

Translate this page into:
Synthesis, in-vivo anti-diabetic & anticancer activities and molecular modelling studies of tetrahydrobenzo[d]thiazole tethered nicotinohydrazide derivatives
⁎Corresponding author. pvrao@ums.edu.my (Visweswara Rao Pasupuleti), dr.b.haribabu@gmail.com (Hari babu Bollikolla)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
A series of thirty new thiazole-pyridine derivatives were synthesized by reaction of 4,4,7,7-tetra-methyl-4,5,6,7-tetrahydrobenzo[d]thiazol-2-amine with 6-chloronicotinate followed by condensing with benzaldehydes and screened for their anti-diabetic activity by in vivo housing Swiss albino mice. All synthesized compounds resulted in reducing the glucose level when compared with reference standard drug glibenclamide. In specific, compound 7 exhibited significant activity in terms of fasting blood glucose level reduction. In addition, the in-silico binding studies of the potential compounds 11f and 11g with human PPAR-γ protein complexed with Retinoid X Receptor (RXR) alpha Nuclear Receptor showed good interactions when compared to the standard drug Rosiglitazone. The newly synthesized drugs may be potential anti-diabetic drugs with possible specific actions.
Keywords
Tetrahydrothiazole
Pyridinehydrazide
Anti-diabetic activity
Molecular modeling

1 Introduction
Diabetes mellitus (DM) is a chronic metabolic condition caused by either insufficient insulin use by the body or insufficient insulin output by the pancreas (WHO, Definition, 1999). It is a major public health hazard around the world, impacting nearly 463 million people and estimated to reach 578 million by 2030 (Mohan et al., 2005). Due to insulin resistance, incorrect insulin secretion, and excessive hepatic glucose production, the majority of Diabetes mellitus patients were classified as Non-insulin-dependent diabetes mellitus (NIDDM) (Olefsky et al., 1985; OlefskyJM, 1995). Accordingly, currently, a number of drugs are in use to combat the disease diabetes mellitus and its complications (Clark, 1998). These drugs show different undesired side effects including hepatic toxicity, flatulence, hyperglycemia, abdominal pain, and diarrhea (Ligthelm et al., 2012). Therefore, the discovery of novel anti-diabetic agents with improved efficacy has been much sought to grant better relief.
In vitro anti-cancer activity of Tetrahydrothiazolyl pyridine (Series 1 and Series 2). IC50 concentrations of the test compounds against MCF7 cell lines after the incubation period of 24 h.
Retinoic acid is a carboxylic acid version of Retinol (Vitamin A) that plays a prime role of morphogen as well as known to contribute towards the developmental process of multicellular organism differentiation, modulation of amino acid, glucose, cholesterol, and fatty acid metabolism (Kam et al., 2012). Retinoid X Receptor (RXR) is an important part of the thyroid family of nuclear receptors. They perform as transcription factors with a specific role in the development and cell death (Dawson and Xia, 2012). RXR’s forms homodimer with themselves apart from being heterodimeric members of various receptors like liver receptors, vitamin D receptors thyroid hormone receptors, and peroxisome proliferator-activated receptors (PPAR) (Plutzky, 2011). The heterodimer of RXR with PPAR-gamma is an interesting molecular target for diabetes (Lenhard, 2001). A recent study reported that the thiazole-bearing moieties especially thiazolidinediones are the potential to act as anti-diabetic agents (Davidson et al., 2017). Further, the thiazolidinedione derivatives were also reported as PPAR‐γ modulators (JavedNaim et al., 2018).
Synthetic drugs belong to the thiazole scaffold family consist of antimicrobial agents such as acinitrazole and sulfathiazole (Borisenko et al., 2006), antidepressant pramipexole (Maj et al., 1997), antibiotic penicillin (Maulard et al., 1993), antineoplastic agents Tiazofurin and Bleomycin (Milne and Gower; Ashgate, 2000), the antiasthmatic drug cinalukast (Markus et al., 2004), anti-HIV drug Ritonavir (Desouza and Dealmeida, 2003) and antiulcer agent Nizatidine (Knadler et al., 1986). Additionally, extensively used thiazole derivatives are the non-steroidal immunomodulatory drug Fanetizole (Lednicer et al., 1990) and the anti-inflammatory drug Meloxicam (Rehman et al., 2005). Similarly, Thiazole scaffold derivatives with polyoxygenated phenyl module were exhibited encouraging anti-fungal activity (Beuchet et al., 1999). Thiazoles found from microbial, and marine ancestries reveal antitumor and antiviral activities. Furthermore, thiazole was recognized as a ligand of estrogen receptors (Fink et al., 1999) and as a unique kind of antagonist for adenosine receptors (Muijlwijk-Koezen et al., 2001).
With the existing biological significance of thiazoles as well as the importance of RXR as an interesting anti-diabetic target, herein we report the synthesis of two series of new tetra hydro thiazolyl conjugated nicotinyl hydrazide analogs as shown in Schemes 1 and 2 (Scheme 1-11 series(11a-o) and Scheme 2-12 series (12a-o)) and evaluated their in-vivo antidiabetic potential and also the molecular modeling studies were also performed to gain more insight into the binding pattern of the synthesized ligands with the protein.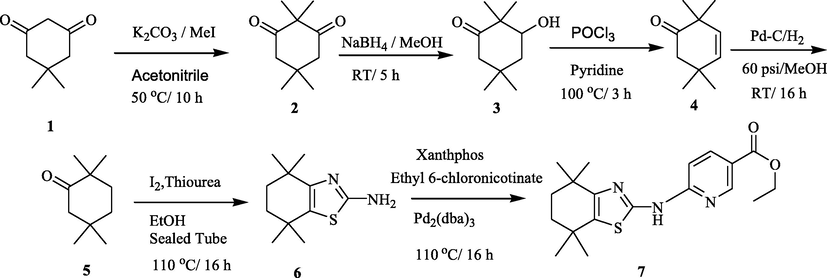
Synthesis of key intermediate 7.
