

Translate this page into:
Synthesis, molecular docking and ADMET studies of bis-benzimidazole-based thiadiazole derivatives as potent inhibitors, in vitro α-amylase and α-glucosidase
⁎Corresponding authors. shahidiqbal@hzu.edu.cn (Shahid Iqbal), sono_waj@gmail.com (Wajid Rehman), ikaeed@mcst.edu.sa (Eslam B. Elkaeed)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Different research synthetic methods have been developed recently for the synthesis of bis-benzimidazole analogs to investigate various biological significances. In this present study, an attempt was made to synthesize a new series of bis-benzimidazole analogs in a fast and efficient method. A variety of spectroscopic techniques, including 13C NMR, 1H NMR, and HREI-MS, were used to establish the existence of every synthesized scaffold. Molecular docking profiles were also carried out to ascertain the binding interactions of the compounds. All derivatives (1–18) were evaluated for their biological potential to investigate the inhibitory activity of α-amylase and α-glucosidase through SAR study. Almost all derivatives were found to be engaged in a highly promising activity when compared to referenced drug acarbose (IC50 = 8.24 ± 0.08 µM), in this regard among the tested series analog 9 (IC50 = 0.10 ± 0.50 and 0.20 ± 0.50 µM respectively), showed excellent activity. Moreover, ADME predictions were also studied for potent compounds, exhibited drug like properties.
Keywords
Bis-benzimidazole
Thiadiazole
α-amylase
α-glucosidase
SAR ADMET
Molecular docking

1 Introduction
A chronic condition called diabetes mellitus prevents the body from properly metabolizing proteins, carbs, and lipids. It also causes several issues in the body that lead to hyperglycemia (American Diabetes Association, 2013). Diabetes presently affects 4.4% of the global population, up from 2.8% in 2000. Diabetes is going to impact approximately 366 million people globally by 2030 (Wild et al., 2004; Sacks, 1997). A range of synthetic oral hypoglycemic medicines, such as sulfonylureas, biguanides, and alpha-glucosidase inhibitors, are used to decrease elevated blood glucose levels. Unfortunately, their longer usage has shown more of its different negative effects (Edwin et al., 2006) such as hypoglycemia, headache, nausea, and dizziness (Chaudhury et al., 2017). The negative effects made it necessary to discover innovative, efficient, and safer substitutes that have the highest priority (Es-Safi et al., 2020). Researchers are actively focusing on heterocyclic compounds because to its potential for being efficient, accessible, and having fewer adverse effects. The current study also based on heterocyclic compounds such benzimidazole-based thiadiazole derivatives. The primary aim of this study was to investigate the inhibitory potential of synthesized scaffolds in contrast to both α-amylase and α-glucosidase enzymes. In compared to other heterocyclic compounds, the benzimidazole ring got considerably more attention. Its ring is commonly referred to as “privileged” due to its wide range of biological significance. The structure of benzimidazole and the chemistry of their ligands are of great interest to researchers (Mechchate et al., 2020; Bonnett, 1963). Similar products, including thiabendazole, misonidazole, omeprazole, astemizole, clotrimazole, and cimetidine were a significant source of biologically active drugs as well as they have some other positive effects in agricultural and veterinary fields (Al-Muhaimeed, 1997). Due to the presence of various groups on the benzimidazole ring, drugs containing benzimidazole moiety exhibit a wide range of pharmacological profiles such as Bactericidal (Carcanague et al., 2002), analgesic (Aghatabay et al., 2007; Demirayak et al., 2005; Gaba et al., 2014; Sondhi et al., 2006; Achar et al., 2010), fungicidal (Lezcano et al., 2002; Sun et al., 2021; Sun et al., 2021), antiviral (Tewari and Mishra, 2006; Basha, 2022; Ibba et al., 2022; Huo et al., 2021), and HIV-1 infectivity inhibition (Gardiner et al., 1995). Benzimidazole and thiadiazole moieties have been extensively studied and found with better inhibitory potential against varied infections, especially diabetes mellitus. Therefore in the project, the aim was to synthesize and evaluate bis-benzimidazole-based thiadiazole analogs and screen against α-amylase and α-glucosidase. They were also found effective inhibitors against both enzymes. Moreover, binding interactions were confirmed through molecular docking studies. Besides, rationales of the current analog-A (Aroua et al., 2021) and analog-B (Zawawi et al., 2016) have been mentioned in the comparison activity of synthesized analogs with previously reported compounds as shown in Fig. 1.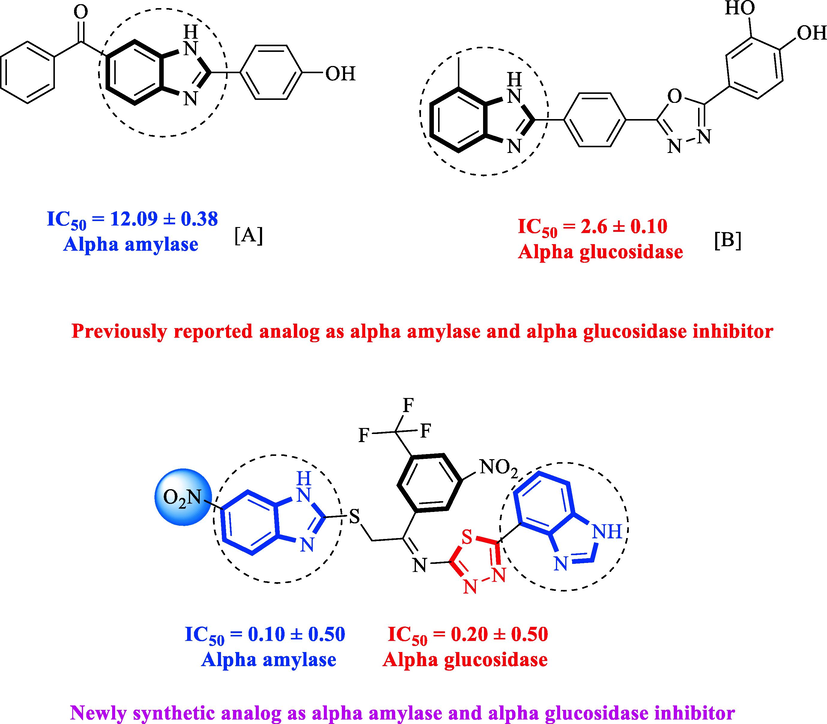
Rationale of the current study.
