

Translate this page into:
Synthesis of new seven membered heterocyclic rings: An easy access to indeno-benzo[1,4]diazepines
⁎Corresponding author. raza2005communications@gmail.com (Raza M. Ghalib)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
One-pot synthesis
Green synthesis
Crystal structures
Up to 80% yield
Abstract
An easy access has been demonstrated for seven membered diazepines and a six membered quinoxaline. The reaction proceeds rapidly with low-cost and readily available chemicals at mild temperature and provides straight forward access to diazepines and quinoxaline. All final products are new and confirmed by spectral analysis and by single crystal X-ray analysis. Two crystal structures of the diazepines have very similar compositions, exhibit similar conformations where four condensed and coplanar rings are placed in the same orientation to the rest of molecule.
Keywords
Synthesis
Crystal structures
Ninhydrin
Diazepine
Benzoimidazole

1 Introduction
Benzodiazepine derivatives have significant pharmacological and biological importance as potential tranquilizing, sedative agents, anti-inflammatory, Central Nervous System depressant, anticonvulsant, muscular relaxant, antispasmodic, antipsychotic drugs and hypnotics (Vida et al., 1981; Vogel, and Gunter, 1967; Petrascheck et al., 2007). Moreover, benzodiazepine drugs as chlorodiazepoxide and diazepam treat anxiety (Dulawa and Hen, 2005; Inada et al., 2003) and clozapines mainly used for schizophrenia (Nawrocka et al., 2001; Miyanaga et al., 2010). The biosynthesis of benzodiazepines is still unknown and assumed that they may be of plant origin since they have been found in plants in trace amounts (Klotz, 1991). Kavvadias et al., in 2000 reported, delorazepam and temazepam benzodiazepines in amounts of about 100–200 ng/g cell tissue of Artemisia dracunculus while temazepam and diazepam in amounts of 70–450 ng/g cell tissue of sterile potato herb (Kavvadias et al., 2000). Three new benzodiazepine alkaloids (Circumdatins D, E, and F) were isolated as minor constituents of culture extracts of a terrestrial strain of the fungus Aspergillus ochraceus (Rahbaek and Breinholt, 1999). Identification of benzodiazepines in Aspergillus ochraceus, Artemisia dracunculus and Solanum tuberosum rationalizing their endogenous formation in plant tissue.
The synthesis of all compounds proceeded regioselectively, and all the derivatives were obtained as crystals suitable for X-ray diffraction analysis, which, combined with spectral studies provided the full characterization of final products. Crystallographic data for the structural analysis have been deposited with the Cambridge Crystallographic Data Centre, CCDC Nos. 1994949, 1994950 and 1994951 for compounds 1, 2 and 3, respectively. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif.
2 Experimental section
2.1 Materials
All reagents and solvents commercially supplied were used as received. Thin layer chromatography was performed on silica gel 60 F254 on aluminium foil, Merck. NMR spectra were recorded on a Bruker DPX 300 spectrometer (300 MHz for 1H NMR and 75 MHz for 13C NMR) in DMSO‑d6 and chemical shifts are reported in ppm. The multiplicity of the signal is indicated as s - singlet, d - doublet, m - multiplet, etc. Coupling constants (J) are quoted in Hz. Infrared spectra were recorded on a Perkin-Elmer Model 1320 spectrometer (KBr disk, 400–4000 cm−1). The mass spectra were recorded on JEOL GCmate mass spectrometer in ionization mode (EI+). The melting points were taken on Stuart SMP30 digital melting point apparatus by open capillary method and is uncorrected.
2.2 General synthesis procedure
Ninhydrin (1.78 g; 1 mmol) and cyclo-1,3-diketone (1 mmol), were refluxed in 50 ml of acetic acid (AcOH) for 30 min. The conversion was monitored by thin layer chromatography. After maximum conversion, 1, 2-phenylenediamine (2.16 g, 2 mmol) was added to reaction mixture and further refluxed for 30 min. The formation of final product was monitored by thin layer chromatography at the interval of 10 min. The completed reaction mixture was left open in a beaker for slow evaporation, resulting crystalline solid (yields 80–65%). Crystals suitable for X-ray diffraction were obtained by recrystallization from acetone/hexane and acetone/chloroform solutions.