![Synthesis of tetrahydro benzo[d]thiazole tethered nicotinohydrazide derivatives.](/content/184/2022/15/2/img/10.1016_j.arabjc.2021.103546-fig3.png)
Synthesis of tetrahydro benzo[d]thiazole tethered nicotinohydrazide derivatives.
2 Materials and methods
2.1 Chemistry methods
Melting points were determined using the Shital-digital programmable melting point apparatus. The IR spectra were recorded on a VERTEX 70 Brucker by using KBr. A Bruker spectrometer 400 and 100 MHz was employed for recording 1H NMR and 13C NMR spectra respectively and CDCl3 and DMSO‑d6 were used as solvent and TMS as an internal standard. Mass spectra were recorded on an Agilent-LCMS instrument.
Synthesis of 2,2,5,5-tetramethyl cyclohexane-1,3-dione (2): To a solution of 5,5-dimethyl cyclohexane-1,3-dione 1 (300 g, 2142 mmol) in acetonitrile (2.4 L) solvent and added K2CO3 (888.0 g, 6428.5 mmol) at 0 °C and stirred 20 min. Then, to the reaction mass added methyl iodide (760 g, 5357.14 mmol) and heated at 50 °C for l0 h. The reaction progress was monitored by TLC. After completion of the reaction, the reaction mixture was concentrated, diluted with water, and extracted into ethyl acetate. The combined organic layer was back washed with water, brine solution, dried over Na2SO4, and concentrated below 40 °C under reduced pressure. The crude product was treated with n-hexane and the solid thus precipitated was filtrated to give the title compound as a white solid 2 (180.0 g, yield: 50%).
Synthesis of 3-hydroxy-2, 2, 5, 5-tetramethyl cyclohexanone (3): To a solution of 2 (200 g, 1190.4 mmol) in MeOH (2 L) at room temperature, cooled to 0 °C followed by addition of NaBH4 (16.6 g, 440.4 mmol. 0.3 eq). The reaction mixture was stirred under nitrogen for 5 h at 0–10 °C. The reaction progress was monitored by TLC. After completion of the reaction, the mixture was quenched with saturated NH4Cl solution. Then, the solvent was evaporated under reduced pressure and the residue was extracted with EtOAc. The combined organic layer was washed with water, brine solution, dried over Na2SO4 and concentrated under vacuum. The crude product obtained was purified by silica column chromatography to give compound 3 as a white solid (l2l g. yield: 60%).
Synthesis of 2,2,5,5-tetramethyl cyclohex-3-enone (4): The solution of 3 (95 g, 558.82 mmol) in pyridine (300 mL) at room temperature, was cooled to 0 °C followed by addition of POCl3 (427.5 g, 2794.11 mmol) and stirred at 100 °C for 3 h. The reaction progress was monitored by TLC. After completion of the reaction, the reaction mixture was cooled to room temperature and poured into crushed ice (highly exothermic), and diluted with diethyl ether. The separated ether layer was neutralized with l N HCI and washed with water, brine solution. The ether layer was dried over Na2SO4 and concentrated below 30 °C in a vacuum. The crude product was purified by silica column chromatography using 100% hexane as eluent to give compound 4 as a light green liquid (37 g, 42%).
Synthesis of 2,2,5,5-tetramethyl cyclohexanone (5): The solution of 4 (35 g, 30.263 mmol) was taken in ethyl acetate (250 mL) and methanol (250 mL) solvent mixture in Parr shaker vessel. The above solution was purged with nitrogen for l0 min. Prior to the addition 10% Pd/C (10 g) to the reaction mixture was kept under hydrogen atmosphere (60 psi pressure) for 16 h. The reaction progress was monitored by TLC. After completion of the reaction, the reaction mixture was filtered through celite pad and dried over anhydrous Na2SO4 and concentrated below 30 °C in a vacuum to give a green liquid 5. The crude product was used in the next step without further purification (27 g. yield-76%).
Synthesis of 4,4,7,7-tetramethyl-4,5,6,7-tetrahydrobenzo[d]thiazol-2-amine (6): To a solution of 5 (33 g, 213.9 mmol) in ethanol followed by taken into a sealed tube and added iodine (65 g, 256.7 mmol). Then, the reaction mixture was maintained at 60 °C for 2 h. The reaction mixture was cooled to room temperature. Then, added thiourea (40 g, 525.97 mmol) and maintained at 110 °C for 12 h. The reaction mixture was cooled to room temperature and poured into a cold aqueous NaOH solution and extracted with ethyl acetate. The organic layer was washed with water and brine and dried over anhydrous Na2SO4 and concentrated under a vacuum to get the crude product. The crude product was purified by silica column chromatography (EtOAc/ Pet ether) to give the compound 6 as a yellow solid (15.5 g, yield: 34.5%).
Synthesis of ethyl 6-((4,4,7,7-tetramethyl-4,5,6,7-tetrahydrobenzo[d]thiazol-2-yl)amino)nicotinate (7): To a solution of 6 (10 g, 47.61 mmol) in toluene (100 mL) and added ethyl 6-chloronicotinate (8.8 g, 47.619 mmol), Na2CO3 (10.6 g, 99.9 mmol) and Xantphos (1 0.4 g, 2.38 mmol) under argon over 10 min. Pd2(dba)3 (0.87 g, 0.95 mmol) was added to the reaction mixture and stirred at 110 °C for 8 h. 'I'he reaction mixture was cooled to room temperature and concentrated under vacuum and the residue was poured into water and extracted with dichloromethane. The combined extracts were washed with water and brine solution. The organic layer was dried over anhydrous Na2SO4 and concentrated under a vacuum. The obtained solid was purified with diethyl ether/hexane to give the title compound as a yellow solid (10 g, yield: 50–53 %). M.P: 220–224 °C.
