

Translate this page into:
Synthesis of novel 5-HT1A arylpiperazine ligands: Binding data and computer-aided analysis of pharmacological potency
⁎Corresponding authors. deanad@chem.bg.ac.rs (Deana B. Andrić),
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Serotonin receptors modulate numerous behavioral and neuropsychological processes. Therefore, they are the target for the action of many drugs, such as antipsychotics, antidepressants, antiemetics, migraine remedies, and many others. The 5-HT1A receptors have been involved in the pathogenesis and treatment of anxiety and depression and represent a promising target for new drugs with reduced extrapyramidal side effects. In most antidepressants, a piperazine-based structural motif can be identified as a common moiety. Here we describe the synthesis, pharmacological, and in silico characterization of a novel arylpiperazines series with excellent 5-HT1A affinity. The final compounds, 4a, 8a, and 8b, were selected according to predictions of in silico pharmacokinetics, docking analysis, and molecular dynamics in conjunction with physical properties, and metabolic stability. The accentuated molecules could serve as a lead compound for developing 5-HT1A drug-like molecules for depression treatment.
Keywords
Synthesis
Heterocycles
Serotonin-1A receptor
Binding assay
Computer-aided drug analysis
Pharmacokinetics

1 Introduction
Depressive disorder is one of the crucial illnesses characterized by pathological depressed mood, loss of interest and satisfaction, decreased energy, guilt, helplessness, low self-esteem, sleep and appetite disorders, poor concentration, and dark thoughts. The causes of depression are very complex since biological and psychosocial factors are intertwined and are the roots of depressive disorder. Until 2021, more than 300 million people worldwide were affected, and it is alarming that it is becoming more prevalent among adolescents (Faquih et al., 2019; Mojtabai et al., 2016; Mojtabai and Olfson, 2020). The COVID-19 global pandemic has negatively affected the mental health of many individuals, causing depressive levels to rise to devastating heights (CMDC, 2021; Mazza et al., 2020). Fear of disease and survival, social restrictions, lockdowns, school and business closures, loss of livelihood, decreases in economic activity and shifting priorities of governments in their attempt to control COVID-19 outbreaks all have the potential to substantially affect the mental health of the population. The need for up-to-date investigations on new antidepressants has never been more urgent.
Serotonin (5-HT) is a monoamine neurotransmitter involved in the regulation of many physiological functions of the body, such as blood circulation, heart functions, sexual behavior, sleep, memory, pain, emotional states, and food intake (Jacobs and Azmitia, 1992). The 5-HT1A receptor is the most investigated and best-characterized subtype among all the serotonin receptor family members. It is an attractive target for pharmacotherapy based on the belief that one of the primary causes of depression is a disturbance in the functioning of the serotonin system (Babb et al., 2018; Chilmonczyk et al., 2015). Suppression of 5-HT1A receptors increases the neuronal activity of serotonin and enhances stress resistance and antidepressant response. 5-HT1A overexpression has been implicated in the reduction of serotonergic neurotransmission and consequently associated with major depression and suicidal tendencies (Zaręba et al., 2019). Until today, numerous high-affinity 5-HT1A receptor ligands were developed, classified as full to partial agonists, antagonists, and inverse agonists.
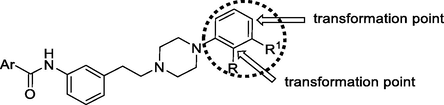
Various antipsychotics are arylpiperazine derivatives showing efficiency as antidepressants and are increasingly popular as supportive medicines in the clinical treatment of depression (Staroń et al., 2018). N1-substituted N4-arylpiperazines, known as long-chain arylpiperazines, have been extensively studied as 5-HT1A receptor ligands. The general structure of these compounds presents a 1-arylpiperazines linked through the alkyl chain of variable length to a terminal fragment (imides, amides, alkyl, arylalkyl, or heteroarylalkyl derivatives) (Lacivita et al., 2012; Kumar et al., 2021). The main goal of the presented investigation was to derive new 5-HT1A ligands with an enhanced affinity and pharmacokinetic properties compared with hitherto investigated compounds. Known piperazine-based therapeutics aripiprazole, cariprazine, and brilaroxazine were referent molecules in the study (Fig. 1) (Pignon et al., 2017).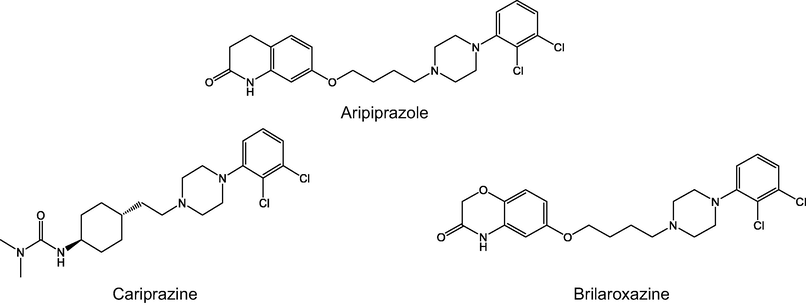
Chemical structures of piperazine-based therapeutics.
N-phenylpiperazine derivatives are known to possess significant biological activity (Chen et al., 2013). Based on our previous findings, the meta-substituted phenyl part of the linker gave the best candidates for further improvement of 5-HT1A affinity (Fig. 2) (Sukalovic et al., 2012, 2013).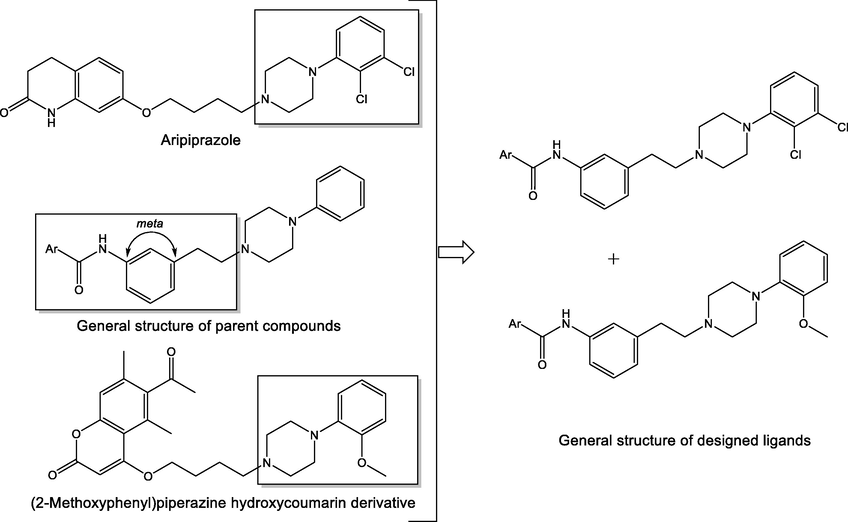
Design and general structures of new arylpiperazine ligands.
The base of the synthetic strategy relied on the fact that a substituted two-carbon linker is optimal for high activity towards 5-HT1A receptors. Substitution on the N-phenylpiperazine part of the molecule affects the increase of affinity, although the chemical nature of the substituent itself is less pertinent because electron-withdrawing and electron-donating groups have an influence. The introduction of electron-withdrawing substituents into the arylpiperazine part of the molecule is favorable for antidepressant activity (Kumar et al., 2021). However, the characteristic of ligands with an introduced methoxy group in the ortho-position (relative to meta and para) is a marked increase in affinity, which is also the case with 2,3-dichloro derivatives. Substitution in the aryl amide (“head part” of the ligand) or coumarin part of the ligand favourably affects the affinity of the ligand if additional interactions with amino acid residues in the receptor's active site are realized. Sometimes, due to the length of the molecule, the N4 piperazine atom is not in a favourable position for the realization of the crucial salt bridge. In that case, the other basic nitrogen atom from the molecule can take its role. It will not establish a salt bridge but stabilize the ligand-receptor complex by realizing additional interactions (Sukalovic et al., 2012, 2013; Chen et al., 2013; Ostrowska et al., 2017, 2018, 2020; Zaręba et al., 2019). In the design of new compounds, the focus was on o-OCH3 and 2,3-dichloro groups incorporation into the skeleton of the parent compounds (Fig. 2).
Designed molecules should satisfy certain physical and chemical properties in the early stages of drug design and development. Rejection of potential drugs at later stages of drug development may cause substantial financial loss for pharmaceutical companies. Therefore, it is reasonable that ADMET (absorption, distribution, metabolism, excretion, and toxicity) pharmacokinetic studies have been highlighted as early predictors of important potential drug parameters. Solubility is the principal physicochemical parameter for a drug administrated orally. As a result, dissolved drugs pass from the digestive tract through biological membranes, reach the bloodstream, and are ultimately transported to the site of action, causing a pharmacological response. If solubility is not satisfactory, there is no absorption and consequently no therapeutic effect of the drug. Also, insufficient solubility entails developmental problems with drug formulation and determination of application doses. Another important parameter is the toxicity of the drug. It is related to the toxicity of active pharmaceutical ingredients (API) and drug impurities (Krüger et al., 2020; Guan et al., 2019).
Although bioinformatics techniques have replaced many in vitro tests, in vitro and in vivo validation and standardization are necessary to guarantee trustable predictions and make a go-no-go decision regarding a drug selection as a potential drug candidate (Li, 2001). Therefore, the affinity of the synthesized ligands towards 5-HT1A was determined in radioligand competition binding assays.
Newly synthesized substances should have structure and activities comparable to adequate commercial substances while maintaining a favourable ADMET profile. Consequently, new substances should present suitable candidates for further evaluation and modification in the rational drug design process.
