

Tetracyclic triterpenoids as inhibitors of cytochrome P450 3A4 and their quantitative structure activity relationship analysis
⁎Corresponding authors at: College of Pharmacy, Second Affiliated Hospital, Dalian Medical University, 9 Western Section, Lvshun South Road, Dalian 116044, China. ningjing0626@163.com (Jing Ning), maxc1978@163.com (Xiao-Chi Ma)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
The interaction between herbs and clinical drugs is one of the risk factors for adverse drug reactions. In the present study, the inhibitory effects of 47 tetracyclic triterpenoids toward the main drug-metabolizing CYPs in humans were investigated. Most of the evaluated tetracyclic triterpenoids (TT) exhibited strong inhibitory effects toward CYP3A4 compared with other CYP subtypes. The inhibition kinetics of compounds 9 (23-acetyl alisol C), 44 (hemslecin A), and 47 (cucurbitacin E) against CYP3A4 were studied, and inhibition constant (Ki) was determined as 2.12, 0.196, and 0.162 µM, respectively. Some representative TT exhibited significant inhibitory effects toward the metabolism of gefitinib, atorvastatin, and quetiapine, indicating a potential interaction between TT derivatives and clinical drugs. Additionally, a quantitative structure–activity relationship (QSAR) study of a series of TT as inhibitors of CYP3A4 was performed. The present study provided key information to guide the rational use of herbs rich in TT.
Keywords
Tetracyclic triterpenoids
Cytochrome P450 3A4
Structure-activity relationship
Herbal-drug interactions
Inhibition mechanism

- 3D-QSAR
-
three-dimensional quantitative structure–activity relationship
- AAC
-
23-acetyl alisol C
- CBE
-
cucurbitacin E
- CoMFA
-
comparative molecular field method
- CYP
-
Cytochrome P450
- HSA
-
hemslecin A
- MDZ
-
midazolam
- PLS
-
partial least squares
- PPD
-
protopanaxadiol
- PPT
-
protopanaxatriol
- PT
-
panaxatriol
- TT
-
tetracyclic triterpenoids
Abbreviations
1 Introduction
Tetracyclic triterpenoids (TT) are widely distributed natural compounds characterized by 30 carbon atoms containing A, B, C, and D rings. The major class of TT can be categorized into dammarane, lanostane, protostane, cucurbitane, and cycloartane group, which are mainly distributed in Cucurbitaceae, Scrophulariaceae, Ranunculaceae, Araliaceae, and so on (Hill and Connolly, 2011, 2020). Many traditional valuable healthcare medicines such as Panax ginseng and Ganoderma lucidum have TT as their main active ingredients (Hamid et al., 2015). For the past few years, a growing body of research indicated that such compounds possessed significant pharmacological effects including anti-cancer (Wang and Fang, 2009), anti-inflammatory (Braca et al., 2011), anti-diabetic (Alqahtani et al., 2013), and anti-Alzheimer activities (Yoo and Park, 2012). The antitumor activities of cucurbitacins have received widespread attention, and their antiproliferation and pro-apoptosis effects were found associated with JAK2/STAT3, P13K/Akt, and MAPK signaling pathways (Zheng et al., 2014; Wang et al., 2016). The marked effects of cucurbitacin E and B on cancer cell lines were evaluated down to the nanomolar level (Duncan et al., 1996). Additionally, the health benefits of TT including anti-obesity, and ameliorating effects on vascular dysfunctions (Sheng and Sun, 2011) have been widely recognized. Taking into consideration the broad applicability and high intake of such compounds in our daily life, the potential interactions of TT with market drugs should be more concerned and evaluated.
Cytochrome P450 (CYP) superfamily is a group of heme-containing proteins, that mainly participates in the oxidative metabolism of many foreign chemicals in mammalian species (Galetin et al., 2008). CYP3A4 mediates the metabolism of about 60% of therapeutic drugs, and is regarded as one of the most important drug-metabolizing enzymes in humans (Rendic, 2002; Zhou et al., 2005). Therefore, any potential inhibitory effects of chemicals including natural products may impose a metabolic disturbance on clinical drugs that mainly undergo CYP3A4-mediated metabolism (Rendic, 2002; Zhou et al., 2005; Guo and Yamazoe, 2004; Li et al., 2013).
In the previous study, there were a few investigations reported that the in vivo metabolism of TT in human were mediated by CYP3A4, indicating the wide range of interactions of TT with CYP3A4 (Hao et al., 2010; Yu et al., 2013). Notably, the activity inhibitory effects of ginsenosides and ganoderic acids toward CYP3A4 had been reported, indicating an interaction risk of TT on clinical drugs that undergoing CYP3A4-mediated metabolism (Liu et al., 2006; Xu et al., 2020). It was found that intestinal metabolites of ginsenosides including protopanaxatriol (PPT) and protopanaxadiol (PPD) both exhibited potent inhibitory effects toward CYP3A4 (Liu et al., 2006). However, the interaction and relationship between TT and this key drug-metabolizing enzyme have not been fully elucidated.