2 Results and discussion
2.1 Chemistry
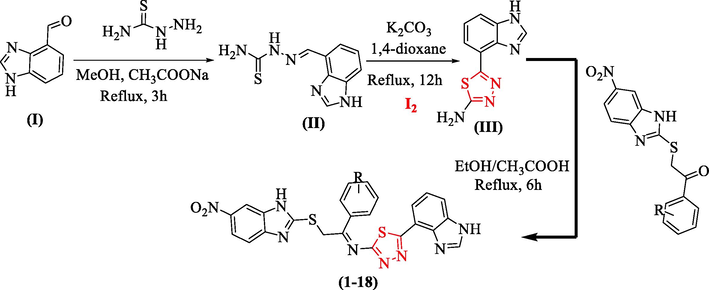
The synthesis of bis-benzimidazole-containing thiadiazole derivatives was conducted using a stepwise reactions technique (1–18). Initially, benzimidazole bearing aldehyde group (I) and thiosemicarbazide were mixed in methanol and refluxed in the reaction mixture for about 3 h in the presence of sodium acetate to obtain Schiff base an intermediate (yields = 88% II). Upon addition of iodine in 1,4-dioxane in the presence of potassium carbonate, the reaction was refluxed for about 12 h, afforded benzimidazole-based thiadiazole containing an amine group (yields = 72% III). Compound (III) was further refluxed for about 6 h with s-substituted benzimidazole-based ketone derivatives in ethanol followed by the addition of acetic acid gives bis-benzimidazole bearing thiadiazole derivatives as shown in the Scheme 1.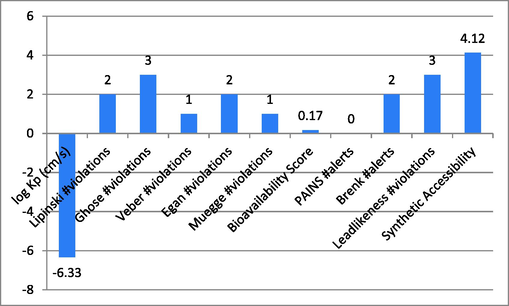
Thiadiazole derivatives based on the synthesis of bis-benzimidazoles (1–18).
2.2 Spectral analysis
A structure interpretation of the represented compound-10 was identified and their proton, as well as carbon peaks, were identified through NMR spectroscopic technique. The proton value of the represented compound has appeared at different ppm which indicates the shielded and de-shielded region of the proton, carbon as well as HREI-MS.
1H NMR (600 MHz, DMSO‑d6): δ. The first proton appeared at 11.87 showing a singlet for NH of benzimidazole ring bearing nitro group similarly, another proton appeared at 11.62 also showing a singlet for NH of second benzimidazole ring. –OH proton appeared at 9.57 showing singlet while other aromatic proton appeared at 8.78 showing doublet with coupling constant (J) 7.1, and another proton appeared at 8.51 showing doublet with J = 6.8 Hz. Similarly, 8.36 shows singlet for Benzimidazole-H and 7.57 showing doublet, with J = 6.7 Hz, of Benzimidazole-H, 7.33 showing singlet for 2H of Benzimidazole-H and 7.10 doublet with J = 8.1 Hz, of Benzimidazole-H, 6.86 displayed as a doublet with J = 8.1 Hz, for 2H of Benzimidazole-H, 6.59 doublet with J = 7.1 Hz of Benzimidazole-H and 3.83 singlets for 2H of -SCH2 and 13C NMR (150 MHz, DMSO‑d6): showing in descending order δ 162.2, 159.9, 149.6, 148.9, 148.0, 147.1, 145.0, 144.3, 142.0, 140.9, 139.8, 139.4, 134.1, 128.7, 128.4, 127.6, 125.9, 123.4, 121.5, 118.6, 115.5, 109.4, 100.9, 55.6 as well as HR EI-MS: m/z calcd for C24H14Cl2N8O3S2 [M]+ 596.0630; Found: 596.0518. The detailed spectral analysis of all the compounds (1–18) has been incorporated in the supplementary information.
3 Structure-activity relationship (SAR)
3.1 α-amylase and α-glucosidase inhibitory profile
Biological profiles of synthesized analogs were compared with one another and the standard drug acarbose. Most of the analogs were found with good to moderate activity when compared to the standard drug. Variation in biological activity might be the position, numbers and type of substituents attached among trifluoro, nitro and hydroxyl-containing analogs were found with excellent potential. Comparisons of analogs based on their substituents and position are given below.
Nitro and trifluoro-substituted analogs were arranged for comparison study due to their excellent biological profile when compared to their potential with standard drug acarbose (IC50 = 3.66 ± 0.12 and 4.85 ± 0.11 µM). These analogs showed a varied range of inhibitory profiles such as 1 (IC50 = 1.10 ± 0.10 and 1.80 ± 0.10 µM), 4 (IC50 = 1.20 ± 0.20 and 2.0 ± 0.20 µM), 9 (IC50 = 0.10 ± 0.50 and 0.20 ± 0.50 µM) and 11 (3.40 ± 0.10 and 4.70 ± 0.10). Among the screens analog 9 was found with remarkable potential while others showed good to moderate activity might be the presence of the trifluoro group which makes strong hydrogen bonding and nitro moiety also dominate the negative charge at meta-position therefore the over effect was found greater as compared to other analogs.