2 Material and methods
2.1 Experimental
The reagents and solvents used in this work were obtained from Alfa Aesar or Sigma-Aldrich and used without further purification. Solvents were routinely dried over anhydrous Na2SO4 before evaporation. 1H- and 13C NMR spectra were recorded on: 1H NMR (200 MHz) and 13C NMR (50 MHz), Gemini 2000 spectrometer; 1H NMR (400 MHz) and 13C NMR (101 MHz), Varian/Agilent spectrometer; and 1H NMR (500 MHz) and 13C NMR (126 MHz), Bruker Avance III spectrometer. Chemical shifts (δ) are reported in parts per million (ppm) from tetramethylsilane (TMS) as an internal standard in deuterated chloroform (CDCl3); all coupling constants (J values) are reported in Hz; high-resolution mass spectra (HRMS) were obtained with a heated ESI (HESI)-LTQ Orbitrap XL spectrometer. Melting points were obtained by a Boetius PHMK apparatus (VEB Analytic, Dresden, Germany) at a heating rate of 4 °C/min and are uncorrected. IR spectra were recorded using a Thermo Scientific Nicolet 6700 Fourier-transform spectrometer operated in the ATR mode. For analytical thin-layer chromatography (TLC), Polygram SIL G/UV254 plastic-backed thin-layer silica gel plates were used (Macherey-Nagel, Germany).
2.1.1 General procedure for the synthesis of 2-(3-nitrophenyl)-1-(4-arylpiperazin-1-yl)ethan-1-ones (1a,b)
Arylpiperazine (52 mmol), 3-nitrophenylacetic acid, (52 mmol) and triethylamine (57.2 mmol) were dissolved in 170 mL DMF dry. The obtained solution was cooled to 10 °C, and PPAA solution (72.7 mmol, 50 % solution in DMF) was added dropwise, and the reaction mixture was stirred for 24 h at room temperature. The reaction was monitored by TLC. The reaction mixture was poured onto ice/water. The pH of the suspension was adjusted to 8 with a 10 % Na2CO3 solution (from the acidic range). The precipitate was filtrated, washed with water, and crystallized from acetone to give the expected product.
2.1.1.1 1-[4-(2-methoxyphenyl)piperazin-1-yl]-2-(3-nitrophenyl)ethan-1-one (1a)
Yield: 78 %, oil. 1H NMR (200 MHz, Chloroform-d): δ 8.34–8.26 (m, 2H, ArH), 8.08–8.06 (m, 1H, ArH), 7.88–7.82 (m, 1H, ArH), 7.74–7.52 (m, 2H, ArH), 7.28–7.24 (m, 1H, ArH), 7.01–6.93(m, 1H, ArH), 3.69–3.55 (m, 4H, piperazine), 3.39 (s, 3H, OCH3), 3.00–2.92 (m, 2H, CH2), 2.50–1.89 (m, 4H, piperazine); 13C NMR (50 MHz, Chloroform-d): δ 168.09, 151.02, 147.75, 141.32, 136.11, 132.75, 129.74, 129.40, 126.58, 125.67, 122.87, 122.31, 121.81, 56.98, 54.10 (2C), 51.21 (2C), 48.46.
2.1.1.2 1-[4-(2,3-dichlorophenyl)piperazin-1-yl]-2-(3-nitrophenyl)ethan-1-one (1b)
Yield: 74 %, oil. 1H NMR (200 MHz, Chloroform-d): δ 8.26–8.12 (m, 2H, Ar), 7.73–7.48 (m, 1H, Ar), 7.46 (m, 2H, Ar), 7.21–7.13 (m, 1H, Ar), 6.99–6.82 (m, 1H, Ar), 3.93–3.77 (m, 4H, piperazine), 3.81–3.56 (m, 2H, CH2), 3.32–2.88 (m, 4H, piperazine); 13C NMR (50 MHz, Chloroform-d): δ 168.07, 150.25, 142.50, 139.76, 134.15, 130.26, 129.86, 127.51, 125.33, 123.79, 123.56, 123.42, 118.68, 42.06 (2C), 40.19 (2C), 34.25.
2.1.2 General procedure for synthesis 1-(3-nitrophenethyl)-4-arylpiperazines (2a,b)
To a suspension of 1a or 1b (46.1 mmol) in 300 mL dry THF, a diborane solution (1 M in THF, 118 mL, 118 mmol) was added dropwise at 0 °C. When the addition of diborane was completed reaction mixture was progressively heated. After the spontaneous boiling stop, the reaction mixture was refluxed for an additional 2 h. After cooling to room temperature, water (35 mL) was slowly added, followed by 5.5 M HCl (70 mL). The reaction mixture was refluxed for another 60 min and was left to cool down to 25 °C. The reaction mixture was evaporated, and the resulting fluid was treated with 10 % NaHCO3 solution to pH 8. The product was extracted with ethyl acetate, washed with water and brine, and evaporated. The resulting 3-nitrophenethyl-piperazines were purified by silica gel column chromatography using a gradient of methanol (0–5 %) in dichloromethane.
2.1.2.1 1-(2-methoxyphenyl)-4-(3-nitrophenethyl)piperazine (2a)
Yield: 69 %, oil. 1H NMR (500 MHz, Chloroform-d): δ 7.08 (t, J = 7.7 Hz, 1H, ArH), 7.00 (td, J1 = 7.6, J2 = 2.0 Hz, 1H, ArH), 6.98–6.90 (m, 2H, ArH), 6.86 (dd, J1 = 7.9, J2 = 1.4 Hz, 1H, ArH), 6.79 (dd, J1 = 7.8, J2 = 2.0 Hz, 1H, ArH), 6.65–6.60 (m, 1H, ArH), 6.57–6.50 (m, 1H, ArH), 3.86 (s, 3H, OCH3), 3.14 (s, 4H, piperazine), 2.83–2.70 (m, 6H, 4H piperazine and 2H, CH2), 2.68–2.63 (m, 2H CH2); 13C NMR (126 MHz, Chloroform-d): δ 152.26, 146.43, 141.55, 141.31, 129.30, 122.90, 120.98, 119.03, 118.21, 115.49, 112.93, 111.16, 60.53, 55.33, 53.41(2C), 50.60 (2C), 33.55.
2.1.2.2 1-(2,3-dichlorophenyl)-4-(3-nitrophenethyl)piperazine (2b)
Yield: 72 %, oil. 1H NMR (500 MHz, Chloroform-d): δ 8.12 (s, 1H, ArH), 8.08 (d, J = 6.1 Hz, 1H, ArH), 7.56 (d, J = 7.6 Hz, 1H, ArH), 7.46 (t, J = 7.9 Hz, 1H, ArH), 7.18–7.11 (m, 2H, ArH), 6.97 (dd, J1 = 6.9, J2 = 2.7 Hz, 1H, ArH), 3.09 (s, 4H, piperazine), 2.93 (s, 2H, CH2), 2.83–2.64 (m, 6H, 4H piperazine and 2H CH2). 13C NMR (126 MHz, Chloroform-d): δ 151.00, 148.15,142.20, 134.85, 133.87, 129.03, 127.32, 127.28, 124.47, 123.46, 121.12, 118.44, 59.31, 53.05 (2C), 51.12 (2C), 32.93.
2.1.3 General procedure for synthesis 3-[2-(4-arylpiperazin-1-yl)ethyl]anilines (3a,b)
Raney/Ni (195 mg) was added, in portions, to a stirred mixture of hydrazine hydrate (32.5 mm), ethanol (26 mL), (6.5 mm) of compound 2a or 2b (6.5 mm), and 1,2-dichloroethane (12 mL). Stirring was continued at room temperature until the mixture became colorless and then was heated at 50 °C for 40 min. The resulting mixture was filtrated through Celite, and the solvent was removed under reduced pressure. The resulting 3-[2-(4-arylpiperazin-1-yl)ethyl]anilines were purified by silica gel column chromatography using a gradient of methanol (0–5 %) in dichloromethane.
2.1.3.1 3-{2-[4-(2-methoxyphenyl)piperazin-1-yl]ethyl}aniline (3a)
Yield: 93 %, oil. 1H NMR (500 MHz, Chloroform-d): δ 7.08 (t, J = 7.7 Hz, 1H, ArH), 7.00 (td, J1 = 7.6, J2 = 2.0 Hz, 1H, ArH), 6.98–6.90 (m, 2H, ArH), 6.86 (dd, J1 = 7.9, J2 = 1.4 Hz, 1H, ArH), 6.79 (dd, J1 = 7.8, J2 = 2.0 Hz, 1H, ArH), 6.65–6.60 (m, 1H, ArH), 6.57–6.50 (m, 1H, ArH), 5.29 (s, 2H, NH2), 3.86 (s, 3H, OCH3), 3.14 (s, 4H, piperazine), 2.83–2.70 (m, 6H, 4H piperazine and 2H, CH2), 2.68–2.63 (m, 2H CH2). 13C NMR (126 MHz, Chloroform-d): δ 152.26, 146.43, 141.55, 141.31, 129.30, 122.90, 120.98, 119.03, 118.21, 115.49, 112.93, 111.16, 60.53, 55.33, 53.41(2C), 50.60 (2C), 33.55.
2.1.3.2 3-{2-[4-(2,3-dichlorophenyl)piperazin-1-yl]ethyl}aniline (3b)
Yield: 89 %, oil. 1H NMR (500 MHz, Chloroform-d): δ 7.50–7.40 (m, 1H, ArH), 7.20–7.12 (m, 2H, ArH), 7.09 (t, J = 7.7 Hz, 1H, ArH), 7,01–6,93 (m, 1H, ArH), 6.67–6.61 (m, 1H, ArH), 6.60–6.53 (m, 1H, ArH), 5.30 (s, 2H, NH2), 3.11 (s, 4H, piperazine), 3.00–2.92 (m, 1H, CH2), 2.80–2.66 (m, 7H, 4H piperazine and 3H CH2). 13C NMR (126 MHz, Chloroform-d): δ 151.22, 146.45, 141.40, 133.97, 129.31, 127.43 (2C), 124.53, 118.97, 118.59, 115.44, 112.94, 60.36, 53.24 (2C), 51.27 (2C), 33.56.
2.1.4 General procedure for synthesis N-{3-[2-(4-aryl-piperazin-1-yl)ethyl]phenyl}arylamides (4a,b-10a,b)
A solution of 3a or 3b (1.77 mmol), triethylamine (4.44 mmol, 0.62 mL), the corresponding carboxylic acid (1.77 mmol), and PPAA (1.95 mmol, 1.14 mL, 50 % solution in DMF) in dry DMF (10 mL) was stirred at room temperature overnight. The mixture was poured into ice/water. Organic layers were separated and evaporated. The resulting N-{3-[2-(4-arylpiperazin-1-yl)ethyl]phenyl}arylamides were purified by silica gel column chromatography using a gradient of methanol (0–5 %) in dichloromethane.