Accordingly, and given the importance of inhibitory effects of TT derivatives against CYP3A4, our present study aims to: 1) measure the potential inhibitory effects of TT toward CYP3A4 and other main drug metabolizing CYP subtypes; 2) explore the inhibition characteristics and mechanism of TT toward CYP3A4; 3) construct a quantitative structure–activity relationship (QSAR) analysis between CYP3A4 inhibiting effects and TT. The results could provide some useful information for elaborating a relationship between the natural products and CYP3A4, facilitating the prediction of the potential herbal-drug interaction to guide the rational use of herbal medicine with chemical drugs.
2 Material and methods
2.1 Chemicals and reagents
The compounds including toosendanin (42), cucurbitacin B (43), hemslecin A (44, HSA), cucurbitacin Ⅱb (45), pachymic acid (46), cucurbitacin E (47, CBE) were purchased from Herbest Biotechnology Co., Ltd (Baoji, China). (24E)-coccinic acid (23), kadpolysperin I (24), schisanlactone B (25), kadcoccinone N (26), schisandronic acid (27), heteroclitalactone B (28), heteroclitalactone E (29), were kindly provided by Prof. Wei Wang (Hunan University of Chinese Medicine, Wang et al., 2006). The other compounds were isolated and identified in our laboratory (Mai et al., 2015; Wei et al., 2017; Zhang et al., 2017; Wei et al., 2018; Sun et al., 2020; Luan et al., 2019). NADPNa2, glucose-6-phosphate dehydrogenase, glucose-6-phosphate, and midazolam (MDZ) were obtained from Sigma-Aldrich (St. Louis, MO). The human liver microsomes were obtained from Bioreclamation IVT (MD, USA) and stored at −80 °C until use.
2.2 Screening of the inhibitory effects of 47 tetracyclic triterpenoids against CYP3A4
The inhibitory effects of 47 TT against CYP3A4 mediated midazolam 1′-hydroxylation were evaluated. The NADPH-generating incubation system (1 mM NADPNa2, 4 mM MgCl2, 1 unit/mL glucose-6-phosphate dehydrogenase, 10 mM glucose-6-phosphate), HLM (0.05 mg/mL), and substrate MDZ incubated in 100 mM potassium phosphate buffer (pH 7.4) in the presence (inhibition samples) or absence (control samples) of various TT compounds (10 µM). The incubation samples were initiated by adding NADPNa2 after 3 min preincubation at 37 °C. Continuous incubation at 37 °C for 7.5 min, adding 100 μL acetonitrile to stop the enzymatic reaction. The incubation samples were centrifugated at 20,000 g for 20 min, and the supernatants were analyzed by LC-MS. The residual activity was calculated by the relative value of the formation rate of 1′-hydroxylated metabolite of MDZ of inhibition samples and control samples.
2.3 Evaluation of the inhibitory effects of 47 tetracyclic triterpenoids toward various CYP subtypes
The inhibition of 47 TT toward other mainly drug metabolizing CYP subtypes distributed in HLM, including CYP1A2, 2B6, 2C8, 2C9, 2C19 and 2D6, was evaluated using respective probe substrate. The functionality of each CYP isoforms was assessed by determining the catalytic activities, that is, phenacetin O-deethylation for CYP1A2, bupropion 4-hydroxylation for CYP2B6, taxol 6-hydroxylation for CYP2C8, diclofenac 4′-hydroxylation for CYP2C9, S-Mephenytoin 4′-hydroxylation for CYP2C19, dextromethorphan O-demethylation for CYP2D6. The incubation conditions were consistent with the previous studies (He et al., 2015; Li et al., 2018).
2.4 IC50 determination
Varying concentrations of 47 TT (25 nM-150 µM) were incubated with MDZ (5 µM) for 7.5 min. The inhibitory dose–response curves were obtained to fit the data with the program Prism (Version 8.0, GraphPad, San Diego, CA). Other conditions were consistent with the described above.
2.5 Inhibition kinetic characterization
Varying concentrations of 23-acetyl alisol C (AAC), HSA, CBE (0.1–25 µM) were incubated for 7.5 min with varying concentrations of MDZ (1–25 µM). The Lineweaver–Burk and Dixon plots were obtained to fit the data with the program Prism (Version 8.0, GraphPad, San Diego, CA). The inhibition constant (Ki) values were determined using the following equations:
When α is very large (a greater than 1), the mixed-model becomes identical to competitive inhibition.
2.6 LC–MS assay
A standard ExionLC AD HPLC system was equipped with a Luna Omega PS C18 (2.1 × 100 mm, 3 μm) analytical column. The mobile phase consisted of 0.1% formic acid aqueous solution (A) and acetonitrile (B), were conducted with the 0.30 mL/min flow rate as follows: 0–1.5 min (10–10% B), 1.5–3.0 min (10–45% B), and 3.0–5.0 min (45–90% B). An Applied Biosystems SCIEX QTRAP 5500 with electrospray ionization (ESI) source was used to analyze target metabolites. The analyses were performed in positive mode, and used the following values: 1′-hydroxylated MDZ (342.0 → 324.0), deethylated phenacetin (152.1 → 110.0), 4-hydroxylated bupropion (256.0 → 238.0), 6-hydroxylated paclitaxel (892.4 → 607.2), 4-hydroxylated diclofenac (312.1 → 231.0), 4′-hydroxylated S-mephenytoin (235.0 → 150.0), demethylated dextromethorphan (258.1 → 157.0), defluorinated gefitinib (445.0 → 128.0), 2- and 4-hydroxylated atorvastatin (575.0 → 440.0), S-oxidated quetiapine (400.1 → 177.9). Related parameters for the quantification assay setting refer to Table S1 in the supplementary materials.