Synthesis of ethyl 6-((cyclopropylmethyl)(4,4,7,7-tetramethyl-4,5,6,7-tetrahydrobenzo[d]thiazol −2-yl) amino)nicotinate (8): To a solution of 7 (10 g, 27.85 mmol) in DMF (100 mL) followed by addition of cesium carbonate (27.2 g, 83.56 mmol) and methyl cyclopropyl bromide (5.8 g, 43.17 mmol) at room temperature. The reaction mixture was maintained at 100 °C for 3 h. The reaction mixture was cooled to room temperature, poured into water, and extracted with ethyl acetate. The organic layer was washed with water and brine solution, dried over anhydrous Na2SO4, and concentrated under vacuum to get a crude compound. The crude product was purified by silica column chromatography to give compound 8 as a light yellow liquid (10.0 g, yield: 87%).
Procedure for the synthesis of 9 & 10: Charged ethyl ester of carboxylic acid 7 & 8 (0.01 mol.) in ethanol (100 mL) and added hydrazine monohydrate (0.011 mol). Then refluxed the reaction mixture for 2 h. After that, the solution was cooled to room temperature and the precipitate formed was then filtered off, dried, and recrystallized from ethanol. (yield: 65–80%).
General procedure for the synthesis of series-1 (11a-11o) & Series 2 (12a-12o): 0.01 mol of previously obtained carboxylic acid hydrazides (9&10)were dissolved in 10–20 mL of ethanol and then 0.011 mol of aldehyde was added. The mixture was heated under reflux for 3 h. After that, the solution was allowed to cool at room temperature and filtered off and recrystallized from ethanol. (yield: 65–70%).
3 In-vivo antidiabetic activity
3.1 Experimental animals
Swiss albino mice weighing 25–30 g were used for this study. Forty-eight animals were grouped into eight groups with series 11 derivatives while fifty-four mice were grouped into nine groups with series 12 derivatives and housed for one week to acclimatize to laboratory conditions before starting the experiment. Animals were fed with a standard diet and water ad libitum, but 12 h prior to inducing diabetes to the mice; the animals were deprived of food but not water. All the experimental procedures were carried out in accordance with CPCSEA guidelines (IAEC approved number is I/IAEC/AGI/027/2017 SM). The Institutional Animal Ethics Committee approved the experimental protocol.
3.2 Induction of diabetes
Overnight-fasted Swiss albino mice were injected with alloxan (150 mg/kg) in saline buffer intraperitoneally (Sekiou et al., 2021). To prevent hypoglycemia animals were fed with 20% (w/v) glucose solution. After 72 h fasting blood glucose levels (FBG) were determined by using ONETOUCH select simple (J&J) Glucometer strips on Day one, day seven, and day 21, and day 28. Glibenclamide was used as the standard 10 mg/kg BW.
3.3 Group design
Mice with < 250 mg/dl were excluded from the experiment and the rest were divided into eight groups with series 11 derivatives and into nine groups with series twelve derivatives. In series eleven, Group 1 (control) was administered with distilled water orally. Group II was treated as diabetic control (Alloxan 150 mg/kg, i.p), Group III,IV,V,VI,VII were treated with 11d, 11f, 11g, 11j, and 11o compounds respectively and group VIII was treated with glibenclamide 10 mg/kg. In the same manner for series twelve compounds, Group I (control) was administered with distilled water orally. Group II was treated as diabetic control (Alloxan 150 mg/kg, i.p), Group III,IV,V,VI,VII,VIII were treated with 12g, 12h, 12i, 12j, 12n, 12o compounds respectively, and group IX was treated with glibenclamide 10 mg/kg.
3.4 Evaluation of cytotoxicity by MTT assay
In 96 well microtiter plates, MCF7 cells were seeded at 1x105 cells/mL in minimum essential medium with fetal bovine serum. For adhesion, the cells were cultured overnight. Drug concentrations were added in triplicates in repeated three-fold dilutions and incubated for 48 h at 5 percent CO2 at 37 °C (see list of drugs and corresponding cell line used in respective results Table). After that, the cells were given 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) treatment (Sigma Chemical Co., St. Louis, MO). All of the medium, including the MTT solution (5 mg/mL), was evacuated from the wells four hours later. The absorbance was measured at 570 nm using a 96 well microplate reader after the residual formazan crystals were dissolved in DMSO (SynergyTM HT, Bio-Tek Instruments, Inc.). As a negative control, untreated cells were used to calculate the cytotoxicity index. The background-corrected absorbance was used to compute the percentage of cytotoxicity (Datta et al., 2014). The IC50 was calculated using the dose–response curve as a guide. By visualizing duplicate data points across a concentration range and computing values using the PRISM program's regression analysis, the drug concentration that reduced cell viability by 50% (IC50) was determined. Based on the reduction of the yellow-colored water-soluble tetrazolium dye MTT to formazan crystals, the MTT assay is a colorimetric assay used to determine cell growth and cytotoxicity. MTT is reduced to insoluble formazan crystals by mitochondrial lactate dehydrogenase, which when dissolved in a suitable solvent exhibits a purple hue whose intensity is proportional to the number of viable cells and can be quantified spectrophotometrically at 570 nm (Alley et al., 1988; Mosmann, 1983). A linear regression equation, Y = Mx + C, was used to calculate the IC50 value. M, and C values were taken from the viability graph (Ganesh et al., 2015), with Y = 50.