2.1.4.1 N-(3-{2-[4-(2-methoxyphenyl)piperazin-1-yl]ethyl}phenyl)benzamide (4a)
Yield: 69 %, oil. IR (ATR, cm−1): 3307.8, 2941.2, 2818.1, 1655.0, 1497.9, 1241.4, 1027.3, 748.2. 1H NMR (400 MHz, Chloroform-d): δ 7.91–7.74 (m, 3H, ArH), 7.54–7.42 (m, 2H, ArH), 7.42–7.30(m, 2H, ArH), 7.23–7.15 (m, 1H, ArH), 7.01–6.74 (m, 5H, ArH), 3.79 (s, 3H, OCH3), 3.06 (s, 4H, piperazine), 2.94–2.72 (m, 2H, CH2), 2.75–2.59–2.56 (m, 6H, 4H piperazine and 2H CH2). 13C NMR (101 MHz, Chloroform-d): δ 165.74, 152.30, 141.50, 141.34, 138.05, 135.04, 131.82, 129.10, 128.75 (2C), 127.03 (2C), 125.03, 122.93, 121.02, 120.50, 118.25, 118.01, 111.24, 60.36, 55.38, 53.42 (2C), 50.64 (2C), 32.56. HRMS (m/z): [M + H]+ C26H29N3O2 Calcd: 416.23325; Found: 416.23268.
2.1.4.2 N-(3-{2-[4-(2,3-dichlorophenyl)piperazin-1-yl]ethyl}phenyl)benzamide (4b)
Yield: 88 %, oil. IR (ATR, cm−1): 3271.8, 2822.6, 1644.7, 1446.6, 961.9, 736.9. 1H NMR (500 MHz, Chloroform-d): δ 7.91–7.82 (m, 2H, ArH), 7.62–7.38 (m, 5H, ArH), 7.29 (t, J = 7.8 Hz, 1H, ArH), 7.20–7.10 (m, 2H, ArH), 7.08–6.99 (m, 1H, ArH), 7.00–6.93 (m, 1H, ArH), 3.09 (s, 4H, piperazine), 2.90–2.82 (m, 2H, CH2), 2.77–2.66 (m, 6H, 4H piperazine and 2H CH2). 13C NMR (126 MHz, Chloroform-d): δ 165.91, 151.45, 141.59, 138.24, 135.21, 134.23, 132.05, 129.31, 129.00 (2C), 127.65 (2C), 127.20 (2C), 125.21, 124.78, 120.66, 118.82, 118.18, 60.41, 53.45 (2C), 51.48 (2C), 33.75. HRMS (m/z): [M + H] + C25H25Cl2N3O Calcd: 454.14474; Found: 454.14488.
2.1.4.3 2-hydroxy-N-(3-{2-[4-(2-methoxyphenyl)piperazin-1-yl]ethyl}phenyl)benzamide (5a)
Yield: 86 %, oil. IR (ATR,cm−1): 3355.2, 2924.5, 1591.3, 1456.6, 1245.5, 1025.2, 757.3. 1H NMR (500 MHz, Chloroform-d): δ 7.94 (dd, J1 = 7.8, J2 = 1.8 Hz, 1H, ArH), 7.43–7.29 (m, 1H, ArH), 7.09–7.00 (m, 2H, ArH), 6.98–6.78 (m, 6H, ArH), 6.58–6.50 (m, 2H, ArH), 3.85 (s, 3H, OCH3), 3.39 (s, 6H, 4H piperazine, 2H, CH2), 3.29–3.11 (m, 3H, piperazine), 3.03–2.94 (m, 3H, 1H piperazine and 2H, CH2). 13C NMR (126 MHz, Chloroform-d): δ 174.65, 161.68, 151.99, 146.88, 139.11, 137.64, 133.66, 130.62, 129.69, 124.17, 121.13 (2C), 118.69, 118.51, 118.21, 116.69, 115.30, 113.77, 111.25, 58.25, 55.37, 52.01 (2C), 47.75 (2C), 30.35. HRMS (m/z): [M + H]+ C26H29N3O3 Calcd: 432.22817; Found: 432.22839.
2.1.4.4 N-(3-{2-[4-(2,3-dichlorophenyl)piperazin-1-yl]ethyl}phenyl)-2-hydroxybenzamide (5b)
Yield: 92 %, oil. IR (ATR, cm−1): 3321.0, 2942.0, 1643.5, 1448.6, 1240.1, 1134.4, 960.4, 780.5. 1H NMR (500 MHz, Chloroform-d): δ 8.26 (s, OH), 7.58 (dd, J1 = 8.0, J2 = 1.6 Hz, 1H, ArH), 7.46–7.35 (m, 1H, ArH), 7.32–7.22 (m, 1H, ArH), 7.14 (s, 1H, ArH), 7.18–7.07 (m, 3H, ArH), 7.04 (d, J = 7.5 Hz, 1H, ArH), 7.02–6.98 (m, 1H, ArH), 6.97–6.94 (m, 1H, ArH), 6.91–6.85 (m, 1H, ArH), 3.15–3.00 (m, 4H, piperazine), 2.99–2.81 (m, 2H, CH2), 2.81–2.63 (m, 6H, 4H piperazine and 2H CH2). 13C NMR (126 MHz, Chloroform-d): δ 168.38, 161.72, 151.41, 141.67, 137.14, 134.74, 134.23, 129.36 (2C), 127.67 (2C), 126.14, 125.84, 124.81, 121.67, 119.19, 118.83 (2C), 115.19, 60.35, 53.46 (2C), 51.47 (2C), 33.72. HRMS (m/z): [M + H]+ C25H25Cl2N3O2 Calcd: 470.13832; Found: 470.13986.
2.1.4.5 N-(3-{2-[4-(2-methoxyphenyl)piperazin-1-yl]ethyl}phenyl)picolinamide (6a)
Yield: 91 %, oil. IR (ATR, cm−1): 3298.1, 2815.1,1682.2, 1498.5, 1240.6, 1025.8, 750.1. 1H NMR (500 MHz, Chloroform-d): δ 10.03 (s, 1H, NH), 8.66–8.58 (m, 1H, ArH), 8.32–8.30 (m, 1H, ArH), 7.92 (td, J1 = 7.8 Hz, J2 = 1.7 Hz, 1H, ArH), 7.67–7.60 (m, 1H, ArH), 7.52–7.39 (m, 1H, ArH), 7.33 (t, J = 7.8 Hz, 2H, ArH), 7.10–6.85 (m, 5H, ArH), 3.88 (s, 3H, OCH3), 3.16 (s, 4H, piperazine), 2.96–2.86 (m, 2H, CH2), 2.85–2.72 (m, 6H, 4H piperazine and 2H CH2). 13C NMR (126 MHz, Chloroform-d): δ 162.18, 152.49, 150.04, 148.17, 141.64, 141.52, 138.02, 137.90, 129.31, 126.65, 125.02, 123.12, 122.59, 121.21, 120.14, 118.44, 117.71, 111.40, 60.61, 55.57, 53.62 (2C), 50.84 (2C), 33.79. HRMS (m/z): [M + H]+ C25H28N4O2 Calcd: 417.22850; Found: 417.22840.
2.1.4.6 N-(3-{2-[4-(2,3-dichlorophenyl)piperazin-1-yl]ethyl}phenyl)picolinamide (6b)
Yield: 88 %, oil. IR (ATR, cm−1): 3337.1, 2949.9, 2819.4, 1687.1, 1536.6, 1447.6, 1239.6, 961.0, 783.9. 1H NMR (500 MHz, Chloroform-d): δ 8.22–8.04 (m, 2H, ArH), 8.02 (s, 2H, ArH), 7.61–7.40 (m, 2H, ArH), 7.36–7.24 (m, 1H, ArH), 7.19–7.10 (m, 2H, ArH), 7.05–6.92 (m, 2H, ArH), 3.14–3.06 (m, 4H, piperazine), 2.95–2.89 (m, 2H, CH2), 2.77–2.71(m, 6H, 4H piperazine and 2H CH2). 13C NMR (126 MHz, Chloroform-d): δ 162.52, 151.29, 147.97, 141.37, 137.83, 137.70, 134.01, 129.09, 127.44, 126.45, 124.77, 124.54, 122.38, 119.94, 118.63, 117.52, 60.26, 53.27 (2C), 51.31 (2C), 36.46, 33.62, 31.42. HRMS (m/z): [M + H]+ C24H24Cl2N4O Calcd: 455.13999; Found: 455.14004.
2.1.4.7 N-(3-{2-[4-(2-methoxyphenyl)piperazin-1-yl]ethyl}phenyl)nicotinamide (7a)
Yield: 75 %, oil. IR (ATR, cm-1): 3296.0, 2939.0, 2815.2, 1669.4, 1499,2, 1241.3, 1025.6, 748.6. 1H NMR (500 MHz, Chloroform-d): δ 9.10 (s, 1H, ArH), 8.72 (d, J = 4,7 Hz, 1H, ArH), 8.55–8.49 (m, 1H, ArH), 8.26–8.11 (m, 1H, ArH), 7.62–7.47 (m, 1H, ArH), 7.46–7.36 (m, 1H, ArH), 7.32–7.23 (m, 1H, ArH), 7.08–6.77 (m, 5H, ArH), 3.86 (s, 3H, OCH3), 3.13 (s, 4H, piperazine), 3.02–2.46 (m, 8H, 4H piperazine and 4H CH2). 13C NMR (126 MHz, Chloroform-d): δ 163.91, 152.17 (2C), 147.89, 141.29, 141.09, 137.67, 135.39, 130.80, 129.05, 125.32, 123.57, 122.92, 120.92, 120.75, 118.31, 118.15, 111.12, 60.14, 55.28, 53.25 (2C), 50.41 (2C), 33.31. HRMS (m/z): [M + H]+ C25H28N4O2 Calcd: 417.22850; Found: 417.22832.
