp) is an ATP-binding transporter and evaluates the active efflux by biological membranes. The substrate for P-gp indicates increased excretion, reduced distribution, and absorption. The compounds 5j and 5 l were predicted to be substrates for P-gp. The interaction with the cytochrome P450 (CYP) involves the excretion after metabolic biotransformation. The compounds were metabolized by CYP3A4 inhibitors enzymes. The enzymes facilitate excretion by the oxidation process (Jeyaraman et al., 2020). All the 5 compounds were predicted to be hepatotoxic, probably due to the presence of sulfur atom in the structures. The compounds except 5j, showed no AMES toxicity (see Table 4). Caco-2 Human colorectal carcinoma (logPapp in 10–6 cm/s); HIA – human intestinal absorption (% absorbed); SP – Skin permeation (log Kp); BBB- blood–brain-barrier (logBB); hFU – Fraction unbound human; P-Gp Subs – P-glycoprotein substrate; CYP3A4 inhibitor.
Compd.
Caco-2
(cm/sec)
HIA
SP
BBB
hFU
P-Gp
CYP3A4 inhibitor
AMES toxicity
Hepatotoxicity
5b
1.245
95.83
−2.734
0.209
0.294
No
No
No
Yes
5c
1.212
97.94
−2.735
−0.155
0.298
No
Yes
No
Yes
5i
1.139
96.84
−2.72
0.484
0.263
No
Yes
No
Yes
5j
1.098
98.09
−2.69
0.071
0.190
Yes
Yes
Yes
Yes
5 l
0.958
92.11
−2.73
0.142
0.225
Yes
Yes
No
Yes
3.4 MD simulation study
The MD simulation studies explains the alterations in the protein's 3D structure upon ligand binding and predict the ligand–protein stability. The RMSD profiles of the docked complexes of compounds 5b, 5c, 5i, 5j and 5 l with 3CLpro as predicted by molecular docking were generated with GROMACS 2018.1 package. The reference was the co-crystallized inhibitor VR4. As shown in Fig. 5A, the trajectories of 50 ns simulation were studied for 3CLpro protein and ligand complex. The RMSD of the backbone atom for 50 ns showed that 3CLpro protein reached stability after 25 ns whereas, the quinazolinones compounds reached stability in less than 25 ns. The reference complex 3CLpro-VR4 complex and the 3CLpro-quinazolinone complex are stabilized until the end of the MD trajectory 50 ns except complex 5c which is stable up to 45 ns and then showed a little fluctuation at the end of the production run. The average RMSD value for the 3CLpro and the 3CLpro-quinazolinone complex were found to be less than 3 Å, which indicates quite rigid and similar trend of the trajectory. All compounds showed little flexibility at initial phase of stimulation and reached stability after 10 ns. From the Fig. 5B, it was observed that compounds 5b, 5c, and 5l has lower RMSD value as compared to 5i, 5j and reference compound VR4. Thus, the compounds 5b, 5c and 5l are well located within the active sites of the receptor 3CLpro. The