Entry
Aldehydes
Product
M.P (°C)
Yield %
1
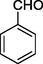
11a
240–244
77
2
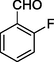
11b
254–258
68
3
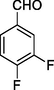
11c
309–312
67
4
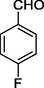
11d
262–264
70
5
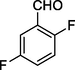
11e
324–326
69
6
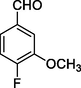
11f
320–322
65
7
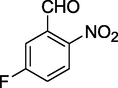
11g
333–335
69
8
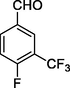
11h
229–232
70
9
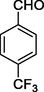
11i
128–132
70
10
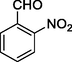
11j
280–284
66
11
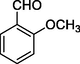
11k
245–249
67
12
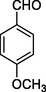
11l
270–274
61
13
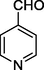
11m
306–310
65
14
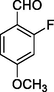
11n
268–272
68
15
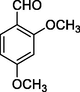
11o
271–274
66
3.5 Molecular docking study
Molecular docking studies of the synthesized molecules 11f and 11 g with human PPAR-γ protein complexed with RXR alpha Nuclear Receptor (PDB ID: 3DZY) were carried out using AutoDock Tools (Ghanbari-Ardestani et al., 2019). Chem3D Ultra 16.0 software was used for the generation of ligand structures. Further, the ligand energies were minimized by using MOPAC (semi-empirical quantum mechanics) tool. The PDB structure of hPPAR-γ protein-RXR alpha nuclear receptor complex (PDB ID: 3DZY) with co-crystal ligand Rosiglitazone was downloaded and imported to the workspace. The protein was processed, and the grid box was generated in a rectangular shape with the value of X: 2.97; Y: 32.82; Z: 11.6 coordination. PyMol visualization tool was employed for the analysis of output files generated from docking. Later, the docking results were validated with the docked co-crystal ligand Rosiglitazone BRL(Expand this for the first time). Each pose was examined manually, and the best pose was taken for per docked ligand. The hydrogen and hydrophobic interactions of the docked ligands with target protein were studied using LIGPLOT (Wallace et al., 1995).
4 Results and discussion
4.1 Chemistry
The synthesis of title hybrid compounds 11 and 12 were designed and synthesized by adopting an easy synthetic route as established in Schemes 1&2. In brief, the commercially accessible starting compound 5,5-dimethylcyclohexane-1,3-dione (1) could react with methyl iodide in the presence of potassium carbonate as a catalyst in acetonitrile as solvent at 50 °C for 10 h to form 2,2,5,5-tetramethylcyclohexane-1,3-dione (2). Further, compound 2 has undergone selective reduction with sodium borohydride in methanol at room temperature for 5 h to give 3-hydroxy-2,2,5,5-tetramethylcyclohexanone (3). The resulting compound 3 was treated with POCl3 in pyridine at 100 °C for chlorination followed by dehydrochlorination to afford 2,2,5,5-tetramethylcyclohex-3-enone (4). Later, compound 4 has undergone hydrogenation with Pd/C and H2 at 60 psi at room temperature for 16 h to get 2,2,5,5-tetramethylcyclohexanone (5). The second part of the synthesis deals with the development of structural fragments required for the generation of diversity to the core scaffold. This step corresponds to the synthesis of 4,4,7,7-tetramethyl-4,5,6,7-tetrahydrobenzo[d]thiazol-2-amine (6) with good yield by reaction of thiourea with 5 in the presence of I2 in ethanol at 110 °C for 16 h with help of a sealed tube. Further, 6 was reacted with ethyl 6-chloronicotinate in the presence of Na2CO3, Xantphos and Pd2(dba)3 under argon at 110 °C for 16 h to afford ethyl 6-((4,4,7,7-tetramethyl-4,5,6,7-tetrahydrobenzo[d]thiazol-2-yl)amino)nicotinate (7) key intermediate.
Compound 7 was reacted with (bromomethyl)cyclopropane in the presence of Cs2CO3 as base in DMF as solvent for 3 h at 110 °C to form ethyl 6-((cyclopropylmethyl)(4,4,7,7-tetramethyl-4,5,6,7-tetrahydrobenzo[d]thiazol-2-yl)amino)nicotinate (8). Compound 7 and 8 were condensed with hydrazine hydrate in refluxing ethanol for 1 h to form 6-((4,4,7,7-tetramethyl-4,5,6,7-tetrahydro-benzo[d]thiazol-2-yl)amino)nicotinohydrazide (9) and 6-((cyclopropylmethyl)(4,4,7,7-tetramethyl-4,5,6,7-tetrahydrobenzo[d]thiazol-2-yl)amino)nicotinohydrazide (10) respectively. The various substituted benzaldehydes were further allowed to treat with the previously prepared 9 and 10 in ethanol at 60 °C for 2 h to afford desired (Z)-N'-benzylidene-6-((4,4,7,7-tetramethyl-4,5,6,7-tetrahydrobenzo[d]thiazol-2-yl)amino)nicotinohydrazide (11) and (Z)-N'-benzylidene-6-((cyclopropylmethyl) (4,4,7,7–tetramethyl-4,5,6,7-tetrahydrobenzo [d] thiazol-2-yl) amino)nicotinohydrazide (12) derivatives respectively in an efficient manner with good yields. The details of the analogs of 11(a-o) series were included in Table 1 and 12(a-o) are given in Table 2. Data are expressed as mean ± S.E.M. (n = 4) and analyzed by one-way ANOVA followed by Tukey's multiple comparison test. ***P < 0.001as compared to the diabetic control group.
