

Translate this page into:
The novel 4-hydroxyphenylpyruvate dioxygenase inhibitors in vivo and in silico approach: 3D-QSAR analysis, molecular docking, bioassay and molecular dynamics
⁎Corresponding authors at: Department of Chemistry, College of Arts and Sciences, Northeast Agricultural University, Harbin 150030, China. yefei@neau.edu.cn (Fei Ye), fuying@neau.edu.cn (Ying Fu) yefei@neau.edu.cn (Ying Fu) fuying@neau.edu.cn (Ying Fu)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
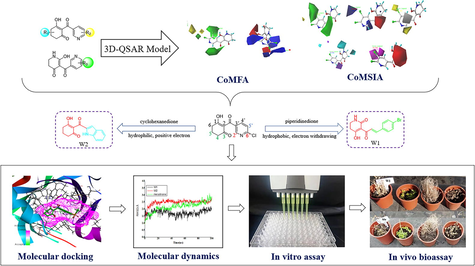
A comprehensive in silico screen approach was set up for design 4-hydroxyphenylpyruvate dioxygenase herbicides. Synthesized compound W1 displayed excellent herbicidal activity. This work paves the way for further design and synthesis of novel HPPD inhibitors.
Abstract
4-Hydroxyphenylpyruvate dioxygenase (HPPD) is not only an important target enzyme for the treatment of type I tyrosinemia, but also a new target for design bleaching herbicides, and it plays key role in the biosynthesis of tocopherol and plastoquinone. Thirty-six known active pyridine derivatives were collected, and comparative molecular field analysis (CoMFA) and comparative molecular similarity index analysis (CoMSIA) models based on common skeleton were constructed to obtain novel HPPD herbicides with higher activity. Two new HPPD inhibitors were rationally designed and synthesized according to the CoMFA and CoMSIA models and verified by enzyme activity, biological assays, and molecular docking. The promising compound W1 ((E)-5-(3-(4-bromophenyl)acryloyl)-6-hydroxy-2,3-dihydropyridin-4(1H)-one) showed better AtHPPD inhibitory activity, and the bioassay results revealed that some weeds showed bleaching symptoms. The good binding stability of W1 and protein was confirmed by molecular dynamics simulation in 100 ns. These results would be highly useful in the progress of new HPPD inhibitors discovery.
Keywords
HPPD inhibitors
3D-QSAR
Molecular docking
Molecular dynamics
Bioassay

1 Introduction
4-Hydroxyphenylpyruvate dioxygenase (HPPD) is an important enzyme in the course of tyrosine metabolism in organisms, and it exists in almost all aerobic organisms (Liang et al., 2020). HPPD is not only an important target remedy for the treatment of type I tyrosinemia, but also an effective target for design herbicides (Hu et al., 2022). HPPD is an oxygenase with iron-dependent nonheme in the 2-His-1-carboxylate facial triad family (Lin et al., 2019a; Raspail et al., 2011; He et al., 2019). HPPD catalyzes the complex conversion of 4-hydroxyphenylpyruvate (HPPA) to homogenate acid (HGA) and plays an important role in the biosynthesis of cofactors plasmoquinone and tocopherol in plants (Li et al., 2021; Wang et al., 2021; Moran, 2014). Photosynthetic damage is due to the inhibition of HPPD-catalyzed reactions in chloroplasts, which leads to leaf bleaching followed by weed necrosis and death (Neidig et al., 2004; Fu et al., 2019a). HPPD inhibitors mainly include pyrazoles, triketones, isoxazoles and others (Ndikuryayo et al., 2017). In particular, the novel pyrazoline-quinazoline-2, 4-diketone hybrids showed good herbicidal activity (He et al., 2020). The newly developed 3-hydroxy-2-(3,5,6-trichloro-4–((4-isopropylbenzyl)amino)picolinoyl) cyclohexyl-2-en-1-one is 5.8-fold more active than the commercially available mesotrione (Nan et al., 2021). The advantages of HPPD inhibitors are their excellent bioactivity, low residue accumulation, broad-spectrum weed control, environmental friendliness, low toxicity, and safe application (Aouidate et al., 2018; Shaner et al., 2004; Wang et al., 2020a; Jia et al., 2019). Hence, the study of novel HPPD inhibitors has been recognized as one of the most promising targets in the field of herbicides (Qu et al., 2021; Lin et al., 2021).
Based on the known crystal structure of HPPD and molecular information of HPPD inhibitors, potential inhibitors can be better validated by molecular docking and molecular dynamics (Sabine et al., 2020; Fu et al., 2020; Singh et al., 2018), which exhibits key features in ligand-receptor binding interactions and enables virtual screening of many compound databases (Xing et al., 2020; Ganesan et al., 2018). These workflows are efficient to identify suitable and potent inhibitors by considering the mechanism of activity prior to the experiment (Sushil et al., 2021). In the past few decades, quantitative structure activity relationship (QSAR) models are an effective calculation tool in drug design and have been widely applied to predict the bioactive compounds (Zhang et al., 2010; Tang et al., 2016; Wang et al., 2017; Dong et al., 2017; Fu et al., 2019b; Mohd et al., 2019). Comparative molecular field analysis (CoMFA) and comparative molecular similarity index analysis (CoMSIA) models were established based on a series of sulfa derivatives, and 35 potential inhibitors against tomato chlorosis virus were successfully developed (Jiang et al., 2021). Cloud 3D-QSAR provides a one-stop solution for molecular interaction field (MIF) calculation and result analysis of integrated molecular structure generation, alignment, and molecular interactions (Wang et al., 2020c).
QSAR combined with molecular docking and molecular dynamics (MD) simulation is a valid means of obtaining highly accurate information about the interactions between ligands and receptors (Liu et al., 2019; Singh et al., 2016; Kothandan et al., 2011). Molecular dynamics helps optimize the understanding of protein function to accelerate drug discovery (Yang et al., 2019). It also provides powerful insights to develop novel herbicides for target enzymes (Huang et al., 2018; Wang et al., 2020b).
In the current study, a group of potential HPPD inhibitors with common skeletons were selected to establish CoMFA and CoMSIA models. Contour maps were employed to guide the design and synthesis of two potential compounds with improved predictive activity. The two synthesized compounds were tested in vivo, and bioassays were performed to verify their bioactivity. Molecular docking and MD studies were employed to analyze and confirm the stability of the receptor–ligand complex and to provide more messages at the binding site.
2 Materials and methods
2.1 Information collection and generation of 3D-QSAR models
In the present study, 36 reported pyridine derivatives were collected as AtHPPD inhibitors (Table 1) (Fu et al., 2019c; Fu et al., 2021). Out of 36 compounds, 29 were randomly chosen as the training set to build the model, and the surplus 6 compounds served as the test set to assess the model. These compounds were selected to cover the entire range of biological activities. Note: a pIC50 = -lg (IC50), calculated with the average value of IC50, IC50: Half maximal inhibitory concentration toward AtHPPD. *Compounds were considered as the test set.