compound 5b and 5c has lower RMSD as compared to the reference, approximately 2 Å and stable after 10 ns. However, compound 5i resides within the catalytic dyad as it has very similar RMSD as that of reference and do not exceed 3 Å. The complex 5l is flexible in the start of the run and becomes stable after 25 ns and was in the active sites like the molecular dynamic studies of the reference compound. The study confirms that upon binding of the synthesized quinazolinone compounds, there were no major variation or conformational changes observed in protein 3CLpro. The root means square fluctuation profile as shown in Fig. 6A of the potent quinazolinones with 3CLpro were also generated. The RMSF analyzed the portion of the protein that diverges from the overall structure. A higher RMSF value shows more flexibility as compared to the lower RMSF values. The compound 5i showed lesser fluctuation in Glu166 and Gln189, and has more stability as compared to 5a, 5b, 5j and 5l. The binding with Gln189 residue showed less flexibility as compared to the other complexes. Comparing all the RMSF plots of the quinazolinones showed the compound 5c to be more flexible than the others. The average RMSF values for the quinazolinones derivatives are <2 Å and are lower than 3CLpro-VR4 complex and were stable enough to be considered as potential drug against SARS-CoV 2. The hydrogen bonds are important for stability and drug specificity. The pattern of the H-bonds expresses the stability of the complex within the protein structure. The results from the Fig. 6B showed that maximum number of hydrogen bonds formed is three with the reference compound and maximum four hydrogen bonds for the potent quinazolinone derivatives was observed with compound 5j. As indicated earlier the binding to the domain I and II of the 3CLpro is important for the compounds to be active against SARS-CoV-2. The compounds showed intermolecular hydrogen bonds with amino acids of the domain I and II. It was observed that maximum of the 2 intermolecular hydrogen bonds interactions exists for 5l and 1 intermolecular hydrogen each with 5b, 5c and 5i complexed with 3CLpro. The hydrogen bonds with the quinazolinone derivatives confirmed binding with protein 3CLpro effectively, like VR4-CLpro complex.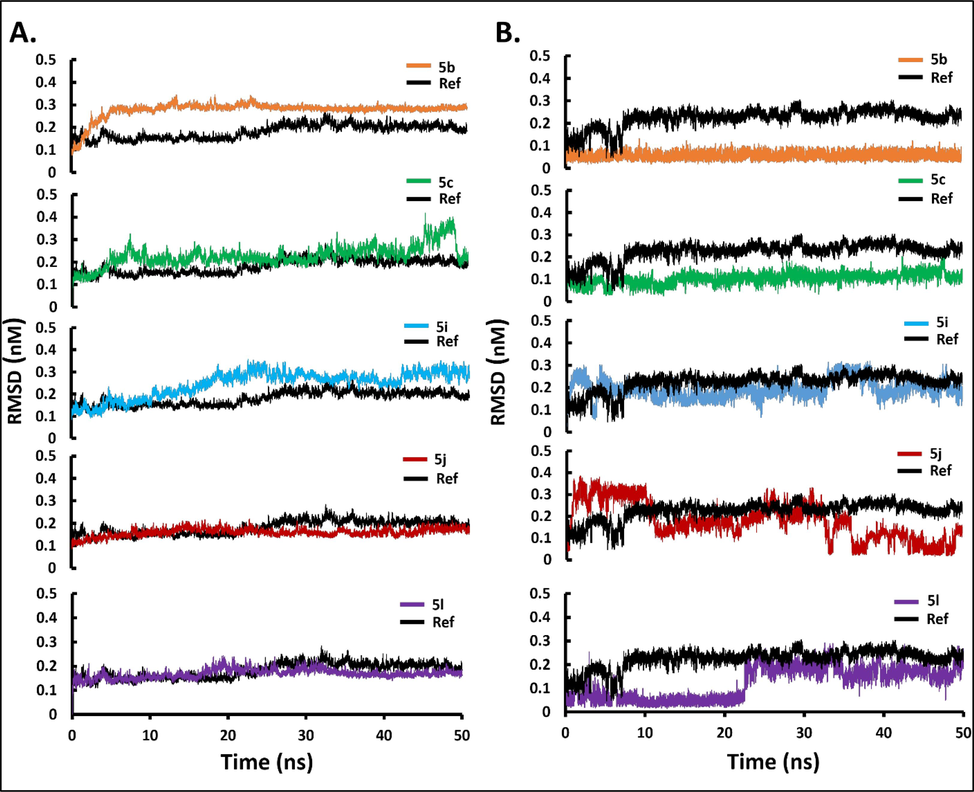
Stability of main protease 3CLpro in complex with top binding quinazolinone derivatives with respect to reference; co-crystalized ligand (VR4) (shown in black). (A) Backbone-RMSD plots of main protease 3CLpro in complex with top binding quinazolinone derivatives over 50 ns MD production run; (B) Ligand-RMSD plots of top binding quinazolinone derivatives over 50 ns MD production run.