2.2.1 2,3,4,5,10-Pentahydrospiro[dibenzo[b,e][1,4]diazepine-11–11′-indeno[1,2-b]quinoxalin]-1(2H)-one (1)
Yield: 3.4 g (80%); white solid; mp 231 °C. IR (KBr): 3429, 3261, 3057, 2885, 2818, 1652, 1542, 1483, 1403, 1284, 1227, 1130, 1081, 999, 955, 872, 749 cm−1. 1H NMR (300 MHz, DMSO-d6): δ = 9.29 (s, 1H), 8.08 (d, J = 5.6 Hz, 1H), 8.01 (d, J = 4.5 Hz, 1H), 7.87 (d, J = 5.6 Hz, 1H), 7.72 (t, J = 4.1 Hz, 1H), 7.67 (t, J = 8.2 Hz, 1H), 7.35 (t, J = 4.5 Hz, 1H), 7.23–7.19 (m, 2H), 6.88 (t, J = 4.9 Hz, 1H), 6.71 (t, J = 4.6 Hz, 1H), 6.66 (d, J = 4.6 Hz, 1H), 6.46 (d, J = 4.7 Hz, 1H), 5.54 (s, 1H), 2.84–2.79 (m, 2H), 1.90–1.86 (m, 4H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 192.2, 165.1, 157.0, 154.7, 153.0, 141.3, 140.7, 137.8, 136.5, 133.2, 130.4, 128.8, 128.4, 128.1, 127.6, 123.4, 122.7, 122.4, 121.6, 121.4, 120.0, 109.6, 67.7, 36.4, 31.9, 20.4 ppm. MS (EI): m/z calcd for C27H20N4O: 416.47; found: 416.39 (M+) (34.9%).
2.2.2 3,3-Dimethyl-2,4,5,10-tetrahydrospiro[dibenzo[b,e][1,4]diazepine-11–11′-indeno[1,2-b]quinoxalin]-1(2H)-one (2)
Yield: 3.1 g (70%); white solid; mp 218 °C. IR (KBr): 3478, 3297, 3238, 3185, 3054, 2958, 1702, 1603, 1586, 1505, 1424, 1373, 1329, 1282, 1210, 1161, 1125, 1072, 1042, 1010, 945, 894, 759 cm−1. 1H NMR (300 MHz, DMSO-d6): δ = 9.22 (s, 1H), 8.08 (d, J = 4.9 Hz, 1H), 8.02 (d, J = 4.5 Hz, 1H), 7.85 (d, J = 4.9 Hz, 1H), 7.71 (t, J = 4.9 Hz, 1H), 7.67 (t, J = 4.9 Hz, 1H), 7.35 (t, J = 4.5 Hz, 1H), 7.21 (t, J = 5.7 Hz, 2H), 6.87 (t, J = 5.7 Hz, 1H), 6.72 (t, J = 4.7 Hz, 1H), 6.64 (d, J = 4.6 Hz, 1H), 6.47 (d, J = 4.7 Hz, 1H), 5.57 (s, 1H), 2.68 (q, J = 20.2, 8.6 Hz, 2H), 1.74 (q, J = 23.5, 9.6 Hz, 2H), 1.11 (s, 3H), 0.96 (s, 3H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 192.0, 165.1, 155.1, 154.7, 153.0, 141.3, 140.8, 137.7, 136.5, 133.3, 130.4, 128.8, 128.4, 128.1, 127.6, 123.5, 122.5, 122.4, 121.5, 120.0, 108.4, 79.1, 68.0, 49.9, 45.2, 31.3, 30.6, 27.5, 27.4 ppm. MS (EI): m/z calcd for C29H24N4O: 444.53; found: 444.31(M+) (39.9%).
2.2.3 3-[2-(1H-Benzoimidazol-2-yl)-phenyl]-1H-quinoxalin-2-one (3)
Yield: 2.2 g (65%); white solid; mp 187 °C. IR (KBr): 3438, 3286, 3151, 3061, 2978, 1706, 1601, 1596 cm−1. 1H NMR (300 MHz, DMSO-d6): δ = 12.82 (s, 1H), 12.07 (s, 1H), 7.94 (d, J = 4.5, 1H), 7.80 (d, J = 4.3 Hz, 1H), 7.71–7.64 (m, 3H), 7.54–7.48 (m, 2H), 7.34–7.27 (m, 3H), 7.15 (t, J = 4.9, 1H), 7.05 (t, J = 4.3, 1H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 161.1, 155.3, 152.6, 144.4, 137.1, 135.5, 133.3, 133.1, 131.4, 131.3, 130.5, 130.1, 129.4, 128.5, 123.7, 123.1, 122.0, 119.6, 115.9, 112.0 ppm. MS (EI): m/z calcd for C21H14N4O: 338.36; found: 338.29 (M+) (54.2%).