Compound
Experimental value
Predicted value (pIC50)
R1
R2
IC50 (μM)
pIC50a
CoMFA
CoMSIA
1
H
6′-Cl
0.262
6.582
6.573
6.567
2
5-CH3
6′-Cl
3.083
5.511
5.607
5.655
*3
5,5-diCH3
6′-Cl
3.280
5.484
5.475
5.465
4
5-Ph
6′-Cl
4.498
5.347
5.362
5.258
5
H
2′-CF3
2.215
5.655
5.676
5.698
6
5-CH3
2′-CF3
3.451
5.462
5.421
5.392
7
5,5-diCH3
2′-CF3
4.368
5.360
5.362
5.258
*8
5-Ph
2′-CF3
10.473
4.980
4.926
4.894
9
H
5′-Br
2.691
5.570
5.59
5.516
10
5-CH3
5′-Br
4.404
5.356
5.485
5.55
11
5,5-diCH3
5′-Br
13.985
4.854
4.817
4.814
12
H
6′-F
3.489
5.457
5.437
5.456
13
5-CH3
6′-F
8.755
5.058
5.074
5.097
14
5-Ph
6′-F
1.905
5.720
5.698
5.787
*15
H
6′-CF3
0.704
6.152
6.128
6.141
16
5-CH3
6′-CF3
0.791
6.102
6.059
6.044
17
5,5-diCH3
6′-CF3
4.009
5.397
5.33
5.434
18
5-Ph
6′-CF3
0.961
6.017
5.912
6.025
19
H
6′-Br
1.731
5.762
5.716
5.759
20
5-CH3
6′-Br
2.729
5.564
5.593
5.579
21
5,5-diCH3
6′-Br
11.315
4.946
4.926
4.894
*22
5-Ph
6′-Br
0.920
6.036
6.047
6.026
23
H
5′-Cl, 6′-Cl
1.024
5.990
5.953
5.893
24
5-CH3
5′-Cl, 6′-Cl
1.616
5.792
5.78
5.698
25
5,5-diCH3
5′-Cl, 6′-Cl
1.940
5.712
5.698
5.787
Experimental value
Predicted value (pIC50)
R3
IC50 (μM)
pIC50
CoMFA
CoMSIA
26
6′-Br
1.157
5.937
6.014
5.944
27
5′-Cl, 6′-Cl
0.959
6.018
5.912
6.025
28
5′- Br, 6′-Cl
0.470
6.328
6.244
6.282
*29
5′-F
1.720
5.764
5.707
5.782
30
5′-Cl
1.590
5.799
5.834
5.775
31
5′-Br
1.347
5.871
5.836
5.982
32
2′-CF3
2.238
5.650
5.698
5.731
*33
6′-CF3
1.423
5.847
5.721
5.634
34
H
8.556
5.068
5.088
5.104
35
6′-F
1.318
5.880
5.853
5.863
36
6′-Cl
1.267
5.897
5.952
5.909
All 36 compounds were generated the 3D conformations with SYBYL-X 2.0 software. Energy minimization was achieved by a Tripos force field and by applying Gasteiger-Hückel charges. The dataset was imported into the molecular spreadsheet. The maximum number of iterations was 1000, and the Powell gradient energy convergence value was set to 0.005 kcal/mol Å. To obtain good 3D-QSAR model, multiple search method was used to acquire the molecular conformation. Each compound performed 1000 maximum cycles and produced 200 maximum conformations. For the sake of getting an orderly alignment, the lowest energy conformation of the most active compound 1 (2-(6-chloronicotinoyl)-3-hydroxycyclohex-2-en-1-one) was selected as the superposition template. Remainder molecules of the training set were aligned with the common skeleton. The alignment results were shown in Fig. 1.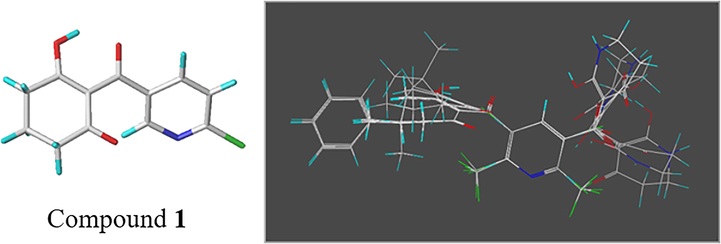
3D-QSAR molecular superposition of the training and test sets using compound 1 as the template.
The CoMFA and CoMSIA models were constructed with 29 compounds as the training set. pIC50 values were selected as the dependent variable for model research. Partial least squares (PLS) regression analysis was applied to generate valid and definite CoMFA and CoMSIA models. The dimensions of the CoMFA fields were calculated by a sp3 carbon with a + 1.0 charge in the process of steric and electrostatic fielding. The regression analysis was performed using the leave-one-out (LOO) cross-validation method. The optimum number of components (ONC), cross-validation coefficient (q2), coefficient of determination (r2), standard error of estimate (SEE) and F value were calculated to evaluate the model. Low SEE values and high q2, r2 and F values were necessary for a favorable model.
2.2 Docking study and physicochemical properties
The preparation of the ligands is the same as that of the dataset in the data collection. The X-ray crystal structure of AtHPPD for molecular docking was gained from the Protein Data Bank (PDB ID: 5YWG) (Lin et al., 2019b). Protein preparation was accomplished by applying the “Prepare Protein” module in Discovery Studio (DS) (Biovia Inc. San Diego, CA, USA, 2020). All water and heteroatoms were deleted from the crystal structure, supplementing missing atoms in residues. The CHARMM force field was used to protonate the structure of HPPD and optimized the conformation of the side chain residues with missing atoms. The binding sites were determined by the cavity position of the co-crystallized ligand in the incipient crystal structure of HPPD.
The co-crystalline ligand mesotrione was redocked to determine whether the CDOCKER program matched the system. The physicochemical properties of the compounds were calculated by the “Calculate Molecular Properties” module of DS.
2.3 Chemical synthesis
The synthetic routes to the compounds (W1 and W2) are depicted in Schemes 1 and 2. The detailed synthetic methods are provided in the Supporting Information.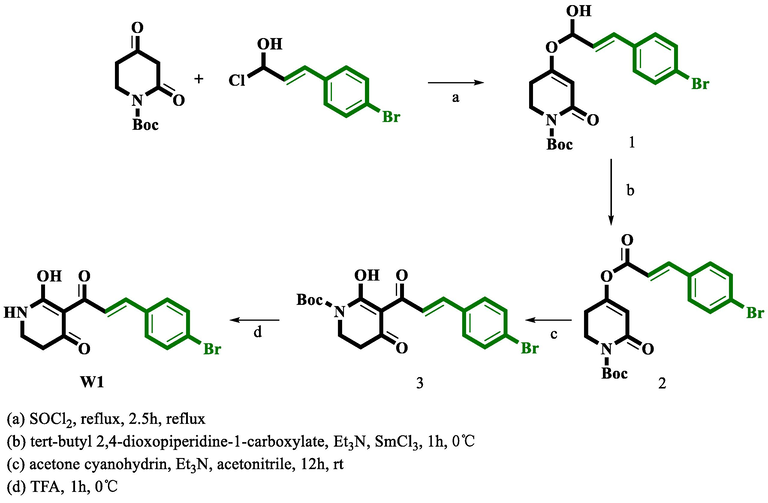
Route for the synthesis of compound W1.
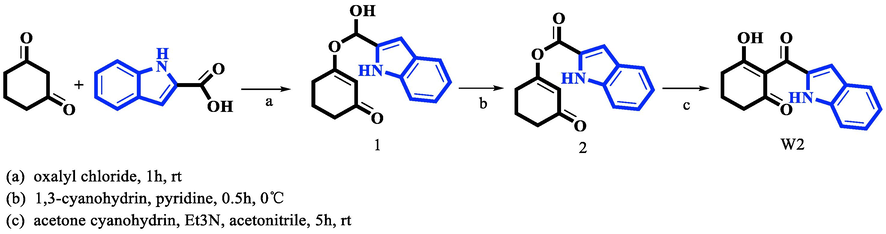
Route for the synthesis of compound W2.