Entry
Aldehydes
Product
M.P (°C)
Yield %
1
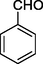
12a
149–151
77
2
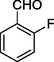
12b
186–188
69
3
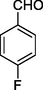
12c
130–132
68
4
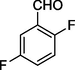
12d
199–203
68
5
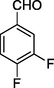
12e
132–136
69
6
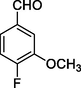
12f
174–178
69
7
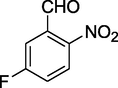
12g
137–141
68
8
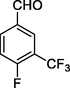
12h
190–194
70
9
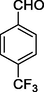
12i
174–178
74
10
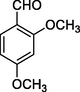
12j
177–180
69
11
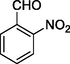
12k
222–224
68
12
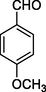
12l
228–232
65
13
12m
188–192
65
14
12n
185–189
68
15
12o
214–216
68
S. No
Groups
Day 1
Day 7
Day 28
1
Control
89.00 ± 2.449
91.25 ± 2.016
92.25 ± 2.056
2
Diabetic control
295.3 ± 6.408
331.0 ± 4.848
348.0 ± 4.416
3
11d
246.3 ± 4.905***
243.5 ± 3.227***
163.0 ± 7.990***
4
11f
214.3 ± 2.926***
196.0 ± 10.98***
118.8 ± 6.210***
5
11g
226.8 ± 5.105***
205.0 ± 3.894***
115.5 ± 3.096***
6
11j
240.0 ± 5.958***
218.8 ± 5.822***
118.8 ± 5.893***
7
11o
245.0 ± 9.678***
245.0 ± 9.678***
130.0 ± 4.601***
8
Glibenclamide
119.3 ± 1.652***
123.5 ± 4.555***
102.5 ± 2.102***
In 11a-o series, the compounds were obtained in the yield range 61–77%. The compound 11a with no substitution on the aromatic ring that is the product with benzaldehyde was found to be obtained in the highest yield in the series. Further, followed by 70% yields of compounds 11d with para-fluoro and 11h with m-CF3 along with para-fluoro substitutions. The compound 11l with para-methoxy substitution was obtained with the lowest yield (61%).
In 12a-o series, the compounds were obtained in the yield range 65–77%. The compound 12a with no substitution on the aromatic ring that is the product with benzaldehyde was found to be again obtained in the highest yield in the series. Further, followed by 74% yields of compounds 12i with para-CF3 and 11h with m-CF3 along with para-fluoro substitutions were obtained with 70% yield. The compounds 12l and 12m with para-methoxy substitution and p-methoxy with Ortho-fluoro substitutions respectively were obtained with the lowest yield (65%).
IR, NMR, and mass spectroscopy were used to determine the structure of the produced title compounds. Spectral data were used to describe all of the tetrahydro thiazolyl conjugated nicotinyl hydrazide analogs (11a-11o) and (12a-12o). Compounds (11a–11o) had a significant C=O stretching band at around 1728 cm−1 and a strong –NH stretching band at around 3326 cm−1, according to IR spectral data. IR studies revealed a strong C=O stretching band of about 1739 cm−1 and a strong –NH stretching band at 3256–3544 cm−1 for additional isomers (12a-12o). The presence of certain functional groups in the molecules was also confirmed by the IR spectra. The 1H NMR values of 11a in the aromatic area are δH 7.0–8.9 and two –NH protons region are 11.5–12.0 in the first step. Similarly, 12a proton NMR values range from 7.2 to 8.9, with one NH proton at 10.3. The cyclopropylmethyl group removed one –NH proton. The authors confirmed the substitution reaction of the cyclopropylmethyl group based on this reason.