2.3 Single crystal X-ray diffraction
The single-crystal X-ray diffraction data for all three compounds were collected at 100(1) K on a Bruker AXS diffractometer with APEX2 CCD area-detector using Mo-Kα radiation. The Lorentz-polarization correction and the integration of the diffracted intensities were performed with the SAINT software package (SAINT, 2006). The absorption effects were corrected on a semiempirical basis using multiple scans (SADABS) (Sheldrick, 1996; Krause et al., 2015). The crystallographic data, the data collection parameters, and the refinement parameters for compounds 1, 2 and 3 are summarized in Table 1S. The structures were solved by direct methods and refined by full-matrix least-squares calculations against F2 (Sheldrick, 2008). All non-hydrogen atoms were refined with anisotropic displacement parameters. The H atoms attached to O and N atoms were refined isotropically, whereas other H atoms were placed in calculated positions and refined as riding atoms. Figures were produced using ORTEP-3 (Farrugia, 1997, 2012) and MERCURY, Version 2.4 (Macrae et al., 2006). The software used for the preparation of the materials for publication: WinGX (Farrugia, 1999, 2012), PLATON (Spek, 2003), PARST (Nardelli, 1995).
3 Results and discussion
3.1 Chemistry
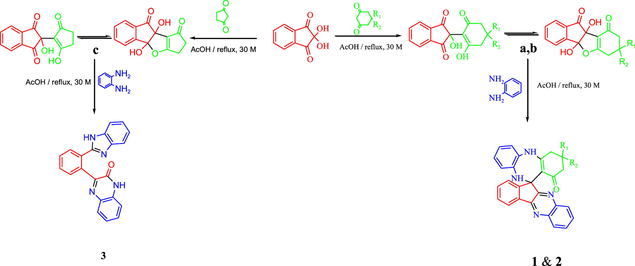
In order to optimize the reaction conditions, the reaction was repeated in THF, dioxane, CH2Cl2, CHCl3, ethanol, methanol, ethylene glycol, acetic acid and Isopropanol. The best results were obtained in acetic acid on refluxing (Ghalib et al., 2017). At the onset of our work ninhydrin (1 mmol) and cyclo 1,3-diketones (1 mmol), were employed as model substrates in acetic acid (AcOH) with heating under reflux. In Cyclohexane-1,3-dione, Dimedone and Cyclopentane-1,3-dione, H atoms are adjacent to 2 carbonyl groups and easily form enolate anions which attacks on electrophilic C-2 position of ninhydrin (Ghalib et al., 2017, 2011; Fun