2.4 HPPD inhibitory in vitro assays and herbicidal assays
The HPPD inhibitory activity in vitro was tested by the coupling method. Measurements were executed in 96-well plates (30 ℃) using a microplate reader, and the UV absorption peak was set to 318 nm. Each group of experiments was performed in three replicates and was averaged. The total reaction mixture volume of 200 μL contained 1 mM (20 μL) HPPA, 20 mM (20 μL) sodium ascorbate, 1 mM (20 μL) FeCl2, 20 mM HEPES buffer (NaOH adjusted pH = 7.0, 4-Hydroxyethylpiperazine ethanesulfonic acid), HGD (40 μL), and HPPD (40 μL). The quantity of HGD added was larger than the amount of HPPD during the experiment to ensure that the reaction was completed. Before performing the measurement, all reactants were hatched at 30 °C for no>15 min. IC50 was obtained from the activity curve of the substrate-inhibitor concentration (Song et al., 2021).
The following broad-leaved weeds were selected to assay the in vivo activity: Abutilon theophrasti (Abutilon theophrasti Medik, AJ), Amaranthus retroflexus (Amaranthus retroflexus L., AR), Eclipta prostrata (Eclipta prostrata L., EP), and gramineous weeds: Setaria viridis (Setaria viridis (L.) Beauv., SF), Digitaria sanguinalis (DigitariaviolascensLink, DS), and Echinochloa crus-galli (Echinochloa crusgalli (L.) Beauv., EC). The seeds (85% or higher) germination rate was evenly spread in the potting soil and grown in natural conditions. When the dicotyledonous weeds grew to the 3–4 leaf stage and the monocotyledonous weeds grew to the 1–2 leaf stage, W1 was dissolved in N,N-dimethylformamide containing 1% Tween-80 emulsifier, the concentrate was diluted with deionized water, and plants were sprayed on their stems and leaves after budding in the concentration of 150 g ai/ha. On the third day after treatment, visual evaluations of the plant stems or roots and other factors were performed. 20 Seed of rice (Oryza sativa L.) and wheat (Triticum aestivum L.) were chosen for crop safety test. The commercial herbicides mesotrione was regarded as positive contrast. When the crop entered the four-leaf stage, the safety test of leaf spray crops was carried out at a concentration of 150 g ai/ha. The visual impairment and growth status of individual plants were regularly observed. After 20 days, the crop safety was evaluated (Wang et al., 2014).
2.5 MD simulations
The Amber 16 package was used for MD simulations, and the Amber ff14SB force field was applied to generate the force field of the protein and ligand. Cobalt ions were processed in a metal center parameter generator, and a side chain model containing cobalt ion with His226, His308 and Glu394 composite was created. The obtained structure was immersed in a TIP3P water cube with a distance of at least 10 Å around the box, and an appropriate amount of counter ions was added to the system to neutralize its charge. The energy minimization, heating and balance of each system were realized by the “Sander” program. In each energy minimum step, the steepest descent algorithm was used for the first 2500 steps, and the conjugate gradient algorithm was used for the last 2500 steps. Then the system was gradually heated to 298 K before reaching 1 ns equilibrium in an isobaric-isothermal (NPT) integrated simulation. In the end, the system was subjected to 100 ns of PMEMD program processing in the NPT at a 2 fs time step. Reference values of root mean square deviation (RMSD) was employed to evaluate the stability of the simulation system.
Molecular mechanics and the Poisson-Boltzmann solvation area (MM-PBSA) were applied to analyze the complex affinity. Prime MM-PBSA calculations were executed using the initial and MD-optimized receptor − ligand complexes. The more negative the value of the binding free energy ΔGbind, the stronger the binding force, which was calculated by applying the MM-PBSA method. Prime MM-PBSA ΔGbind, the binding free energy, was calculated with the formula:
2.6 ADMET
ADMET prediction for small molecules was performed in DS software. The following parameters were predicted: Solubility Level, PPB# Prediction, Hepatotoxicity, Aerobic Biodegradability, Mutagenicity, Carcinogenicity, Skin irritancy, and DTP Prediction.
3 Results and discussion
3.1 3D-QSAR models
The experimental and predicted pIC50 values of the CoMFA and CoMSIA models for the training and test sets are shown in Table 1, and their calculated statistical parameters were listed in Table 2. The ONC in the CoMFA model was 7, and the q2 was 0.707 (>0.5), which certified that the CoMFA model had good predictive ability. In the CoMFA model, SEE was 0.128 (less than0.95), and F was 224.565, and r2 was 0.954 (>0.9), suggesting that the model is capable of fine fitting. The contribution of the steric field was 47.3%, slightly lower than the contribution of the electrostatic field of 52.7%. The CoMSIA model displayed good parameters of r2 (0.944), F test value (172.455), and q2 (0.657) and low SEE (0.184), and the ONC was 7. The contributions of steric, electrostatic, hydrophobic, H-bond donor and H-bond acceptor fields were 12.7%, 26.4%, 23.1%, 19.2% and 18.6%, respectively.
Parameter
CoMFA
CoMSIA
Threshold
q2
0.707
0.657
>0.5
ONC
7
7
–
r2
0.954
0.944
>0.6
SEE
0.128
0.184
Low value
F
224.565
172.455
High value
r2 pred
0.865
0.853
>0.6
Steric
47.3
12.7
–
Electrostatic
52.7
26.4
–
Hydrophobic
–
23.1
–
H-bond Donor
–
19.2
–
H-bond Acceptor
–
18.6
–
Electrostatic field made the greatest contribution in CoMFA model. The electrostatic and hydrophobic fields were found to play major roles in CoMSIA model. In Fig. 2, the black squares and red dots were scattered on both sides of the line Y = X, further confirmed that the experimental values of the entire dataset were very close to the predicted values, suggesting the model has superior predictive ability.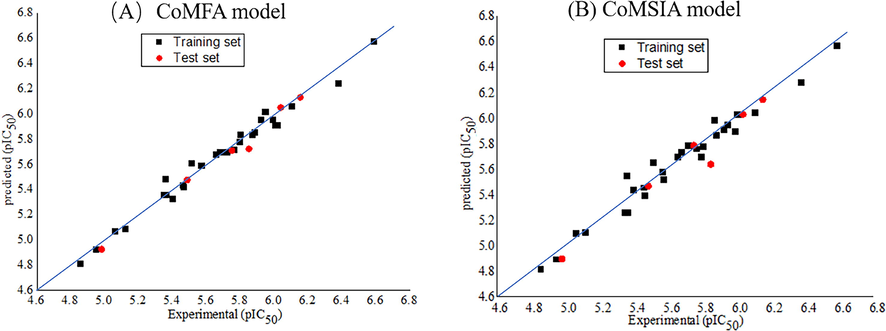
Experimental and predictive activities of molecules in training and test sets. (A) CoMFA model and (B) CoMSIA model.