2.1.4.8 N-(3-{2-[4-(2,3-dichlorophenyl)piperazin-1-yl]ethyl}phenyl)nicotinamide (7b)
Yield: 69 %, M.p. 72 °C. IR (ATR, cm−1): 3286.4, 2938.3, 2820.1, 1666.5, 1446.2, 1204.7, 959.8, 780.7. 1H NMR (500 MHz, Chloroform-d): δ 9.09 (s, 1H, NH), 8.73 (d, J = 3.8 Hz, 1H, ArH), 8.25–8.17 (m, 2H, ArH), 7.59 (s, 1H, ArH), 7.46 (d, J = 7.9 Hz, 1H, ArH), 7.43–7.38 (m, 1H, ArH), 7.29 (t, J = 7.8 Hz, 1H, ArH), 7.18–7.09 (m, 2H, ArH), 7.04 (d, J = 7.6 Hz, 1H, ArH), 6.96 (dd, J1 = 6.9, J2 = 2.6 Hz, 1H, ArH), 3.09 (s, 4H, piperazine), 2.91–2.81 (m, 2H, CH2), 2.78–2.63 (m, 6H, 4H piperazine and 2H CH2). 13C NMR (126 MHz, Chloroform-d): δ 163.83, 152.35, 151.13, 147.79, 141.42, 137.56, 135.33, 133.94, 130.77, 129.09, 127.37 (2C), 125.38, 124.51, 123.63, 120.68, 118.53, 118.19, 60.09, 53.18 (2C), 51.20 (2C), 33.47. HRMS (m/z): [M + H]+ C24H24Cl2N4O Calcd: 455.13999; Found: 455.14018.
2.1.4.9 N-(3-{2-[4-(2-methoxyphenyl)piperazin-1-yl]ethyl}phenyl)isonicotinamide (8a)
Yield: 78 %, oil. IR (ATR, cm−1): 3330.0, 2936.3, 2920.86, 1674.6, 1498.7, 1241.0, 1135.2, 750.4. 1H NMR (500 MHz, Chloroform-d): δ 8.79–8.72 (m, 2H, ArH), 8.59–8.52 (m, 1H, ArH), 7.78–7.70 (m, 1H, ArH), 7.57 (s, 1H, ArH), 7.53 (d, J = 8.1 Hz, 1H, ArH), 7.30 (t, J = 7.7 Hz, 1H, ArH), 7.09–7.03 (m, 1H, ArH), 7.01–6.99 (m, 1H, ArH), 6.97–6.90 (m, 2H, ArH), 6.87 (dd, J1 = 8.1, J2 = 1.4 Hz, 1H, ArH), 3.86 (s, 3H, OCH3), 3.15 (s, 4H, piperazine), 2.91–2.83 (m, 2H, CH2), 2.82–2.65 (m, 6H, 4H piperazine and 2H CH2). 13C NMR (126 MHz, Chloroform-d): δ 165.38, 153.71 (2C), 152.01, 143.68, 142.78, 142.57, 139.07, 130.63, 127.01, 124.53, 122.56, 122.48, 122.28, 119.86, 119.71, 116.97, 112.68, 61.65, 56.82, 54.79 (2C), 51.91 (2C), 32.38. HRMS (m/z): [M + H]+ C25H28N4O2 Calcd: 417.22850; Found: 417.22831.
2.1.4.10 N-(3-{2-[4-(2,3-dichlorophenyl)piperazin-1-yl]ethyl}phenyl)isonicotinamide (8b)
Yield: 70 %, M.p. 88 °C. IR (ATR, cm−1): 3300.0, 2936.5, 2820.8, 1674.6, 1498.7, 1241.3, 126.0, 750.4. 1H NMR (500 MHz, Chloroform-d): δ 8.75 (d, J = 5.7 Hz, 2H, ArH), 8.25 (s, 1H, NH), 7.70 (d, J = 5.6 Hz, 2H, ArH), 7.58 (s, 1H, ArH), 7.46 (d, J = 7.8 Hz, 1H, ArH), 7.30 (t, J = 7.8 Hz, 1H, ArH), 7.20–7.10 (m, 2H, ArH), 7.06 (d, J = 7.6 Hz, 1H, ArH), 6.96 (dd, J1 = 6.9, J2 = 2.5 Hz, 1H, ArH), 3.09 (s, 4H, piperazine), 2.90–2.79 (m, 2H, CH2), 2.77–2.64 (m, 6H, 4H piperazine and 2H CH2). 13C NMR (126 MHz, Chloroform-d): δ 163.75, 151.10, 150.58 (2C), 142.05, 141.46, 137.33, 133.94, 129.13 (2C), 127.39, 125.59, 124.53, 120.87 (2C), 120.66, 118.53, 118.18, 60.05, 53.16 (2C), 51.17 (2C), 33.43. HRMS (m/z): [M + H]+ C24H24Cl2N4O Calcd: 455.13999; Found: 455.14032.
2.1.4.11 N-(3-{2-[4-(2-methoxyphenyl)piperazin-1-yl]ethyl}phenyl)pyridazine-4-carboxamide (9a)
Yield: 77 %, oil. IR (ATR, cm−1): 3343.4, 2939.7, 2815.5, 1685.5, 1240.9, 1023.1, 751.7. 1H NMR (500 MHz, Chloroform-d): δ 9.65 (s, 1H, NH), 9.51 (d, J = 1.5 Hz, 1H, ArH), 8.83–8.77 (m, 1H, ArH), 8.61–8.54 (m, 1H, ArH), 7.63–7.54 (m, 1H, ArH), 7.32 (t, J = 7.8 Hz, 1H, ArH), 7.10–7.03 (m, 1H, ArH), 7.03–6.89 (m, 4H, ArH), 6.89–6.83 (m, 1H, ArH), 3.86 (d, J = 2.9 Hz, 3H, OCH3), 3.13 (s, 4H, piperazine), 2.95–2.86 (m, 2H, CH2), 2.80–2.63 (m, 6H, 4H piperazine and 2H CH2). 13C NMR (126 MHz, Chloroform-d): δ 162.18, 153.84, 149.08, 146.22, 145.98, 143.91, 143.21, 142.87, 138.84, 130.76, 126.90, 124.49, 122.56, 121.65, 119.78, 119.18, 112.76, 61.92, 56.92, 54.99 (2C), 52.21 (2C), 35.15. HRMS (m/z): [M + H]+ C24H27N5O2 Calcd: 418.22375; Found: 418.22381.
2.1.4.12 N-(3-{2-[4-(2,3-dichlorophenyl)piperazin-1-yl]ethyl}phenyl)pyridazine-4-carboxamide (9b)
Yield: 69 %, M.p. 109 °C. IR (ATR, cm−1): 3266.6, 2941.8, 2819.2, 1676.5, 1446.4, 1240.7, 960.4, 782.2. 1H NMR (500 MHz, Chloroform-d): δ 9.62 (s, 1H, NH), 9.36–9.29 (m, 1H, ArH), 9.20 (s, 1H, ArH), 7.97–7.93 (m, 1H, ArH), 7.49 (d, J = 8.2 Hz, 1H, ArH), 7.34–7.20 (m, 1H, ArH), 7.20–7.11 (m, 3H, ArH), 7.11–7.03 (m, 1H, ArH), 7.01–6.90 (m, 1H, ArH), 3.08 (s, 4H, piperazine), 2.89–2.82 (m, 2H, CH2), 2.77–2.63 (m, 6H, 4H piperazine and 2H CH2). 13C NMR (126 MHz, Chloroform-d): δ 161.80, 151.85, 151.08, 148.41, 141.50, 137.20, 133.95, 132.81, 129.14 (2C), 127.39 (2C), 125.92, 124.54 (2C), 120.95, 118.52, 60.03, 53.17 (2C), 51.18 (2C), 33.44. HRMS (m/z): [M + H]+ C23H23Cl2N5O Calcd: 456.13524; Found: 456.13569.
2.1.4.13 N-(3-{2-[4-(2-methoxyphenyl)piperazin-1-yl]ethyl}phenyl)pyrazine-2-carboxamide (10a)
Yield: 75 %, oil. IR (ATR, cm−1): 3354.9, 2939.3, 2812.1, 1686.4, 1497.3, 1239.2, 1022.4, 747.9. 1H NMR (400 MHz, Chloroform-d): δ 9.65 (s, 1H, NH), 9.52 (d, J = 1.5 Hz, 1H, ArH), 8.81 (d, J = 2.5 Hz, 1H, ArH), 8.62–8.55 (m, 1H, ArH), 8.21–8.09 (m, 1H, ArH), 8.08–8.01 (m, 1H, ArH), 7.64–7.56 (m, 1H, ArH), 7.45–7.39 (m, 1H, ArH), 7.37–7.22 (m, 1H, ArH), 7.09–6.85 (m, 3H, ArH), 3.87 (s, 3H, OCH3), 3.15 (s, 4H, piperazine), 3.01–2.87 (m, 2H, CH2), 2.83–2.68 (m, 6H, 4H piperazine and 2H CH2). 13C NMR (101 MHz, Chloroform-d): δ 160.61, 147.52, 144.70, 142.34, 137.30, 132.06, 130.11, 129.21, 128.79, 128.71, 125.34, 122.92, 121.01, 120.08, 118.23, 117.61, 111.23, 60.37, 55.38, 53.45 (2C), 50.68 (2C), 33.62. HRMS (m/z): [M + H]+ C24H27N5O2 Calcd: 418.22375; Found: 418.22384.
2.1.4.14 N-(3-{2-[4-(2,3-dichlorophenyl)piperazin-1-yl]ethyl}phenyl)pyrazine-2-carboxamide (10b)
Yield: 74 %,.oil. IR (ATR, cm−1): 3353.4, 2939.9, 2819.5, 1694.6, 1541.2, 1446.8, 1245.6, 960.4, 795.3. 1H NMR (400 MHz, Chloroform-d): 9.66 (s, 1H, NH), 9.52 (d, J = 1.5 Hz, 1H, ArH), 8.80 (d, J = 2.5 Hz, 1H, ArH), 8.60–8.57 (m, 1H, ArH), 7.70 (s, 1H, ArH), 7.61–7.54 (m, 1H, ArH), 7.33 (t, J = 7.8 Hz, 1H, ArH), 7.21–7.10 (m, 2H, ArH), 7.09–7.04 (m, 1H, ArH), 6.97 (dd, J1 = 6.2, J2 = 3.3 Hz, 1H, ArH), 3.11 (s, 4H, piperazine), 2.93–2.85 (m, 2H, CH2), 2.80–2.66 (m, 6H, 4H piperazine and 2H CH2). 13C NMR (101 MHz, Chloroform-d): 160.61, 151.29, 147.53, 144.68, 142.34, 141.58, 137.32, 134.02, 129.20 (2C), 127.45, 125.30, 124.56, 120.08, 118.62 (2C), 117.63, 60.22, 53.28 (2C), 51.34 (2C), 33.63. HRMS (m/z): [M + H]+ C23H23Cl2N5O Calcd: 456.13658; Found: 456.13502.