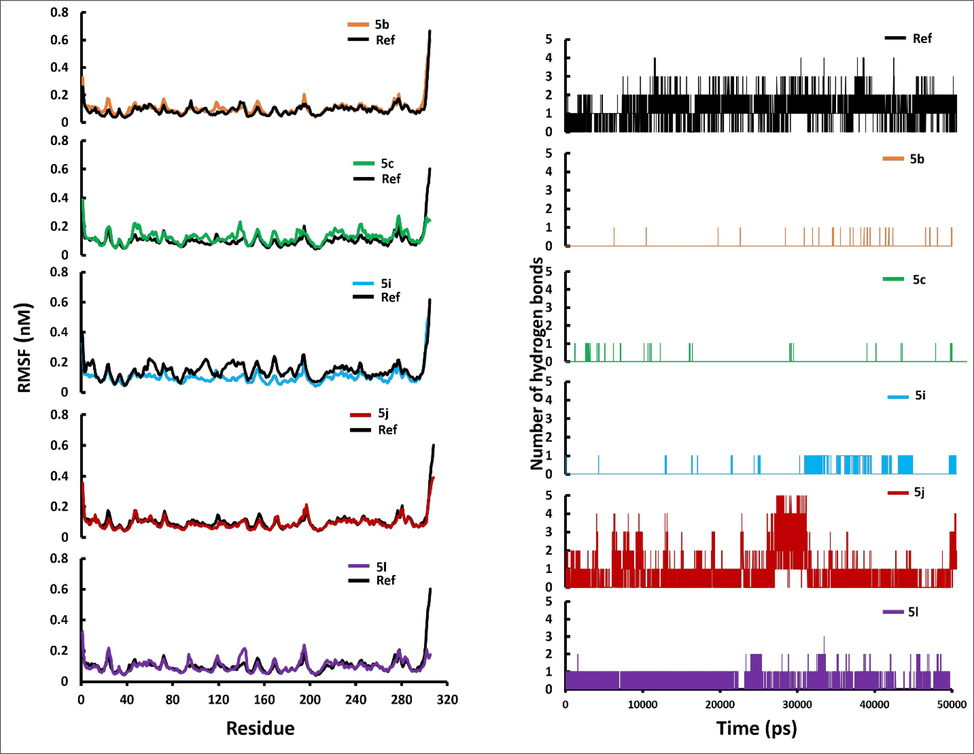
(A) Backbone-RMSF plots of main protease 3CLpro in complex with top binding quinazolinone derivatives over 50 ns MD; (B) The number of hydrogen bond between main protease 3CLpro and quinazolinone derivatives over 50 ns MD production run.
3.5 Effect of compounds on VeroE6 cells
To find out any toxic effect of compounds (5b, 5c, 5i, 5j and 5l) on mammalian cells, cell viability test by MTT assay was performed. It was observed that all the five selected compounds did not showed cytotoxicity against the VeroE6 cell line even at higher dose (Fig. 7). The assay results confirmed that the tested compounds (5b, 5c, 5i, 5j and 5l) do not have any cytotoxic effect on VeroE6 cells at a concentration of 20 μM after 24 h of duration.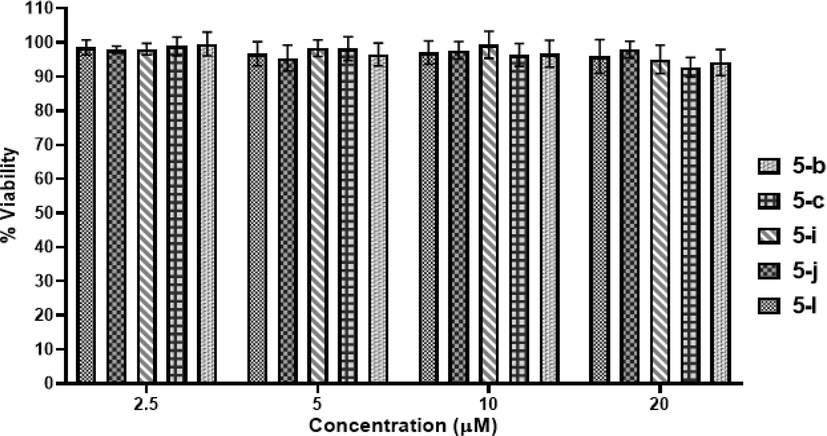
Cell viability assay of compounds 5b, 5c, 5i, 5j and 5l at varying concentrations (2.5, 5, 10 and 20 μM) on VeroE6 cells. A representation of three individual experiments with triplicate values is presented graphically.