et al., 2009) to form condensation products a, b, and c in 100% yields by subsequent elimination of a water molecule (Scheme 1) (Kaupp et al 2002). In further step of reaction, a, b and c (1 mmol) react with 1, 2-phenylenediamine (2 mmol) with same reaction conditions. a and b reacts with same reaction mechanism to give seven membered diazepine derivatives. While, c reacts differently to an unexpected product as quinoxaline derivative. First step products a, b and c formed during the reaction processes have been successfully crystallized and unambiguously confirmed by spectral data and X-ray diffraction analysis of single crystal of c (Mehdi et al., 2011). The mechanisms of formation of 1, 2 and 3 can be proposed as shown in Schemes 2 and 3. The Scheme 2 involves the formation of a and b; and diazepine ring formation steps, that consists of intramolecular shifting of cyclohexyl-1,3-dione moiety from C-2 to C-1 position in ninhydrin moiety with the condensations of two 1, 2-phenylenediamine molecules, catalyzed by acetic acid (De et al., 2018; Cheng et al., 2011). Scheme 3 depicts the nucleophilic attack of one 1, 2-phenylenediamine molecule on c and ring fission in ninhydrin moiety between C-2 and C-3. The second 1, 2-phenylenediamine molecule attacks on C-3 position with the elimination of water molecule and formation C⚌N, and C—N functions in pentacycle (Ziarani et al., 2015). Nucleophilic attack of nitrogen on C-2 caused the release of cyclopentyl moiety and formation of hexacycle with C⚌O, C⚌N, and C-N functions. Compound 1, 2, and 3 were identified by FT-IR, NMR spectroscopy (Figs. S5–S10), mass spectroscopy and finally by single crystal X-ray analysis. Full assignment of the spectral data for all final products can be found in supplementary materials.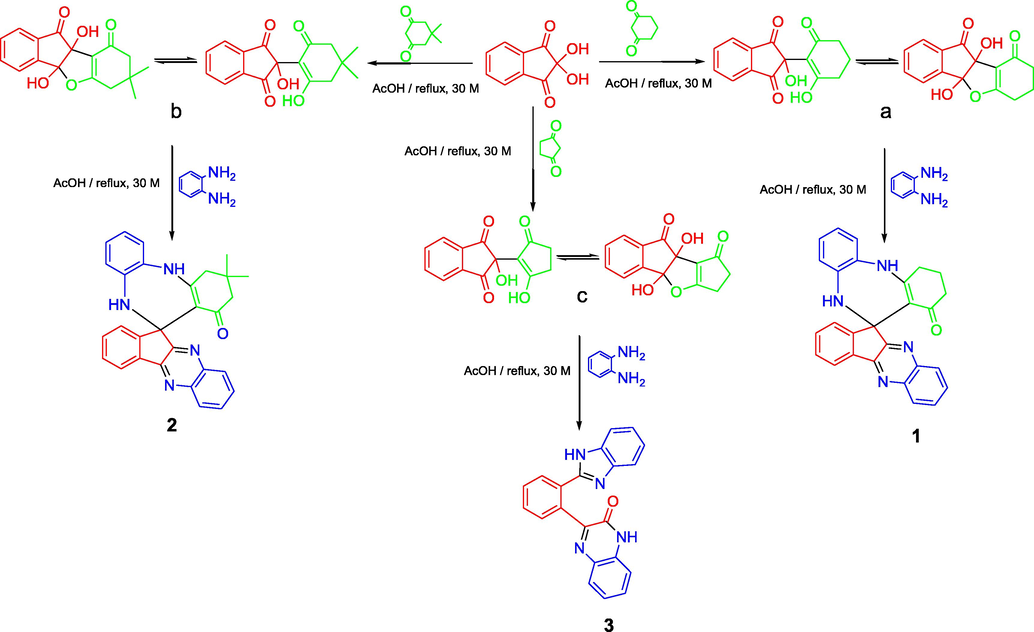
Route for the synthesis of 1, 2, 3.
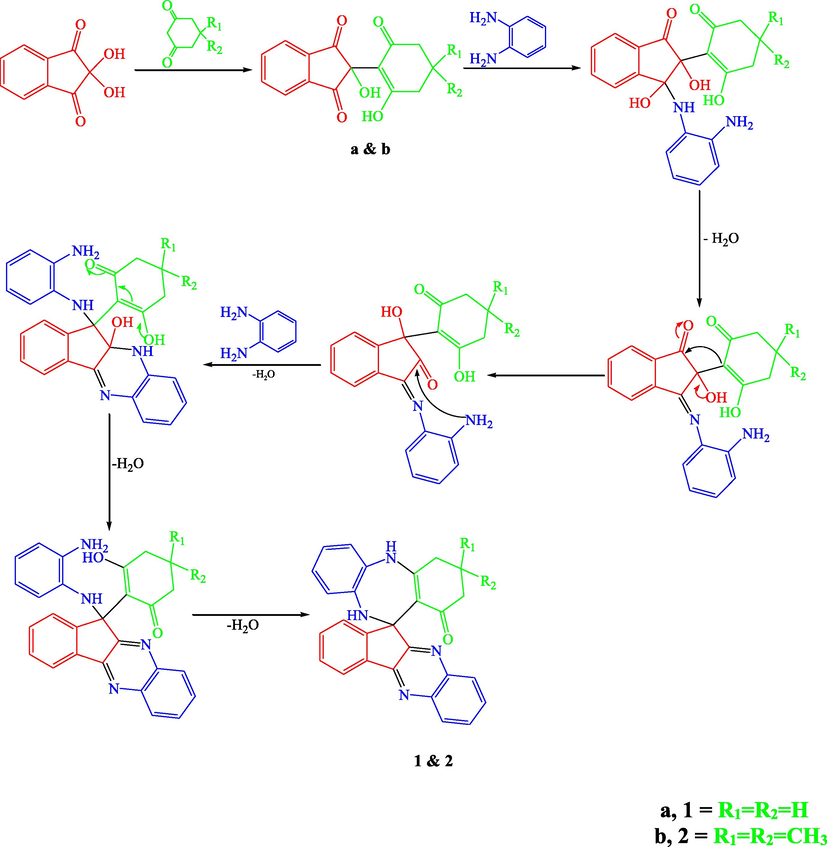
A reasonable reaction mechanism for the synthesis of 1 and 2.