3.2 Contour maps analysis of CoMFA models
Compound 1 with the best activity was placed in the contour plot as a reference to explain the effects of various fields on activity. The steric field is shown in Fig. 3A, and the green proved that the bulky group was beneficial to the bioactivity of the HPPD inhibitors. In contrast, the small substituents marked in yellow were beneficial for bioactivity. Compounds 1 to 25 were cyclohexanedione derivatives. A small yellow area close to the 5-position of the cyclohexanedione recommended that a small size group at this position had a beneficial effect on inhibition. Compounds exhibited activity when there was no substituent on cyclohexanedione (compounds 1, 5, 9, 12, 15, 19, 23), especially compound 1, which displayed the good activity (Beaudegnies et al., 2009). With the introduction of the methyl group at R1, the inhibitory activity of the compound was obviously decreased. For example, comparing compounds 1 and 3, the inhibitory activity was obviously reduced because the hydrogen atom at the 5-position of compound 1 (IC50 = 0.262 μM) was displayed by a methyl (compound 3, IC50 = 3.083 μM). Fig. 3B depicts the contour map of the electrostatic descriptor. The electropositive group was favorable for improving the bioactivity of the compounds, as proven by the blue area; in contrast, the electronegative group was favorable for enhancing the bioactivity of the compounds, as indicated by the red area. The red area at the 6-position of the pyridine ring indicated that the electronegative substituents would contribute to the bioactivity of the compounds. Comparing compound 34 with compounds 33, 35 and 36, it was found that the activity was increased when the hydrogen atom at the 6-position of the pyridine ring was substituted by trifluoromethyl, fluorine, or chlorine atoms. These maps were informative in designing new potent and selective HPPD inhibitors.
CoMFA StDev*Coeff contour maps. (A) Steric and (B) Electrostatic.
3.3 Contour maps analysis of CoMSIA models
Steric and electrostatic contour maps of the CoMSIA model were shown in Fig. 4A and B, which were greatly analogous to those of the CoMFA model. In Fig. 4C, yellow regions with hydrophobic groups were favorable for activity, while white regions corresponding to hydrophilic substitutes were preferred. The R1-location observation to white, it was advantageous with hydrophilic substitution. A yellow region covered R2 at the 2-position of the pyridine ring, representing the favorability of hydrophobic features at this position. The introduction of hydrophobic groups provided more potent HPPD inhibitors. For example, the IC50 of compounds 1 (6-Cl) and 9(5-Br) were 0.262 μM and 2.69 μM. In the H-bond donor contour map (Fig. 4D), the cyan outline indicated a location where the H-bond donor group favored better activity. In contrast, the position of the gray area indicated that H-bond donor experience for lower activity. A small cyan outline was observed near 3-position on cyclohexanedione ring. This suggested that the introducing H-bond donor here was beneficial for increasing activity. The H-bond receptor contour map was shown in Fig. 4E, purple and magenta indicated areas where the replacement of H-bond receptor was positive and negative for inhibition. The magenta polyhedron at R2 on the pyridine ring indicated that the introduction of a H-bond acceptor was unfavorable for the activity. This was consistent with the observation between compounds 12 (6-F, IC50 = 3.489 μM) and 19 (6-Br, IC50 = 0.731 μM). The H-bond donor and acceptor formed a positive H-bond interaction with the protein, greatly affecting the inhibition of the compound. The IC50 of the piperidinedione derivatives (compounds 5, 9, 12, 19 and 23) were lower than those of cyclohexanedione derivatives (compounds 32, 31, 35, 26 and 27), indicating that compounds with piperidinedione subunits possessed better activity than those with cyclohexanedione subunits.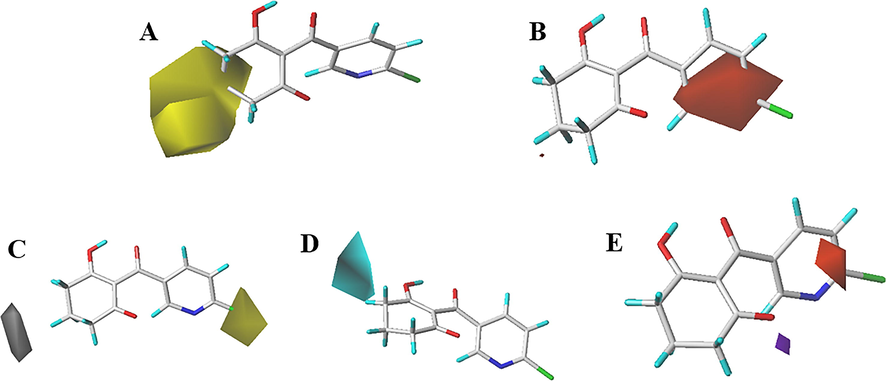
CoMSIA StDev*Coeff contour maps. (A) Steric, (B) Electrostatic, (C) Hydrophobic, (D) H-bond donor and (E) H-bond acceptor.
3.4 Design compounds based on CoMFA and CoMSIA models
The structural features favorable for improving the activity are summarized in Fig. 5 on the basis of the information gained by the contour maps. Compounds with piperidinedione as the subunit were more active than those with the cyclohexanedione fragment. It was favorable for the activity by introduction of small sizes, hydrophilic and electropositive substitutions at the 3-, 4- and 5-positions of piperidinedione or cyclohexanedione, and H-bond donors at this district would magnify the activity. The activity was also enhanced with the import of bulky, electronegative and hydrophobic substituents at the 2′-, 5′- and 6′-positions of the pyridine ring. Furthermore, the effect of substituents at the 5′ or 6′ position was better than that at the 2′-position.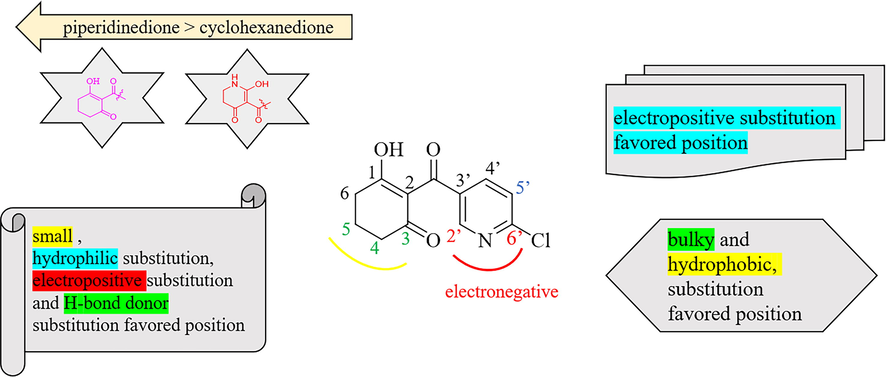
Summary of the structure–activity relationship.
In short, bulky and electronegative substitutes of the pyridine ring were favorable, and derivatives of piperidinedione and cyclohexanedione were synthesized. Therefore, four compounds (W1, W2) were designed in terms of the CoMFA and CoMSIA models. The physicochemical properties of the compounds markedly influenced the biological activity and its interaction with the herbicide target enzyme. Generally, the numbers of H-bond acceptors (HBAs), H-bond donors (HBDs) and aromatic rings (ARs) were positively correlated with the biological activity. Placing emphasis on comparing the physicochemical property of the compounds W1, W2 and mesotrione, it is worth noting that the HBAs, surface area (SA), number of ARs and rotatable bonds (RB), and electronegativity of compounds W1 and W2 were quite similar to those of mesotrione (Table 3). Notes: a CS Chemdraw drawing for predicting molecular weight (MW); b DS for predicting Logp, -CDOCKER_ ENERGY (kcal mol−1), pKa, rotatable bonds (RBS), aromatic rings (ARS), surface area, hydrogen bond acceptors (HBAs) and donors (HBDs); c Sybyl-X 6.9 for predicting electronegativity.