3.6 3CL protease inhibitory activity
The compounds 5b, 5c, 5i, 5j and 5l were investigated for their potential inhibitory effect on the activity of 3CLpro, by testing them at varied concentrations of 10, 5, 2.5, 1.25 and 0.625 µM. It was observed that all of the tested five compounds significantly inhibited 3CL-protease activity in a dose dependent manner (Fig. 8) and 5j showed extremely significant inhibitory effect against SARS-CoV-2 3CLpro. The 50 % inhibitory concentration (IC50) of 5b, 5c, 5i, 5j and 5l were found at 1.58, 1.25, 1.97, 0.44 and 2.56 µM concentrations, respectively. The IC50 value of positive control (GC376) against 3CLpro was achieved at 1.61 nM. After molecular docking studies, the five high scoring derivatives (5b, 5c, 5i, 5j and 5l) having Glide XP score −7.4, −8.3, −7.8, −7.5 and −8.2 respectively, were selected for further selected for ADMET analysis, MD simulation, cellular viability assay and in vitro 3CLpro inhibition assay studies. However, in 3CLpro inhibition assay the IC50 value for the respective compounds were found to be 1.58, 1.25, 1.97, 0.44 and 2.56 µM. Although, all the high scoring compounds were found to be active in inhibiting SARS-CoV-2 3CL protease, compound 5j was found to be the most potent having IC50 value 0.44 µM. This lack of correlation between the docking score and IC50 value can be attributed to the presence of cyano group in compound 5j. In the molecular docking studies, we observed that the cyano group interacted with Cys44 with a hydrogen bond, while no other derivatives have hydrogen bonding with Cys44. The high potency of compound 5j can be attributed to the covalent bond formation between the cyano group and Cys44 during in vitro experiments.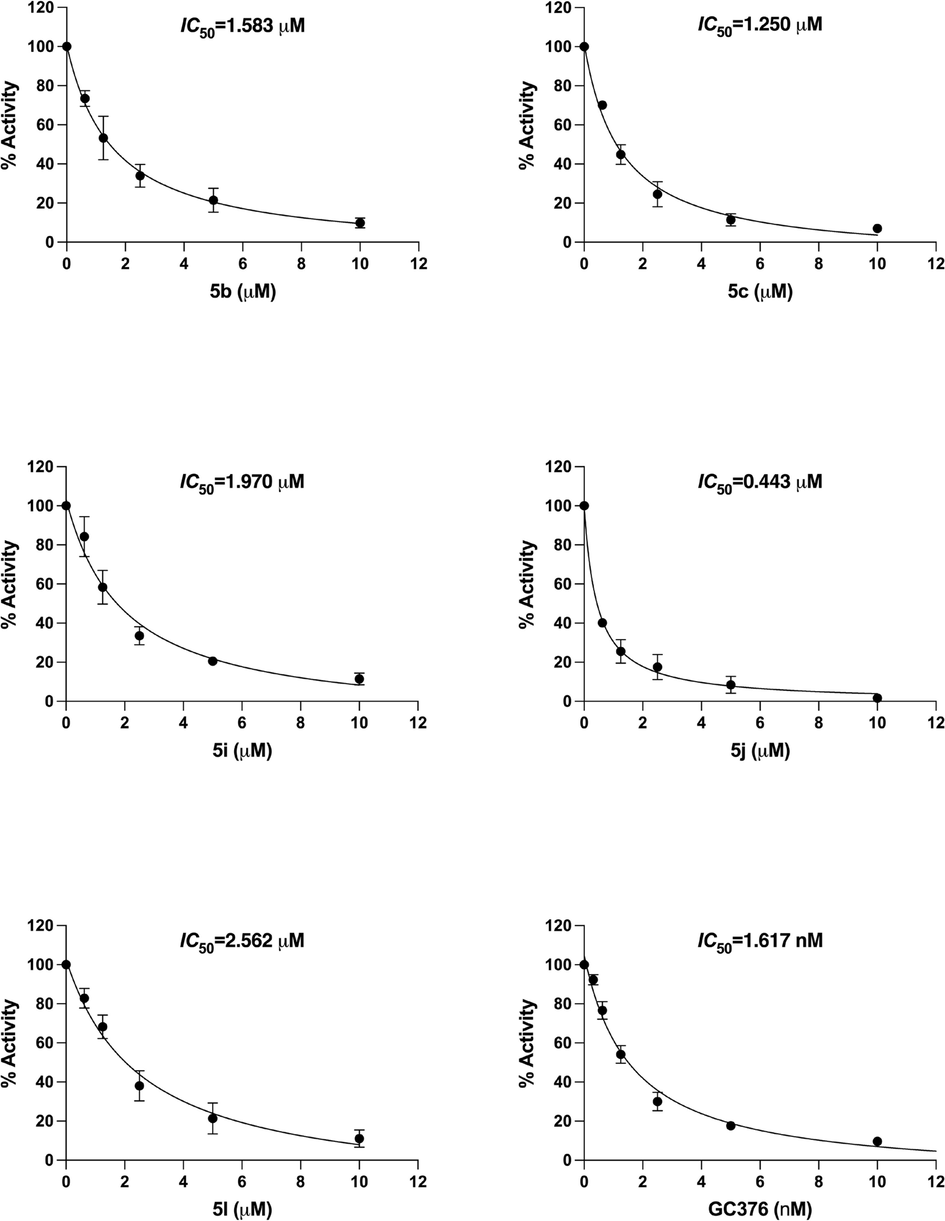
3CLpro inhibition by compounds 5b, 5c, 5i, 5j, 5l and GC376. The statistical analysis of the experimental data was presented as mean ± SEM from three independent experiments in triplicate.