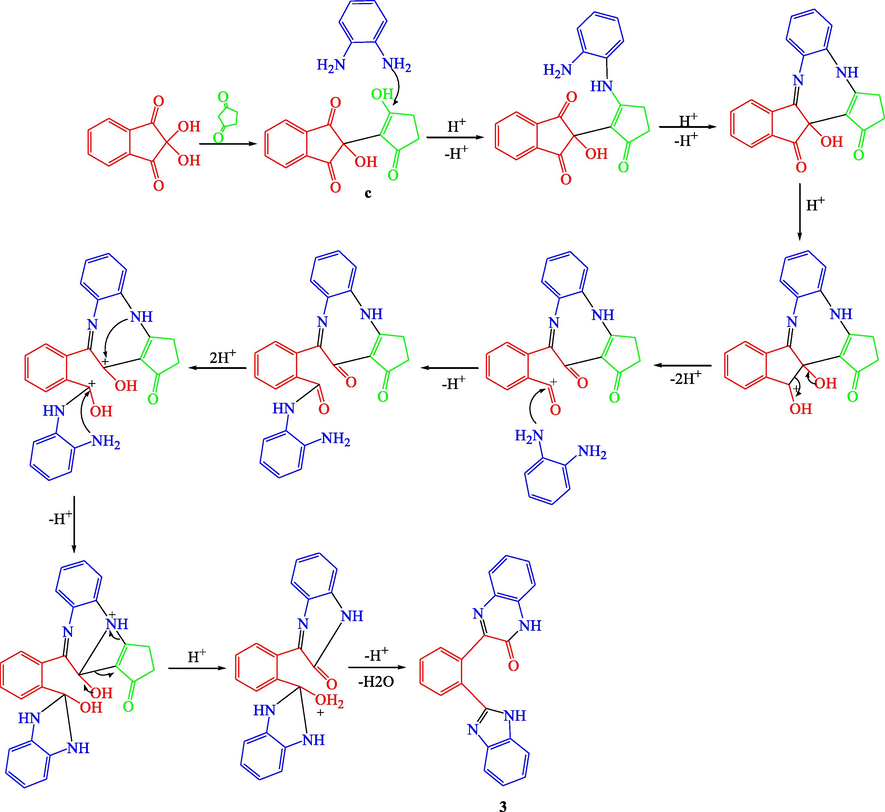
A reasonable reaction mechanism for the synthesis of 3.
3.2 Single crystal X-ray study
The single-crystal X-ray analysis shows that both compounds 1 and 2 contain seven rings labelled in Fig. 1 from Rg1 to Rg7. These rings are equivalent in composition and manner of mutual connection. Only structural difference between two molecules is in the C19 atom which is in the case of compound 2 bonded to two methyl groups (C28 and C29) instead of two hydrogen atoms in 1. Both compounds crystallize with one molecule of acetone but the crystal lattice of 1 in addition includes a water molecule of crystallization instead of CHCl3 in the case of 2 (but formulas are 1·O⚌C(CH3)2·H2O and 2·O⚌C(CH3)2·CHCl3).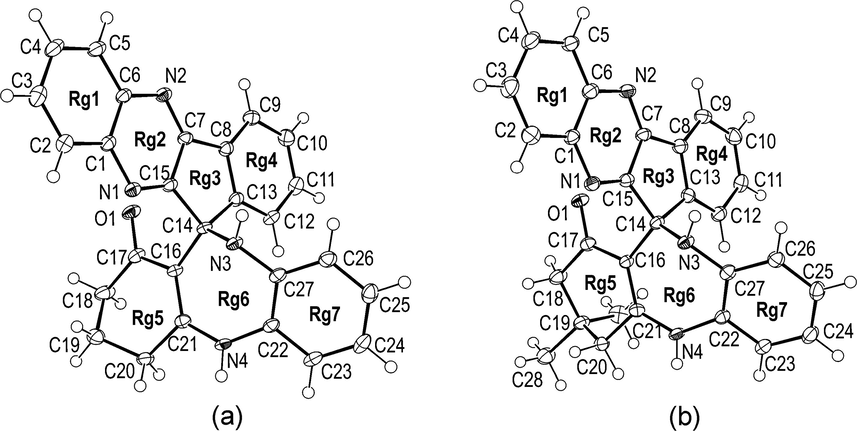
The molecular structure of 1 (a) and 2 (b) with atom numbering scheme. Displacement ellipsoids are drawn at the 50% probability level and solvents molecules have been omitted for clarity. A complete contents of the asymmetric unit for both crystal structures is shown in the Supplementary Material (Fig. S1).