2.7 3D-QSAR model studies
The comparative molecular field method (CoMFA) has been widely used in the discovery and optimization of lead compounds (Melo-Filho et al., 2014; Yang et al., 2011). In this study, the CoMFA method was used to construct a three-dimensional quantitative structure–activity relationship (3D-QSAR) model based on pIC50.
During the modeling process, all molecules were prepared in SYBYL X1.1 by minimizing in the standard force field (Tripos), adding Gasteiger-Huckel charge and searching all molecules to produce the lowest energy conformation of small molecules. Then, the lowest energy conformation was used as the initial structure of the CoMFA model. In order to build the CoMFA model and verify its predictive ability, we divide all molecules into a training set and a test set. Considering the distribution of biological activity data and the diversity of chemical structures, 4, 6, 15, 24, 30, 37 and 44 were selected from 47 compounds as the test set, and the remaining 40 compounds as the training set. In order to modeling the biologically active conformations, compounds in the training set were superimposed based on a common scaffold, and 49 (HSA) with the largest pIC50 value was set as template molecule of superimposition. During the construction of CoMFA, the steric field and electrostatic field are calculated. Then, Partial Least Squares (PLS) in SYBYL-X 1.1 was applied to perform CoMFA modeling. The pIC50 value of the training set in the CoMFA model is set as Y, and the CoMFA parameter values as X. The optimal number of components (ONC) was obtained by leave-one-out (LOO) cross-validation analysis. After the model is built, internal and external validation are used to evaluate the robustness (R2 and Q2) and predictive ability. The statistical parameters of the CoMFA model are listed in Table S2 in the supplementary materials.
| Compound | Inhibition type | Ki (µM) | ɑ* | The goodness of fit (R2) |
|---|---|---|---|---|
| CBE | Mixed inhibition | 0.162 | 8.97 | 0.9828 |
| AAC | Mixed inhibition | 2.12 | 10.5 | 0.9046 |
| HSA | Mixed inhibition | 0.196 | 55.9 | 0.9292 |
2.8 Interactions between tetracyclic triterpenoid derivatives and clinical drugs
In the present study, quetiapine, atorvastatin and gefitinib that mainly undergoing CYP3A4 mediated metabolism were used as potential interacting clinical drugs with TT derivatives. The inhibitory effects of five representative TT derivatives (0.1–100 µM) toward the oxidation metabolism of the above-mentioned clinical drugs were evaluated. The concentration of clinical drugs in incubation system was seated according to the corresponding reported Km values of probe substrates. The incubation conditions were consistent with the previous studies (Li et al., 2018).
3 Results and discussions
3.1 Inhibitory effects of tetracyclic triterpenes derivatives toward CYP3A4
The inhibitory effects of 47 TT toward CYP3A4 were first evaluated (Scheme 1). It was found that, at the concentration of 10 μM, alismanin D (2), (23S)-11β,23-dihydroxy-8α,9β,14β-dammar-13(17)-ene-3,24-dione (3), 23-acetyl alisol C (AAC, 9), alisol B 23-acetate (10), alisol B (AB, 11), kadpolysperin I (24), kadcoccinone N (26), panaxatriol (PT, 32), (20S)-protopanaxdiol (S-PPT, 34), hemslecin A (HSA, 44) and cucurbitacin E (CBE, 47) exhibited a strong inhibitory effects toward CYP3A4 mediated MDZ 1′-hydroxylation, and the percentage remaining activity were all less than 25% (Fig. 1). The compound 47 (cucurbitacin E, CBE), a major component of Curcurbitaceae plants, was a potential inhibitor toward CYP3A4 with the percentage remaining activity determined as 5.3%. In contrast, the activity of CYP3A4 was slightly modulated by compounds including 7-oxo-ganoderlactone D (12), Ganolactone B (14), ganoderic acid H (15), banoderic acid B (16), 12β-hydroxyganoderenic F (17), ganoderic AM1 (18), ganoderic acid C (19), ganoderic acid F (20), ganoderic acid C6 (21), schisandronic acid (27), ginsenoside Rh1 (39), toosendanin (42) and pachymic acid (46) with the percentage remaining activity more than 75% at the same screening concentration. It can be found that ganoderic acid all exhibited weak inhibitory effects toward CYP3A4.