Name
W1
W2
Mesotrione
Structure a
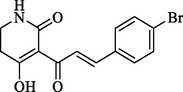
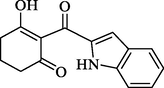
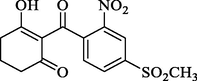
-CDOCKER_ ENERGY b
49.02
48.62
42.64
-Interactive energy
58.33
13.14
42.78
Log p b
2.46
2.28
1.40
pKa b
3.58
3.13
5.12
MW a
322.15
255.27
339.32
HBAs b
3
3
4
HBDs b
2
1
2
RBs b
3
2
4
ARs b
1
2
1
SA b
322.15
244.92
339.32
Electronegativity c
Compared with mesotrione, these compounds also showed a good docking score (-CDOCKER_ENERGY). In order to further demonstrate the reliability of molecular docking, the interactive energy of W1 was significantly higher than commercial herbicides, indicating the reliability of the model. The predicted pKa of the obtained compound W1 was less than 6.0 and even lower than that of mesotrione. Weak acid and low Log p were conducive to the spread of plants and absorption.
All of these results indicated that compound W1 might be the leading candidate for the generation of novel HPPD inhibitors. Then, synthesis and AtHPPD inhibition assays were carried out.
3.5 Chemical synthesis and enzyme inhibitory in vitro and Bioassay experiments
The inhibitory activities of compounds W1, W2 and mesotrione on HPPD from different sources are shown in Table 4. The IC50 against AtHPPD in vitro of W1 was 0.201 µM, analogous to mesotrione (IC50 = 0.221 µM). The activity of compound W1 was greatly increased than the previous screened structure with the pyranone replaced by piperidone (Fu et al., 2018). The IC50 of W2 was 5.221 µM, which was significantly greater than that of mesotrione and compound W1. The inhibition of rice HPPD and wheat HPPD showed that the IC50 value of compound W1 was>10 µM, almost same as mesotrione, which indicated that compound W1 might be safe for rice HPPD and wheat HPPD. Furthermore, compound W1 did not exhibit the potential to inhibit hHPPD activity. Compound W2 showed good safety in rice HPPD, but the estimate was not optimistic for wheat HPPD, and it did not have the potential to inhibit hHPPD activity.
Compound
AtHPPD
IC50 (µM) pIC50
hHPPD
IC50 (µM) pIC50
Rice HPPD
IC50 (µM) pIC50
Wheat HPPD
IC50 (µM) pIC50
W1
0.201 ± 0.001
6.697
18.394 ± 0.139
4.735
14.877 ± 0.062
4.827
15.631 ± 0.020
4.806
W2
5.221 ± 0.075
5.282
5.067 ± 0.145
5.295
>100
–
6.582 ± 0.146
5.182
Mesotrione
0.231 ± 0.002
6.636
12.165 ± 0.082
4.915
13.460 ± 0.009
4.871
10.000 ± 0.306
5.000
Therefore, after further structural modification, it was determined that compound W2 could be developed into HPPD-inhibiting herbicide for rice fields, and compound W1 may serve as a good template for designing effective HPPD inhibitors. After spraying the weeds with mesotrione and the leaves with W1 for three days, most of the weeds exhibited bleaching symptoms. The leaves, stems and roots of SF and EC dried up and lost water, accompanied by obvious bleaching symptoms; leaf bleaching also occurs for AJ and AR, and DS and AR grow slowly under the same environmental conditions (Fig. 6A). Judging from the degree of whitening of AR, AJ and DS, the herbicidal activity of mesotrione was lower than that of W1. In vivo safety tests on crops of compound W1 on rice (Fig. 6B) and wheat (Fig. 6C) show that it produces a very invulnerable effect for crops.
Bioassay experiment of compound W1. (A) Bioassay results of compound W1 on weeds: AJ, EP, AR, SF, DS, and EC, (B) Bioassay results of compound W1 on rice and (C) Bioassay results of compound W1 on wheat.
3.6 Docking analysis
Molecular docking is the process of studying molecular interactions in drug design. Through the asteroid plot of ligands and residues (Fig. 7A) (https://www.mrc-lmb.cam.ac.uk/rajini/index.html), key amino acid interactions were initially obtained. The large circles on the main chain corresponding to Phe424 and Phe381 indicated that the contributions of amino acids Phe424 and Phe381 were substantial. The native and redocked ligands had identical conformations for docking with the binding site, with RMSD being of 0.74 (Fig. 7B). The re-docked ligand could be well aligned with the ligands in the cocrystallized complex; therefore, the method demonstrated the accuracy and reliability of the docking.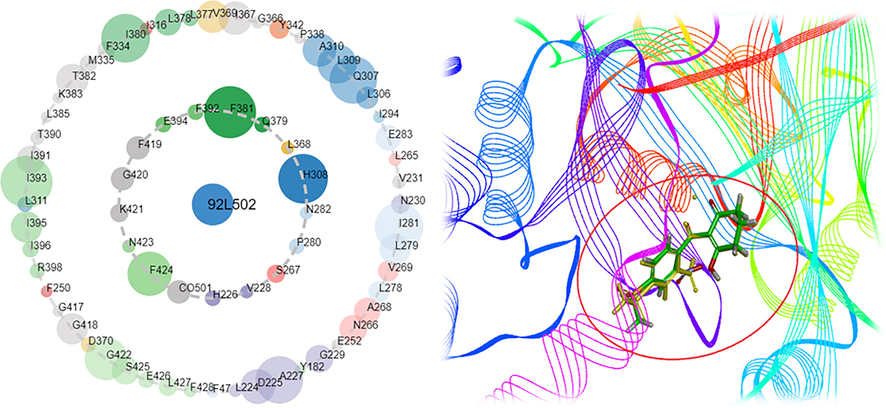
(A) The asteroid plot of ligands and residues and (B) Ligand docking diagram in crystal complex.
According to the contribution of steric field, compound 3 with bulk substituents (5,5-diCH3) at the 5-positions (R1) of the cyclohexanol diketone showed less the pi-alkyl with Lys421 than compound 1 (Fig. 8 A and B), which had higher -CDOCKER_ ENERGY and interactive energy than compound 3 (Table 5). The same rule was also observed in compounds 5 and 6. The hydrogen bond of Glu379 between compound 5 and enzyme was not found in compound 6 (Fig. 8. C and D).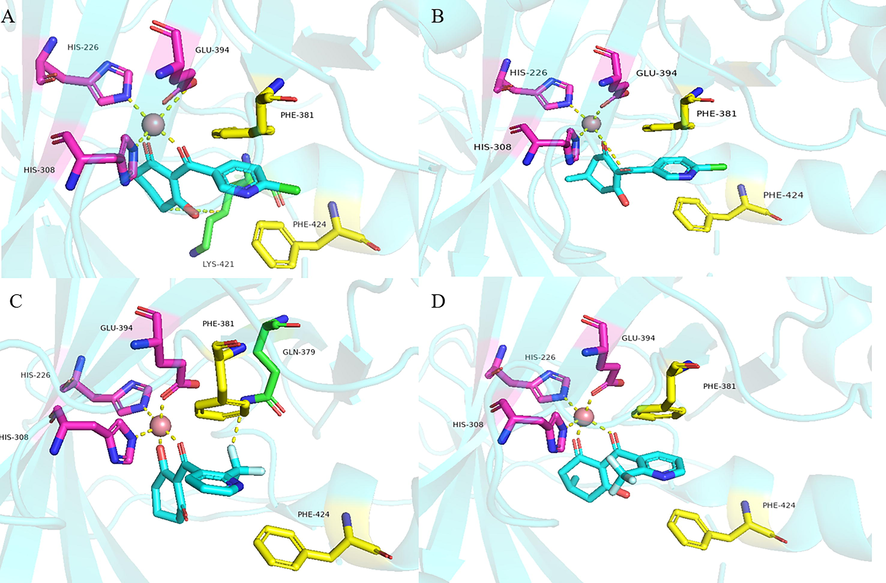
The QSAR dataset compounds molecular docking in active site (A) compound 1, (B) compound 3, (C) compound 5 and (D) compound 6.