Having in mind the above similarities and differences between crystal structures of 1 and 2, it is very interesting to compare their molecular structures and geometrical parameters. In both compounds, seven rings are mutually fused in two groups. First four rings (Rg1-4) are condensed in one group while another three rings (Rg5-7) constitute second group. These two ring systems are connected in C14 atom (Fig. 1). Although the C14 atom is not only one sp3 carbon atom in 1 and 2 it forms two the longest bonds in both crystal structures, the C13-C14 and C14-C15 (Table 1). It is worth to mention that C13 and C15 are sp2. All other corresponding bond lengths for two compounds are also very similar (Table 1). There are four nitrogen atoms and eight N-C bonds in both molecules but only the N3 is sp3 and has a tetrahedral arrangement of bonds. As expected this nitrogen has the longest N-C bond (N3-C14) while the shortest two N-C bonds are formed by N1 and N2 in both compounds (N1-C15 and N2-C7, Table 1).
1
2
3
O1-C17
N1-C15
N1-C1
N2-C7
N2-C6
N3-C27
N3-C14
N4-C21
N4-C22
C1-C6
C7-C15
C7-C8
C8-C13
C13-C14
C14-C16
C14-C15
C16-C21
C16-C17
C17-C18
C18-C19
C19-C20
C20-C21
C22-C271.2451(18)
1.2991(19)
1.3846(19)
1.3109(19)
1.3809(19)
1.4049(19)
1.4785(19)
1.3481(19)
1.4080(19)
1.421(2)
1.432(2)
1.463(2)
1.409(2)
1.536(2)
1.515(2)
1.532(2)
1.3880(19)
1.447(2)
1.509(2)
1.521(2)
1.512(2)
1.516(2)
1.400(2)1.250(2)
1.301(3)
1.391(2)
1.305(2)
1.382(3)
1.408(3)
1.487(2)
1.344(3)
1.412(2)
1.412(3)
1.434(3)
1.462(3)
1.402(3)
1.538(3)
1.520(3)
1.529(3)
1.381(3)
1.428(3)
1.515(3)
1.526(3)
1.520(3)
1.517(3)
1.394(3)O1-C8
N1-C8
N1-C1
N2-C7
N2-C6
N3-C15
N3-C16
N4-C15
N4-C21
C1-C6
C7-C8
C7-C9
C14-C15
C16-C211.237(2)
1.365(3)
1.388(3)
1.297(3)
1.394(3)
1.367(3)
1.377(3)
1.328(3)
1.398(3)
1.395(3)
1.485(3)
1.492(3)
1.468(3)
1.399(3)
All four rings which constitute the ring system Rg1-4 are planar and mutually coplanar. Nevertheless the Rg1-4 system is somewhat less planar in 2 than in 1. Thus root-mean-square deviation of 17 constituent atoms from corresponding mean plane of the Rg1-4 system is 0.024 and 0.048 Å for 1 and 2 respectively. Dihedral angle between terminal Rg1 and Rg2 phenyl rings is 1.42(10) and 5.25(12)° in 1 and 2 respectively. The largest difference in values for corresponding torsion angles is for the N2-C7-C8-C9 [−0.6(3) and −5.5(4)° for 1 and 2 respectively].
The conformation of corresponding rings in Rg5-7 system is also very similar for both compounds. The Rg6 is only one seven-membered ring in the molecules and adopts an non-planar form. The N3, N4, C21, C22 and C27 atoms are approximately coplanar while the C14 and C16 significantly deviate from the mean plane of N3-N4-C21-C22-C27 [displacement of C14/C16 is 0.823(2)/0.723(2) and 0.861(3)/0.641(3) Å for 1 and 2 respectively]. The largest torsion angle within the Rg6 ring is the C27-N3-C14-C16 [71.4(2) and 75.6(2)° for 1 and 2 respectively]. The Rg5 ring in both molecules clearly adopts an envelope conformation with C19 significantly displaced from the C16-C17-C18-C20-C21 mean plane [displacement of C19 is 0.678(2) and 0.643(3)Å for 1 and 2 respectively]. The orientation of the Rg5-7 system regarding to the Rg1-4 system is similar for both molecules. Thus a dihedral angle between Rg7 phenyl ring and mean plane of Rg1-4 is 60.96(3) and 66.33(5)° for 1 and 2 respectively.