Chloro-substituted analogues 3 (IC50 = 3.40 ± 0.10 and 5.80 ± 0.10 µM), 12 (IC50 = 6.10 ± 0.10 and 7.70 ± 0.10 µM), and 17 (IC50 = 2.80 ± 0.10 and 3.70 ± 0.10 µM), were shown to have action similar to that of the medication acarbose (IC50 = µM). The position of substituents also significantly affects the inhibitory potential of analogs, as demonstrated by this measurement, where para-Chloro (17) was found to be much more potent than ortho and meta. In this case, the chloro group is attached to a different position of an aromatic ring, which has a significant impact. The strong inhibitory potential of analogs might be the presence of halogen atom at para-position dominantly increases the ring charge which produces greater effects for inhibitions.
Similarly, hydroxyl group-containing analogs (10, 14, and 16) were found with excellent α-amylase and α-glucosidase inhibitory profiles. The creation of powerful hydrogen bonds with the active sites of enzymes may be the cause of the –OH-bearing analogs' inhibitory potentials. In this regard position and the number of the –OH group on the aromatic ring are also crucial for greater interactions therefore analog 14 (IC50 = 0.70 ± 0.50 and 1.20 ± 0.10 µM) has two –OH groups at meta-position. The presence of one –OH 16 (IC50 = 2.50 ± 0.10 and 3.50 ± 0.10 µM) activity profile was found lower and analog 10 (IC50 = 1.30 ± 0.10 and 1.70 ± 0.10 µM) showed somewhat better activity than analog 16 might be the presence of two Chloro group on both meta-position of the ring. The activity profile of –OH analogs was found potent when comparing their biological potential to standard drug acarbose (IC50 = 3.66 ± 0.12 and 4.85 ± 0.11 µM).
Methyl-substituted analogs (2 and 8) were also discovered to have an action similar to that of the medication acarbose. Here, in this comparison position of attached substituents also matters to the biological profile of the analog. The Methyl group attached to the aromatic ring at para and ortho-position respectively. The activity profile of the competitive analog was found better in the case of para-substituted analog 2 (IC50 = 4.60 ± 0.10 and 5.90 ± 0.10 µM) than analog 8 (IC50 = 5.30 ± 0.20 and 7.20 ± 0.10 µM).
3.1.1 The general structure of the molecule
The general structure represents the different parts of the molecule and varied substitutions attached to the aromatic ring. These substituents play a key role in the biological nature of molecules as discussed above. General representation of molecule as displayed in Fig. 2.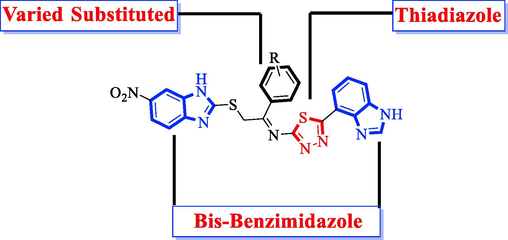
General representation of the molecule.
All of the screen compounds were discovered to have similar activity, however, certain analogs were discovered to have a few folds better results in both amylase and glucosidase than the conventional medication acarbose. Compounds 1, 4, 9, and 14 had impressive potential in this respect, suggesting that the improved activity may be caused by the presence of connected functional groups, which improves ring activity for better interaction.
4 Molecular docking study
A molecular docking study was conducted for subjected (synthesized molecules having much potential against α-amylase and α-glucosidase) analogs which were found with excellent interactions in a superimposed complex. Analogues' potent binding abilities are caused by the functional group that is connected at a variety of positions on the aromatic ring. Grater hydrogen bonding was observed in the case of triflouro, nitro and hydroxyl-containing moieties. Most of the analogs were found with good to poor interactions this may be the attached substituents but 1, 4, 9 and 14 were the most active analogs with the greater number of interactions (see Table 1).
S.NO
R
α‐amylase inhibition IC50
(µM)
α‐glucosidase inhibition IC50
(µM)
1
1.10 ± 0.10
1.80 ± 0.10
2
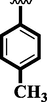
4.60 ± 0.10
5.90 ± 0.10
3
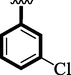
3.40 ± 0.10
5.80 ± 0.10
4
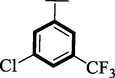
1.20 ± 0.20
2.0 ± 0.20
5
19.80 ± 0.20
21.50 ± 0.20
6
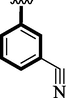
17.40 ± 0.05
18.10 ± 0.05
7
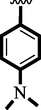
15.60 ± 0.30
16.30 ± 0.30
8
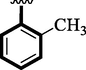
5.30 ± 0.20
7.20 ± 0.10
9
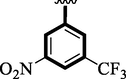
0.10 ± 0.50
0.20 ± 0.50
10
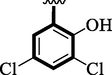
1.30 ± 0.10
1.70 ± 0.10
11
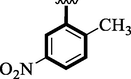
3.40 ± 0.10
4.70 ± 0.10
12
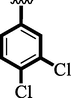
6.10 ± 0.10
7.70 ± 0.10
13
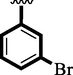
23.20 ± 0.50
24.30 ± 0.50
14
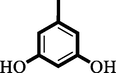
0.70 ± 0.50
1.20 ± 0.10
15

6.10 ± 0.10
8.30 ± 0.10
16
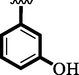
2.50 ± 0.10
3.50 ± 0.10
17
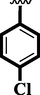
2.80 ± 0.10
3.70 ± 0.10
18
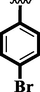
22.70 ± 0.20
23.70 ± 0.10
Standard drug Acarbose
3.66 ± 0.12 4.85 ± 0.11
The binding interaction of a molecule with an enzyme's active site is revealed by molecular docking research, which depends on the kind of attached substituents and how they affect the interactive features. Molecular docking was performed by using varied software such as Auto dock vina (1.5.7), Molecular operational environment (MOE-2015) and discovery studio visualizer (DSV-2021) (Kharb et al., 2012; Khan et al., 2022; Li et al., 2015; Rao et al., 2021; Khan et al., 2022; Khan et al., 2022). Molecular docking studies were performed in three steps, in the first step both protein (retrieved from RCSD protein data bank by thorough codes 1b2y and 3w37 for α-amylase and α-glucosidase respectively) and ligand were prepared and energy was minimized in MOE. In the next step, both protein and ligand were transferred to the auto dock where water was removed and polar hydrogen, as well as Kollman and Gasteiger charges, were added. After completion, the protein, ligand and their X, Y and Z coordinate were saved in PDBQT and text format respectively. In the last step, the location of the docking folder was set and using command prompt to carry out the molecular docking study. Finally, DSV was used to explore the binding interaction (Table 2) in the form of 2D and 3D structures as shown in Figs. 3-10.