2.2 Binding assays
The affinity of the synthesized compounds towards 5-HT1A was determined in radioligand competition binding assays. The assays were performed using crude cell membrane homogenates obtained from HEK cells stably transfected with human 5-HT1A receptor and the 5-HT1A specific radioligand [3H]–OH-DPAT (obtained from PerkinElmer, NET929250UC). Membrane suspension was incubated with 1 nM [3H]–OH-DPAT and various concentrations of the test compound in 25 mM Tris-HCl, pH 7.4 buffer containing 120 mM NaCl, 5 mM KCl at room temperature for 60 min. Non-specific binding was determined in the presence of 10 µM of serotonin. The reaction was terminated by rapid filtration using Whatman GF/B glass-fibre filters, pre-soaked in 0.3 % polyethyleneimine, and a 48-channel harvester (Biomedical Research and Development Laboratories, Gaithersburg, MD, USA) followed by washing four times with ice-cold TRIS-HCl buffer. Filter-bound radioactivity was quantified by liquid scintillation counting. At least three separate experiments in triplicate were performed to determine inhibitory constant (Ki) values. The data were analyzed by GraphPad Prism, Version 4.1 (GraphPad Inc., La Jolla, CA). The Ki was calculated according to the Cheng-Prusoff equation:
Ki = IC50/(1 + ([L]/KD)) where Ki is the inhibition constant, IC50 is the concentration of competitive inhibitor that displaces 50 % of the specifically bound labelled ligand, [L] is the concentration of the labelled ligand, and KD = the affinity constant of the labelled ligand.
2.3 Absorption, distribution, metabolism, excretion and toxicity (ADMET) analysis
To predict absorption, distribution, metabolism, and excretion (ADME) qualities of tested ligands, we used the SwissADME webserver (www.swissadme.ch) (Daina et al., 2017). Toxicology prediction was done through Pro Tox-II virtual lab server for the prediction of toxicities of small molecules (https://tox-new.charite.de/protox_II/index.php?site=home) (Drwal et al., 2014). Ligand structures were provided as SMILES using ChemDraw.
2.4 Docking simulations
Docking simulations were done in Maestro Suite software (Schrödinger, 2018). 3D model of the 5-HT1A receptor with bound aripiprazole (PDB Code 7E2Z) was obtained from the GPCR database (Xu et al., 2021). 2D structures of ligands were drawn in ChemDraw software, and prepared for docking in Maestro software using default LigPrep procedures.
Induced fit docking (IFD) simulation using standard sampling protocol and default values, was carried out for prepared receptor model and selected ligands (Sherman et al., 2006). Bind site was defined, based on bound aripiprazole and centered on ASP 116 residue. The inner grid box was set to 10x10x10 Å and the outer box size according to the size of each tested ligand. Grid spacing was set to 1 Å. Obtained docking structures were examined and selected for further analysis based on the number of receptor-ligand interactions and calculated post docking MM-GBSA energy.
2.4.1 Molecular dynamics simulations
Molecular dynamics (MD) simulations were performed using the Schrodinger Desmond software package (Schrödinger, 2018). Docking poses selected for MD were prepared for simulation by embedding the protein–ligand complex into the POPC membrane bilayer using the Desmond system builder module. Protein was oriented in the membrane according to the data from the Orientations of Proteins in Membranes (OPM) server (http://opm.phar.umich.edu/). The entire system was solvated with a TIP3P explicit water model, and neutralized via counter ions and salt solution of 0.15 M KCl. We used OPLS 2003 forcefield to calculate the interactions between all the atoms. For the calculation of the long-range Coulombic interactions, particle-mesh Ewald (PME) method was used, with the cut-off radius of 9 Å for the short-range Van der Waals (VdW) and electrostatic interactions.
During the course of the simulation, a constant temperature of 310 K and a pressure of 1.01235 bars were maintained, using the Nose-Hoover thermostat, and the Martyna Tobias Klein method. 100 ns MD simulation with 2.0 fs step for each complex was performed and the collected trajectory was used in the MD analysis to assess the docking pose and protein–ligand interactions stability.
2.4.2 Molecular electrostatic potential calculation
Molecular Electrostatic Potential (MEP) calculation was completed in the Jaguar module, using a 6–311*+ basis set (Schrödinger, 2018). Molecule geometry was optimized using the same basis set and ab initio DFT B3LYP method. After optimal geometry was achieved, MEP calculation was performed and ESP values of −0.25 to 0.25 were mapped on the electron density surface.
2.5 Cytochrome P450 site of metabolism analysis
In addition to basic sites of metabolism analysis (SoMs) prepared with NERDD (https://nerdd.univie.ac.at/) FAME3 (Šícho et al., 2019) / GLORY (de Bruyn Kops et al., 2019) modules, Maestro Suite (Schrödinger, 2018) P450 Site of Metabolism workflow was used to predict the metabolic stability of tested ligands. All three cytochrome available isoforms (PDB code: 2C9, 2D6 and 3A4) were used for the analysis. All calculation parameters were left at default values.
3 Results and discussion
3.1 Chemistry
The general synthetic route and chemical structures of the compounds synthesized in the present study are shown in Scheme 1.![Synthetic pathways for N-{3-[2-(4-aryl-piperazin-1-yl)ethyl]phenyl}arylamides and tabelar representation of aryl part of new and parent compounds. Reagents and conditions for the synthesis of 4a,b-10a,b: (i) Arylpiperazine, Et3N, 10 °C, propylphosphonic acid anhydride (PPAA), N,N-dimethylformamide (DMF), r.t.; (ii) B2H6, THF, 0 °C for 6 h, r.t. for 1 h, then reflux for 2 h; (iii) Ra/Ni, NH2NH2, EtOH, 1,2-dichloroethane; (iv) ArCO2H, Et3N, PPAA, DMF, r.t. over night. Parent ligands 4ca-10ca (R = R1 = H) were previously synthesized (Sukalovic et al., 2013).](/content/184/2023/16/4/img/10.1016_j.arabjc.2023.104636-fig3.png)
Synthetic pathways for N-{3-[2-(4-aryl-piperazin-1-yl)ethyl]phenyl}arylamides and tabelar representation of aryl part of new and parent compounds. Reagents and conditions for the synthesis of 4a,b-10a,b: (i) Arylpiperazine, Et3N, 10 °C, propylphosphonic acid anhydride (PPAA), N,N-dimethylformamide (DMF), r.t.; (ii) B2H6, THF, 0 °C for 6 h, r.t. for 1 h, then reflux for 2 h; (iii) Ra/Ni, NH2NH2, EtOH, 1,2-dichloroethane; (iv) ArCO2H, Et3N, PPAA, DMF, r.t. over night. Parent ligands 4ca-10ca (R = R1 = H) were previously synthesized (Sukalovic et al., 2013).
2-Methoxy and 2,3-dichloro substituents were introduced into the phenylpiperazine part of the already active parent compounds (4c-10c, R = R1 = H, Scheme 1, Fig. 2) (Sukalovic et al., 2013) to improve affinity toward 5-HT1A receptors. In brief, N-{3-[2-(4-aryl-piperazin-1-yl)ethyl]phenyl}arylamides were obtained: by acylation of N-arylpiperazine using 3-nitrophenylacetic acid and gave rise to 2-(3-nitrophenyl)-1-(4-arylpiperazin-1-yl)ethan-1-ones (1a,b), followed by reduction of amides 1a,b using diborane in tetrahydrofuran (THF). 1-(3-nitrophenethyl)-4-arylpiperazines (2a,b) were reduced by Raney/Ni to provide 3-[2-(4-arylpiperazin-1-yl)ethyl]anilines (3a,b). Target arylmides 4a,b-10a,b were obtained by condensation of anilines 3a,b with corresponding aryl acid in the presence of propylphosphonic acid anhydride (PPAA) in N,N-dimethylformamide (DMF). All the compounds were characterized by 1H and 13C NMR spectroscopy and mass spectroscopy (for HRMS spectra of new ligands 4a,b-10a,b please, see Suppl. S1).
3.2 5-HT1A receptor affinity
As presented in Table 1, eight (4a-b; 5a-b; 6b; 7a; 8a-b) of the 14 newly synthesized ligands had an enhancement in affinity for 5-HT1A receptors compared to parent compounds (4c-8c). Analysis of the obtained experimental affinity values of the newly synthesized ligands and the affinities of the parent compounds shows that the conspicuous increase was observed with ligands 5a, followed by 8a, 8b, and 5b. The decreasing order of increase of other ligands is 4a > 4b > 6b > 7a (from moderate to mild). In the case of the other newly synthesized ligands, the desired, pronounced increase in affinity was absent (for a diagram processing the results, please see Suppl. S2). Also, in the analysis of the relationship between the experimental affinity values of the newly synthesized ligands and aripiprazole, the most pronounced increase was observed for ligands 8a and 8b (2.1 and 1.8-fold, respectively). Obtained experimental data showed that in the case of starting structures 9c and 10c, neither methoxy nor 2,3-dichloro substitution did not lead to an affinity increment towards the 5-HT1A receptors. The same is true for resulted compounds 6a and 7b. The proposed modification did not result in a significant increase in affinity. Designation of: Tox.-Toxicity; Total number of violations (L.-Lipinski; M.- Muegge; G.- Ghose); C*logP-Consensus logP. Designation of toxicity: *Immunotoxicity active, **Carcinogenicity active. Values of previously synthesized and commercial compounds adapted from: a(Sukalovic et al., 2013), b(Keck and McElroy, 2003), c(Baek et al., 2015), d(Kiss et al., 2010), e(Citrome, 2013), f(Cantillon et al., 2017), g(Bhat et al., 2018).