5 Biology
5.1 In-vivo antidiabetic studies
Effect of Chemically Synthetic Compounds on blood Glucose Levels: Alloxan administration leads to a significant increase in blood glucose levels (295.3 ± 6.40 mg/dL) when compared to normal control mice. Treatment with chemically synthetic compounds for seven days significantly reduced the glucose levels when compared to diabetic mice. Among the synthetic compounds, compounds 11f and 11g showed significant reduction in the blood glucose levels from 214.3 ± 2.92 to 118.8 ± 6.21 mg/dL and 226.8 ± 5.10 to 115.5 ± 3.09 mg/dL, respectively. Whereas the compounds 11d, 11j and 11o showed a moderate decrease in the blood glucose level as compared to standard glibenclamide treated mice (Table 3, Table 4). The antidiabetic action of these molecules was significant and the mechanism needs to be known clearly as the mechanistic studies could be through insulin pathways or through the biochemical alterations in the mice which leads to lower blood glucose levels in the mice. For 28 days, the change in the blood glucose levels was clearly evident and the raised blood glucose levels were normalized with the use of the tested compounds. All the results were compared with the diabetic control to see the change in the parameters.
5.2 In-vitro anti-cancer studies
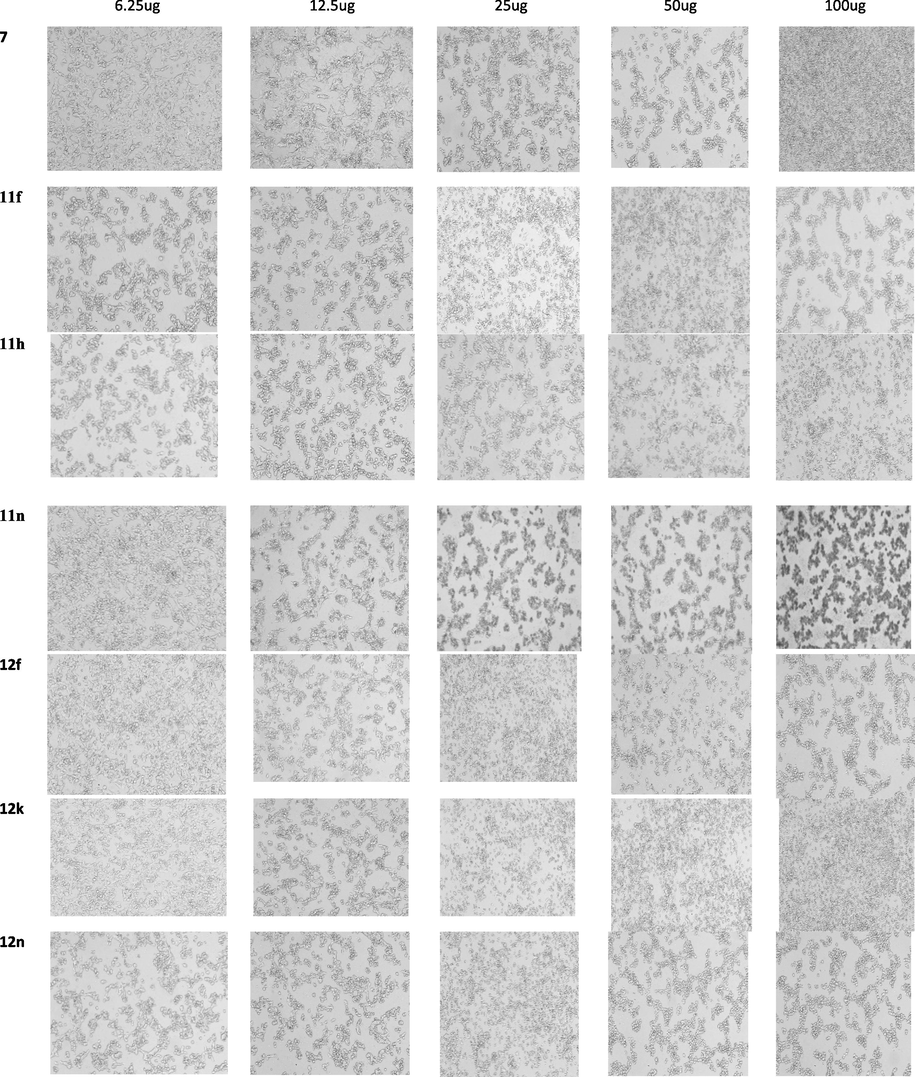
The results of the MTT test Fig. 1 demonstrated that all the compounds did not inhibit the proliferation of MCF7 cells effectively (Table 5). Compounds 11f, 11h, and 11n showed the potential inhibitory effects based on the IC50 values. In the 12 series the compounds 12f, 12k, and 12n have shown the potential activity. The anticancer activity (IC50) of compound 7 and comparison with the standard drug oxaliplatin has been shown in Fig. 2.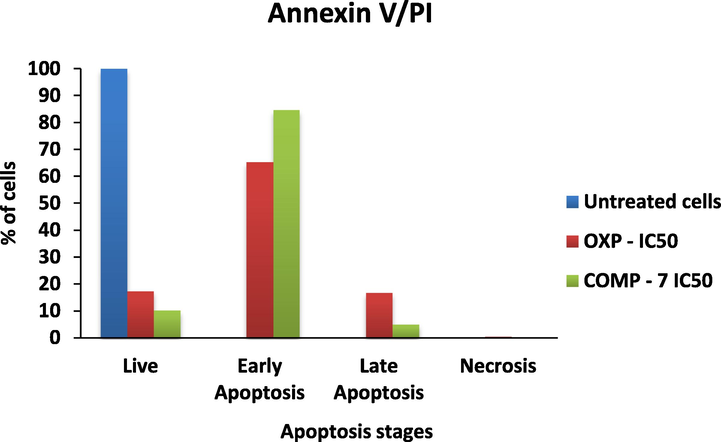
The apoptosis stages tested with Oxaliplatin (OXP) and COMP-7.
Comparison of the dose–response of selected compounds of series1 showing lower IC50 with MCF 7cell lines and response of increasing dose concentration of the compounds of series1 added to the cells in culture.
MTT measures cell respiration and the amount of formazan produced is proportional to the number of living cells present in the culture. An increase or decrease in cell number results in a concomitant change in the amount of formazan formed, indicating the degree of cytotoxicity caused by the drug. IC50 is the concentration of the tested drug able to cause the death of 50% of the cells and can be predictive of the degree of cytotoxic effect. The lower the value, the more cytotoxic is the substance. Fig. 3 shows the comparison of the selected compounds of series 11 showing the lowest IC50 value against MCF7 cancer cell lines. Our results revealed that these selected compounds of series 11f, 11h, 11n, 12f, 12k, 12n are potential anticancer drug properties compared to compound 7 (Ulukaya et al., 2004).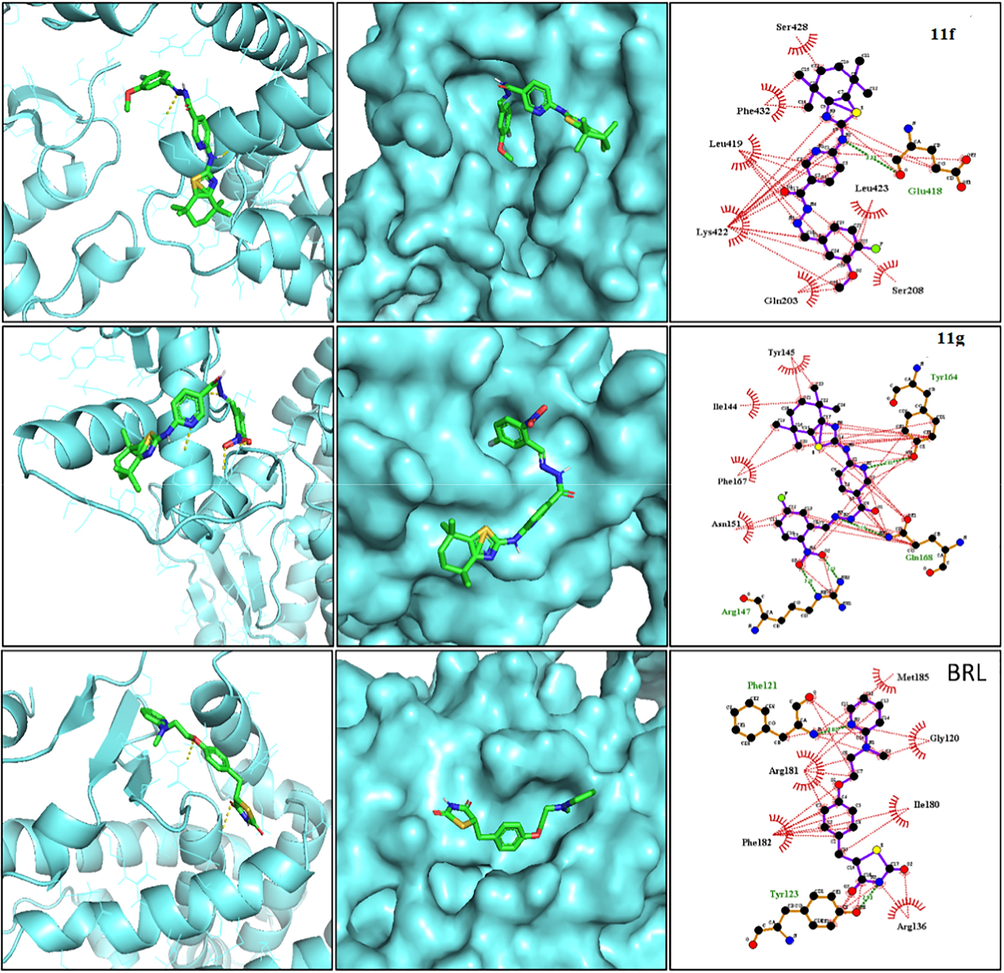
(A) Binding interactions and best poses for the synthesized compounds against hPPAR-γ protein active site (PDB ID: 3DZY). The best docked pose for active compounds 11f and 11g was represented in the image. The co-crystal ligand Rosiglitazone (BRL) was used as positive control.