Compound
Receptor
Interaction
Distance
Docking Score
Analog 1(A) against α-Amylase
TYR-A-334
Pi-Sulphur
4.54A°
-12.9
TYP-A-334
Pi-Pi-Stacked
4.50A°
TRP-A-279
Pi-Pi-T-Shaped
5.76A°
SER-A-286
Hydrogen-Bond
4.35A°
GLY-A-117
Hydrogen-Bond
3.35A°
TYR-A-116
Vander wall
6.86A°
TRP-A-84
Pi-Pi Stacked
5.48A°
PHE-A-330
Pi-Pi Stacked
4.58A°
TYR-A-121
Hydrogen-Bond
7.74A°
SER-A-81
Carbon-Hydrogen
3.54 A°
Analog 1(B) against α-Glucosidase
ALA-A-328
Pi-Alkyl
6.33A°
-11.4
TRP-A-82
Conventional H-B
5.13A°
GLY-A-116
Pi-Pi Stacked
4.30A°
HIS-A-438
Pi-Cation
6.19A°
GLU-A-197
Pi. Hydrogen-Bond
6.14A°
SER-A-287
Hydrogen-Bond
3.00A°
ASP-A-70
Hydrogen-Bond
3.78A°
TYR-A-332
Amide Pi-stacked
4.92A°
Analog 4(C) against α-Amylase
SER-A-287
Hydrogen-Flouride
4.58A°
-11.7
SER-A-287
Hydrogen-Flouride
5.39A°
TYR-A-322
Hydrogen-Bond
5.78A°
ASP-A-70
Conventional H-B
4.34A°
PHE-A-329
Pi-Pi- T-shaped
5.22A°
TRP-A-430
Vander wall
5.31A°
TRP-A-440
Vander wall
5.96A°
ALA-A328
Pi-Alkyl
5.25A°
TRP-A-82
Conventional H-B
4.37A°
GLU-A-197
Conventional H-B
4.19A°
Analog 4(D) against α-Glucosidase
ASN-A-85
Hydrogen-Flouride
3.53A°
-12.5
TYR-A-84
Hydrogen-Flouride
6.17A°
TYR-A-70
Hydrogen-Flouride
6.71A°
ASP-A-72
Pi-Anion
4.33A°
TYR-A-121
Vander wall
7.13A°
PHE-A-330
Pi-Pi T-shaped
4.58A°
TYR-A-130
Vander wall
6.85A°
GLY-A-117
Hydrogen-Bond
3.41A°
PHE-A-331
Pi-Pi T-shaped
5.65A°
TYR-A-334
Pi-Pi Sacked
4.57A°
TRP-A-279
Pi-Pi T-shaped
6.69A
Analog 9(E) against α-Amylase
TRP-A-334
Hydrogen-Flouride
4.95A°
-12.8
TRP-A-279
Pi-Alkyl
5.82A°
TRP-A-334
Pi-Sulphur
4.11A°
TRP-A-121
Vander wall
5.77A°
ASP-A-72
Pi-Sulphur
4.35A°
GLY-A-123
Hydrogen-Bond
3.96A°
ASN-A-85
Carbon-Hydrogen bond
3.91A°
PRO-A-86
Carbon-Hydrogen bond
4.46A°
GLU-A-199
Pi-Anion
7.85A°
GLY-A-118
Pi- Hydrogen-Bond
3.76A°
ARG-A-289
Conventional H-B
4.85A°
Analog 9(F) against α-Glucosidase
TRP-A-231
Pi-Alkyl
4.33A°
-12.3
PHE-A-398