Compound
Structure
-CDOCKER_ ENERGY (kcal mol−1)
-Interactive energy
(kcal mol−1)
1
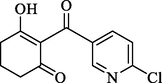
40.27
55.23
3
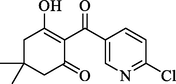
39.72
54.61
5
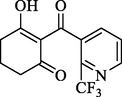
34.95
54.24
6
34.21
54.12
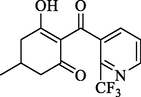
All three compounds were well docked into the binding pocket and showed that all the ligands bound to cobalt ions (Fig. 9). The -CDOCKER_ENERGY values of compounds W1 and W2 were greater than that of mesotrione, which proved that compounds W1 and W2 were expected to be potential HPPD inhibitors. As shown in Fig. 9A, mesotrione was entirely embedded in the active pocket and coordinated with Co2+ together with His226, His308 and Glu394. Moreover, the benzene ring also formed a sandwich π-π interaction with Phe381 and Phe424, which was conducive to stabilizing the binding protein. The binding mode of compound W1 is shown in Fig. 9B. W1 bonded with the protein through His226, His308, Glu394, Phe381, Phe392 and Phe424. Compared with the structure of mesotrione, the carbon chain of W1 was extended by inserting an ethenyl, which made Phe392 and the benzene ring form a new stable π-π interaction, which was more conducive to the structure. Compound W2 was also well inserted into the active site of the protein (Fig. 9C). Compared with mesotrione, compound W2 introduced an indole ring via bioisosterism of the benzene. The phenyl part of the indole ring formed a sandwich π-π interaction with Phe381 and Phe424, and His308 and the pyrrole subunit of the indole ring also formed a π-π interaction. For the sake of investigating the factors affecting inhibitor activity, MD simulations were carried out.
The receptor–ligand interactions of (A) mesotrione, (B) compound W1 and (C) compound W2 at the HPPD binding site.
3.7 MD analysis
MD simulations were performed to analyze the stability of ligand-receptor interactions. The overall stability of the system was monitored by RMSD of backbone atoms (C, Cα, N, and O) relative to the original docking structure. RMSD value of the backbone Cα atoms was the major parameter used to assess the stability of the system. As Fig. 10A shown, the two complexes for HPPD-W1 and HPPD-mesotrione showed slight fluctuations at the start, and then the main chain Cα atoms of the proteins tended to equilibrate after 20 ns. RMSD value of the main chain Cα atoms of compound W1 was smaller than that of mesotrione, which proved that the skeleton structure of compound W1 was more stable. Fig. 10B illustrates the RMSD value of the protein active pocket with residues 5 Å around the ligand. In the entire MD simulation process, all compounds were relatively stable. Fig. 10C showed that all the heavy atoms in the ligand balanced within the final 5 ns, indicating that all systems reached the steady state in the course of simulation.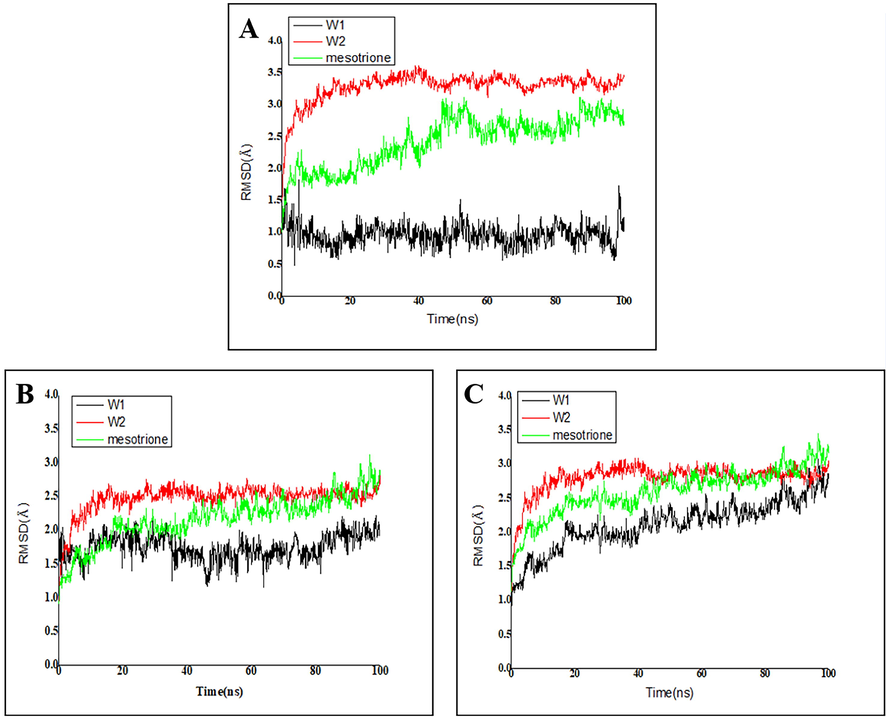
Protein-ligand RMSD of (A) Cα atoms. (B) Side chains and (C) heavy atoms.
The calculated MM-PBSA was given in Table 6. ΔGbind reflected the degree of binding between the compound and the protein. The ΔGbind values of the hit compounds W1, W2 and mesotrione were −44.590, −28.001 and −35.560 kcal mol−1, respectively. The calculated results indicated that the binding ability of W1 with protein was better than that of mesotrione. The ΔEvdw, ΔEele and ΔGSA values calculated by MM-PBSA were the positive contributions to ΔGbind, while ΔGPB weakened the binding process. It was found that the ΔEvdw value of compound W1 (-37.491 kcal mol−1) was comparatively lower than that of mesotrione (-36.423 kcal mol−1). The nonpolar solvation of compound W1 (ΔEvdw + ΔGSA = -38.885 kcal mol−1) was slightly better than that of mesotrione (-37.054 kcal mol−1). Therefore, the above results confirmed that nonpolar interactions made a lot of sense to strengthen binding affinities of molecules with the protein system. ΔEvdw, van der Waals energy; ΔEele, electrostatic energy; ΔGPB, polar solvation energy with the PB model; ΔGSA, nonpolar solvation energy with the PB model; ΔEMM, interaction energy; ΔGsol, solvation contribution; ΔEMM = ΔEvdw + ΔEele; ΔGsol = ΔGPB + ΔGSA; ΔGbind = ΔEvdw + ΔEele + ΔGPB + ΔGSA.