5.3 Molecular docking
The active compounds (11f and 11g) from the preliminary results were studied for in silico binding analysis with human PPAR-γ protein complexed with RXR alpha Nuclear Receptor (Fig. 4). The glitazone-based anti-diabetics which are derivatives of Thiazolidinediones have been widely used as PPAR-γ agonists given their ability to induce activation of the said protein (Staels and Fruchart, 2005). Since the core scaffold of the newly synthesized (thiazole) is like that of thiazolidinediones, a representative drug from that category Rosiglitazone is taken as a reference to the perform docking studies. The analysis of molecular docking results revealed that all the compounds are bound to the active site of proteins by hydrogen bonds and hydrophobic bonds. Ten different conformations per each ligand were generated and examined. The docked ligands showed good binding energies and RMSD scores with the target protein (Table 6). It was determined that the compound 11g was the best docked ligand as compared to other ligands. The binding energies for the compounds 11f and 11g in the protein active site were −8.4 and −8.2 kcal/mol, respectively. The co-crystal ligand rosiglitazone (BRL) showed −6.1 kcal/mol. Both the compounds formed hydrogen bonds with the target protein. PPAR gamma is the main target of thiazolidinediones used in diabetes mellitus characterized by insulin resistance. Thiazolidinediones, acting via PPARγ, influence free fatty acid flux and thus reduce insulin resistance and blood glucose levels [28/33]. The binding interactions among protein and ligands were majorly influenced by hydrogen bonds and hydrophobic interactions (Fig. 4). The binding energies, RMSD score and the amino acids of the target protein interacted with the ligands were shown in Table 6. Data are expressed as mean ± S.E.M. (n = 4) and analysed by one way ANOVA followed by Tukey's multiple comparison test. ***P < 0.001, **P < 0.01, *P < 0.05as compared to the diabetic control group.
S. No
Groups
Day 1
Day 7
Day 28
1
Control
89.00 ± 2.449
91.25 ± 2.016
92.25 ± 2.056
2
Diabetic control
295.3 ± 6.408
331.0 ± 4.848
348.0 ± 4.416
3
12g
279.0 ± 3.536
300.5 ± 2.217
325.5 ± 8.088
4
12h
245.8 ± 7.983***
287.3 ± 3.092*
312.5 ± 4.664
5
12i
302.8 ± 8.469
238.8 ± 5.935***
124.3 ± 4.029
6
12j
286.0 ± 4.637
299.8 ± 1.652
306.8 ± 2.562
7
12n
234.8 ± 4.479***
219.0 ± 6.721***
114.5 ± 3.775***
8
12o
256.3 ± 3.945**
267.3 ± 4.973***
235.8 ± 50.33***
9
Glibenclamide
119.3 ± 1.652***
123.5 ± 4.555***
102.5 ± 2.102***
Test Compounds
IC50 µg/mL
11a
40.6
11c
97.32
11d
26.08
11e
68.67
11f
7.24
11g
73.26
11h
10.35
11i
20.39
11j
278.49
11k
88.73
11l
16.19
11m
43.03
11n
9.02
11o
22.15
12a
96.43
12c
71.75
12d
75.73
12e
53.29
12f
11.33
12i
12.38
12k
9.11
12m
36.79
12n
8.89
12o
51.38
Aag
78.09
Aap
24.21
Aas
19.08
7
18.86
8
54.31
9
19.84
Ligand
Binding energy (Kcal/mol)
RMSD
H-bond/s
Protein–Ligand interactions
Compound 11f
−8.2
2.265
Glu418
Ser208, Gln203, Lys422, Leu419, Phe432, Ser428, Leu423
Compound11g
−8.4
4.106
Arg147, Gln168,
Tyr164Asn151, Phe167, Ile144, Tyr145
BRL
(Co-crystal ligand)−6.1
2.867
Phe121, Tyr123
Arg136, Ile180, Arg181, Phe182, Gly120, Met185
6 Conclusion
In conclusion, thetetrahydrobenzo[d]thiazole fused nicotinohydrazide derivatives were synthesized using dimedone as starting material. Upon synthesizing the key intermediate ethyl 6-((4,4,7,7-tetramethyl-4,5,6,7-tetrahydrobenzo[d]thiazol-2-yl) amino)nicotinate(7), it was further transformed to two different series of hydrazides. A total of 30 derivatives were synthesized in order to study the effect of various functional substitutions such as electron-donating and withdrawing on biological activity including antidiabetic activity. Compounds 11f and 11g showed a significant reduction in the blood glucose levels of diabetic mice. Further, molecular studies have given insight into the binding modes of synthesized ligands with the target protein. Among the tested compounds, two compounds (11f and 11g) exhibited promising binding affinities with human PPAR-γ protein complexed with RXR alpha nuclear receptor as compared to standard drug Rosiglitazone. Upon further, structural optimization and detailed biological target-based mechanistic studies the compounds can be accelerated to the next level of the drug discovery process.