Ligand
No
Ki (nM)
Tox.
ADME
L.
M.
G.
C*logP
M.W.
(g/mo)
ESOL Class
4a
8.8
Inactive
0
0
1
3.36
415.53
Moderately soluble
4b
17.3
Inactive
1
1
1
4.39
454.39
Poorly soluble
4c
37.9a
Inactive
0
0
0
3.71
385.5
Moderately soluble
5a
2.6
0.54*
0
0
1
2.81
431.53
Moderately soluble
5b
6.1
Inactive
0
1
1
3.83
470.39
Poorly soluble
5c
51.3a
Inactive
0
0
0
3.15
401.5
Moderately soluble
6a
4.6
0.87*
0
0
1
2.34
416.52
Moderately soluble
6b
1.5
Inactive
0
1
1
3.36
455.38
Poorly soluble
6c
2.4a
Inactive
0
0
0
2.68
386.49
Moderately soluble
7a
1.7
0.65*
0
0
1
2.34
416.52
Moderately soluble
7b
7.6
Inactive
0
0
1
3.36
455.38
Moderately soluble
7c
2.5a
Inactive
0
0
0
2.68
386.49
Moderately soluble
8a
0.8
Inactive
0
0
1
2.34
416.52
Moderately soluble
8b
0.9
Inactive
0
0
1
3.36
455.38
Moderately soluble
8c
8.5a
Inactive
0
0
0
2.68
386.49
Moderately soluble
9a
2.6
0.61**
0
0
0
2.15
417.5
Soluble
9b
1.3
Inactive
0
0
1
3.15
456.37
Moderately soluble
9c
0.6a
0.61**
0
0
0
2.47
387.48
Soluble
10a
12.7
0.87*
0
0
0
1.33
417.5
Moderately soluble
10b
15.4
Inactive
0
0
1
2.34
456.37
Moderately soluble
10c
2.7a
Inactive
0
0
0
1.65
387.48
Moderately soluble
Aripiprazole
1.7b
Inactivec
0
0
1
3.65
448.39
Moderately soluble
Cariprazine
2.6d
Inactivee
0
0
0
3.75
427.41
Moderately soluble
Brilaroxazine
1.5f
Inactiveg
0
0
0
2.90
450.36
Moderately soluble
Two compounds stood out as representatives of the successful approach, N-(3-{2-[4-(2-methoxyphenyl)piperazin-1-yl]ethyl}phenyl)isonicotinamide (8a), and N-(3-{2-[4-(2,3-dichlorophenyl)piperazin-1-yl]ethyl}phenyl)isonicotinamide (8b), with Ki values of 0.8 nM and 0.9 nM, respectively. Presented Ki values correspond to commercial, structurally resembling drugs.
3.3 ADME prediction and toxicity
Determining a compound’s toxicity and absorption, distribution, metabolism, and excretion characteristics is a material step in the drug development process.
Investigating ADME and toxicology through animal experiments is expensive and time-consuming. On the other hand, the prediction of ADMET in silico is a fast and affordable alternative (Banerjee et al., 2018a; Daina et al., 2017). The method relies on existing databases to construct a capable model for the ADMET prediction of novel compounds (Table 1), where results of Lipinski, Muegge and Ghose rules are presented as the total number of violations (for the detailed ADMET profile of all tested ligands, and criteria for violation of Lipinski, Muegge and Ghose rules, please see Suppl. S3 and S4) (Banerjee et al., 2018b; Drwal et al., 2014). Results of toxicology prediction suggested that substances 5a and 7a have the potential to be immunotoxic. Performed comparative study of two different formulas of aripiprazole (free base crystal or cocrystal formula) suggested that aripiprazole, in prescribed doses, is not cytotoxic or immunotoxic. The same is true in the case of cariprazine and brilaroxazine (Baek et al., 2015; Citrome, 2013; Bhat et al., 2018).
ADME analysis pointed out that several newly synthesized compounds suffer from high molecular weight and low solubility (4b, 5b, and 6b). Both parameters are substantial for bio-oral availability (substance ability to be administrated and transported through the body). High molecular weight, by itself, doesn’t have to be eliminating factor. Some commercially available substances fall into this category but, combined with low solubility, rule out ligand as a desirable drug candidate. Betterment of solubility was not the subject of this research. Therefore substances 4b, 5b, and 6b were excluded from a further survey (Table 1). After ADMET analysis, ligands 4a, 8a, and 8b emerged as possible candidates for the additional analysis (for the ADMET profile of all tested ligands, please see Suppl. S3 and S4).
3.4 Docking simulations, molecular dynamics, and electrostatic potential calculations
Ligands 4a, 8a, and 8b were subject to docking simulations, molecular dynamics (MD), and electrostatic potential calculations (ESP) (Schrödinger, 2018; Bowers et al., 2006). A potential drug candidate should make a stable receptor-ligand complex, forming interactions comparable to commercial substances with a similar structure (Table 1).
Since the 5-HT1A receptor model with bound aripiprazole is a readily available crystal structure (PDB code 7E2Z) (Xu et al., 2021), all tested substances were docked into the receptor using the Induced Fit docking protocol (IFD) (Sherman et al., 2006). IFD protocol can take into account both flexible ligand and flexible binding site to closely simulate natural conditions.
All docked substances share the same bind cavity and number of key interactions with aripiprazole (and other selected commercial substances). The superposition of docked ligand 8a and aripiprazole inside the receptor bind site was shown in Fig. 3 (for the interactions of all superposed ligands, please see Suppl. S5).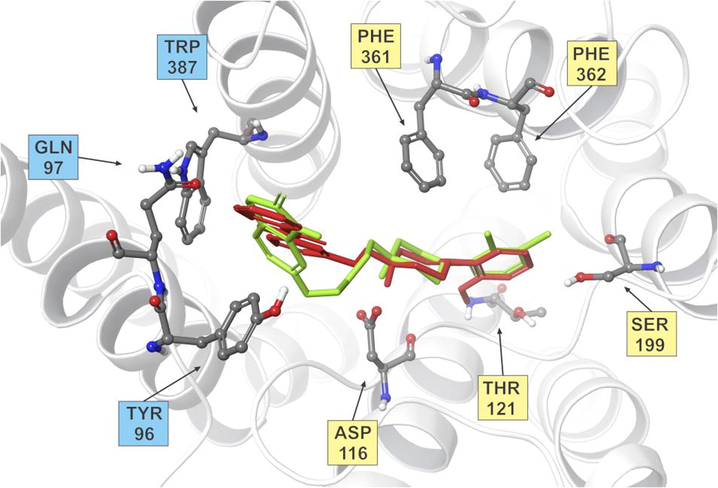
Superposition of docked ligand 8a (red) and aripiprazole (green) inside the receptor bind site with interacting residues forming OBS (yellow) and EBS (blue). Only key amino acid residues are shown for clarity.
The formation of a short salt bridge with Asp 116, the first requirement for high ligand affinity, was observed in all docking runs. Arylpiperazine part of the ligand has correct orientation inside the orthosteric bind site (OBS), formed by TM 3, TM 5, and TM 6 helices. The remaining part of the ligand binds into the extended bind site (EBS), made in part by TM 2, TM 7 and extracellular loop 3 (ECL3) (Fig. 3).
ESP calculations show compatible positive and negative areas on ligands that correspond to key amino acid residues on the receptor. The positive area around protonated nitrogen atom orients ligand toward negatively charged Asp 116. The negative area inside the aryl ring of the ligands arylpiperazine part forms edge-to-face interactions with Phe 361 and 362. The introduction of the methoxy group in position 2 of the aryl part enhances mentioned negative area and facilitates hydrogen bonding with Thr 121 (4a and 8a). Much the same is achieved with the introduction of 2,3-dichloro substitution in compound 8b, but this time hydrogen bond is formed with Ser 199 (Table 2).