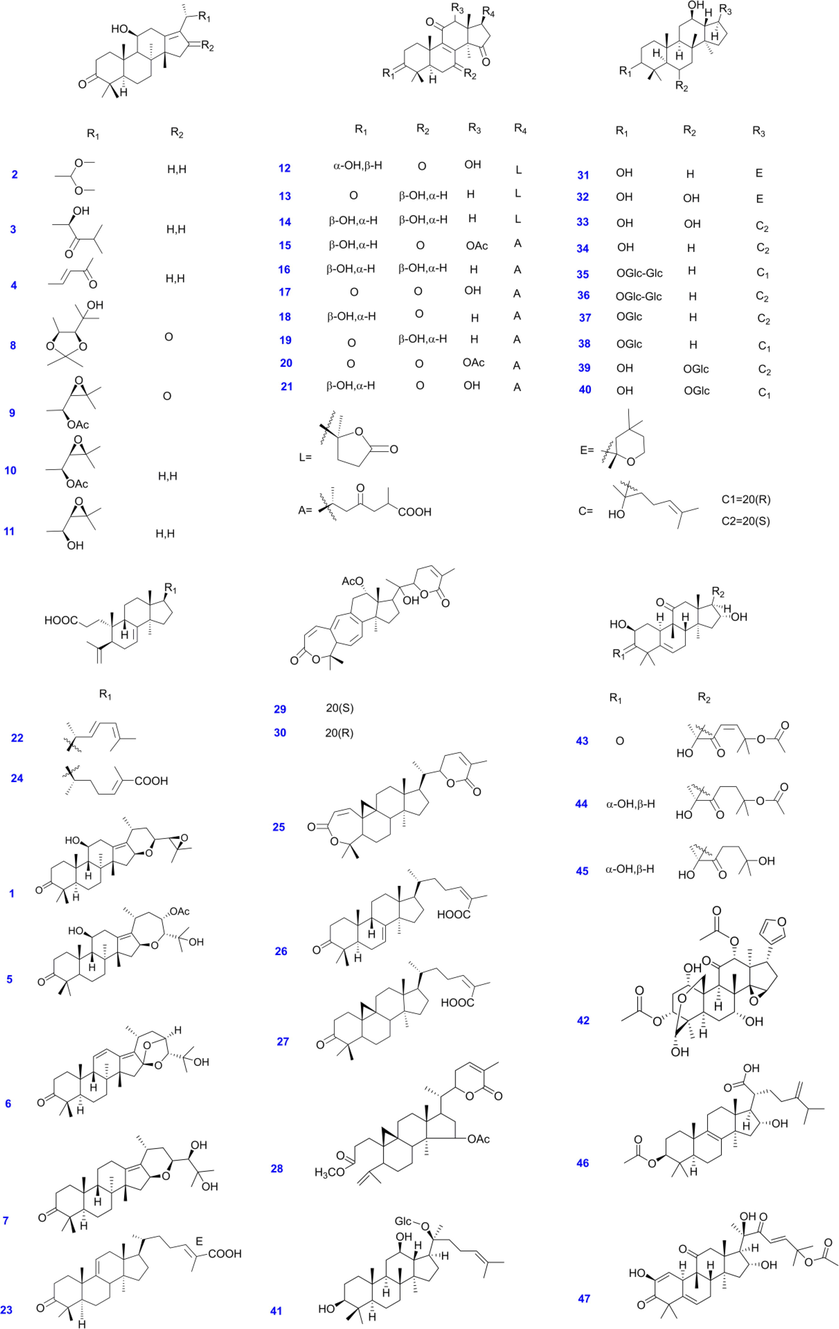
- The structures of tetracyclic triterpenoids (1–47) are used.
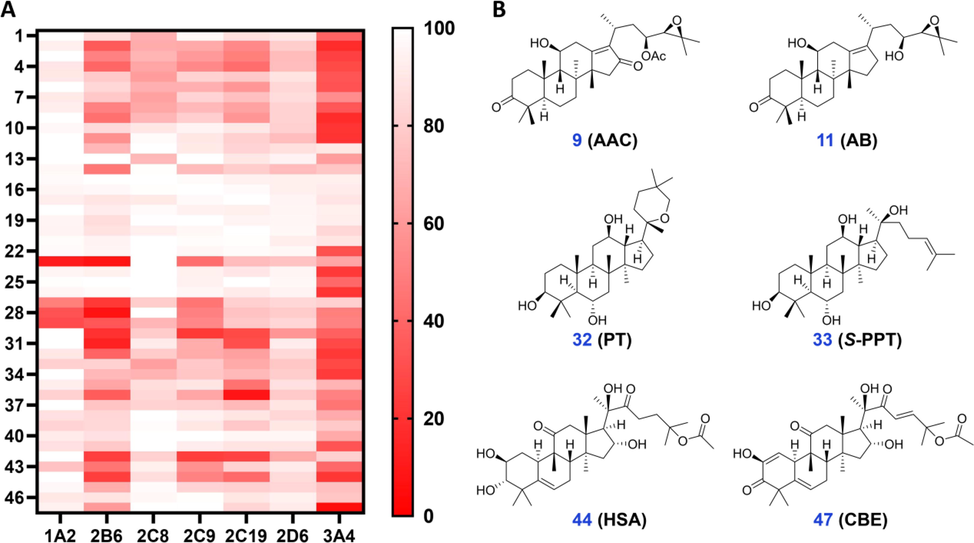
- (A) A brief profile of inhibitory effects of tetracyclic triterpenoids (1–47) toward major human CYP isoforms in the human liver. (B) The representative tetracyclic triterpenoids exhibited strong inhibitory effects toward CYP3A4.
The dose-dependent inhibition behaviors of representative TT toward CYP3A4 were evaluated (Fig. 2). The inhibitory activities of compounds including 9 (AAC), 11 (AB), 32 (PT), 33 (S-PPT), 44 (HSA), and 47 (CBE) expressed as IC50 values and determined as 1.42, 1.27, 1.80, 4.94, 0.65 and 0.42 µM, respectively.