Pi-Alkyl
6.69A°
GLY-A-117
Carbon-Hydrogen bond
3.83A°
PHE-A-329
Pi-Pi-Stacked
6.88A°
HIS-A-438
Vander wall
5.11A°
SER-A-198
Conventional H-B
4.40A°
TYR-A-332
Pi-Pi T-shaped
5.48A°
TRP-A-82
Conventional H-B
5.13A°
TYR-A-440
Conventional H-B
5.54A°
ALA-A-328
Pi-Alkyl
6.39A°
ALA-A-277
Pi-Alkyl
4.18A°
Analog 14 (G) against α-Amylase
TYR-A-130
Conventional H-B
6.75A°
-12.2
GLY-A-117
Hydrogen-Bond
3.36A°
PHE-A-330
Pi-Pi T-shaped
4.59A°
TRP-A-84
Carbon-Hydrogen bond
5.46A°
TYR-A-121
Conventional H-B
7.17A°
SER-A-81
Hydrogen-Bond
2.76A°
TYR-A-334
Pi-Pi Stacked
4.64A°
TRP-A-279
Pi-Pi T-shaped
5.76A°
PHE-A-331
Carbon-Hydrogen bond
4.51A°
SER-A-286
Conventional H-B
4.23A°
Analog 14 (H) against α-Glucosidase
TYR-A-128
Conventional H-B
6.06A°
-11.5
TRP-A-82
Pi-Pi T-shaped
4.19A°
TRP-A-82
Pi-Pi T-shaped
6.17A°
GLY-A-117
Hydrogen-Bond
3.50A°
SER-A-198
Carbon-Hydrogen bond
4.16A°
PHE-A-329
Pi-Pi Stacked
5.94A°
PRO-A-285
Conventional H-B
6.06A°
ASP-A-70
Pi-Anion
3.90A°
Acarbose in α-amylase complex
ASP-A-300
Conventional H-B
4.69A°
-70.5
ASP-A-300
Conventional H-B
4.94A°
GLU-A-233
Conventional H-B
4.62A°
GLU-A-233
Conventional H-B
4.63A°
HIS-A-201
Carbon H-B
4.63A°
GLU-A-240
Conventional H-B
5.50A°
Acarbose in α-glucosidase
ASP-A-232
Conventional H-B
4.50A°
-91.5
LYS-A-506
Conventional H-B
3.71A°
ASN-A-496
Conventional H-B
4.81A°
SER-A-505
Conventional H-B
3.72A°
GLU-A-603
Conventional H-B
4.08A°
ALA-A-232
Carbon H-B
5.29A°
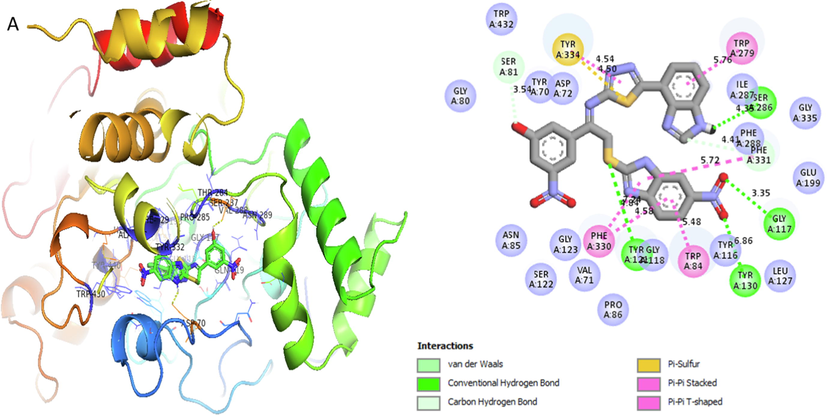
Represent 2D and 3D structure for analogue1-A in α-amylase complex.
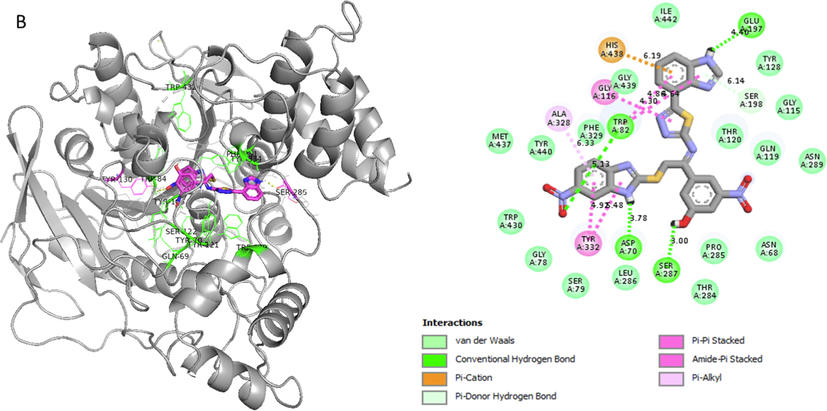
Represent 2D and 3D structure for analogue1-B in α-glucosidase complex.
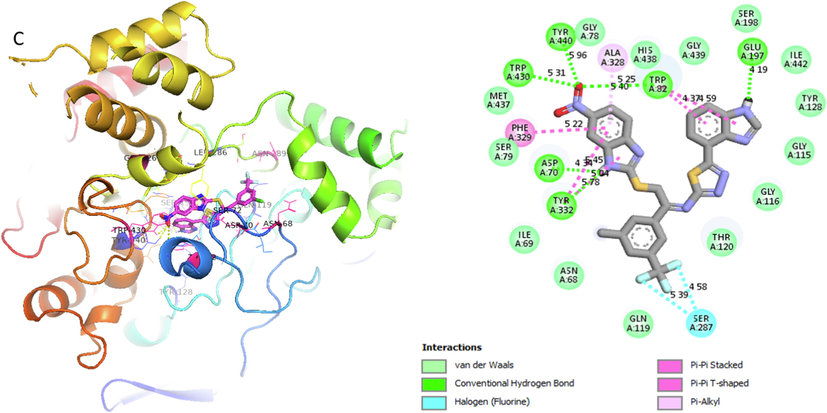
Represent 2D and 3D structure for analog 4-C in α-amylase complex.
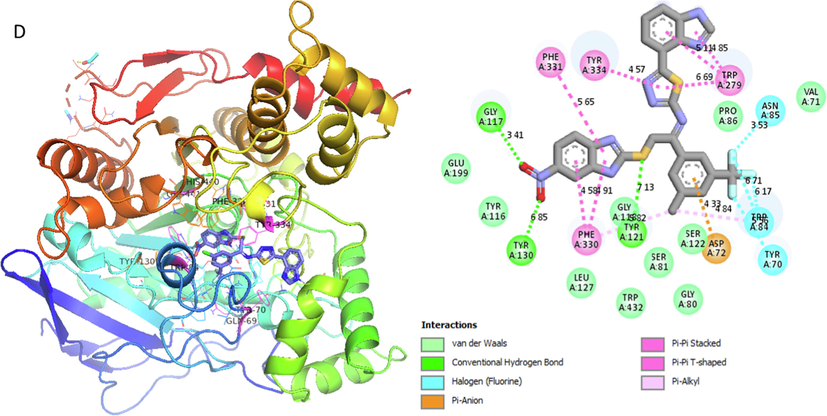
Represent 2D and 3D structure for analog 4-D in α-glucosidase complex.