4 Conclusions
The present study describes design, synthesis, molecular docking and dynamics simulations study of a new series of 2-sulfanylsubstituted-3-p-fluorophenyl-6-fluoro-quinazolinones derivatives and the toxicity study of potent compounds on VeroE6 Cells and their inhibitory potential against SARS-CoV-2 3CL protease. The designed 2-quinazolinones derivatives, based on a previously reported potent inhibitor, were synthesized by S-alkylation/arylation of an intermediate, 6-fluoro-3-(4-fluorophenyl)-2-mercaptoquinazolin-4(3H)-one and their successful synthesis was established by analytical methods, viz. IR, 1H NMR, & 13C NMR spectroscopy. Compounds 5b, 5c, 5i, 5j and 5l exhibited strong binding within the active sites having high score in molecular docking study. The catalytic dyad (Cys-His) in the active sites of the SARS-CoV-2 3CLPro protease were examined to explore the interaction and stability of the compounds (5b, 5c, 5i, 5j and 5l) using 50 ns molecular dynamics simulation studies. The compounds studied showed stable interaction over 50 ns production run and well located inside the active site with lower RMSD values. The RMSF values were found to be lower with compound 5i and maximum numbers of hydrogen bonds was observed with compound 5j. The ADMET data obtained by Swiss ADME and pkCSM web tools softwares confirms the quinazolinone derivatives to be potential biologically active molecules. Compounds 5b, 5c, 5i, 5j and 5l were found to inhibit the 3CLpro activity in vitro and compound 5j showed significant inhibitory activity with IC50 value of 0.44 µM. Thus, in silico and in vitro inhibitory properties of quinazolinones derivatives against SARS-CoV-2 3CLpro might pave the way for the development of more potent antiviral compounds against coronaviruses and its emerging variants.