Ligand
NoMM-GBSA energy [kcal/mol]
Key interactions in EBS and OBS
Type of interacion
4a
−31.4
Tyr 96
Aromatic
Gln 97
Hydrogen bond
Trp 387
Aromatic
Asp 116
Salt bridge
Phe 361/362
Aromatic
Thr 121
Hydrogen bond
8a
−37.6
Tyr 96
Aromatic
Gln 97
Hydrogen bond
Trp 387
Aromatic
Asp 116
Salt bridge
Phe 361/362
Aromatic
Thr 121
Hydrogen bond
8b
−37.8
Tyr 96
Aromatic
Gln 97
Hydrogen bond
Trp 387
Aromatic
Asp 116
Salt bridge
Phe 361/362
Aromatic
Ser 199
Hydrogen bond
Aripiprazole
−38.1
Tyr 96
Aromatic
Gln 97
Hydrogen bond
Asn 386
Aromatic
Asp 116
Salt bridge
Phe 361/362
Aromatic
Ser 199
Hydrogen bond
Additional affinity gains come from ligand interactions with EBS. Here, the optimal orientation of corresponding ligand functional groups is of the utmost importance. Hydrogen bond with Gln 97 and aromatic interactions with Tyr 96 and Trp 387 are needed to further enhance affinity (Table 2). MD simulations, carried over 100 ns, showed that all obtained docking poses are stable and that crucial interactions hold more than 20 % of the overall interaction time (Fig. 4).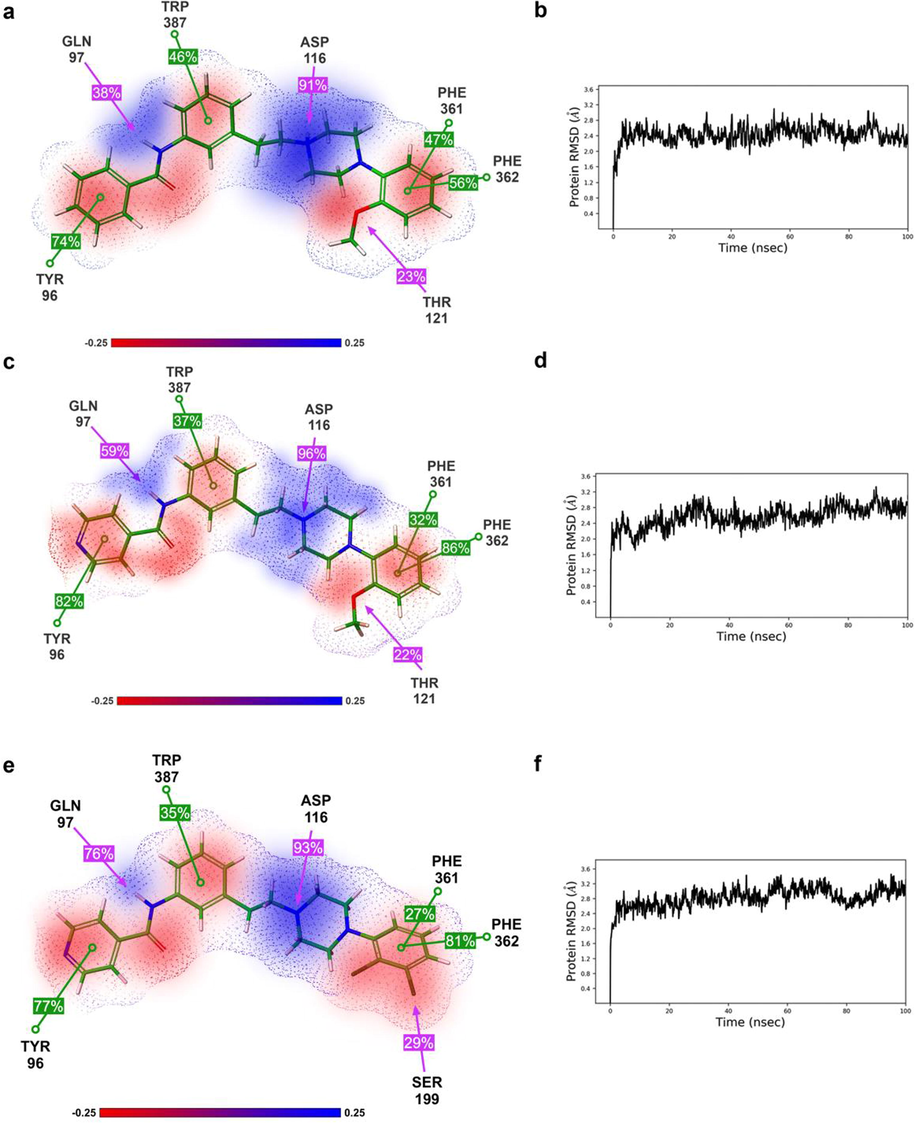
2D Diagrams of selected ligands key receptor-ligand interactions with 3D ESP map (a, c, e) and RMSD profiles throughout the 100 ns MD simulation (b, d, f): for ligand 4a (a and b); for ligand 8a (c and d); for ligand 8b (e and f). Only key amino acid residues are shown for clarity. Aromatic interactions are shown as green and electrostatic interactions are pink lines. Numbers in square denote the percentage of time interactions are observed during the 100 ns MD run. Negative ESP is red, while positive is blue.
3.5 Prediction of drug metabolism
In vivo interaction of drug molecules with Cytochromes P450 superfamily of metabolic enzymes can cause the breakdown or alteration of their structure (Tyzack et al., 2017; Tyzack and Kirchmair, 2019). The change in character or composition, typically in a comparatively small but significant way, happens at the sites of metabolism (SoMs) and results in a modified, usually more polar structure. Consequently, formed metabolites have different pharmacological and toxicological properties regarding the parent compound (Kirchmair et al., 2013; Tyzack and Glen, 2014).
Prediction of SoMs and metabolites was completed using the NERDD server and FAME3 (Šícho et al., 2019) / GLORY (de Bruyn Kops et al., 2019) protocol. Due to the chemical nature of compounds 4a, 8a and 8b, the location of their major SOM is in the arylpiperzine part of the molecule and foreseen metabolites correspond to metabolic modifications in this part of the molecule. Although another SOM is predicted in the aryl amide part, this metabolic modification is minor and less likely (for the complete list of ranked metabolites, please see Suppl. S6). Predicted metabolites are summarized in Table 3 and their modifications are comparable to known metabolites of commercial substances, namely aripiprazole. Aripiprazole is metabolized in the liver by the cytochrome P450 isoenzymes CYP3A4 and CYP2D6. Dehydroaripiprazole is a predominant metabolite, along with products of N-dealkylation, dehydrogenation, and hydroxylation (in the same manner as shown for 8b, Table 3) (Kirschbaum et al., 2008). Designation of: Predict. Score-Prediction Score; Tox.-Toxicity; Total number of violations (L.-Lipinski; M.- Muegge; G.- Ghose); C*logP-Consensus logP. Only the most probable rank 1 metabolites are included, as well as the most probable products according to GLORY prediction. For a complete predicted metabolite list, please see Suppl. S6 and S7.
Ligand
No
Metabolite
structure
Predict.
Score
Tox.
ADME
MM-GBSA energy (kcal/mol)
L.
M.
G.
C*logP
M.W. (g/mo)
ESOL Class
4a
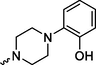
4.12
inactive
0
0
0
3.54
401.50
Moderately soluble
−30.15
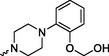
4.12
inactive
0
0
1
3.34
431.53
Moderately soluble
−31.21
8a
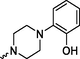
4.24
inactive
0
0
0
2.80
402.49
Moderately soluble
−36.54
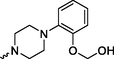
4.24
inactive
0
0
0
2.66
418.49
Moderately soluble
−37.60
8b

2.77
inacitve
0
0
1
3.84
471.38
Moderately soluble
−34.16
Structurally, formed metabolites include the addition of –OH group in position 4 on arylpiperazine part in case of ligand 8b, and for ligands, 8a and 4a, methoxy group transformation into –OH or -O-CH2-OH group. Their ADMET profile is very much the same compared to parent compounds. To determine the mode of metabolite interaction with 5-HT1A, we performed the IFD docking of listed metabolites (Table 3) and compared them with the initial docking results. All tested metabolites can bind to the 5-HT1A receptor. Interaction with the 5-HT1A receptor takes place in the same or altered way, compared to the parent substances.
In the case of metabolites of ligands 8a and 4a, all key interactions formed by the parent substances and metabolites are identical (Fig. 5).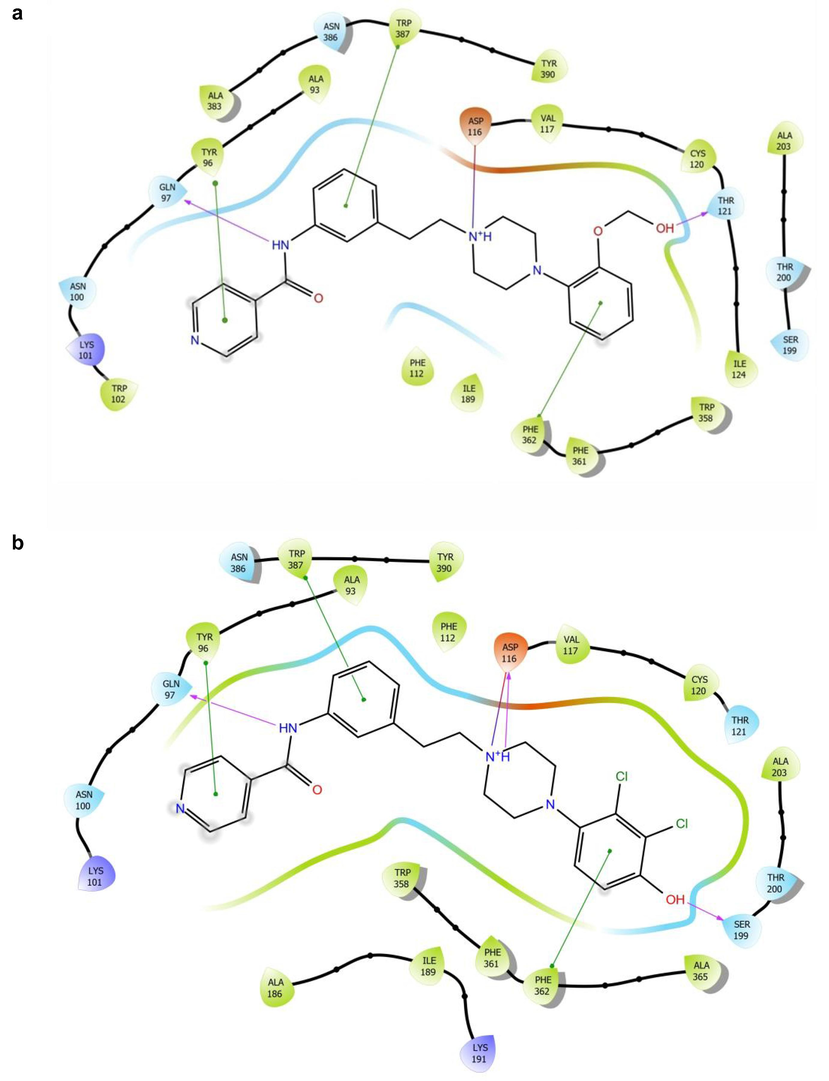
2D docking schematics of selected metabolite products of a) 8a and b) 8b, with the key receptor-ligand interactions (salt bridge–red, hydrogen bonds–blue, and aromatic interactions–green).
Ligand 8b metabolite with –OH group at position 4 of the arylpiperazine ring can form a stronger hydrogen bond with Ser 199 compared to the parent compound. Thus, 8b metabolite may benefit from this interaction. Predicted MM-GBSA energies, put all tested metabolites affinities in the similar range as parent compounds (for the ADME profile and 2D diagrams of all tested metabolites, please see Suppl. S7 and S8).
4 Conclusion
In summary, out of 14 synthesized compounds with the desired increase in 5-HT1A affinity, eight were noteworthy. The most pronounced increase in activity was in the case of ligands 4a-b, 5a-b, and 8a-b. However, ADMET calculations eliminated ligands 4b, 5a-b, and 6b as potential drug candidates. Suspected toxicity was crucial for compound 5a elimination; the low solubility of 4b and 5b was reason enough for exclusion from further analysis.