Compound
ΔEvdw
ΔEele
ΔGPB
ΔGSA
ΔEMM
ΔGsol
ΔGbind
W1
−37.491
−31.226
25.521
−1.394
−68.717
24.127
−44.590
W2
−41.861
−26.801
41.770
−1.109
−68.662
40.661
−28.001
Mesotrione
−36.423
–32.947
33.179
−0.631
−69.370
33.810
−35.560
3.8 ADMET
The obtained compounds W1 and W2 were analyzed using ADMET (Table 7). The ADMET properties for the two compounds were all within the safety ranges for human beings., The compound would be a lower possibility of liver toxicity if the Bayesian score in the hepatotoxicity model was less than −0.4095. Therefore, the three compounds were determined to possess very low hepatotoxicity. The PPB model was a significant indicator of drug distribution. However, the three compounds met the critical fraction of −2.209 for highly combined use with plasma proteins. The CYP2D6_Applicability parameter indicated mesotrione = 18.140, W1 = 12.745, and W2 = 10.122. All properties and three components were within expected ranges. The premise of a medicine is to satisfy the absorptive, distributional, metabolic, excretory and, most importantly, nontoxic requirements in vivo. For pesticides, nontoxicity is the key consideration, but the nature of ADME in the body is still not negligible. Solubility Level: Categorical solubility level. 2: Yes, low; 3: Yes, good. Hepatotoxic: <−0.4095: nontoxic; >−0.4095: toxic. PPB: Plasma Protein Binding ability. < − 2.209: ≥90%, false; >−2.209: ≤90%, true.
No.
W1
W2
Mesotrione
Solubility Level
3
3
3
PPB# Prediction
True
True
True
Hepatotoxic
−2.89
−1.25
−6.39
Aerobic Biodegradability
Degradable
Degradable
Degradable
Mutagenicity
Non-Mutagen
Non-Mutagen
Non-Mutagen
Carcinogenicity
Non-Carcinogen
Non-Carcinogen
Non-Carcinogen
Skin irritancy
None
None
None
CYP2D6_Applicability
12.74
10.12
18.14
DTP Prediction
Non-Toxic
Non-Toxic
Non-Toxic
Compounds W1, W2 and mesotrione were determined to be relatively safe through assessments of toxicity prediction, carcinogenicity, mutagenicity and skin irritation and were found to be aerobically and biodegradable. All in all, compounds W1 and W2 are expected to become new HPPD inhibitors due to their excellent solubility, degradability, and lack of mutagenicity, carcinogenicity, and skin irritation.
4 Conclusion
CoMFA and CoMSIA models with perfect cross-validated correlation coefficient values were built based on 36 pyridine derivatives. The effects associated with each substituent were predicted in the molecular skeleton, leading to better insights for designing new HPPD inhibitors. Two newly designed compounds, W1 and W2, were synthesized and subjected to AtHPPD inhibition and bioassay verification. Molecular docking experiments verified the binding mode of the novel compounds at the HPPD active pocket. Compound W1 showed satisfactory inhibitory activity with IC50 being 0.201 μM. AJ, AR, SF and EC showed bleaching symptoms after treatment with leaf spray. MD simulation and MM-PBSA energy calculation confirmed that the binding efficiency of compound W1 to the enzyme was high. ADMET prediction showed that these two newly designed compounds had excellent physicochemical properties, low toxicity and weak side effects. The above work supplied theoretical guidance and practical application for designing novel HPPD inhibitors with powerful activity.
CRediT authorship contribution statement
Juan Shi: Methodology, Software, Formal analysis, Investigation, Writing – original draft. Li-Xia Zhao: Project administration. Jia-Yu Wang: Formal analysis, Visualization, Investigation. Tong Ye: Resources, Data curation. Meng Wang: Validation. Shuang Gao: Visualization, Investigation. Fei Ye: Supervision, Conceptualization, Writing – review & editing. Ying Fu: Supervision, Funding acquisition, Writing – review & editing.
Acknowledgement
This work was supported by the National Nature Science Foundation of China (22077014). The authors are grateful to Prof. Jian Wang (Shenyang Pharmaceutical University) for helping in the application of SYBYL software.
References
- Furanone derivatives as new inhibitors of CDC7 kinase: development of structure activity relationship model using 3D QSAR, molecular docking, and in silico ADMET. Struct Chem.. 2018;29:1031-1043.
- [CrossRef] [Google Scholar]
- 4-Hydroxyphenylpyruvate dioxygenase inhibitors-a review of the triketone chemistry story from a syngenta perspective. Bioorgan. Med. Chem.. 2009;17:4134-4152.
- [CrossRef] [Google Scholar]
- Combining molecular docking and QSAR studies for modeling the anti-tyrosinase activity of aromatic heterocycle thiosemicarbazone analogues. J. Mol. Struct.. 2017;1151:353-365.
- [CrossRef] [Google Scholar]
- Identification of novel inhibitors of p-hydroxyphenylpyruvate dioxygenase using receptor-based virtual screening. J. Taiwan. Inst. Chem. Eng.. 2019;103:33-43.
- [CrossRef] [Google Scholar]
- Quantitative structure activity relationship studies and molecular dynamics simulations of 2-(aryloxyacetyl) cyclohexane-1,3-diones derivatives as 4-hydroxyphenylpyruvate dioxygenase inhibitors. Front. Chem.. 2019;7:556.
- [CrossRef] [Google Scholar]
- Fu, Y., Sun, Y.N., Yi, K.H., Li, M.Q., Cao, H.F., Li, J.Z., F, Ye., 2018. Combination of virtual screening protocol by in silico toward the discovery of novel 4-hydroxyphenylpyruvate dioxygenase inhibitors. Front. Chem. 6, 14. https://doi.org/10.3389/fchem.2018.00014.
- Design, synthesis, herbicidal activity and CoMFA of novel aryl-formyl piperidinone HPPD inhibitors. Pestic Biochem Physiol.. 2021;174:104811
- [CrossRef] [Google Scholar]
- Based on the virtual screening of multiple pharmacophores, docking and molecular dynamics simulation approaches toward the discovery of novel hppd inhibitors. Int. J. Mol. Sci.. 2020;21(15):5546.
- [CrossRef] [Google Scholar]
- Design, synthesis, SAR and molecular docking of novel green niacin-triketone HPPD inhibitor. Ind. Crops Prod.. 2019;137:566-575.
- [CrossRef] [Google Scholar]
- Exploring the DNA interactions, FGF growth receptor interaction and biological screening of metal (II) complexes of NNN donor ligand derived from 2-(aminomethyl)benzimidazole. J. Biol. Macromol.. 2018;126:1303-1317.
- [CrossRef] [Google Scholar]
- Pyrazole-isoindoline-1,3-dione hybrid: a promising scaffold for 4-hydroxyphenylpyruvate dioxygenase inhibitors. J. Agric. Food Chem.. 2019;67:10844-10852.
- [CrossRef] [Google Scholar]
- Discovery of novel pyrazole-quinazoline-2,4-dione hybrids as 4-hydroxyphenylpyruvate dioxygenase inhibitors. J. Agric. Food Chem.. 2020;68:5059-5067.
- [CrossRef] [Google Scholar]
- 3D-QSAR, molecular docking and molecular dynamics simulations of oxazepane amidoacetonitrile derivatives as novel DPPI inhibitors. J. Mol. Struct.. 2018;1168:223-233.
- [CrossRef] [Google Scholar]
- Design, synthesis and biological activity of novel triketone-containing quinoxaline as hppd inhibitor. Pest Manag. Sci.. 2022;78:938-946.
- [CrossRef] [Google Scholar]
- A drug-likeness toolbox facilitates ADMET study in drug discovery. Drug Discov. Today.. 2019;25:248-258.
- [CrossRef] [Google Scholar]
- Discovery of novel chromone derivatives containing a sulfonamide moiety as anti-ToCV agents through the tomato chlorosis virus coat protein-oriented screening method. J. Agric. Food Chem.. 2021;41:12126-12134.