Author contributions
All authors contributed significantly and agreed to the published version of the manuscript.
Funding
The Deanship of Scientific Research (DSR) at King AbdulAziz University, Jeddah, Saudi Arabia has funded this project, under grant no. (KEP-24–166-42).
Acknowledgments
The Deanship of Scientific Research (DSR) at King AbdulAziz University, Jeddah, Saudi Arabia has funded this project, under grant no. (KEP-24-166-42). Therefore, the authors gratefully acknowledge technical and financial support provided by the DSR, King Abdulaziz University, Jeddah, Saudi Arabia.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Discovery of novel thioquinazoline-N-aryl-acetamide/N-arylacetohydrazide hybrids as anti-SARS-CoV-2 agents: synthesis, in vitro biological evaluation, and molecular docking studies. J. Mol. Struct.. 2023;1276:134690
- [Google Scholar]
- Structural stability of SARS-CoV-2 3CLpro and identification of quercetin as an inhibitor by experimental screening. Int. J. Biol. Macromol.. 2020;164:1693-1703.
- [Google Scholar]
- pH profiles of 3-chymotrypsin-like protease (3CLpro) from SARS-CoV-2 elucidate its catalytic mechanism and a histidine residue critical for activity. J. Biol. Chem.. 2023;299(2):102790
- [Google Scholar]
- Pharmacoinformatics and molecular dynamic simulation studies to identify potential small-molecule inhibitors of WNK-SPAK/OSR1 signaling that mimic the RFQV motifs of WNK kinases. Arab. J. Chem.. 2020;13(4):5107-5117.
- [Google Scholar]
- A Guide to COVID-19: a global pandemic caused by the novel coronavirus SARS-CoV-2. FEBS J.. 2020;287(17):3633-3650.
- [Google Scholar]
- Targeting SARS-CoV-2 proteases and polymerase for COVID-19 treatment: state of the art and future opportunities. J. Med. Chem.. 2020;65(4):2716-2746.
- [Google Scholar]
- A highly sensitive cell-based luciferase assay for high-throughput automated screening of SARS-CoV-2 nsp5/3CLpro inhibitors. Antiviral Res.. 2022;201:105272
- [Google Scholar]
- Merbromin is a mixed-type inhibitor of 3-chyomotrypsin like protease of SARS-CoV-2. Biochem. Biophys. Res. Commun.. 2022;591:118-123.
- [Google Scholar]
- SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep.. 2017;7:42717.
- [Google Scholar]
- Postinfection treatment with a protease inhibitor increases survival of mice with a fatal SARS-CoV-2 infection. PNAS. 2021;118(29):e2101555118
- [Google Scholar]
- Applying linear and non-linear methods for parallel prediction of volume of distribution and fraction of unbound drug. PLoS One. 2013;8(10):e74758.
- [Google Scholar]
- Key dimer interface residues impact the catalytic activity of 3CLpro, the main protease of SARS-CoV-2. J. Biol. Chem.. 2022;298(6):102023
- [Google Scholar]
- Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J. Med. Chem.. 2006;49(21):6177-6196.
- [Google Scholar]
- Structure-based design, synthesis, and biological evaluation of peptidomimetic SARS-CoV 3CLpro inhibitors. Bioorg. Med. Chem. Lett.. 2007;17(21):5876-5880.
- [Google Scholar]
- Targeting the SARS-CoV-2 RNA genome with small molecule binders and ribonuclease targeting chimera (RIBOTAC) degraders. ACS Cent. Sci.. 2020;6(10):1713-1721.