It is interesting to note that both molecules form 1D-chains using the same N4-H…O1 hydrogen bond (Fig. S2, see Supplementary Material file). This bond is the strongest hydrogen bond in both crystal structures (Table 2). In the case of 1 the neighbouring molecules within the chain are additionally connected via water molecules (O1w—H2w…N1 and O1w—H1w…N2 hydrogen bonds, Table 2). The N3-H donor group in both crystal structures forms hydrogen bonds to the O2 oxygen atom from acetone (Table 2). There is no other important hydrogen bonds in 1 and 2.
D—H…A
D—H
D…A
H…A
D—H…A
1
O1w—H2w…N1i
0.95(3)
2.979(2)
2.03(3)
175(2)
O1w—H1w…N2ii
0.83(3)
2.951(2)
2.12(3)
175(2)
N3—H1n3…O2i
0.86(2)
3.063(2)
2.21(2)
172(2)
N4—H1n4…O1ii
0.83(2)
2.857(2)
2.03(2)
176(2)
Symmetry codes: (i) x, y, z; (ii) x + 1/2, −y + 1/2, z
2
N3—H1n3…O2i
0.88(2)
3.094(2)
2.23(2)
167(2)
N4—H1n4…O1ii
0.84(2)
2.807(2)
1.98(2)
173(2)
Symmetry codes: (i) x, y, z; (ii) x − 1/2, y, −z + 1/2
3
N1—H1n1…N4i
0.93(3)
2.922(2)
2.02(3)
165(2)
N3—H1n3…O1ii
0.90(3)
2.927(2)
2.08(3)
156(2)
Symmetry codes: (i) x, −y + 1/2, z + 1/2; (ii) − x, −y, −z + 1/2
Compound 3 contains five rings labelled in Fig. 2 from Rg1 to Rg5. Six-membered rings Rg1 and Rg2 are almost ideally coplanar [Dihedral angle between Rg1 and Rg2 is 1.29(10)°]. Root-mean-square deviation of 10 constituent atoms from mean plane of the Rg1-2 system is 0.016 Å. Rings Rg4 and Rg5 are also fused and almost coplanar [Rg4/Rg5 dihedral angle is 2.09(14)°]. Interplanar angle between Rg3 phenyl ring and the mean plane of two ring systems Rg1-2 and Rg4-5 is 55.03(4) and 25.86(7)° respectively. Dihedral angle between Rg1-2 and Rg4-5 is 62.09(3)°.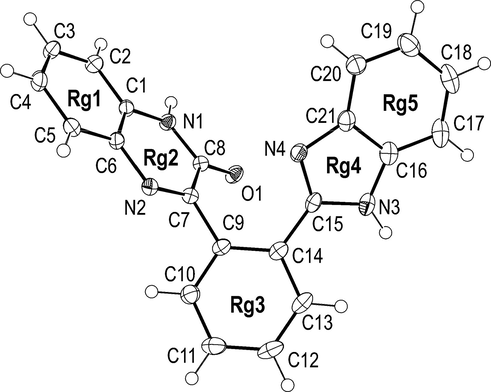
The molecular structure of 3 with atom numbering scheme. Displacement ellipsoids are drawn at the 50% probability level.
Compound 3 contains four nitrogen atoms but, unlike to 1 and 2, all nitrogens are sp2. The N2 atom forms the shortest N—C bond in the molecule (Table 1). This bond, N2-C7, with length of 1.297(3) Å could be accepted as localised N—H double bond. The Rg3 phenyl ring is connected to the Rg2 ring through the longest bond in the molecule, C7-C9 (Table 1). Molecules of 3 crystallize without solvents. Using N3-H…O1 hydrogen bonds (Table 2) they form centrosymmetric dimers (See Fig. S3 in Supplementary Material file). These dimers are interconnected via the N1-H…N4 hydrogen bonds into bulky 2D (two-dimensional) network which extends in the (1 0 0) crystallographic plane (Fig. S4).
4 Conclusion
In summary, we have developed an experimentally and operationally simple procedure. The multicomponent reaction offers high yield, short reaction time. Single-crystal X-ray analysis showed that two molecules (1 and 2) contain seven rings. Four rings are mutually fused and form a large nearly planar system with only one sp3 hybridized atom. In both compounds mean deviation of 17 constituent atoms does not exceed 0.05 Å. Two of the remaining three rings adopt non-planar conformation where some of torsion angles are larger than 70°.