Acknowledgements
The authors are very thankful to Acharya Nagarjuna University, for permitting the research work and for constant encouragement. The authors also thank Universiti Malaysia Sabah, Kota Kinabalu, Sabah, Malaysia for monitory support to this work.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- WHO, Definition, diagnosis and classification of diabetes mellitus and its complications? Geneva: 1999.
- Mohan, D., Raj, D., Shanthirani, C.S., Datta, M., Unwin, N.C., Kapur, A., 2005. J. Assoc. Physicians India 53, 283–287.
- Olefsky, J.M., Revers, R.R., Prince, M., Henry, R.R., Garvey, W.T., Scarlett,J.A., Kolterman, O.G., 1985. Adv Exp Med Biol 189: 176-205.
- Am. J. Clin. Nutr.. 1995;61:980-986.
- Jr Diabetes Care. 1998;21:32-34.
- J. Am. Geriatr. Soc.. 2012;60:1564.
- Retinoic acid synthesis and functions in early embryonic development. Cell Biosci.. 2012;2:11.
- [Google Scholar]
- The PPAR-RXR transcriptional complex in the vasculature: energy in the balance. Cric. Res.. 2011;108:1002-1016.
- [Google Scholar]
- Thiazolidinedione drugs in the treatment of type 2 diabetes mellitus: past, present and future. J. Crit. Rev. Toxicol.. 2017;48
- [Google Scholar]
- Mohd. JavedNaim, OzairAlam , Md. Jahangir Alam , Mohammad Shaquiquzzaman, Md. Mumtaz Alam, Vegi Ganga Modi Naidu, Synthesis, docking, in vitro and in vivoantidiabetic activity of pyrazole‐based 2,4‐thiazolidinedione derivatives as PPAR‐γ modulators,Arch Pharm Chem Life Sci.2018;e1700223
- Hydrogen bonds of 2-aminothiazolesin intermolecular complexes (1:1 and 1:2) with proton acceptors in solutions. J. Mol. Struct.. 2006;783:101-115.
- [Google Scholar]
- Antidepressant effects of pramipexole, a novel dopamine receptor agonist. J. Neural Transm.. 1997;104:525-533.
- [Google Scholar]
- Biological Evaluation of Compounds with –NCSGroup or derived from thiazole and imidazole, activity on prostaglandin synthetase complex. J. Pharm. Pharmacol.. 1993;45:731-735.
- [Google Scholar]
- Milne, G.W.A., 2000. Handbook of Antineoplastic Agents, Gower; Ashgate, Ed., London, UK.
- Markus, B., Salomevon, G., Christoph, B., Werner, J.P., 2004. Molecular aspects of drug recogni tionby specific T cells. Curr Drug Tar 4: 1 – 11.
- Knadler, M.P., Bergstrom, R.F., Callaghan, J.T., Rubin, A., Nizatidine, 1986. An H2-blocker. Its metabolism and disposition in man, Drug MetabDispos 14, 175–182.
- D. Lednicer, L.A. Mitscher, G.I. George, Organic Chemistry of Drug Synthesis, Wiley, New York, 4, 1990, 95-97.
- An efficient synthesis of 2-Alkyl-4-hydroxy-2H-1,2-benzothiazine-3-carboxamide-1,1-dioxides. Bull. Korean Chem. Soc.. 2005;26:1771-1775.
- [Google Scholar]
- New 2-sulfonamidothiazoles substituted at C-4: synthesis of polyoxygenated aryl derivatives and in vitro evaluation of antifungal activity. Eur. J. Med. Chem.. 1999;34:773-779.
- [Google Scholar]
- Novelstruc tural templates for estrogen-receptor ligands and prospects for combinatorialsynthesis of es trogens. Chem. Biol.. 1999;6:205-219.
- [Google Scholar]
- Muijlwijk-Koezen, J.E., Timmerman, H., Vollinga, R.C., DrabbeKunzel, J.F., Groote, M., Visser, S., IJzerman, A.P., 2001. Thiazole and Thiadiazole Analogues as a NovelClass of Adenosine Re ceptor Antagonists. J. Med. Chem. 44, 749–762.
- Sekiou, O., Boumendjel, M., Taibi, F., Tichati, L., Boumendjel, A., Messarah, M., 2021. Nephroprotective effect of Artemisia herba alba aqueous extract in alloxan-induced diabetic rats. J. Tradit. Complement. Med. 11(1), 53–61.26.
- In-vitro antioxidant activities of Plumbago zeylanica Linn. and Plumbago rosea Linn: a comparative study. Int. J. Pharmacognosy Phytochem. Res.. 2014;6(3):531-535.
- [Google Scholar]
- Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Can. Res. 1988;48(3):589-601.
- [Google Scholar]
- Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. Immunol. Methods. 1983;65(1–2):55-63.
- [Google Scholar]
- Metal based imaging probes of DO3A-Act-Met for LAT1 mediated methionine specific tumors : synthesis and preclinical evaluation. Pharm. Res.. 2015;32:955-967.
- [Google Scholar]
- The effect of different percentages of triethanolammoniumbu tyrate ionic liquid on the structure and activity of urate oxidase: molecular docking, molecular dynamics simulation, and experimental study. J. Mol. Liq.. 2019;292:111318
- [Google Scholar]
- LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel.. 1995;8:127-134.
- [Google Scholar]
- Interference by anti-cancer chemotherapeutic agents in the MTT-tumor assay. Chemotherapy. 2004;50:43-50.
- [Google Scholar]
- Therapeutic roles of peroxisome proliferator–activated receptor agonists. Diabetes. 2005;54(8):2460-2470.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2021.103546.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