- [Google Scholar]
- Biflavonoid as potential 3-chymotrypsin-like protease (3CLpro) inhibitor of SARS-Coronavirus. Results Chem.. 2021;3:100087
- [Google Scholar]
- Inhibition of Chikungunya Virus Infection by 4-Hydroxy-1-Methyl-3-(3-morpholinopropanoyl)quinoline-2(1H)-one (QVIR) Targeting nsP2 and E2 Proteins. ACS Omega. 2021;6(14):9791-9803.
- [Google Scholar]
- Synthesis, characterization, ADMET, in vitro and in vivo studies of mixed ligand metal complexes from a curcumin Schiff base and lawsone. Nucleosides Nucleotides Nucleic Acids. 2020;40(3):242-263.
- [Google Scholar]
- Design, synthesis and biological evaluation of 2-aminoquinazolin-4 (3H)-one derivatives as potential SARS-CoV-2 and MERS-CoV treatments. Bioorg. Med. Chem. Lett.. 2021;39:127885
- [Google Scholar]
- Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev.. 2001;46(1–3):3-26.
- [Google Scholar]
- The development of Coronavirus 3C-Like protease (3CLpro) inhibitors from 2010 to 2020. Eur. J. Med. Chem.. 2020;206:112711
- [Google Scholar]
- Natural product-derived phytochemicals as potential agents against coronaviruses: a review. Virus Res.. 2020;284:197989
- [Google Scholar]
- Ultrasound assisted Cu-catalyzed Ullmann-Goldberg type coupling-cyclization in a single pot: Synthesis and in silico evaluation of 11H-pyrido [2, 1-b] quinazolin-11-ones against SARS-CoV-2 RdRp. J. Mol. Struct.. 2023;1280:135044
- [Google Scholar]
- Anti-SARS-CoV-2 activity of various PET-bottled Japanese green teas and tea compounds in vitro. Arch. Virol. 2022;167(7):1547-1557.
- [Google Scholar]
- An overview of severe acute respiratory syndrome–coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J. Med. Chem.. 2016;59(14):6595-6628.
- [Google Scholar]
- Small-molecule thioesters as SARS-CoV-2 main protease inhibitors: enzyme inhibition, structure–activity relationships, antiviral activity, and X-ray structure determination. J. Med. Chem.. 2022;65(13):9376-9395.
- [Google Scholar]
- pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem.. 2015;58(9):4066-4072.
- [Google Scholar]
- Synthesis, characterization, biological evaluation and molecular docking of a new quinazolinone-based derivative as a potent dual inhibitor for VEGFR-2 and EGFR tyrosine kinases. J. Biomol. Struct. Dyn.. 2022;40(15):6810-6816.
- [Google Scholar]
- Cell-based high-throughput screening protocol for discovering antiviral inhibitors against SARS-COV-2 main protease (3CLpro) Mol. Biotechnol.. 2021;63:240-248.
- [Google Scholar]
- Genomics approaches to synthesize plant-based biomolecules for therapeutic applications to combat SARS-CoV-2. Genomics. 2020;112(6):4322-4331.
- [Google Scholar]
- Fused benzo[1,3]thiazine-1,2,3-triazole hybrids: Microwave-assisted one-pot synthesis, in vitro antibacterial, antibiofilm, and in silico ADME studies. Bioorg. Med. Chem. Lett.. 2021;47:128201
- [Google Scholar]
- Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem.. 2002;45(12):2615-2623.
- [Google Scholar]
- Design, synthesis and in vitro anti-influenza A virus evaluation of novel quinazoline derivatives containing S-acetamide and NH-acetamide moieties at C-4. Eur. J. Med. Chem.. 2020;206:112706
- [Google Scholar]
- SwissParam: a fast force field generation tool for small organic molecules. J. Comput. Chem.. 2011;32(11):2359-2368.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.105384.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