Third compound (3) characterized by X-ray crystallography contains five rings. Molecules of this compound form in solid state H-bonded (N—H…O) centrosymmetric dimers which are interconnected (via N—H…N interactions) into bulky layers.
Acknowledgements
This work was funded by the Deanship of Scientific Research (DSR), University of Jeddah, Jeddah, under grant No. (UJ-02-050-DR). The authors, therefore, acknowledge with thanks DSR technical and financial support. G.A.B thanks to the Ministry of Education, Science and Technological Development of the Republic of Serbia for financial support.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [4+2+1] Domino cyclization in water for chemo- and regioselective synthesis of spiro-substituted benzo[b]furo[3,4-e][1,4]diazepine derivatives. Green Chem.. 2011;13:2107-2115.
- [Google Scholar]
- Zeolite-Y-mediated multicomponent reaction of isatins, cyclic 1,3-diketones, and 1,2-phenylenediamine: easy access to spirodibenzo[1,4]diazepines. ChemCatChem. 2018;10:590-600.
- [Google Scholar]
- Recent advances in animal models of chronic antidepressant effects: the novelty-induced hypophagia test. Neurosci. Biobehav. Rev.. 2005;29:771-783.
- [Google Scholar]
- ORTEP-3 for Windows - a version of ORTEP-III with a Graphical User Interface (GUI) J. Appl. Crystallogr.. 1997;30:565.
- [Google Scholar]
- WinGX suite for small-molecule single-crystal crystallography. J. Appl. Cryst.. 1999;32:837-838.
- [Google Scholar]
- Green synthesis of novel class of benzazocine derivatives, their crystal structures and anticancer activity. Curr. Org. Synth.. 2017;14:127-136.
- [Google Scholar]
- Efficacy of diazepam as an anti-anxiety agent: meta-analysis of double-blind, randomized controlled trials carried out in Japan. Hum. Psychopharmacol. Clin. Exp.. 2003;18:483-487.
- [Google Scholar]
- Quantitative reaction cascades of ninhydrin in the solid state. Chem. Eur. J.. 2002;8:594-600.
- [Google Scholar]
- Identification of benzodiazepines in artemisia dracunculus and solanum tuberosum rationalizing their endogenous formation in plant tissue. Biochem. Biophys. Res. Commun.. 2000;269:290-295.
- [Google Scholar]
- Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr.. 2015;48:3-10.
- [Google Scholar]
- Mercury: visualization and analysis of crystal structures. J. Appl. Crystallogr.. 2006;39:453-457.
- [Google Scholar]
- Synthesis, characterization, antimicrobial and enzymatic activity of 4b,9b-dihydroxy-7,8-dihydro-4bH-indeno[1,2-b]benzofuran-9,10(6H,9bH)-dione. J. Mol. Struct.. 2011;1006:318-323.
- [Google Scholar]
- Anti-invasive and anti-angiogenic activities of naturally occurring dibenzodiazepine BU-4664L and its derivatives. Bioorg. Med. Chem. Lett.. 2010;20:963-965.
- [Google Scholar]
- PARST95 – an update to PARST: a system of Fortran routines for calculating molecular structure parameters from the results of crystal structure analyses. J. Appl. Crystallogr.. 1995;28:659.
- [Google Scholar]
- Synthesis and antiproliferative activity in vitro of novel 1,5-benzodiazepines. Part II Arch. Pharm. Pharm. Med. Chem.. 2001;334:3-10.
- [Google Scholar]
- An antidepressant that extends lifespan in adult Caenorhabditis elegans. Nature. 2007;450:553-556.
- [Google Scholar]
- Circumdatins D, E, and F: further fungal benzodiazepine analogues from aspergillus ochraceus. J. Nat. Prod.. 1999;62(6):904-905.
- [Google Scholar]
- Data Reduction and Frame Integration Program for the CCD Area-Detector System. Madison, Wisconsin, USA: Bruker Analytical X-ray Systems; 2006.
- Program SADABS: Area-detector absorption correction. Germany: University of Göttingen; 1996.
- Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr.. 2003;36:7-13.
- [Google Scholar]
- Vida, J.A., 1981. Medicinal Chemistry, Part-Ill. In: Wolf, M.W., Burger, A. (Eds.), John Wiley & Sons, New York, 1981, 787 and references cited therein.
- Benzene oxide-oxepin valence tautomerism. Angew. Chem. Int. Ed. Engl.. 1967;6:385-476.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2020.08.011.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