The three remaining compounds, 4a and 8a-b, showed the best profile for further computer-aided drug analysis. Docking studies, confirmed by molecular dynamics, revealed the formation of stable receptor-ligand complexes for all remaining ligands; observed interactions were comparable to commercial drugs with similar structures. Prediction of SoMs and metabolites, followed by MM-GBSA binding energy calculations, confirmed that formed metabolites are non-toxic, with an ADME profile comparable to the parent compound and similar binding energies - receptor affinities.
Finally, the best candidates for extended studies are compounds 8a and 8b, followed by ligand 4a. All of the above-stated findings make them promising and leading compounds in further fine-tuned research to examine the profile of potential antidepressants.
CRediT authorship contribution statement
Jelena Z. Penjišević: Investigation, Visualization, Writing – original draft, Writing – review & editing. Vladimir B. Šukalović: Conceptualization, Formal analysis, Visualization, Writing – review & editing. Sladjana Dukic-Stefanovic: Conceptualization, Formal analysis, Visualization, Writing – review & editing. Winnie Deuther-Conrad: Conceptualization, Visualization, Writing – review & editing. Deana B. Andrić: Conceptualization, Visualization, Supervision, Writing – original draft, Writing – review & editing. Slađana V. Kostić-Rajačić: Resources, Project administration, Writing – review & editing.
Acknowledgements
This work was supported by the Ministry of Education, Science and Technological Development of the Republic of Serbia (Grant No. 451-03-68/2022-14/200026, and for D.B. Andric Contract number: 451-03-68/2022-14/200168).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Evidence for intact 5-HT1A receptor-mediated feedback inhibition following sustained antidepressant treatment in a rat model of depression. Neuropharm.. 2018;141:139-147.
- [CrossRef] [Google Scholar]
- Immunotoxicological effects of aripiprazole: in vivo and in vitro studies. Korean J. Physiol. Pharmacol.. 2015;19(4):365-372.
- [CrossRef] [Google Scholar]
- ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res.. 2018;46(W1):W257-W263.
- [CrossRef] [Google Scholar]
- Prediction is a balancing act: Importance of sampling methods to balance sensitivity and specificity of predictive models based on imbalanced chemical data sets. Front. Chem.. 2018;6:362.
- [CrossRef] [Google Scholar]
- Brilaroxazine (RP5063) clinical experience in schizophrenia: A new option to address unmet needs. J. Neurol. Neuromed.. 2018;3(5):39-50.
- [CrossRef] [Google Scholar]
- Bowers, K.J., Chow, E., Xu, H., Dror, R.O., Eastwood, M.P., Gregersen, B.A., Klepeis, J.L., Kolossvary, I., Moraes, M.A., Sacerdoti, F.D., Salmon, J.K., Shan, Y., Shaw, D.E., 2006. Scalable algorithms for molecular dynamics simulations on commodity clusters. In: SC '06: Proceedings of the 2006 ACM/IEEE conference on Supercomputing. November, 84-es. https://doi.org/10.1145/1188455.1188544.
- Dopamine serotonin stabilizer RP5063: a randomized, double-blind, placebo-controlled multicenter trial of safety and efficacy in exacerbation of schizophrenia or schizoaffective disorder. Schizophr. Res.. 2017;189:126-133.
- [CrossRef] [Google Scholar]
- Synthesis and biological investigation of coumarin piperazine (piperidine) derivatives as potential multireceptor atypical antipsychotics. J. Med. Chem.. 2013;56:4671-4690.
- [CrossRef] [Google Scholar]
- Functional selectivity and antidepressant activity of serotonin 1A receptor ligands. Int. J. Mol. Sci.. 2015;16:18474-18506.
- [CrossRef] [Google Scholar]
- Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability. Expert Opin. Drug Metab. Toxicol.. 2013;9(2):193-206.
- [CrossRef] [Google Scholar]
- COVID-19 Mental Disorders Collaborators, 2021. Global prevalence and burden of depressive and anxiety disorders in 204 countries and territories in 2020 due to the COVID-19 pandemic. Lancet. 2021;398:1700-1712.
- [CrossRef] [Google Scholar]
- SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep.. 2017;7:42717.
- [CrossRef] [Google Scholar]
- GLORY: generator of the structures of likely cytochrome P450 metabolites based on predicted sites of metabolism. Front. Chem.. 2019;7:402.
- [CrossRef] [Google Scholar]
- ProTox: a web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res.. 2014;42(Web Server issue):W53-W58.
- [CrossRef] [Google Scholar]
- A review of novel antidepressants: a guide for clinicians. Cureus. 2019;11
- [CrossRef] [Google Scholar]
- ADMET-score - a comprehensive scoring function for evaluation of chemical drug-likeness. Med. Chem. Comm.. 2019;10:148-157.
- [CrossRef] [Google Scholar]
- Structure and function of the brain serotonin system. Physiol. Rev.. 1992;72:165-229.
- [CrossRef] [Google Scholar]
- Aripiprazole: a partial dopamine D2 receptor agonist antipsychotic. Expert Opin. Invest. Drugs. 2003;12(4):655-662.
- [CrossRef] [Google Scholar]
- How do metabolites differ from their parent molecules and how are they excreted? J. Chem. Inf. Model.. 2013;53:354-367.
- [CrossRef] [Google Scholar]
- Serum levels of aripiprazole and dehydroaripiprazole, clinical response and side effects. World J. Biol. Psychiatry.. 2008;9(3):212-218.
- [CrossRef] [Google Scholar]
- Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J. Pharmacol. Exp. Ther.. 2010;333(1):328-340.
- [CrossRef] [Google Scholar]
- Krüger, A., Maltarollo, V.G., Wrenger, C., Kronenberger, T., 2020. ADME profiling in drug discovery and a new path paved on silica, in: Gaitonde, V., Karmakar, P., Trivedi, A. (Eds.), Drug Discovery and Development - New Advances. IntechOpen, Ch.6. https://doi.org/10.5772/intechopen.86174.
- Piperazine, a key substructure for antidepressants: its role in developments and structure-activity relationships. ChemMedChem. 2021;16:1878-1901.
- [CrossRef] [Google Scholar]
- The therapeutic potential of 5-HT1A receptors: a patent review. Expert Opin. Ther. Pat.. 2012;22:887-902.
- [CrossRef] [Google Scholar]
- Screening for human ADME/Tox drug properties in drug discovery. Drug Discov. Today. 2001;6:357-366.
- [CrossRef] [Google Scholar]
- Anxiety and depression in COVID-19 survivors: Role of inflammatory and clinical predictors. Brain Behav. Immun.. 2020;89:594-600.
- [CrossRef] [Google Scholar]
- National trends in the prevalence and treatment of depression in adolescents and young adults. Pediatrics.. 2016;138:e20161878.
- [CrossRef] [Google Scholar]
- National trends in mental health care for US adolescents. JAMA Psychiatry.. 2020;77:703-714.
- [CrossRef] [Google Scholar]
- Synthesis of a new series of aryl/heteroarylpiperazinyl derivatives of 8-acetyl-7-hydroxy-4-methylcoumarin with low nanomolar 5-HT1A affinities. Eur. J. Med. Chem.. 2017;137:108-116.
- [CrossRef] [Google Scholar]
- 5-HT1A and 5-HT2A receptors affinity, docking studies and pharmacological evaluation of a series of 8-acetyl-7-hydroxy-4-methylcoumarin derivatives. Bioorg. Med. Chem.. 2018;26:527-535.
- [CrossRef] [Google Scholar]
- 6-Acetyl-5-hydroxy-4,7-dimethylcoumarin derivatives: Design, synthesis, modeling studies, 5-HT1A, 5-HT2A and D2 receptors affinity. Bioorg. Chem.. 2020;100:103912.
- [CrossRef] [Google Scholar]
- The place of antipsychotics in the therapy of anxiety disorders and obsessive-compulsive disorders. Curr. Psychiatry Rep.. 2017;19:103.
- [CrossRef] [Google Scholar]
- Schrödinger, 2018. Schrödinger Release 2018-4: Maestro, Schrödinger, LLC, New York, NY, 2018.
- Use of an induced fit receptor structure in virtual screening. Chem. Biol. Drug Des.. 2006;67:83-84.
- [CrossRef] [Google Scholar]
- FAME 3: Predicting the sites of metabolism in synthetic compounds and natural products for phase 1 and phase 2 metabolic enzymes. J. Chem. Inf. Model.. 2019;59:3400-3412.
- [CrossRef] [Google Scholar]
- 5-HT1A receptor ligands and their therapeutic applications: Review of new patents. Expert Opin. Ther. Pat.. 2018;28:679-689.
- [CrossRef] [Google Scholar]
- Interactions of N-{[2-(4-phenyl-piperazin-1-yl)-ethyl]-phenyl}-2-aryl-2-yl-acetamides and 1-{[2-(4-phenyl-piperazin-1-yl)-ethyl]-phenyl}-3-aryl-2-yl-ureas with dopamine D2 and 5-hydroxytryptamine 5HT(1A) receptors. Bioorg. Med. Chem. Lett.. 2012;22:3967-3972.
- [CrossRef] [Google Scholar]
- N-{[2-(4-Phenyl-piperazin-1-yl)-ethyl]-phenyl}-arylamides with dopamine D2 and 5-Hydroxytryptamine 5HT(1A) activity: synthesis, testing, and molecular modeling. Arch. Pharm. (Weinheim). 2013;346:708-717.
- [CrossRef] [Google Scholar]
- Understanding enzyme function evolution from a computational perspective. Curr. Opin. Struct. Biol.. 2017;47:131-139.
- [CrossRef] [Google Scholar]
- Investigating and predicting how biology changes molecules and their properties. Mol. Inform.. 2014;33:443-445.
- [CrossRef] [Google Scholar]
- Computational methods and tools to predict cytochrome P450 metabolism for drug discovery. Chem. Biol. Drug Des.. 2019;93:377-386.
- [CrossRef] [Google Scholar]
- Structural insights into the lipid and ligand regulation of serotonin receptors. Nature. 2021;592:469-473.
- [CrossRef] [Google Scholar]
- New dual ligands for the D2 and 5-HT1A receptors from the group of 1,8-naphthyl derivatives of LCAP. Bioorg. Med. Chem. Lett.. 2019;29:2236-2242.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.104636.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