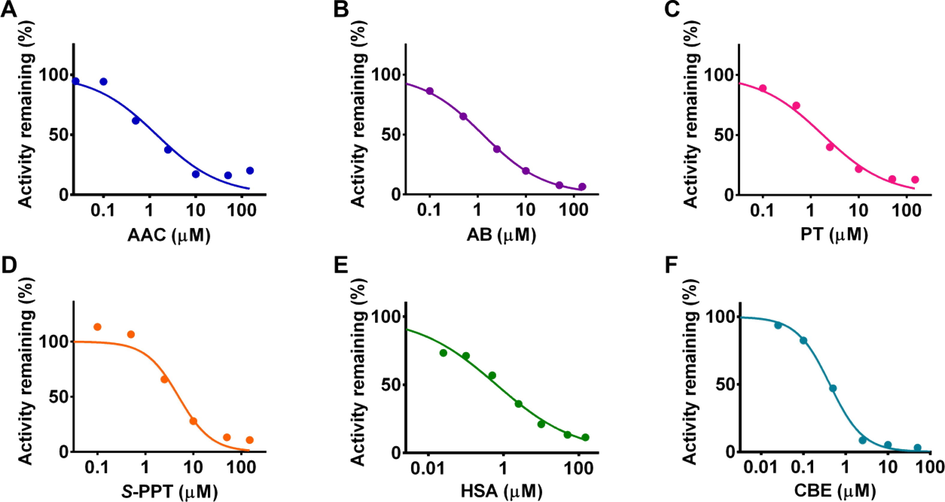
- Dose-dependent inhibition behavior of tetracyclic triterpenoids toward CYP3A4.
3.2 Inhibitory effects of tetracyclic triterpenoids toward other CYP subtypes
To explore the inhibitory selectivity of 47 TT, the inhibitory effects of these compounds toward other CYP subtypes that are mainly involved in drug metabolism were evaluated. Compound 47, which was the potential inhibitor of CYP3A4, exhibited very weak inhibitory effects toward other CYP subtypes at the concentration of 10 μM (Fig. 1). And the extensive strong inhibitory effects toward CYP3A4 of a majority of TT were not observed in other CYP subtypes. It was found that a few TT inhibited CYP2B6 with strong inhibition activity, including (24E)-coccinic acid (23), heteroclitalactone B (28), and panaxadiol (31). However, the TT all exhibited relatively weak inhibitory effects toward CYP2C8 and CYP2D6, with a percentage remaining activity were all more than 75%. Additionally, it was found that compound 23 strongly inhibited CYP1A2; compounds 30 and 42 strongly inhibited CYP2C9; and compound 36 strongly inhibited CYP2C19. Overall, TT derivatives exhibited a strong inhibitory effect toward CYP3A4 in comparison with other CYP subtypes.
3.3 3D-QSAR of tetracyclic triterpenoids by CoMFA
The strong inhibitory effects of TT prompted us to explore the interaction relationship between TT and CYP3A4. Above all, the IC50 values were obtained for the 47 ligands to screen for their inhibitory ability toward CYP3A4. All the data were listed in Table S2, and it was found that the variation of the IC50 values for these TT was very large.
Subsequently, the pIC50 is set as the dependent variable for the construction of the CoMFA model. Considering the distribution of biological activity data and the diversity of chemical structures, compounds 4, 6, 15, 24, 30, 37, and 44 were selected from 47 compounds as the test set, and the remaining 40 compounds as the training set. The results of partial least square (PLS) analysis are listed in Table S3, and plots of the predicted values against experimental values are shown in Figure S1. The CoMFA model for the training set yields statistically significant results with the cross-validated q2 of 0.522 and non-cross-validated r2 of 0.936. The raw experimental and predicted pIC50 values was shown in Table S2, and the correlation is shown in Fig. 3. All these parameters indicate that the CoMFA model is more reliable and the consistency of internal and external data sets is good.
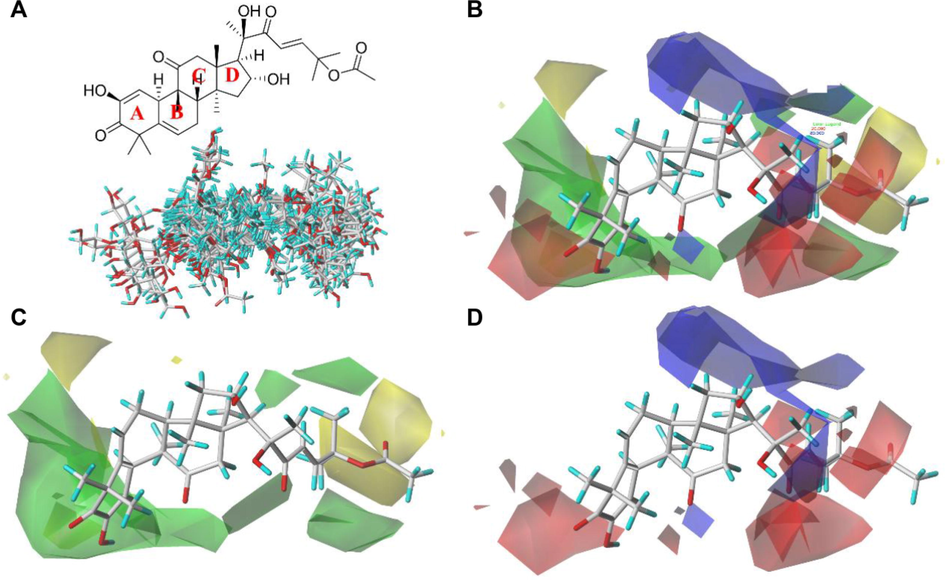
- The 3D quantitative structure–activity relationship of the tetracyclic triterpenoids based on the ligands. (A) The conformational overlap of 47 tetracyclic triterpenoids with compound 47 as a template molecule. (B) Equipotential diagram of CoMFA model based on template molecule 47. (C) Stereo field equipotential map of CoMFA model. The favorable area of the stereo field is shown in green, and the unfavorable area is shown in yellow. (D) The electrostatic field equipotential diagram of the CoMFA model. The positively charged area is shown in blue, and the negatively charged area is shown in red.