- [CrossRef] [Google Scholar]
- Binding site analysis of CCR2 through in silico methodologies: docking, CoMFA, and CoMSIA. Chem. Biol. Drug Des.. 2011;78:161-174.
- [CrossRef] [Google Scholar]
- Cobalt (II) complex as a fluorescent sensing platform for the selective and sensitive detection of triketone HPPD inhibitors. J. Hazard. Mater.. 2021;404:124015
- [CrossRef] [Google Scholar]
- 4-Hydroxyphenylpyruvate dioxygenase from sea cucumber Apostichopus japonicus negatively regulates reactive oxygen species production. Fish Shellfish Immunol.. 2020;101:261-268.
- [CrossRef] [Google Scholar]
- Crystal structure of 4-hydroxyphenylpyruvate dioxygenase in complex with substrate reveals a new starting point for herbicide discovery. Research.. 2019;2019:2602414.
- [CrossRef] [Google Scholar]
- Rational redesign of enzyme via the combination of quantum mechanics/molecular mechanics, molecular dynamics, and structural biology study. J. Am. Chem. Soc.. 2021;143:15674-15687.
- [CrossRef] [Google Scholar]
- Molecular insights into the mechanism of 4-hydroxyphenylpyruvate dioxygenase inhibition: enzyme kinetics, X-ray crystallography and computational simulations. FEBS. J.. 2019;286:975-990.
- [CrossRef] [Google Scholar]
- Virtual identification of novel peroxisome proliferator-activated receptor (PPAR) alpha/delta dual antagonist by 3D-QSAR, molecule docking and molecule dynamics simulation. J. Biomol. Struct. Dyn.. 2019;38:4143-4161.
- [CrossRef] [Google Scholar]
- Structure based identification of novel inhibitors against ATP synthase of mycobacterium tuberculosis: a combined in silico and in vitro study. Int. J. Biol. Macromol.. 2019;135:582-590.
- [CrossRef] [Google Scholar]
- 4-Hydroxyphenylpyruvate dioxygenase and hydroxymandelate synthase: exemplars of the α-keto acid dependent oxygenases. Arch. Biochem. Biophys.. 2014;544:58-68.
- [CrossRef] [Google Scholar]
- Synthesis and herbicidal activity of triketone-aminopyridines as potent p-hydroxyphenylpyruvate dioxygenase inhibitors. J. Agric. Food Chem.. 2021;69:5735-5745.
- [CrossRef] [Google Scholar]
- 4-Hydroxyphenylpyruvate dioxygenase inhibitors: from chemical biology to agrochemicals. J. Agric. Food Chem.. 2017;65:8523-8537.
- [CrossRef] [Google Scholar]
- CD and MCD studies of the non-heme ferrous active site in (4-hydroxyphenyl) pyruvate dioxygenase: correlation between oxygen activation in the extradiol and alpha-KG-dependent dioxygenases. J. Am. Chem. Soc.. 2004;126:4486-4487.
- [CrossRef] [Google Scholar]
- Structure-guided discovery of silicon-containing subnanomolar inhibitor of hydroxyphenylpyruvate dioxygenase as a potential herbicide. J. Agric. Food Chem.. 2021;69:459-473.
- [CrossRef] [Google Scholar]
- 4-Hydroxyphenylpyruvate dioxygenase catalysis: identification of catalytic residues and production of a hydroxylated intermediate shared with a structurally unrelated enzyme. J. Biol. Chem.. 2011;286:26061-26070.
- [CrossRef] [Google Scholar]
- Aclonifen targets solanesyl diphosphate synthase, representing a novel mode of action for herbicides. Pest Manag. Sci.. 2020;76(10):3377-3388.
- [CrossRef] [Google Scholar]
- Herbicide safety relative to common targets in plants and mammals. Pest Manag. Sci.. 2004;60:17-24.
- [CrossRef] [Google Scholar]
- Computational identification of novel piperidine derivatives as potential HDM2 inhibitors designed by fragment-based QSAR, molecular docking and molecular dynamics simulations. Struct. Chem.. 2016;27:993-1003.
- [CrossRef] [Google Scholar]
- Isolation of pyomelanin from bacteria and evidences showing its synthesis by 4-hydroxyphenylpyruvate dioxygenase enzyme encoded by hppd gene. Int. J. Biol. Macromol.. 2018;119:864-873.
- [CrossRef] [Google Scholar]
- Design, synthesis, structure-activity relationship, molecular docking, and herbicidal evaluation of 2-cinnamoyl-3-hydroxycyclohex-2-en-1-one derivatives as novel 4-hydroxyphenylpyruvate dioxygenase inhibitors. J. Agric. Food Chem.. 2021;69:12621-12633.
- [CrossRef] [Google Scholar]
- Spike protein recognizer receptor ACE2 targeted identification of potential natural antiviral drug candidates against SARS-CoV-2. Int. J. Biol. Macromol.. 2021;191:1114-1125.
- [CrossRef] [Google Scholar]
- Molecular modelling studies of 3,5-dipyridyl-1,2,4-triazole derivatives as xanthine oxidoreductase inhibitors using 3D-QSAR, Topomer CoMFA, molecular docking and molecular dynamic simulations. J. Taiwan. Inst. Chem. Eng.. 2016;68:64-73.
- [CrossRef] [Google Scholar]
- Synthesis and herbicidal evaluation of triketone-containing quinazoline-2,4-diones. J. Agric. Food Chem.. 2014;62:11786-11796.
- [CrossRef] [Google Scholar]
- Graph attention convolutional neural network model for chemical poisoning of honey bees’ prediction. Sci. Bull.. 2020;65:1184-1191.
- [CrossRef] [Google Scholar]
- Molecular modeling study of CP-690550 derivatives as JAK3 kinase inhibitors through combined 3D-QSAR, molecular docking, and dynamics simulation techniques. J. Mol. Graph. Model.. 2017;72:178-186.
- [CrossRef] [Google Scholar]
- Synthesis and herbicidal activities of aryloxyacetic acid derivatives as HPPD inhibitors. Beilstein. J. Org. Chem.. 2020;16:233-247.
- [CrossRef] [Google Scholar]
- The structure of 4-hydroxylphenylpyruvate dioxygenase complexed with 4-hydroxylphenylpyruvic acid reveals an unexpected inhibition mechanism. Chin. Chem. Lett.. 2021;32:1920-1924.
- [CrossRef] [Google Scholar]
- Cloud 3D-QSAR: a web tool for the development of quantitative structure–activity relationship models in drug discovery. Brief Bioinform.. 2020;22:276.
- [CrossRef] [Google Scholar]
- Synthesis and bio-evaluation of a novel selective butyrylcholinesterase inhibitor discovered through structure-based virtual screening. Int. J. Biol. Macromol.. 2020;166:1352-1364.
- [CrossRef] [Google Scholar]
- LARMD: integration of bioinformatic resources to profile ligand-driven protein dynamics with a case on the activation of estrogen receptor. Brief Bioinform.. 2019;21:2206-2218.
- [CrossRef] [Google Scholar]
- Design, syntheses and 3D-QSAR studies of novel N-phenyl pyrrolidin-2-ones and N-phenyl-1H-pyrrol-2-ones as protoporphyrinogen oxidase inhibitors. Bioorg. Med. Chem.. 2010;18:7948-7956.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2022.103919.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary Data 1
Supplementary Data 1