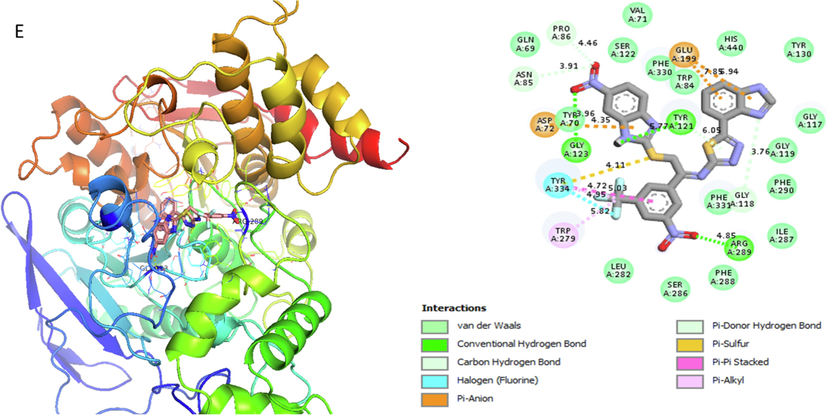
Represent 2D and 3D structure for analog 9-E in α-amylase complex.
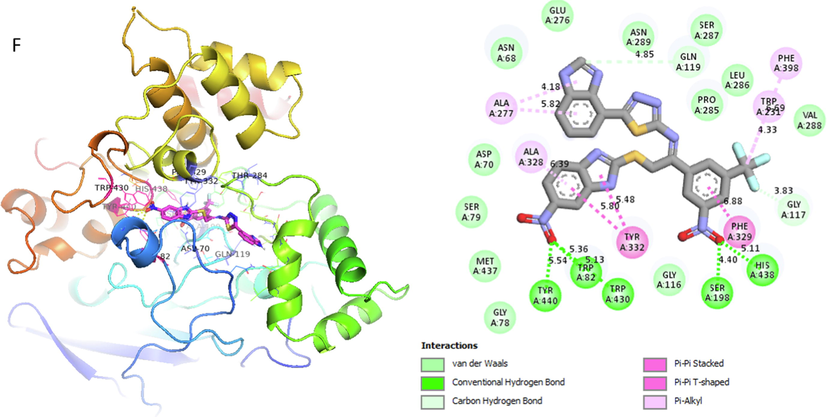
Represent 2D and 3D structure for analog 9-F in α-glucosidase complex.
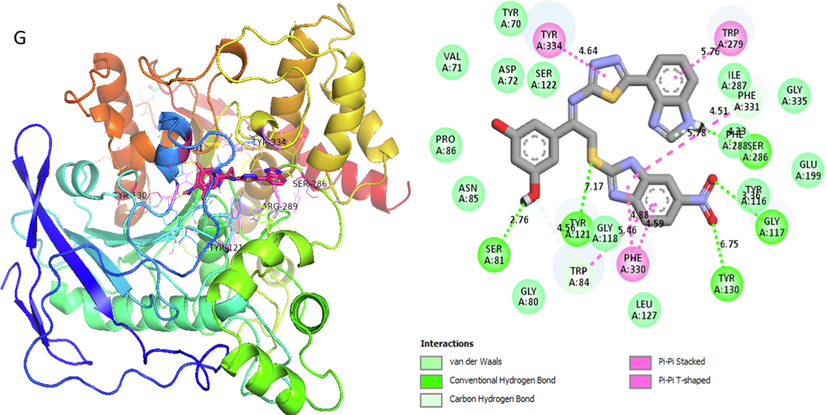
Represent 2D and 3D structure for analog 14-G in α-amylase complex.
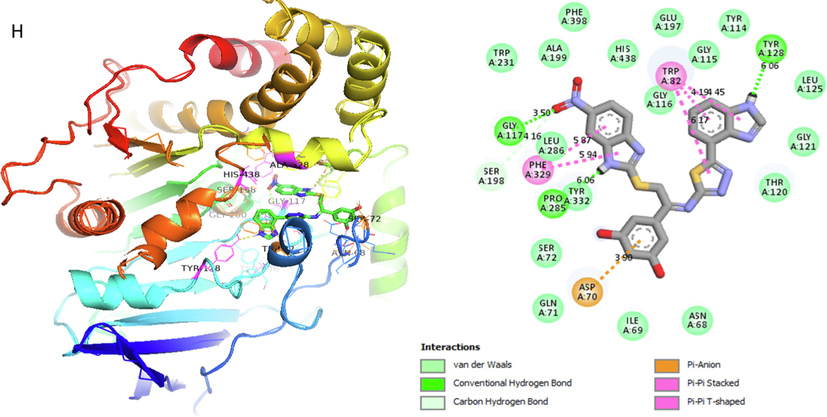
Represent 2D and 3D structure for analog 14-H in α-glucosidase complex.