The equipotential map generated by the PLS model can explain the relationship between the three-dimensional structures and inhibition activities of these compounds. Based on the conformational superposition of 47 TT inhibitors (Fig. 3A), the 3D-QSAR model of the steroid inhibitors was obtained by constructing a COMFA equipotential map (Fig. 3B). It can be seen that the inhibition activities of these TT were simultaneously affected by the stereo field and electrostatic fields, and the electrostatic contribution is nearly two times the contribution of the steric field. The steric field is mainly distributed on two sides of the TT scaffold (the A ring and D ring regions, see Fig. 3C), indicating that the modification of the substituents in these regions has a greater impact on the inhibitory activity of the TT inhibitors. The green contour indicates that the substitution of a larger group can increase the inhibition activity, while the yellow contour indicates that the substitution of a smaller group helps to increase the inhibition activity. As shown in Fig. 3D, the distribution of the electrostatic field is relatively dispersed. Among them, there is a favorable area of negative charge in the area near the A ring, indicating that the appropriate negative group substitution can enhance the activity of the inhibitor. The positively charged and negatively charged regions exist simultaneously around the D ring change, which indicates that increasing the positively charged and negatively charged groups in the corresponding regions can enhance the activity of the inhibitor.
3.4 Inhibition kinetics of representative tetracyclic triterpenes toward CYP3A4
The inhibition kinetics of CYP3A4 by CBE, AAC, and HSA were performed to explore the possible inhibition mechanism of TT. As shown in Fig. 4, the Dixon and Lineweaver–Burk plots of three TT indicated that CYP3A4 inhibition was all fitted in a mixed inhibition manner. Notably, the fitting parameters α were all greater than 1, indicating that the mixed model for CBE, AAC, and HSA was inclined to competitive inhibition (Table 1). The value of Ki for CBE, AAC, and HSA was determined as 0.162, 2.12, and 0.193 µM, respectively.
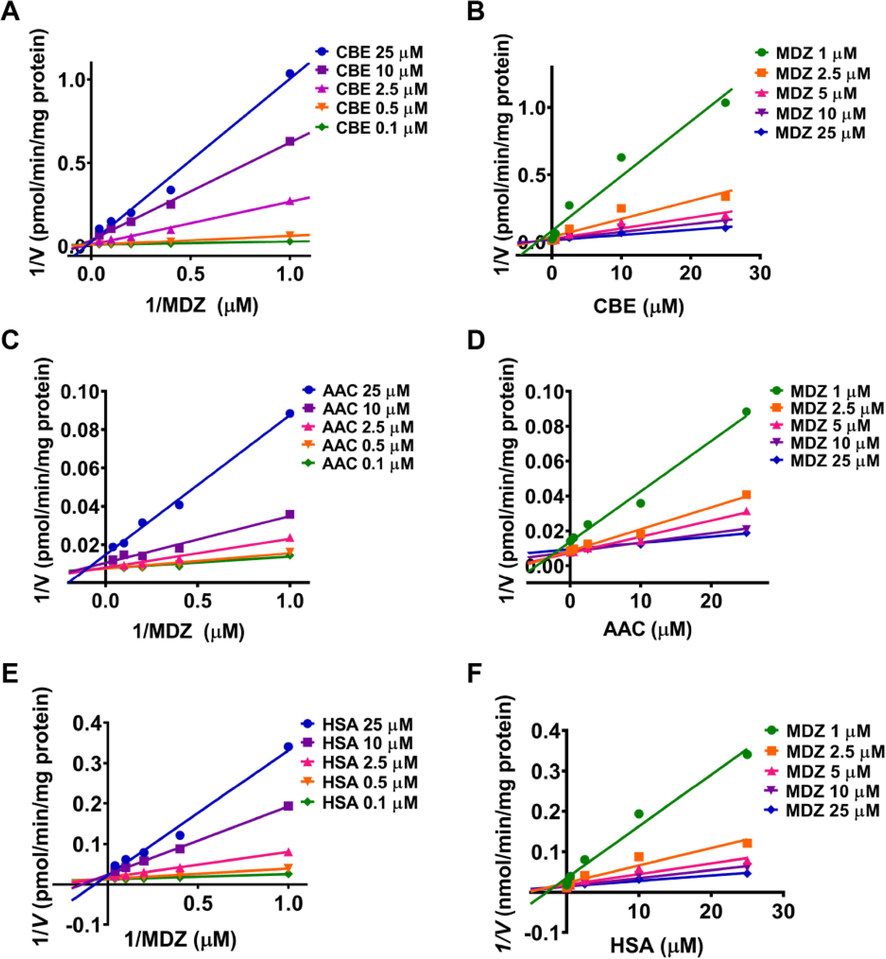
- The Lineweaver-Burk and Dixon plots for CYP3A4 inhibition in the presence of CBE (A and B), AAC (C and D), and HSA (E and F), respectively.