Docking score were calculated for analogs 1, 4, 9 and 14 due to varied functionalities and position produced different affect which also increase or decrease the binding interaction therefore their docking score is different from one another and their interactions ranges also. The potency of these compounds might be the presence of functional group, position and number of substituents which enhance or reduce the binding interactions of molecules in a complex (Table 2) (see Fig. 11).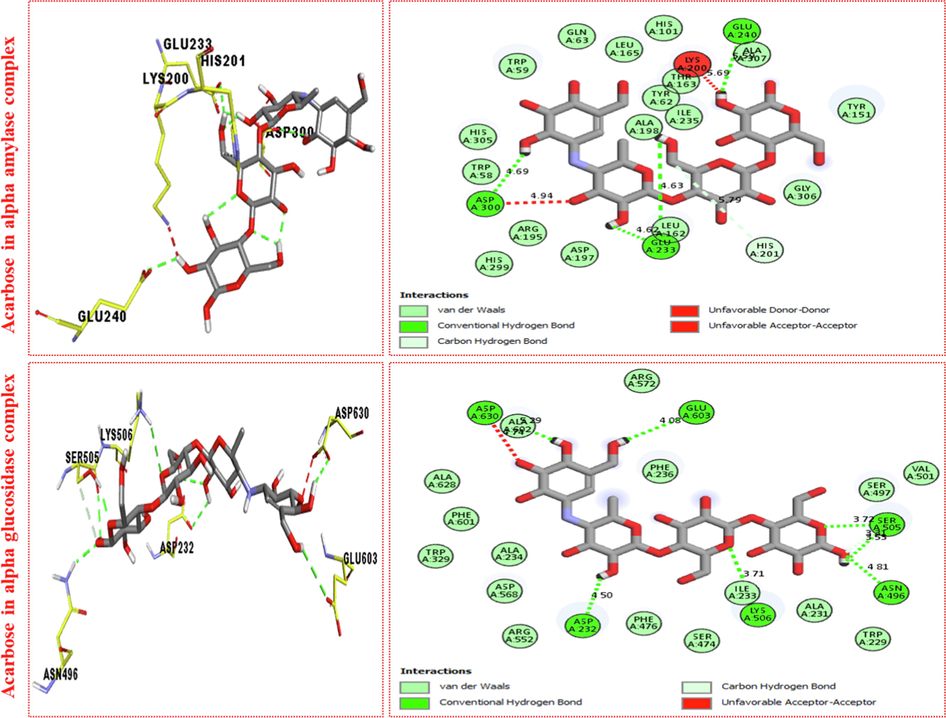
Represent 2D and 3D structure for acarbose in both α-amylase and α-glucosidase complex.
5 ADMET prediction
Through the use of the online tool SwissADME, several features of the compounds used in this research were identified to assure absorption, distribution, metabolism, excretion, and toxicity. This research observed the log Kp, Ghose, Veber, Lipinski, Muegge violations, gan, PAINS, Brenk alerts, Bioavailability score, as well as Leadlikeness violations. The findings for the tested candidates 1, 4, 9 and 14 were significant, as shown in Graph 1-4. The characteristics of the examined substances showed a definite difference.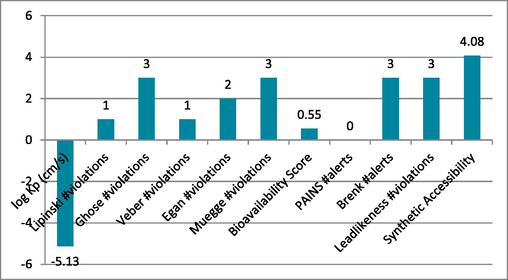
Show compound-1′s ADMET characteristics.
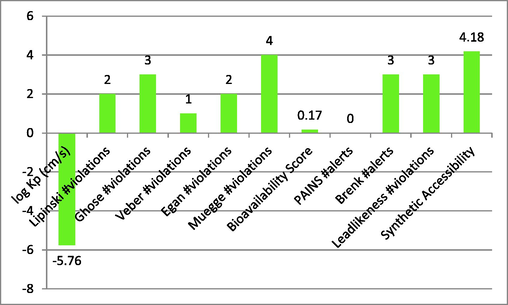
Describe compound-4′s ADMET characteristics.
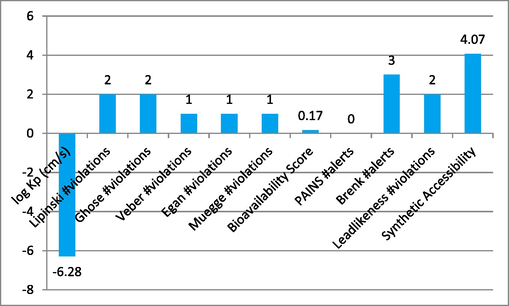
Represent the ADMET properties of compound-9.
Represent the ADMET properties of compound-14.
6 Conclusion
A series of processes were used to create the bis-benzothiazole-based thiadiazole analogs (1–18), and these reactions were verified by employing spectroscopic methods such as 1H NMR, 13C NMR, and HREI-MS. All of the produced analogs were also tested against the enzymes glucosidase and amylase. When compared to the standard medication acarbose, the derivatives 1, 4, 9, and 14 were found to have excellent activities against both -amylase and -glucosidase. The majority of them, which had various substituents at various positions of the aromatic ring, displayed good to moderate inhibitory activity. An analysis of the topic analogs' major binding modes using molecular docking technology was also found. As well as ADMET study also revealed the varied significant properties of the selected compounds. Among the evaluated (E)-N-(5-(1H-benzo[d]imidazol-4-yl)-1,3,4-thiadiazol-2-yl)-2-((6-nitro-1H-benzo[d]imidazol-2-yl)thio)-1-(3-nitro-5-(trifluoromethyl)phenyl)ethan-1-imine 9 (IC50 = 0.10 ± 0.050 and 0.20 ± 0.05 µM respectively) was considered as the most potent one. Inhibitors of -amylase and -glucosidase are found in a novel family of thiadiazole compounds based on bis-benzimidazoles in this investigation.
Author contributions
The manuscript was written with the contributions of all authors. All authors have approved the final version of the manuscript.
Acknowledgment
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University, Saudi Arabia for funding this work through Small Groups Project under Grant Number (RGP.1/248/44). This research was funded by Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2023R95), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Declaration of competing interest
The authors declare no conflict of interest.
References
- In-vivo analgesic and anti-inflammatory activities of newly synthesized benzimidazole derivatives. Eur. J. Med. Chem.. 2010;45(5):2048-2054.
- [Google Scholar]
- Raman, FT-IR, NMR spectroscopic data and antimicrobial activity of bis [μ2-(benzimidazol-2-yl)-2-ethanethiolato-N, S, S-chloro-palladium (II)] dimer,[(μ2-CH2CH2NHNCC6H4) PdCl] 2· C2H5OH complex. Eur. J. Med. Chem.. 2007;42:1069-1075.
- [Google Scholar]
- A parallel-group comparison of astemizole and loratadine for the treatment of perennial allergic rhinitis. J. Int. Med. Res.. 1997;25:175-181.
- [Google Scholar]
- A facile approach synthesis of benzoylaryl benzimidazole as potential α-amylase and α-glucosidase inhibitor with antioxidant activity. Bioorg. Chem.. 2021;114:105073
- [Google Scholar]
- Therapeutic Efficacy of Benzimidazole and Its Analogs: An Update. Poly. Arom. Comp. 2022:1-21.
- [Google Scholar]
- Carcanague, D., Shue, Y.-K., Wuonola, M. A., UriaNickelsen, M., Joubran, C., Abedi, J. K., Jones, J., Kuehler, and T. C. 2002. Novel structures derived from 2-[[(2-pyridyl) methyl] thio]-1 h-benzimidazole as anti-helicobacter p ylori agents, Part 2.J. Med. Chem. 45, 4300–4309.
- Clinical Review of Antidiabetic Drugs: Implications for Type 2 Diabetes Mellitus Management. Front. Endocrinol.. 2017;8
- [Google Scholar]
- Synthesis and analgesic activities of some 2-(benzazolylacetyl) amino-3-ethoxycarbonylthiophene derivatives. Phosphorous Sulfur Silicon Elements.. 2005;180:1841-2184.
- [Google Scholar]
- A comparative study on antihyperglycemic activity of fruits and barks of Ficus bengalensis (L.) Adv. Pharmacol. Toxicol.. 2006;7:69-71.
- [Google Scholar]
- Es-Safi, I., Mechchate, H., Amaghnouje, A., El Moussaoui, A., Cerruti, P., Avella, M., Grafov, A., and Bousta, D. 2020. Marketing and legal status of phytomedicines and food supplements in Morocco. J. Complement. Integr. Med. 2020.
- Benzimidazole: an emerging scaffold for analgesic and anti-inflammatory agents. Eur. J. Med. Chem.. 2014;76:494-505.
- [Google Scholar]
- Mahmood, N. Synthesis and HIV-1 inhibition of novel benzimidazole derivatives. Bioorg. Med. Chem. Lett.. 1995;7:1251-1254.
- [Google Scholar]
- Design, synthesis, in vitro and in vivo anti-respiratory syncytial virus (RSV) activity of novel oxizine fused benzimidazole derivatives. Eur. J. Med. Chem.. 2021;224:113684
- [Google Scholar]
- Benzimidazole-2-Phenyl-Carboxamides as Dual-Target Inhibitors of BVDV Entry and Replication. Viruses. 2022;14(6):1300.
- [Google Scholar]
- New Quinoline-based triazole hybrid analogues as effective inhibitors of α-amylase and αglucosidase: synthesis, in vitro evaluation and molecular docking along with in silico study. Front. Chem. 2022:1099.
- [Google Scholar]
- New biologically potent benzimidazole-based-triazole derivatives as acetylcholinesterase and butyrylcholinesterase inhibitors along with molecular docking study. J. Hetercyc. Chem. 2022
- [Google Scholar]
- Synthesis, in vitro α-amylase, α-glucosidase activities and molecular docking study of new benzimidazole bearing thiazolidinone derivatives. J. Mol. Struct. 2022:133812.
- [Google Scholar]
- Introduction to molecular docking software technique in medicinal chemistry. Int. J. Drug Res. Technol.. 2012;2:189-197.
- [Google Scholar]
- Complexation of several benzimidazole-type fungicides with α-and β-cyclodextrins. J. Agric. Food. Chem.. 2002;50:108-112.
- [Google Scholar]
- Adaptive molecular docking method based on information entropy genetic algorithm. App. Soft Comput.. 2015;26:299-302.
- [Google Scholar]
- Combination of Catechin, Epicatechin, and Rutin:optimization of a novel complete antidiabetic formulation using a mixture design approach. J. Nutr. Biochem.. 2020;108520
- [Google Scholar]
- Molecular docking and dynamic simulations of benzimidazoles with beta-tubulins. Bioinformation. 2021;17(3):404.
- [Google Scholar]
- Implications of the Revised Criteria for Diagnosis and Classification of Diabetes Mellitus. Clin. Chem.. 1997;43:2230-2232.
- [Google Scholar]
- Synthesis, anti-inflammatory, analgesic and kinase (CDK-1, CDK-5 and GSK-3) inhibition activity evaluation of benzimidazole/benzoxazole derivatives and some Schiff’s bases. Bioorg. Med. Chem.. 2006;14(11):3758-3765.
- [Google Scholar]
- Synthesis and fungicidal activity of novel 2-(2-alkylthio-6-phenylpyrimidin-4-yl)-1H-benzimidazoles. Bioorganic & Med. Chem. Lett.. 2021;47:128210
- [Google Scholar]
- Synthesis and fungicidal activity of novel benzimidazole derivatives bearing pyrimidine-thioether moiety against Botrytis cinerea. Pest Manag. Sci.. 2021;77(12):5529-5536.
- [Google Scholar]
- Synthesis and antiviral activities of N-substituted-2-substituted-benzimidazole derivatives. Indian J. Chem Sect.. 2006;45:489-493.
- [Google Scholar]
- Global Prevalence of Diabetes: Estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047-1053.
- [Google Scholar]
- Benzimidazole derivatives as new α-glucosidase inhibitors and in silico studies. Bioorg chem.. 2016;64:29-36.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.104847.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary Data 1
Supplementary Data 1