3.5 Interactions between representative tetracyclic triterpenes and clinical drugs
Some clinical drugs that mainly undergo CYP3A4-mediated metabolism including gefitinib, atorvastatin, and quetiapine were used to evaluate the potential interaction between TT and clinical drugs. The IC50 values of CBE, AAC, HSA, S-PPT, and PT were determined and listed in Table 2. The involving TT exhibited significant inhibitory effects toward the CYP3A4-mediated oxidative metabolism of gefitinib, atorvastatin, and quetiapine. Therein, CBE strongly inhibited the metabolism of gefitinib, atorvastatin, and quetiapine, with the IC50 lying between 0.94 and 3.75 µM.
| Substrate | IC50 (µM) | ||||
|---|---|---|---|---|---|
| CBE | AAC | HSA | S-PPT | PT | |
| Gefitinib | 0.94 | 16.55 | 5.32 | 5.64 | 1.12 |
| Atorvastatin | 1.24 | 2.31 | 13.30 | 14.66 | 12.42 |
| Quetiapine | 3.75 | 12.13 | 12.03 | 11.88 | 5.68 |
CBE and HSA are major components of curcurbitaceae plants including Hemsleya amabilis Diels, Trichosanthes kirilowii Maxim, and Cucurbita moschata Duch that displayed anti-inflammatory, antipyretic, and antibacterial activity (Kaushik et al., 2015). Recently, this type of TT has been proven to have a strong anticancer activity the antiproliferative effect of CBE on PC 3, HL-60, and Bel-7402 lay between 10 and 43 nM, respectively (Chen et al., 2012). Cucurbitacin tablets and other drugs that contain these active ingredients have served as adjutant drugs in tumor therapy (Wang et al., 2017).
In the present study, the potential interaction between CBE and gefitinib was observed, indicating that more attention should be paid to the monitoring of gefitinib blood concentration and side effects when using CBE-containing drugs as adjutant agents. Additionally, S-PPT and PT are important metabolites of ginsenosides that are enriched in commonly used herbs such as Panax notoginseng (Burkill) F. H. Chen ex C. H. and Panax ginseng C. A. Meyer (Liu et al., 2006). The high frequency in the daily life of these herbal medicines may induce herbal-drug interaction due to the strong inhibitory effects of metabolites of ginsenosides toward CYP3A4. Overall, the results indicated that the interactions between TT and clinical drugs may be widespread, and the potential drug interaction induced by TT and they are containing herbal medicine should be taken attention.
4 Conclusions
In the present study, we evaluated the inhibitory effects of a series of TT toward main drug-metabolizing CYP subtypes in humans. The results indicated that TT exhibited strong inhibitory effects toward CYP3A4 compared with other CYP subtypes. Subsequently, a quantitative structure–activity relationship (QSAR) study of a series of TT as inhibitors of CYP3A4 was performed. The results disclosed the key domain and substituents that affect CYP3A4 inhibitory activity. Furthermore, the representative TT also displayed strong inhibitory effects on the metabolism of clinical drugs. Therefore, the present study indicated a potential metabolism-mediated interaction between TT derivatives and clinical drugs and could provide key information to guide the rational use of herbs rich in TT.
CRediT authorship contribution statement
Wei Pan: Investigation, Methodology, Writing – original draft. Lei Feng: Investigation, Methodology, Writing – original draft. Cheng-Peng Sun: Investigation, Methodology, Writing – original draft. Xiang-Ge Tian: Methodology, Software. Chao Shi: Methodology, Software. Chao Wang: Formal analysis. Xia Lv: Methodology, Software. Yan Wang: Methodology, Software. Shan-Shan Huang: Formal analysis. Bao-Jing Zhang: Formal analysis. Jing Ning: Conceptualization, Project administration, Writing – review & editing, Funding acquisition. Xiao-Chi Ma: Conceptualization, Project administration, Writing – review & editing, Funding acquisition.
Acknowledgements
The authors thank the National Natural Science Foundation of China (82174228 and 82004211), National Key R&D program of China (2018YFC1705900), Distinguished professor of Liaoning Province (XLYC2002008), Dalian Science and Technology Leading Talents Project (2019RD15).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- The pentacyclic triterpenoids in herbal medicines and their pharmacological activities in diabetes and diabetic complications. Curr. Med. Chem.. 2013;20:908-931.
- [Google Scholar]
- Triterpene derivatives as inhibitors of protein involved in the inflammatory process: molecules interfering with phospholipase A2, cycloxygenase, and lipoxygenase. Curr. Drug Targets. 2011;12:302-321.
- [Google Scholar]
- Biological activities and potential molecular targets of cucurbitacins: a focus on cancer. Anticancer Drugs. 2012;23:777-787.
- [Google Scholar]
- Cucurbitacin E-induced disruption of the actin and vimentin cytoskeleton in prostate carcinoma cells. Biochem. Pharmacol.. 1996;52:1553-1560.
- [Google Scholar]
- Potential role of intestinal first-pass metabolism in the prediction of drug-drug interactions. Expert Opin. Drug Metab. Toxicol.. 2008;4:909-922.
- [Google Scholar]
- Inhibition of cytochrome P450 by furanocoumarins in grapefruit juice and herbal medicines. Acta Pharmacol. Sin.. 2004;25:129-136.
- [Google Scholar]
- Tetracyclic triterpenoids in herbal medicines and their activities in diabetes and its complications. Curr. Top Med. Chem.. 2015;15:2406-2430.
- [Google Scholar]
- Microsomal cytochrome p450-mediated metabolism of protopanaxatriol ginsenosides: metabolite profile, reaction phenotyping, and structure-metabolism relationship. Drug Metab. Dispos.. 2010;38:1731-1739.
- [Google Scholar]
- Inhibition of human cytochrome P450 enzymes by licochalcone A, a naturally occurring constituent of licorice. Toxicol. In Vitro. 2015;29:1569-1576.
- [Google Scholar]
- Cucurbitacins-An insight into medicinal leads from nature. Pharmacogn. Rev.. 2015;9:12-18.
- [Google Scholar]
- Pungent ginger components modulates human cytochrome P450 enzymes in vitro. Acta Pharmacol. Sin.. 2013;34:1237-1242.
- [Google Scholar]
- Drug interaction study of flavonoids toward CYP3A4 and their quantitative structure activity relationship (QSAR) analysis for predicting potential effects. Toxicol. Lett.. 2018;294:27-36.
- [Google Scholar]
- Ginsenoside metabolites, rather than naturally occurring ginsenosides, lead to inhibition of human cytochrome P450 enzymes. Toxicol. Sci.. 2006;91:356-364.
- [Google Scholar]
- Highly potent non-steroidal FXR agonists protostane-type triterpenoids: Structure-activity relationship and mechanism. Eur. J. Med. Chem.. 2019;182:111652
- [Google Scholar]
- Protostane triterpenoids from the rhizome of alisma orientale exhibit inhibitory effects on human carboxylesterase 2. J. Nat. Prod.. 2015;78:2372-2380.
- [Google Scholar]
- 3D-QSAR approaches in drug design: perspectives to generate reliable CoMFA models. Curr. Comput. Aided Drug Des.. 2014;10:148-159.
- [Google Scholar]
- Summary of information on human CYP enzymes: human P450 metabolism data. Drug Metab. Rev.. 2002;34:83-448.
- [Google Scholar]
- Synthesis, biology and clinical significance of pentacyclic triterpenes: a multi-target approach to prevention and treatment of metabolic and vascular diseases. Nat. Prod. Rep.. 2011;28:543-593.
- [Google Scholar]
- Protostane-type triterpenoids as natural soluble epoxide hydrolase inhibitors: Inhibition potentials and molecular dynamics. Bioorg. Chem.. 2020;96:103637
- [Google Scholar]
- Pentacyclic triterpenoids and their saponins with apoptosis-inducing activity. Curr. Top Med. Chem.. 2009;9:1581-1596.
- [Google Scholar]
- New triterpenoids from Kadsura heteroclita and their cytotoxic activity. Planta Med.. 2006;72:450-457.
- [Google Scholar]
- Cucurbitacin E inhibits osteosarcoma cells proliferation and invasion through attenuation of PI3K/AKT/mTOR signaling. Biosci. Rep.. 2016;36:e00405.
- [Google Scholar]
- Simultaneous determination of cucurbitacin B and cucurbitacin E in rat plasma by UHPLC-MS/MS: A pharmacokinetics study after oral administration of cucurbitacin tablets. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci.. 2017;1065–1066:63-69.
- [Google Scholar]
- C27-Nor lanostane triterpenoids of the fungus Ganoderma lucidum and their inhibitory effects on acetylcholinesteras. Phytochem. Lett.. 2017;20:263-268.
- [Google Scholar]
- Chemical characteristics of the fungus Ganoderma lucidum and their inhibitory effects on acetylcholinesterase. J. Asian Nat. Prod. Res.. 2018;20:992-1001.
- [Google Scholar]
- In vitro inhibitory effects of ganoderic acid A on human liver cytochrome P450 enzymes. Pharm. Biol.. 2020;58:308-313.
- [Google Scholar]
- Molecular dynamics simulation, free energy calculation and structure-based 3D-QSAR studies of B-RAF kinase inhibitors. J. Chem. Inf. Model. 2011;51:680-692.
- [Google Scholar]
- Terpenoids as potential anti-Alzheimer's disease therapeutic. Molecules. 2012;17:3524-3538.
- [Google Scholar]
- In vitro metabolism of alisol A and its metabolites' identification using high-performance liquid chromatography-mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci.. 2013;941:31-37.
- [Google Scholar]
- Novel protostane-type triterpenoids with inhibitory human carboxylesterase 2 activities. RSC Adv.. 2017;7:28702.
- [Google Scholar]
- Cucurbitacin B inhibits growth and induces apoptosis through the JAK2/STAT3 and MAPK pathways in SH-SY5Y human neuroblastoma cells. Mol. Med. Rep.. 2014;10:89-94.
- [Google Scholar]
- Mechanism-based inhibition of cytochrome P450 3A4 by therapeutic drugs. Clin. Pharmacokinet.. 2005;44:279-304.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.105156.
Appendix A
Supplementary material
The following are the Supplementary data to this article:




