

Translate this page into:
The site preference and doping effect on mechanical properties of Ni3Al-based γ′ phase in superalloys by combing first-principles calculations and thermodynamic model
⁎Corresponding authors. wubo@fzu.edu.cn (Bo Wu), asifncp11@yahoo.com (Asif Hayat)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
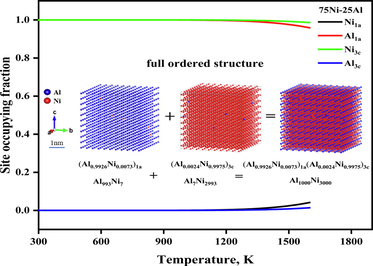
Fig. The temperature- and composition-dependent site occupying fraction of stoichiometric Ni3Al-based γ′ phase intermetallics and visualization of the atomic distributions of the full ordered configuration of the stoichiometric Ni3Al γ′ phase with the real relative size bar on the sublattice 1a and 3c based on a 10×10×10 supercell of the FCC_L12 structure at high temperature (1473 K) are presented graphically.
Abstract
Site preference, mechanical and thermodynamic properties were predicted in the Ni3Al-based γ′ phase. Co, Mn, and Ti atoms always prefer 3c sublattice (Ni site) in the whole temperature range. The site preferences of Cr, Cu, Fe, Mo, Re, Ta, V, and W atoms are affected by the heat treatment temperature. Cr, Re, V doping can strengthen the hardness of Ni3Al alloys, and particularly, the effect of Cr is remarkable. Cr doping increases the Debye temperature of Ni3Al alloys significantly.
Abstract
The fundamental aspects of site preference of alloying elements on sublattice of the strengthen γ′ phase with L12 structure have not been well understood, which hinders the optimized design of advanced Ni-based high-temperature alloys. In this contribution, the temperature- and composition-dependent site occupying preferences of the binary, ternary, and quaternary of Ni3Al-based γ′ phase alloyed with Mi where Mi represents the additional transitional metals Co, Cr, Cu, Fe, Mn, Mo, Re, Ta, Ti, V, or W atoms (arranged in alphabetical order) chosen frequently, were studied using a two-sublattice thermodynamic model (Ni, Al, Mi)1a(Ni, Al, Mi)3c. The site occupying fractions (SOFs) were calculated based on a thermodynamic database established in this work, where the thermodynamic data of the end-members involved were obtained using first-principles calculations and phonon spectrum calculations. The calculated SOFs results show that there is an obvious site preference for stoichiometry binary Ni3Al, and its site configuration changes from (Al)1a(Ni)3c at room temperature to (Al0.9984Ni0.0015)1a (Al0Ni0.9994)3c at 1273 K. For the γ′ phase with the composition 78Ni-26Al-4Mi (atom ratio and xNi/xAl = 3:1), Mo atoms always preferred to occupy the 1a sublattice (Al site), Co, Mn, and Ti atoms always prefer the 3c sublattice (Ni site) in the whole temperature range, while the site preference of Cr, Cu, Fe, Re, Ta, V, or W atom is affected by temperature. For example, when the heat treatment temperature is lower than 700 K, Cr, Cu, Fe, Ta, V, and W atoms occupy the 1a and 3c sublattice randomly, and Re atoms prefer to 3c sublattice, while when the heat treatment temperature is higher than 1273 K, Cr, Cu, and W atoms prefer 3c sublattice, Fe and Ta atoms prefer to 1a sublattice, while all Re atoms occupy the 3c sublattice exclusively, and all V atoms occupy the 1a sublattice exclusively, respectively. Likewise, the site preference of the quaternary system with selective compositions 78Ni-26Al-2 M1-2 M2 was also predicted. Based on calculated SOFs results, the mechanical and thermodynamic properties were studied at the ground state. It has been revealed that Cr, Re, and V doping can improve the microhardness of Ni3Al alloys; in particular, the effect of Cr is extraordinary; and all elements, except Mn, Mo, and Ti, would enhance the bulk modulus of Ni3Al-based γ′ phase, in which Mn have the greatest influence on reducing the bulk and shear modulus, respectively. Furthermore, all the B/G ratios of the computed Ni3Al-based γ′ phase are >1.75, showing inherent ductility. Only Cr doping significantly enhances the Debye temperature of the Ni3Al-based γ′ phase.
Keywords
Ni-based superalloys
Site preference
Sublattice model
Mechanical properties
First-principles calculations
Alloy thermodynamics

1 Introduction
Since their discovery in the 1940 s, Nickel-based superalloys have got significant importance for the design of high-temperature applications such as modern turbine engines, nuclear power, chemical processing plants, and rocket engines based on their outstanding characteristics i.e., corrosion-resistant behaviors, lightweight, and excellent mechanical properties (Brimhall et al., 1983; Hung et al., 1983; Ivanov et al., 1988; Miracle and No, 1993; Noebe et al., 1993; Inoue et al., 1998; Golberg et al., 1999; Lu and Hirohashi, 1999; Pal et al., 2006; Pollock and Tin, 2006; Reed, 2006; Darolia et al., 1993). The γ′-Ni3Al intermetallic compound is incredibly important in science and technology due to its effective high-temperature oxidation resistance and exceptional high-temperature strength. The unique microstructure of the alloy, which normally comprises of a cuboid-shaped strengthening γ′ phase homogeneously dispersed in a γ phase matrix, is responsible for the alloy's extraordinary mechanical characteristics. The γ phase is a solid-solution phase with a disordered face-centered cubic (FCC) structure, and the γ′ phase is an intermetallic compound Ni3Al with an ordered L12 structure. One of the most effective approaches proven for increasing temperature capability and stabilizing the γ′ phase, as well as achieving magnificent mechanical properties and higher-temperature performance under various service conditions, is to add additional alloying elements to superalloys (Rawlings and Staton-Bevan, 1975; Chiba et al., 1991; Chiba et al., 1992; Gleeson et al., 2004). For instance, Hf, Re, Ta, and W are utilized to enhance mechanical strength and the solvus temperature; B and Zr to enhance ductility; Al, Cr, and Ti improve the corrosion resistance and oxidation (Barrett et al., 1983; Caron and Khan, 1999; Blavette et al., 2007), and the strengthening elements e.g. Nb, Mo, Re, Ta, and W significantly enhanced the mechanical behavior of superalloy (Gariboldi et al., 2008; Wang and Wang, 2009; Amouyal et al., 2010; Bensch et al., 2010). Therefore, the knowledge of the strengthening mechanism of site preferences of alloying elements in the γ′ phase is important for understanding the roles of the substitutional atoms in determining the mechanical properties of Ni-based superalloys.
The site occupying information is indispensable data for a theoretical and quantitative design of alloys. However, it is a long-time challenge for both theoretical and experimental investigations to determine the site preference due to the complex configuration as well as the requirement of the single-crystal sample at the phase equilibrium state. Furthermore, the mechanical properties of Ni3Al-based γ′ phase alloys are related to the sublattice site occupying behaviors of alloying elements (Chiba et al., 1991). For example, the ductility and ordering energy of Ni3Al alloys decreases, while its yield strength increases when the Ni site (i.e., 3c sublattice) is substituted by alloying elements. The reliability of γ and γ′ phases was reported by Suzuki (Suzuki and Oya, 1981). If additional elements occupy the 1a sublattice; the γ′-phase will be more stable in the matrix, on the other hand, if elements occupy the 3c sublattice, the γ-phase is more stable, respectively. Therefore, the investigation of the site preference of alloying elements in Ni3Al is of great practical and fundamental interest. Several attempts have been employed to predict the site occupancy of alloying elements in the γ′ phase. In 1959 s, a phenomenological thermodynamic model was proposed to explain alloying behavior first time, proposing that the electronic structure of the ternary addition determined the site substitution behavior of ternary additions to Ni3Al (Guard, 1959). Rawlings and Staton-Bevan proposed a strong correlation between the site preferences and mechanical properties of the γ′ phase of the ternary additions (Rawlings and Staton-Bevan, 1975). Hono et al. (Hono et al., 1992) studied the site preferences of Cu and Ge by using atom probe field ion microscopy (APFIM) with different compositions Ni75-xAl23X1-x, and their results showed that Cu occupied the 3c sublattice and Ge occupies the 1a sublattice. Geng et al. (Geng et al., 2004) used supercell models based on first-principles total-energy calculations at room temperature to explore the site preference of PGM additions in γ′-Ni3Al. Their findings demonstrated that Mo favors the 1a sublattice, while Os, Ru, Rh, Pd, Ir, and Pt atoms prefer the 3c sublattice, in which Os shows a weak site preference tendency for the 3c sublattice. Saito and Harada used the Monte Carlo approach to show that Co favors the 3c sublattice in CMSX-4 and TMS63 alloys, whereas Mo, Ta, and Ti favor the 1a sublattice, and Cr occupies both 1a and 3c sublattice. But, there is insufficient experimental data to support their calculation (Saito and Harada, 1997). Jiang et al. (Jiang et al., 2006) predicted the site preference of Pt, Hf, Cr, and Ir in Ni3Al, as well as the impact of temperature on site preference of ternary alloying elements by utilizing a statistical-mechanical Wagner-Schottky model based on first-principles with different compositions, and calculated the point defect formation enthalpies. By using the atom-probe tomography (APT) and first-principles calculations, Booth and Mao (Booth-Morrison et al., 2008) studied the site preference of Cr and Ta in the Ni3Al (L12)-type γ′-precipitates of a ternary system with different compositions, in which Al, Cr, and Ta occupied the 1a sublattice. Kim et al. (Kim et al., 2010) employed the strain–stress method based on first-principles calculations to study the impact of alloying elements (Cr, Hf, Pt, Y, and Zr) on the elastic characteristics and site preference of Ni3Al alloys at 0 K. Their study revealed that Cr, Hf, Y, and Zr favor the Al sublattice, while Pt prefers the Ni sublattice. Kumar and Chernatynskiy (Kumar et al., 2015) investigated the influence of alloying elements (Cr, Zr, B, Ce, and La) on the site preferences and the elastic properties of Ni3Al alloys at room temperature with different compositions. Zhu et al. (Zhu et al., 2020) employed the first density functional theory to elucidate the mechanical properties and site preference of Ni/Ni3Al alloys doped with Re, Ta, and W at a low temperature of 50 K. Pan et al. employed the first-principles calculations (Chen and Pan, 2022; Pan, 2022; Pan and Chen, 2020; Yu and Pan, 2021; Pan, 2021) to study the site preference, structural stability, electronic, optical, catalytic, thermodynamic, and mechanical properties (Pu and Pan, 2022; Pu and Pan, 2022; Pan and Chen, 2022; Pu and Pan, 2022; Pu and Pan, 2022; Pu and Pan, 2022) of ternary phases as well as high temperature superalloys. According to first-principles calculations (at the ground state) and APT results, Mo and Ti favor the 1a sublattice (Enomoto and Harada, 1989; Mekhrabov et al., 1997; Tu et al., 2012; Raju et al., 1996). The atomic site occupancy of additional elements, including Cr, Co, Re, Ru, Ta, and W, is still elusive based on theoretical predictions (Amouyal et al., 2010; Jiang et al., 2006; Enomoto and Harada, 1989; Mekhrabov et al., 1997; Raju et al., 1996; Wu and Li, 2012; Eriş et al., 2017; Özcan et al., 2009; Amouyal et al., 2014; Amouyal et al., 2009) and experimental data from APT (Amouyal et al., 2010; Saito and Harada, 1997; Booth-Morrison et al., 2008; Tu et al., 2012; Amouyal et al., 2014; Amouyal et al., 2009; Bagot et al., 2017; Blavette and Bostel, 1984; Zhou et al., 2008; Huang et al., 2016), energy dispersive spectroscopy (EDS) (Broderick et al., 2018), and channeling-enhanced microanalysis (Horita et al., 1995; Liebscher et al., 2008).
However, previous theoretical investigations focus only on the site preference at the ground state (0 K) using a supercell model supported with first-principles calculations and calculating the enthalpy of formation of the alloy phase at the ground state. In the previous supercell model, they generally placed the additional alloying element either on the 1a sublattice or on the 3c sublattice, and the corresponding two configurations are (Ni1-xMx)3Al and Ni3(AlxM1-x), respectively. However, the chemical compositions are different for the two configurations, so the site preference of atoms on sublattice judged from the enthalpy of formation of the alloy phase with different compositions may be insufficient. Most importantly, no reasonable theoretical report concerning the effects of heat treatment temperature on the site preference, so it is necessary to thoroughly understand the temperature- and composition-dependent site preference in order to design the Ni-based high-temperature alloys strengthened with γ′ phase rationally.
In our prior study, the sublattice model based on the crystal lattice structure information has been widely applied to investigate the temperature-dependent site preference and ordering behaviors concerning NbCr2-based Lave phases with two sublattices (Wu et al., 2013), Ti2AlNb orthorhombic phase with three sublattices (Wu et al., 2008), ThMn12 structure with four sublattices (Zheng et al., 2010), Co-based superalloys (Ali, 2022), as well as in high entropy alloys (Wu et al., 2022), and also employed the first-principle calculations to studied the crystal lattice structure, thermodynamics and mechanical properties in ZnZrAl2 intermetallic compound (Wei et al., 2016); elastic, electronic and thermodynamic properties of the Ti2AlNb orthorhombic phase (Hu et al., 2017; Wei et al., 2021). In this work, we predicted the temperature- and composition-dependent site occupying preferences of the binary, ternary, and quaternary of Ni3Al-based γ′ phase with an FCC_L12 structure alloyed with Mi where Mi represents the additional transitional metals Co, Cr, Cu, Fe, Mn, Mo, Re, Ta, Ti, V, and W atoms (arranged in alphabetical order), were studied using a two-sublattice thermodynamic model (Ni, Al, Mi)1a(Ni, Al, Mi)3c. The site occupying fractions (SOFs) were calculated using a thermodynamic software package based on a thermodynamic database established in this work, where the temperature-dependent thermodynamic data were obtained using first-principles calculations based on density-functional theory (DFT) and density-functional perturbation theory (DFPT). Based on calculated SOFs results, we studied the mechanical and thermodynamic properties at the ground state (0 K), which agree well with both theoretical and experimental data in the available literature. Our approach is general for any given composition of Ni3Al-based γ′ phase with an FCC_L12 structure alloyed with one or more additional transitional metal Mi. For this purpose, we presented the model computation methodology, and the results of some selective Ni3Al-based γ′ phases with an FCC_L12 structure alloyed with Mi.
2 Theoretical methodology and computational approaches
2.1 Sublattice model
The prototype of the Ni3Al-based γ′ phase with an FCC_L12 structure is the full ordered AuCu3. The crystallographic information is shown in Table 1. The combination of multiplicity and Wyckoff letter of Wyckoff positions are generally named as 1a sublattice and 3c sublattice, thus the Au atoms are localized at the cube corner (1a sublattice) and the Cu atoms are localized at the face centers (3c sublattice).
Prototype
Pearson’s
SymbolStrukturbericht
DesignationSpace group
Wyckoff positions
Symbol
Number
Atom
Multiplicity
Wyckoff letter
AuCu3
cP4
L12
Pm3m
221
Au
1
a
Cu
3
c
In this work, we do not premeditate the site preferences and suppose that each type of alloying element including Ni, Al, and M atoms, can occupy both sublattice 1a and 3c with a probability, i.e., the SOFs. So, the configuration of the FCC_L12 γ′-Ni3Al phase alloying with additional transition metals M can be described with the following general two-sublattice model in Eq. (1),
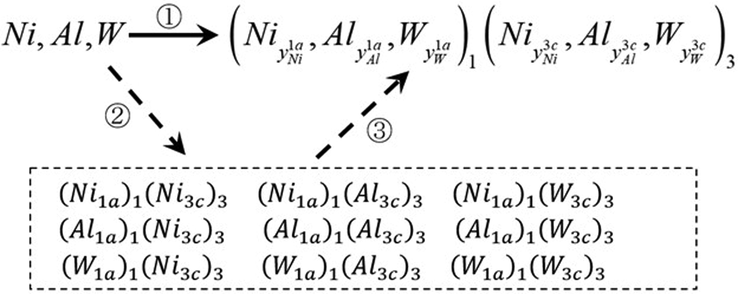
The alternative computation path of the thermodynamic function, i.e., ①=②+③.
Firstly, the hypothetical end-members (i.e., compounds with sublattices fully occupied by only one type of atom) are created from pure elements, and secondly, the γ′ phase formed from the end-members, i.e., ①=②+③. For example, the Gibbs energy of formation of the single-phase FCC_L12 γ′ phase derived from the pure elements can be calculated by Eq. (3),
We got Eq. (5) and Eq. (6) by assuming mass balance and composition normalization as:
Once the value of
at definite temperature is obtained, the relationship of SOFs with temperature and composition can be determined as seen from Eq. (7),
Thus, the site preference and thermodynamic functions of the Ni3Al-based γ′ alloy phase were easily estimated based on the predicted temperature and composition-dependent site occupying fractions.
2.2 Thermodynamic database of Gibbs free energy of formation of end-members
The Gibbs free energies of formation
of the end-members may be determined using Eq. (8),
2.3 Elastic constants
The stress–strain method (Shang et al., 2007) is used to calculate the elastic constants (Cij). During ionic position relaxations, the volume and cell (Pu and Pan, 2022) structure are fixed, and only the forces acting on ions can be relaxed. The elastic constants in terms of Hook’s law are expressed as,
Then we used Voigt-Reuss-Hill (VRH) approximation (Ledbetter and Reed, 1973; Xiong and Gu, 2015) to calculate the polycrystal elastic moduli i.e., bilk (B), shear (G), and Young’s (E) modulus and Poisson ratio ( ), respectively.
3 Results and discussions
3.1 The Gibbs free energy of pure elements at SER and end-members
In this work, there are 13 alloying elements involved, including Al, Co, Cr, Cu, Fe, Mn, Mo, Ni, Re, Ta, Ti, V, and W, thus 169 (13 × 13) end-members with FCC_L12 structure were created. The essential details of the pure element of their stable structure at room temperature employed in this work are summarized in Table S2. The calculated scattered data of Gibbs free energies using the QHA model of all the pure elements at SER and end-members involved in this work were plotted and then fitted using Eq. (9). Both the calculated scattered data of Gibbs free energies and the fitted curve for the representative pure element Ni_FCC were plotted in Fig. S1, and those for the representative end-member (Al)1a (Ni)3c with a full ordered structure were plotted in Fig. S2, while the scattered data of the rest pure elements with SER structure were plotted in Fig. S3, the scattered data of the rest end-members were plotted in Fig. S4. It can be observed that the fitted curve covers the scattered data quite well, all the plots show very smooth, and the fitted deviations are considerably small. To further validate the computation's accuracy and validity, the bulk moduli and lattice constants of pure elements at 0 K derived via phonon calculation are listed in Table S3, which accord very well with the currently available literature. The fitting formula of the temperature-dependent Gibbs free energy of pure element Ni_FCC, and end-member (Al)1a(Ni)3c are given in Eq. (13) and Eq. (14),
3.2 Temperature-dependent site preference and thermodynamic functions
The temperature-dependent SOFs and thermodynamics functions of the Ni3Al-based γ′ phase were calculated and plotted in Fig. 2 to Fig. 8, where the thermodynamic functions include Gibbs free energy of formation, enthalpy of formation, the entropy of formation, as well as configurational entropies. The temperature-dependent SOFs of stoichiometric and non-stoichiometric Ni3Al-based binary γ′ phases are presented in Fig. 2. From Fig. 2(a), It has been observed that the Ni3Al-based γ′ phase shows full order behavior in the whole temperature range, and its site configuration changes from (Al)1a(Ni)3c at room temperature to (Al0.9984Ni0.0015)1a (Al0Ni0.9994)3c at 1273 K, where Al atoms favor sublattice 1a and Ni atoms favor 3c sublattice. Moreover, we have observed that the site preferences of Ni atoms are slightly affected by the heat treatment, for example, at low temperature i.e., 298 K, Ni atoms occupy the 3c sublattice exclusively, but when the heat treatment is higher than 1273 K, then Ni atoms occupied both the sublattices but preferred to 3c, whereas Al atoms always occupied 1a sublattice in the whole temperature range. Furthermore, to deeply understand and verify this, we also plotted the SOFs of the nonstoichiometric Ni3Al-based γ′ phase. From Fig. 2(b) and Fig. 2(c), we found that the nonstoichiometric case also shows the similar behavior which was observed in the stoichiometric case. At low temperature i.e., 298 K, Ni atoms occupy 3c sublattice exclusively, but when the heat treatment is higher than 1273 K, then Ni atoms occupied both the sublattices but preferred to 3c, whereas Al atoms always occupied 1a sublattice in the whole heat treatment process. As a counterpart of the Ni3Al-based γ′ phase, we also investigated the site preference of the stoichiometric Co3Al-based γ′ phase , illustrated in Fig. 3. From Fig. 3, it is seen that there is a strong order–disorder transition for the stoichiometric Co3Al-based binary γ′ phase, the critical temperature of the order–disorder transition is about 1073 K, accompanied by the site configuration changes from (Al)1a(Co)3c at room temperature to (Al0.2590Co0.7409)1a (Al0.2469Co0.7530)3c at 1273 K, where Al atoms occupy 1a sublattice exclusively, and Co atoms occupy 3c sublattice exclusively.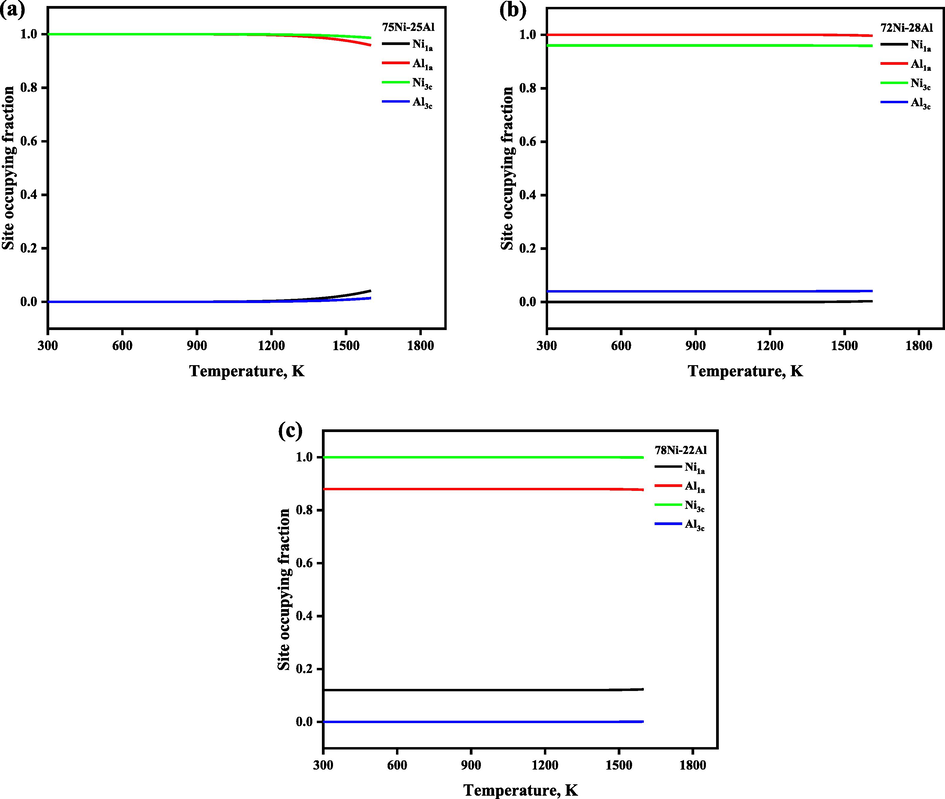
The temperature-dependent site occupying fraction of stoichiometric and nonstoichiometric Ni3Al-based γ′ phase.
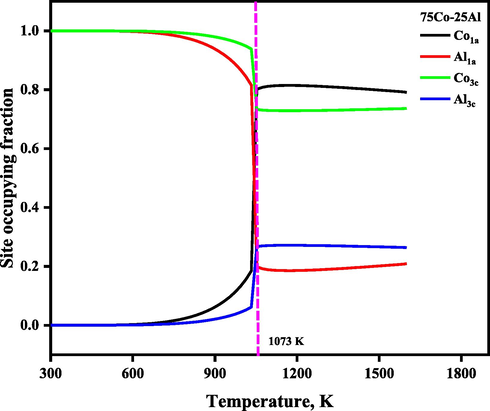
The temperature-dependent site occupying fraction of stoichiometric Co3Al-based γ′ phase.
The temperature-dependent thermodynamic functions of stoichiometric and nonstoichiometric Ni3Al intermetallic are illustrated in Fig. 4 to explore the thermodynamic mechanism of the ordering behavior of the Ni3Al-based binary γ′ phase. It is noted that ΔGmix decreases as temperature increases; the lower the Gibbs energy, the more stable the structure (Wu et al., 2008), and the rest of the thermodynamic functions changes with the temperature considerably, where the plots of configurational entropies were further calculated using Eq. (16) based on the SOFs obtained in this work.
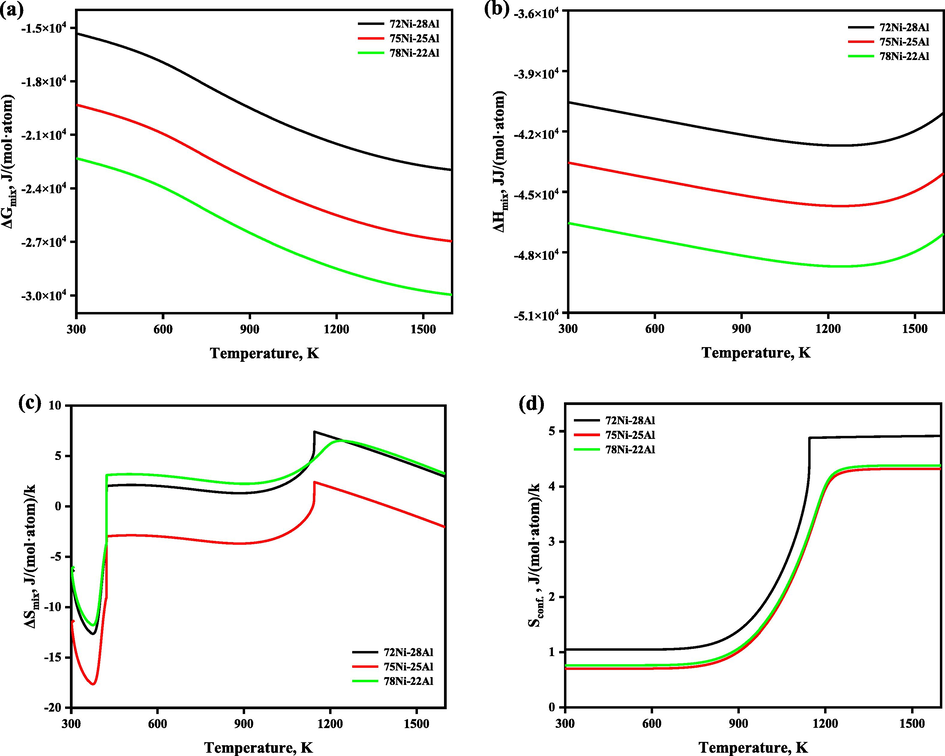
The temperature-dependent thermodynamic functions of stoichiometric and nonstoichiometric Ni3Al-based γ′ phase.
Following the understanding of the ordering behavior of the Ni3Al-based binary γ′ phase, we explored the ordering behaviors of the additional alloying element Mi on the Ni3Al-based γ′ phase further, where eleven additional transition metals Mi, including Co, Cr, Cu, Fe, Mn, Mo, Re, Ta, Ti, V, and W (arranged in alphabetical order) were investigated, which are the commonly used elements in Ni-based superalloy. The sublattice model proposed in this work can be used to predict the SOFs of Ni3Al-based γ′ phase with any composition, however, for simplifying the question to understand the site preference of additional alloying elements, here we fixed the xNi/xAl at 3:1 and set the demonstrated composition as composition 78Ni-26Al-4Mi, which is normalized as 72.22Ni-24.08Al-3.7Mi (at. %). Notice that the reason for the composition choice will be mentioned again later, i.e., to agree with the total atom number 108 for a 3 × 3 × 3 supercell of the AB3_L12 structure. The temperature-dependent SOFs of the alloying element in the Ni3Al-based γ′ phase with the composition 78Ni-26Al-4Mi are shown in Fig. 5, and the corresponding temperature- and composition-dependent thermodynamic functions were plotted together in Fig. 6, and we have observed that ΔGmix, ΔHmix, and ΔSmix are decreasing with the increase of the heat treatment, and configurational entropy Sconf is increasing with the increase of the temperature, which shows that the doping of ternary elements enhances the structural stability, respectively. As seen in Fig. 5, the atoms are not randomly distributed on the lattice but have a specific site preference. Co, Mn, and Ti atoms always prefer 3c sublattice (Ni site), and Mo atoms always occupied both the sublattice, but preferred the 1a sublattice (Al site) in the whole temperature range, while the site preference of the alloying elements such as Cr, Cu, Fe, Re, Ta, V, and W atoms is affected by the heat treatment temperature. For instance, when the heat treatment temperature is lower than 700 K, Cr, Cu, Fe, Ta, V, and W atoms occupy the 1a and 3c sublattice randomly, and Re atoms prefer to 3c sublattice, while when the heat treatment temperature is higher than 1273 K, Cr, Cu, and W atoms prefer 3c sublattice, Fe, Ta atoms prefer to 1a sublattice, while all Re atoms occupy 3c sublattice exclusively, and all V atoms occupy 1a sublattice exclusively, respectively. Our calculated SOFs results (at the ground state) are highly consistent with the theoretical and experimental results in the available literature, except for Ti atoms, which were reported to occupy 1a sublattice at the ground state (Jinlong et al., 1993; Sluiter and Kawazoe, 1995; Ding et al., 2020). The calculated site preferences of alloying elements in the ternary Ni3Al-based γ′ phase compared with the available literature were tabulated in Table 2. N signifies that the element has no site preference in sublattice of Ni3Al.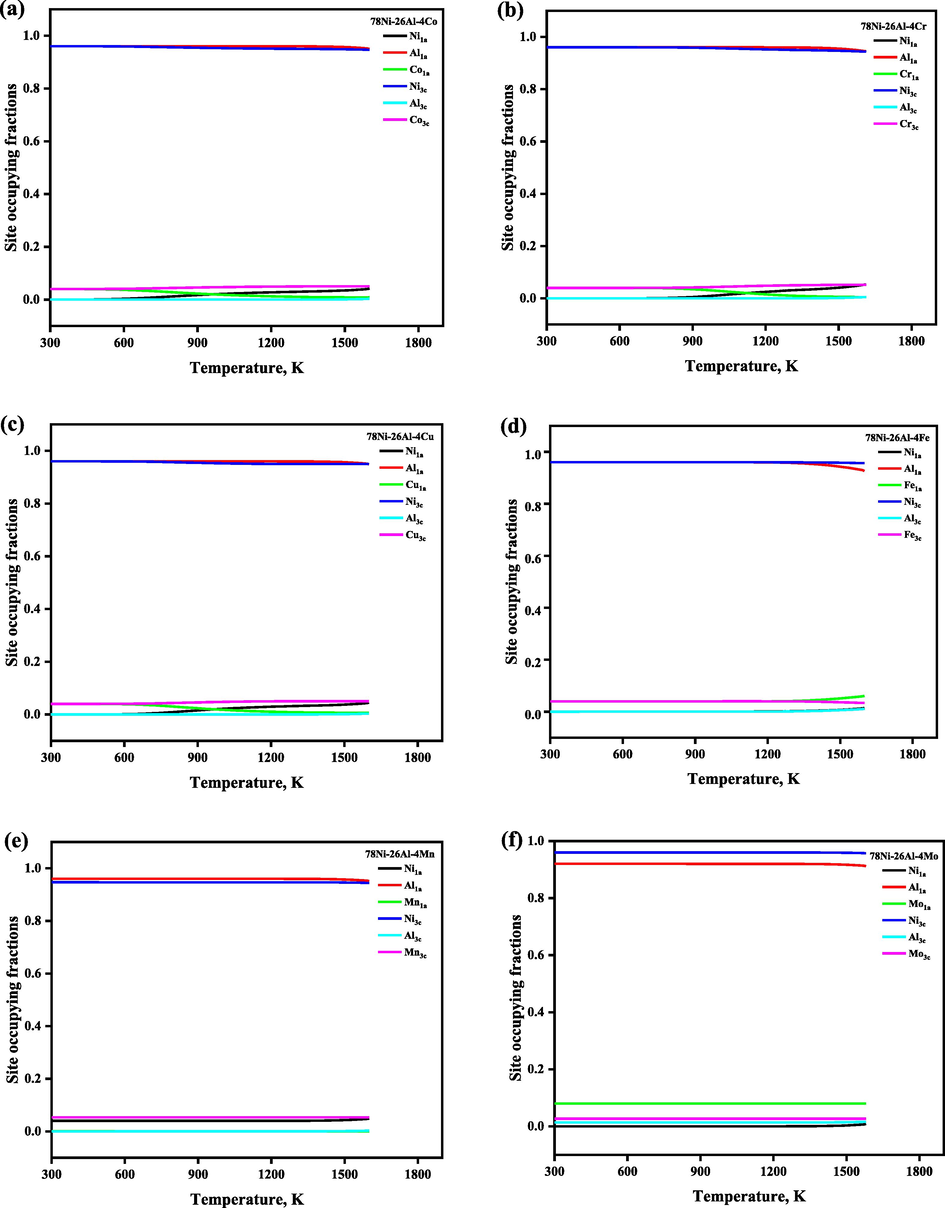
The temperature- and composition-dependent SOFs of the alloying element in Ni3Al-based γ′ phase with the composition 78Ni-26Al-4Mi, which is normalized as 72.22Ni-24.08Al-3.7Mi (at. %).
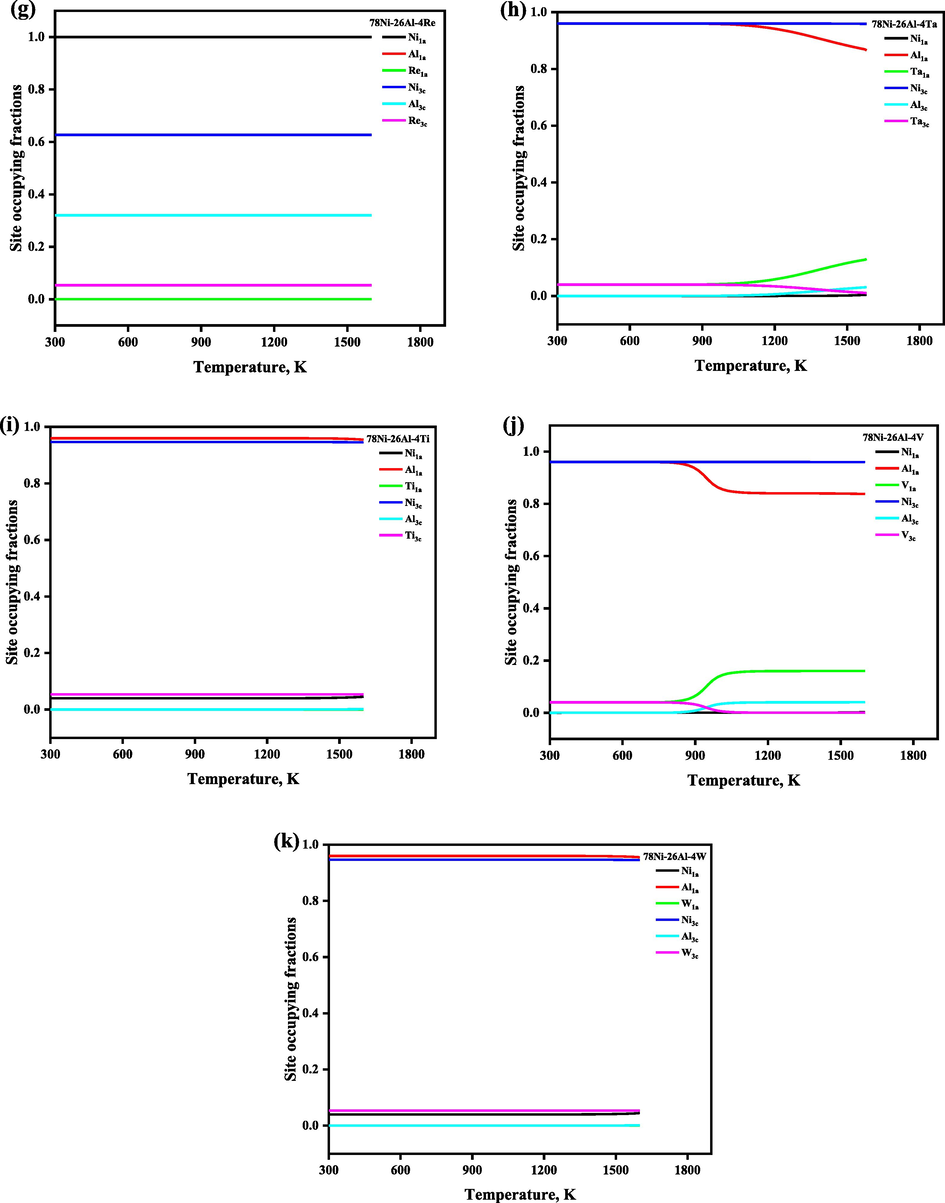
The temperature- and composition-dependent SOFs of the alloying element in Ni3Al-based γ′ phase with the composition 78Ni-26Al-4Mi, which is normalized as 72.22Ni-24.08Al-3.7Mi (at. %).
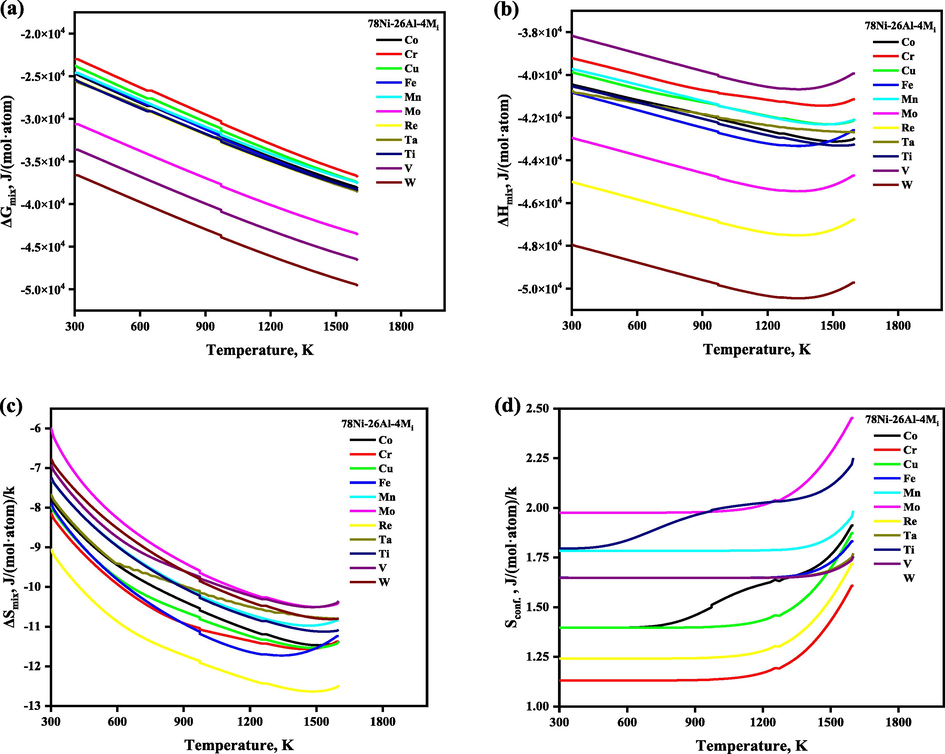
The temperature- and composition-dependent thermodynamic functions of the alloying element in Ni3Al-based γ′ phase with the composition 78Ni-26Al-4Mi, which is normalized as 72.22Ni-24.08Al-3.7Mi (at. %).
The site preference of the quaternary system with a selective composition 78Ni-26Al-2 M1-2 M2, which is normalized as 72.22Ni-24.08Al-1.85 M1-1.85 M2 (at. %) was also investigated, and the SOFs were plotted in Fig. 7. It is revealed that Co, Cu, Mn, Re, Ta, V, and W atoms prefer 3c sublattice (Ni site), Ti prefers 1a sublattice (Al site), in the whole temperature range, while the site preferences of Cr, Fe, and Mo atoms are affected by the heat treatment. For example, when the heat treatment temperature is lower than 700 K, then Cr, Fe, and Mo atoms randomly occupied both the sublattices i.e., 1a and 3c, but preferred to occupy the 1a sublattice. When the heat treatment temperature is higher than 1273 K, then Fe, and Mo atoms preferred the 1a sublattice, and all Cr atoms preferred the 3c sublattice exclusively. The temperature-dependent thermodynamic functions of the quaternary system with the composition 78Ni-26Al-2 M1-2 M2 are shown in Fig. 8, and we have observed that ΔGmix, ΔHmix, and ΔSmix are decreasing with the increase of the heat treatment, and configurational entropy Sconf is increasing with the increase of the temperature, which shows that the doping of quaternary elements also enhances the structural stability, respectively. The calculated site preference for stoichiometric, and nonstoichiometric Ni3Al, stoichiometric Co3Al along with ternary and quaternary systems at selective low and high temperatures was given in Table 3 for comparison conveniently.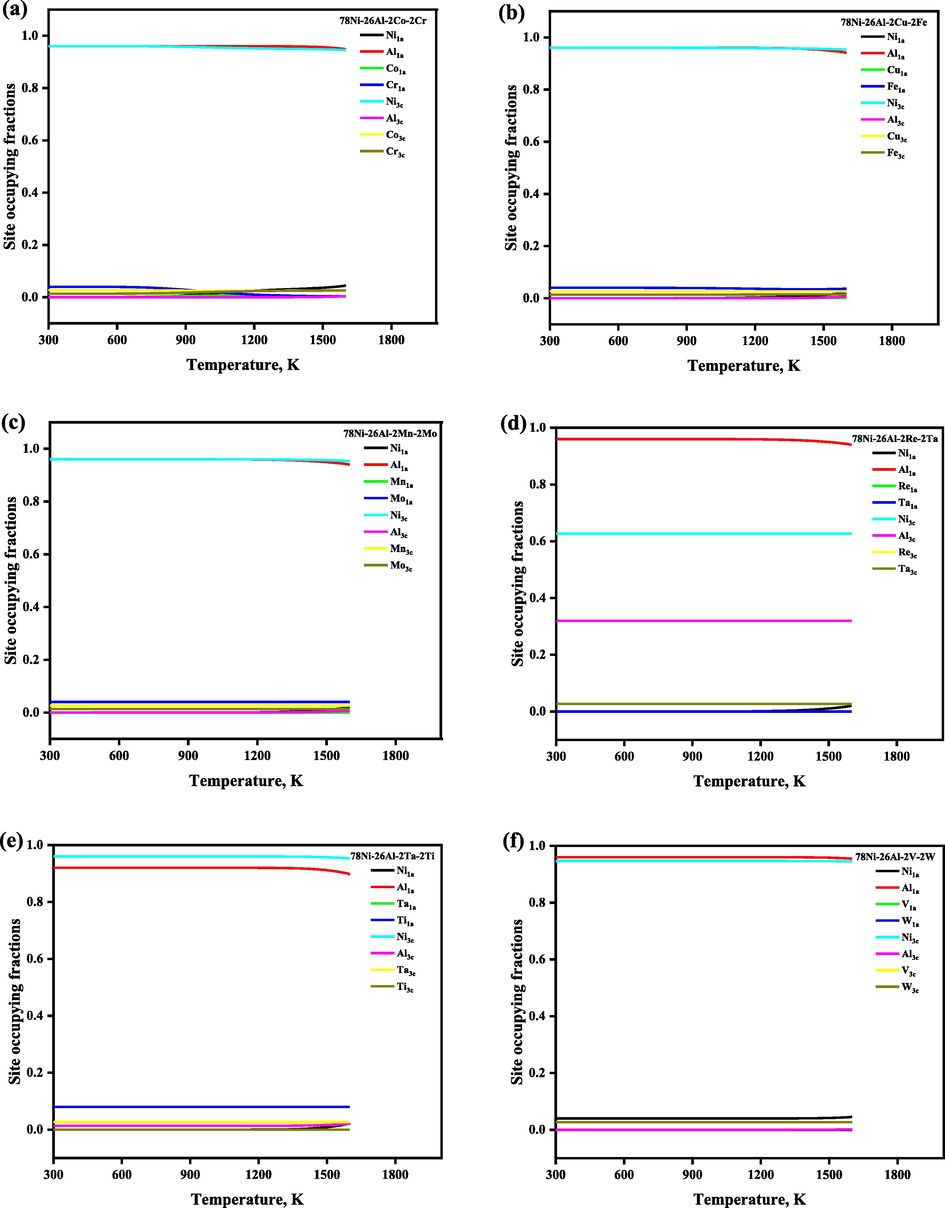
The temperature- and composition-dependent SOFs of the alloying element in Ni3Al-based γ′ phase with the composition 78Ni-26Al-2 M1-2 M2 which is normalized as 72.22Ni-24.08Al-1.85 M1-1.85 M2 (at. %).
Compositions
Elements
Site occupying fractions (SOFs)
at 298 K
at 1273 K
1a
3c
Preferred
1a
3c
Preferred
Ni75Al25
Ni
0
1
3c
0.0015
0.9994
3c
Al
1
0
1a
0.9984
0
1a
Co75Al25
Co
0
1
3c
0.7409
0.7530
3c
Al
1
0
1a
0.2590
0.2469
1a
Ni72Al28
Ni
0
1
3c
0
0.9599
3c
Al
1
0
1a
1
0.0401
1a
Ni78Al22
Ni
0.1199
1
3c
0.1200
0.9999
3c
Al
0.8800
0
1a
0.8799
0
1a
Ni78Al26Co4
Ni
0
0.9600
3c
0.0285
0.9504
3c
Al
1
0
1a
0.9598
0
1a
Co
0
0.0400
3c
0.0115
0.0494
3c
Ni78Al26Cr4
Ni
0
0.9600
3c
0.0285
0.9504
3c
Al
0.9600
0
1a
0.9597
0
1a
Cr
0.0400
0.0400
both
0.0116
0.0494
3c
Ni78Al26Cu4
Ni
0
0.9600
3c
0.0314
0.9495
3c
Al
0.9600
0
1a
0.9598
0
1a
Cu
0.0400
0.0400
both
0.0086
0.0504
3c
Ni78Al26Fe4
Ni
0
0.9600
3c
0.0015
0.9594
3c
Al
0.9600
0
1a
0.9578
0
1a
Fe
0.0400
0.0400
both
0.0405
0.0398
1a
Ni78Al26Mn4
Ni
0.0398
0.9467
3c
0.0401
0.9466
3c
Al
0.9600
0
1a
0.9598
0
1a
Mn
0
0.0532
3c
0
0.0533
3c
Ni78Al26Mo4
Ni
0
0.9600
3c
0
0.9600
3c
Al
0.9202
0.0130
1a
0.9200
0.0133
1a
Mo
0.0798
0.0267
1a
0.0799
0.0267
1a
Ni78Al26Re4
Ni
0.0197
0.9534
3c
0.0401
0.9466
3c
Al
0.9600
0
1a
0.9598
0
1a
Re
0.0202
0.0465
3c
0
0.0533
3c
Ni78Al26Ta4
Ni
0
0.9600
3c
0
0.9599
3c
Al
0.9600
0
1a
0.9298
0.0100
1a
Ta
0.0400
0.0400
both
0.0700
0.0299
1a
Ni78Al26Ti4
Ni
0.2247
0.8753
3c
0.0400
0.9466
3c
Al
0.7752
0.0713
1a
0.9599
0
1a
Ti
0
0.0533
3c
0
0.0533
3c
Ni78Al26V4
Ni
0
0.9600
3c
0
0.9599
3c
Al
0.9600
0
1a
0.8401
0.0399
1a
V
0.0400
0.0400
both
0.1597
0
1a
Ni78Al26W4
Ni
0
0.9599
3c
0.0362
0.9479
3c
Al
0.9600
0
1a
0.9598
0
1a
W
0.0399
0.0400
3c
0.0038
0.0520
3c
Ni78Al26Co2Cr2
Ni
0
0.9600
3c
0.0277
0.9508
3c
Al
0.9600
0
1a
0.9598
0
1a
Co
0
0.0267
3c
0.0050
0.0250
3c
Cr
0.0400
0.0133
1a
0.0075
0.0242
3c
Ni78Al26Cu2Fe2
Ni
0
0.9600
3c
0.0046
0.9585
3c
Al
0.9600
0
1a
0.9592
0
1a
Cu
0
0.0267
3c
0
0.0264
3c
Fe
0.0400
0.0133
1a
0.0354
0.0149
1a
Ni78Al26Mn2Mo2
Ni
0
0.9600
3c
0.0020
0.9593
3c
Al
0.9600
0
1a
0.9581
0
1a
Mn
0
0.0267
3c
0
0.0267
3c
Mo
0.0400
0.0133
1a
0.0399
0.0134
1a
Ni78Al26Re2Ta2
Ni
0
0.6266
3c
0.0019
0.6268
3c
Al
0.9999
0.3200
1a
0.9581
0.33
1a
Re
0
0.0266
3c
0
0.0267
3c
Ta
0
0.0266
3c
0
0.0267
3c
Ni78Al26Ta2Ti2
Ni
0
0.9600
3c
0
0.9598
3c
Al
0.9200
0.0133
1a
0.9295
0.0134
1a
Ta
0
0.0266
3c
0
0.0266
3c
Ti
0.0800
0
1a
0.08
0
1a
Ni78Al26V2W2
Ni
0.0400
0.9466
3c
0.0400
0.9466
3c
Al
0.9600
0
1a
0.9599
0
1a
V
0
0.0266
3c
0
0.0266
3c
W
0
0.0267
3c
0
0.0267
3c
3.3 Graphical demonstration of atoms distribution on the sublattice
Based on the calculated SOFs, the atom distributions on the sublattice and thus on the full lattice were graphically demonstrated for an intuitive understanding of the tendency of site preference. The site occupying fraction of FCC_L12 structure alloys 75Ni-25Al with unit cell and supercell is shown in Fig. 9, where a 10 × 10 × 10 supercell has been built based on AB3_L12 crystal lattice structure, and the distribution of atoms on the sublattice and thus on the full lattice were shown in a statistical sense. The supercell has 4000 atoms, including 1000 atoms on 1a sublattice and 3000 atoms on 3c sublattice. In this study, the estimated SOFs were used to account for the integer numbers of distinct atoms on each sort of sublattice. Here we relatively assumed that the atoms occupy each type of sublattices randomly for the calculated integer numbers, so for a specific configuration, the distribution of atoms in each type of sublattice and whole lattice may be visually represented. Similarly, we have built a 10 × 10 × 10 supercell for a better view of the atom distribution (4000 atoms in total) for the alloying composition 72Ni-24Al-4 W. In order to fulfill further calculations concerning the lattice distortion, thermodynamic properties, and mechanical properties based on the available computing resources, a 3 × 3 × 3 supercell containing 108 atoms was also built, and 1a and 3c sublattices were separated and nested in Fig. 10, thus an applied structure file (POSCAR) was prepared. So, we reasonably described the distribution of the atoms on sublattice and thus on a crystal lattice, which is one of the most essential structural information.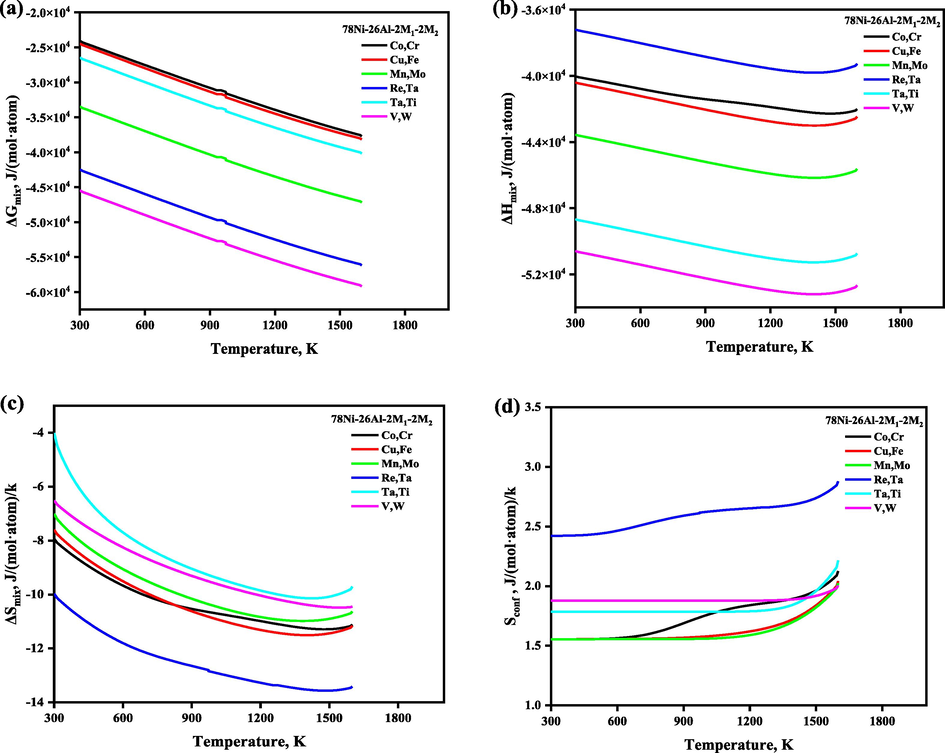
The temperature- and composition-dependent thermodynamic functions of the alloying element in Ni3Al-based γ′ phase with the composition 78Ni-26Al-2 M1-2 M2 which is normalized as 72.22Ni-24.08Al-1.85 M1-1.85 M2 (at. %).
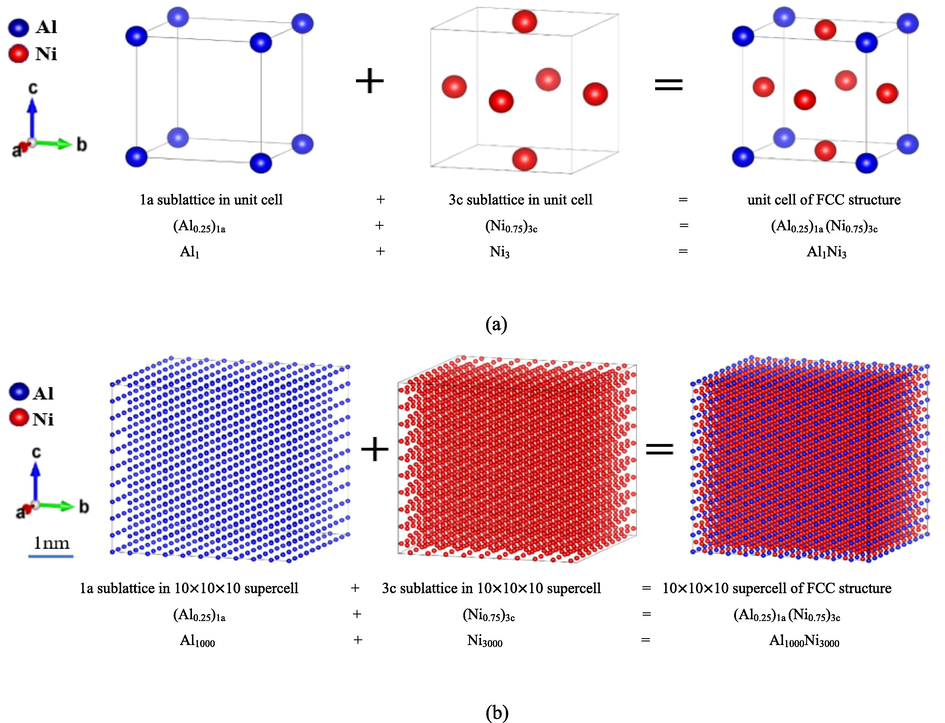
Visualization of the atomic distribution of the stoichiometric Ni3Al γ′ phase with the real relative size bar on the sublattice 1a and 3c based on a 10 × 10 × 10 supercell of the FCC_L12 (a) unit-cell of pure stoichiometric Ni3Al (b) ordered structure at room temperature.
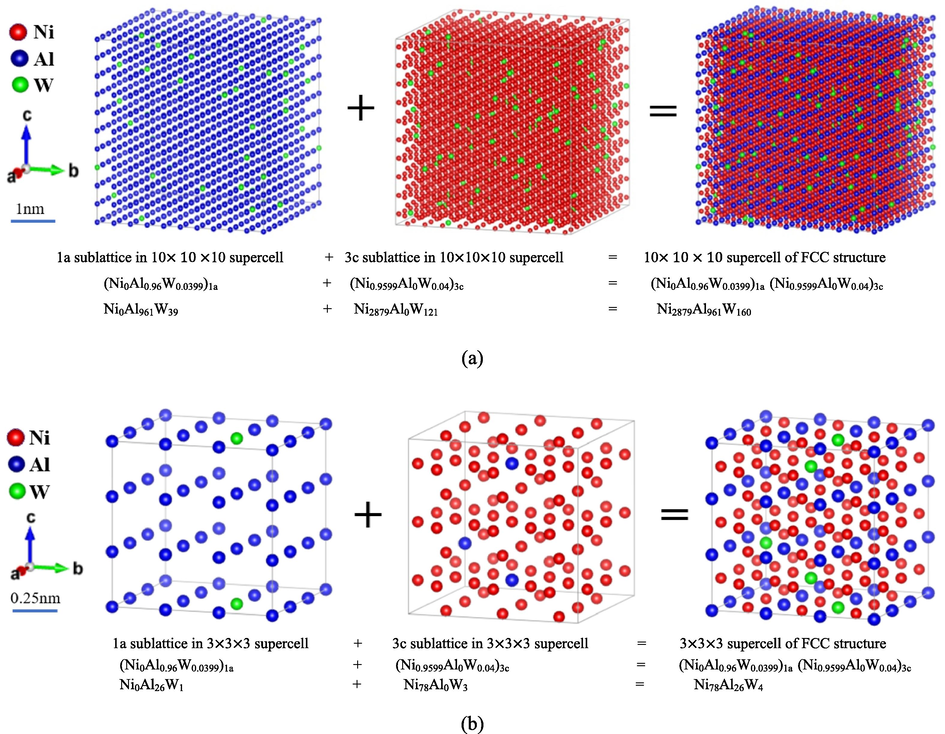
Atomic site occupancy configuration of FCC_L12 structure alloys 72Ni-24Al-4 W with the real relative size bar based on a different dimension of the FCC_L12 supercell at room temperature (a) 10 × 10 × 10 (b) 3 × 3 × 3 supercell.
3.4 Alloying effects on mechanical and thermodynamic properties
To explore the effect of the alloying elements on the mechanical properties, the elasticity was calculated at the ground state i.e., 0 K. Because it relates to the key characteristics of the mechanical properties of materials such as the stability of the mechanical structure, brittleness, stiffness, hardness, strength, fracture toughness (Chen and Bielawski, 2008), and interatomic bonding (Pugh and Xcii., 1954; Nye, 1985). Table 4 summarizes the elastic modulus calculation findings., which reveal that the elastic properties of pure Ni3Al agree well with the theoretical (Kim et al., 2010; Wu and Li, 2012; Xu et al., 2013; Xu et al., 2018) as well as experimental (Kayser and Stassis, 1981; Prikhodko et al., 1999; Chen and Knowles, 2003) data available in the literature. For the reliability of our calculations, the elastic properties of pure Co3Al are also summarized in Table 4 by implementing the same methodology, and the present results agree well with other available calculated (Xu et al., 2013; Xu et al., 2018) and experimental data (Kayser and Stassis, 1981; Prikhodko et al., 1999). The results showed that all the alloying elements would enhance the bulk modulus of polycrystalline Ni3Al alloys except Mn, Mo, and Ti, in which Mn have the greatest influence on reducing the bulk and shear modulus of Ni3Al. Additionally, we employed the mechanical stability criteria (Born, 1939) i.e., C11 > 0, C44 > 0, C11 > |C12|, (C11 + 2C12) > 0 to examine the mechanical stability of superalloys. It is noted that the additional alloying Mi elements have a significant effect on the elastic constants, but they fulfill the mechanical stability criteria which show that they are mechanically stable.
Alloys
C11
C12
C44
C12 − C44
B
G
E
v
H
B/G
Ni3Al
238.85
146.07
126.61
46.39
19.46
176.98
84.68
219.10
0.29
11.65
2.09
Calc. (Kim et al., 2010)
242.2
151.8
125.4
–
–
182.0
83.0
217.
–
–
2.18
Calc. (Wu and Li, 2012)
243.8
148.7
123.4
–
–
180.4
84.2
218.5
–
–
2.14
Calc. (Xu et al., 2013)
231.0
152.2
120.2
–
–
178.5
76.9
201.8
0.31
–
2.32
Calc. (Xu et al., 2018)
232.7
151.4
120.6
–
–
178.5
78.1
204.4
0.31
–
2.29
Expt. (Kayser and Stassis, 1981)
224.0
148.0
125.0
–
–
173.3
77.7
202.9
031
–
2.23
Expt. (Prikhodko et al., 1999)
224.5
148.6
124.4
–
–
173.9
77.5
202.3
0.31
–
2.24
Expt. (Chen and Knowles, 2003)
225
149
124
–
–
174
77
202
–
–
2.26
Co3Al
211.93
196.24
96.23
7.85
100.01
201.47
39.18
110.37
0.41
2.38
5.14
Calc. (Xu et al., 2013)
205.7
179.5
92.7
–
–
188.2
44.0
113.3
0.39
–
4.28
Calc. (Xu et al., 2018)
218.5
194.8
92.1
–
–
202.7
42.4
118.9
0.40
–
4.78
Ni78Al26Co4
235.15
185.91
130.95
24.62
54.96
202.32
68.21
183.97
0.35
6.89
2.97
Ni78Al26Cr4
258.30
137.23
115.48
60.54
21.75
177.58
89.11
229.03
0.29
12.77
1.99
Ni78Al26Cu4
221.20
157.31
98.45
31.95
58.86
178.61
62.78
168.59
0.34
6.58
2.84
Ni78Al26Fe4
224.40
163.59
120.58
30.41
43.01
183.86
69.83
185.95
0.33
7.85
2.63
Ni78Al26Mn4
189.90
159.34
103.13
15.28
56.21
169.53
49.62
135.63
0.37
4.41
3.42
Ni78Al26Mo4
201.84
158.58
111.52
21.63
47.06
173.00
58.73
158.27
0.35
5.97
2.95
Ni78Al26Re4
242.95
172.58
125.94
35.19
46.64
196.04
75.81
201.47
0.33
8.66
2.59
Ni78Al26Ta4
230.59
172.80
122.90
28.90
49.90
192.06
69.35
185.70
0.34
7.45
2.77
Ni78Al26Ti4
218.59
148.20
92.50
35.19
55.70
171.66
62.80
167.92
0.34
6.83
2.73
Ni78Al26V4
227.56
161.87
131.90
32.85
29.97
183.77
76.03
200.45
0.32
9.21
2.42
Ni78Al26W4
216.37
168.87
121.24
23.75
47.63
184.70
64.07
172.28
0.34
6.64
2.88
To study the ductile and brittle behavior of materials, we computed the bulk to shear modulus ratio (B/G ratio) proposed by Pugh (Pugh and Xcii., 1954). A material is ductile if the ratio is higher than 1.75, and if it's lower than 1.75 then the material is brittle, our calculated results showed that all Ni3Al alloys are ductile in nature because their B/G ratios are higher than 1.75. Thus, these alloying elements might be utilized as a precipitate to increase superalloy toughness. Furthermore, we calculated the tetragonal shear modulus and Cauchy pressure (C12 – C44) (Pettifor, 1992) to examine the bonding nature. A positive Cauchy pressure specifies a ductile behavior and metallic bonding, while a negative one implies angular character and covalent bonding. Additionally, the toughness of materials is more stable with higher Cauchy pressure. Evidently, the Cauchy pressures of all additional alloying elements have positive values, suggesting their toughness and metallic characteristics, as shown in Table 4. It can be seen from Table 4 that the addition of impurity elements increases the Cauchy pressure of the alloy, indicating that the addition of some alloy elements to the alloy system can increase the plasticity of the alloy. In addition to Ni78Al26Cr4, the Poisson's ratio and B/G ratio, which judged the ductility of materials, also show the same results, but the addition of impurity elements will reduce the shear modulus of the system. The shear modulus is based on the C44 value and is used to measure the resistance of solids to shape change, the smaller its value, the weaker its resistance to shape change, which results in increased its plasticity, which is consistent with the results of Cauchy pressure, Poisson's ratio, and B/G value. Except for Ni78Al26Cr4, the addition of impurity elements reduces the young's modulus of the system, which is an index to measure the stiffness of materials. We can conclude that the addition of some impurity elements will reduce the stiffness of the system, which also indicates that its resistance to elastic deformation decreases.
Furthermore, we also calculated the microhardness
(Cheng et al., 2013) which is the key characteristic of solid materials that gives information about the wear resistance to permanent or plastic deformation. It is often assumed that the higher the material's hardness, the better the wear resistance. According to our calculations, we have observed that Cr doping can increase the microhardness of Ni3Al alloys, and has a significant effect because its microhardness is the largest (12.77 GPa) among other additional alloying elements, so it possesses the best wear resistance. Additionally, hardness is often correlated with bond energy. It's also worth noting that materials with high hardness have a high bond strength, implying that Cr doping has a highest average bond energy (Sun et al., 2020). To deeply understand the relationship between macroscopic properties and the microscopic mechanism of alloys, we have calculated the bond energy of some selective alloying elements. For this purpose, firstly, we built a 15 × 15 × 15 (Å) cubic cell, then placed atoms into the cube to only optimized the atomic position, and then extract the total energy from the output file as the bond energy. For example, we calculated the bond energies of the pure elements Al (-0.247) and Ni (-0.343) in eV/atom, and the calculated bond energies of all compositions involved in this work are Ni78Al26Co4 (-6.322), Ni78Al26Cr4 (-6.613), Ni78Al26Cu4 (-6.464), Ni78Al26Fe4 (-6.469), Ni78Al26Mn4 (-6.452), Ni78Al26Mo4 (-6.480), Ni78Al26Re4 (-6.483), Ni78Al26Ta4 (-6.470), Ni78Al26Ti4 (-6.472), Ni78Al26V4 (-6.493), and Ni78Al26W4 (-6.446) in eV/atom, respectively. We have observed that the bond energy of Ni78Al26Cr4 is the minimum among other compositions which showed that it has the highest hardness, which corresponds to our calculated results. In addition, the calculated crystal lattice parameters of doped Ni3Al-based γ′ phase alloys involved in this work are summarized in Table 5. *Fully relaxed: Volume, shape, and atom position are fully relaxed by setting ISIF = 3 and NSW = 10 in the INCAR file.
Alloys
a(Å)
b(Å)
c(Å)
α(°)
β(°)
γ(°)
V (Å^3)
Ni3Al
10.73036
10.73036
10.73036
90.0000
90.0000
90.0000
1235.5007
Co3Al
10.77142
10.80456
10.77487
90.1515
89.9816
90.1573
1253.9754
Ni78Al26Co4
10.75576
10.75904
10.70553
89.9976
89.9864
90.0016
1238.8611
Ni78Al26Cr4
10.79226
10.75047
10.72976
89.9533
90.1585
89.9371
1244.8800
Ni78Al26Cu4
10.72994
10.79370
10.76744
90.0969
89.7926
90.0931
1247.0265
Ni78Al26Fe4
10.71747
10.71747
10.71747
90.0000
90.0000
90.0000
1231.0520
Ni78Al26Mn4
10.78957
10.75607
10.75665
90.0458
89.9412
89.9884
1248.3448
Ni78Al26Mo4
10.74301
10.74301
10.74301
90.0000
90.0000
90.0000
1239.8746
Ni78Al26Re4
10.66760
10.82554
10.71324
89.8878
89.8871
89.9994
1237.1876
Ni78Al26Ta4
10.73043
10.76918
10.84262
90.0943
89.9925
89.9590
1252.9491
Ni78Al26Ti4
10.70245
10.70245
10.70245
90.0000
90.0000
90.0000
1225.8830
Ni78Al26V4
10.86879
10.48691
10.86796
89.7902
89.4979
89.9953
1238.6743
Ni78Al26W4
10.75450
10.75450
10.75450
90.0000
90.0000
90.0000
1243.8587
Furthermore, to consider the relationship between the density of states (DOS) and mechanical properties, we have calculated the DOS of pure Ni3Al-based γ′ phase with composition 75Ni-25Al (at. %), pure Co3Al-based γ′ phase with composition 75Co-25Al (at. %), and DOS of Ni3Al doped with Co with composition 72Ni-24Al-4Co (at. %), here we take it as an example. From Fig. 11, it is evident that the range of DOS has been expanded in both the valence and conduction regions. The peak intensities have been decreased to make the DOS near Fermi-level flat slightly for all the compounds. According to the above analyses, the addition of the alloying elements (i.e., TMs) can effectively reduce the covalent nature of Ni3Al-based γ′ phase alloys, resulting in enhanced ductility, which is resembled to our calculated mechanical properties results.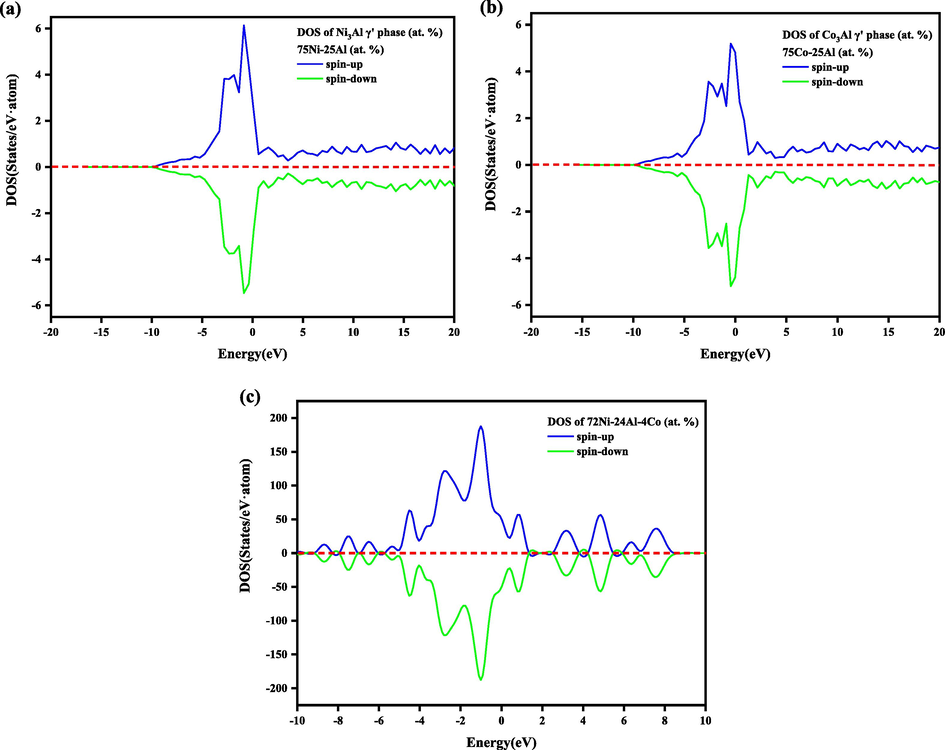
Projected density of states of Ni3Al-based γ′ phase with different composition (at. %) (a) 75Ni-25Al (b) 75Co-25Al (c) 72Ni-24Al-4Co, at 0 GPa pressure. The DOS for spin-up and spin-down are plotted by the blue and green lines, respectively. The Fermi level has been subtracted to zero energy.
Debye temperature
is used to observe the impact of alloying elements on the thermodynamic properties of Ni3Al alloys. Besides indicating a specific shear modulus, elastic parameters, thermal conductivity, and melting temperature, the Debye temperature also signifies the stiffness of lattices. A high Debye temperature indicates strong interactions of atoms. The Debye temperature in terms of average sound velocity
can be obtained by (Lowrie, 1963):
The calculated results are listed in Table 6, which shows that only doping with Cr enhances the Debye temperature of Ni3Al alloys remarkably, resulting in improved crystallographic interaction. Although there is no measured data
for composition 78Ni-26Al-4Mi available in the literature for comparative comparison, but based on the excellent agreement between the current study with other theoretical (Xu et al., 2013) and experimental (Kayser and Stassis, 1981; Stassis et al., 1981) results of
for Ni3Al, it seems reasonable to conclude that the present theoretical results are valid for Ni3Al superalloys alloyed with Mi, respectively.
Alloys
Ni3Al
3235.89
6132.89
3617.69
478.60
Calc. (Xu et al., 2013)
3219.9
6147.4
3601.6
476.8
Expt. (Kayser and Stassis, 1981; Stassis et al., 1981)
3225.3 (Kayser and Stassis, 1981)
6102.6 (Kayser and Stassis, 1981)
3605.4 (Kayser and Stassis, 1981)
470 (Stassis et al., 1981)
Ni78Al26Co4
3126.07
6481.86
3513.96
464.45
Ni78Al26Cr4
3673.93
6700.42
4096.02
540.20
Ni78Al26Cu4
2959.48
6049.35
3324.12
438.65
Ni78Al26Fe4
3195.07
6363.13
3583.38
473.84
Ni78Al26Mn4
2729.24
5948.06
3075.46
403.97
Ni78Al26Mo4
2614.06
5407.51
2938.05
385.77
Ni78Al26Re4
2410.36
4771.75
2702.32
356.50
Ni78Al26Ta4
2326.01
4711.39
2611.25
344.68
Ni78Al26Ti4
3133.10
6318.43
3516.45
463.60
Ni78Al26V4
3395.21
6575.10
3801.24
502.45
Ni78Al26W4
2231.72
4582.54
2507.32
330.16
4 Conclusions
The temperature- and composition-dependent site preferences were predicted concerning the binary, ternary, and quaternary Ni3Al-based γ′ phase with L12 structure alloyed with some frequently adopted transition metals Mi, where Mi represents Co, Cr, Cu, Fe, Mn, Mo, Re, Ta, Ti, V, and W atoms, using a two-sublattice thermodynamic model (Ni, Al, Mi)1a(Ni, Al, Mi)3c. It was revealed that for the stoichiometry binary Ni3Al show a fully ordered structure at all temperatures. For the γ′ phase with the composition 78Ni-26Al-4Mi, where xNi/xAl = 3:1, Mo atoms always preferred to occupy the 1a sublattice (Al site), while Co, Mn, and Ti atoms always prefer the 3c sublattice (Ni site) in the whole temperature range, while the site preference of the alloying elements such as Cr, Cu, Fe, Re, Ta, Vlore W atom is affected by the heat treatment temperature. For example, when the heat treatment temperature is lower than 700 K, Cr, Cu, Fe, Ta, V, and W atoms occupy the 1a and 3c sublattice randomly, and Re atoms prefer to 3c sublattice, while when the heat treatment temperature is higher than 1273 K, Cr, Cu, Mo, and W atoms prefer 3c sublattice, Fe, Ta atoms prefer to 1a sublattice, while all Re atoms occupy 3c sublattice exclusively, and all V atoms occupy 1a sublattice exclusively, respectively. The site preference of the quaternary system with a selective composition 78Ni-26Al-2 M1-2 M2, has been also investigated. It is revealed that Co, Cu, Mn, Re, Ta, V, and W atoms prefer 3c sublattice (Ni site), Ti prefers 1a sublattice (Al site), in the whole temperature range, while the site preferences of Cr, Fe, and Mo atoms are affected by the heat treatment. For example, when the heat treatment temperature is lower than 700 K, Cr, Fe, and Mo atoms have no obvious site preference by randomly occupying both 1a sublattice and 3c sublattice, but slightly prefer to 1a sublattice. When the heat treatment temperature is higher than 1273 K, Fe, and Mo atoms prefer the 1a sublattice, and all Cr atoms occupy the 3c sublattice exclusively. Cr, Re, and V doping can improve the microhardness of Ni3Al alloys; in particular, the effect of Cr is extraordinary; and all elements, except Mn, Mo, and Ti, would enhance the bulk modulus of Ni3Al-based γ′ phase, in which Mn have the greatest influence on reducing the bulk and shear modulus, respectively. All of the computed Ni3Al alloys are all inherent ductile based on the Pugh criterion. Only Cr doping significantly enhances the Debye temperature of the Ni3Al-based γ′ phase. Our calculated results are highly consistent with the available literature. The quantitative understanding of the site preference and strengthening mechanisms are expected to promote the development of the novel Ni-based superalloy.
Acknowledgments
This work is financially supported by the National Natural Science Foundation of China (50971043, 51171046, 21973012), Key Research and Development Program of China (2017YFB0701700, CISRI-21T62450ZD), Natural Science Foundation of Fujian Province (2021J01590, 2014J01176, 2018J01754, 2020J01474), National Key Laboratory of Eco-materials Advanced Technology (Fuzhou University), Student Research and Training Program (SRTP) of Fuzhou University (27297), State Administration for Market Regulation (2021MK050) and Fujian Provincial Department of Science & Technology (2021H6011).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Thermodynamic assessment of the Cu−Pt system. Journal of Phase Equilibria and Diffusion. 2006;27(1):5-13.
- [Google Scholar]
- Materials Today Communications. 2022;33:104447. The ordering behavior of Co3Al-based γ′ phase with L12 structure predicted by the thermodynamic model with support of first-principles calculations
- On the interplay between tungsten and tantalum atoms in Ni-based superalloys: An atom-probe tomographic and first-principles study. Appl. Phys. Lett.. 2009;94(4):041917
- [Google Scholar]
- Effects of tantalum on the partitioning of tungsten between the γ-and γ′-phases in nickel-based superalloys: Linking experimental and computational approaches. Acta Mater.. 2010;58(18):5898-5911.
- [Google Scholar]
- Combined atom probe tomography and first-principles calculations for studying atomistic interactions between tungsten and tantalum in nickel-based alloys. Acta Mater.. 2014;74:296-308.
- [Google Scholar]
- A simplified method for calculating the Debye temperature from elastic constants. J. Phys. Chem. Solids. 1963;24(7):909-917.
- [Google Scholar]
- Thermo-Calc & DICTRA, computational tools for materials science. Calphad. 2002;26(2):273-312.
- [Google Scholar]
- An Atom Probe Tomography study of site preference and partitioning in a nickel-based superalloy. Acta Mater.. 2017;125:156-165.
- [Google Scholar]
- Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001;73:515.
- [Google Scholar]
- Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys.. 2001;73(2):515.
- [Google Scholar]
- Density-functional perturbation theory for quasi-harmonic calculations. Rev. Mineral. Geochem.. 2010;71(1):39-57.
- [Google Scholar]
- The effects of Cr, Al, Ti, Mo, W, Ta, and Cb on the cyclic oxidation behavior of cast Ni-base superalloys at 1100 and 1150 C. Oxid. Met.. 1983;20(5):255-278.
- [Google Scholar]
- Modelling and analysis of the oxidation influence on creep behaviour of thin-walled structures of the single-crystal nickel-base superalloy René N5 at 980 ℃. Acta Mater.. 2010;58(5):1607-1617.
- [Google Scholar]
- Phase composition and long range order in γ′ phase of a nickel base single crystal superalloy CMSX2: An atom probe study. Acta Metall.. 1984;32(5):811-816.
- [Google Scholar]
- Phase transformation and segregation to lattice defects in Ni-base superalloys. Microsc. Microanal.. 2007;13(6):464-483.
- [Google Scholar]
- Chromium and tantalum site substitution patterns in Ni3Al (L12) γ′-precipitates. Appl. Phys. Lett.. 2008;93(3):033103
- [Google Scholar]
- Amorphous phase formation in irradiated intermetallic compounds. Radiation Effects. 1983;77(3–4):273-293.
- [Google Scholar]
- Discovering chemical site occupancy-modulus correlations in Ni based intermetallics via statistical learning methods. Comput. Condens. Matter. 2018;14:8-14.
- [Google Scholar]
- Heat Treatment Microstructure and Properties of Nonferrous Alloys. Metallurgical Industry Press. 1988;32
- [Google Scholar]
- Evolution of Ni-based superalloys for single crystal gas turbine blade applications. Aerosp. Sci. Technol.. 1999;3(8):513-523.
- [Google Scholar]
- Site occupancy of chromium in the γ′-Ni3Al phase of nickel-based superalloys: a combined 3D atom probe and first-principles study. Philos. Mag. Lett.. 2012;92(9):495-506.
- [Google Scholar]
- Site preference and interaction energies of Co and Cr in gamma prime Ni3Al: a first-principles study. Modell. Simul. Mater. Sci. Eng.. 2013;21(5):055006
- [Google Scholar]
- Interfacial fracture toughness of transition metal nitrides. Surf. Coat. Technol.. 2008;203(5–7):598-601.
- [Google Scholar]
- A synergistic reinforcement of Re and W for ideal shear strengths of γ′-Ni3Al phases. J. Phys. Chem. Solids. 2019;131:34-43.
- [Google Scholar]
- Mechanism of< 1 1 2>/3 slip initiation and anisotropy of γ′ phase in CMSX-4 during creep at 750℃ and 750 MPa. Mater. Sci. Eng., A. 2003;356(1–2):352-367.
- [Google Scholar]
- Enhancing catalytic properties of noble metal@ MoS2/WS2 heterojunction for the hydrogen evolution reaction. Appl. Surf. Sci.. 2022;591:153168
- [Google Scholar]
- First-principles density functional calculation of mechanical, thermodynamic and electronic properties of CuIn and Cu2In crystals. J. Alloy. Compd.. 2013;546:286-295.
- [Google Scholar]
- Ductilization of Ni3Al by macroalloying with Pd. Acta Metall.et materialia. 1991;39(8):1799-1805.
- [Google Scholar]
- Effect of γ and γ′ former doping on ductility of Ni3Al. Scr. Metall.et materialia. 1991;25(2):303-307.
- [Google Scholar]
- Ductilization of Ni3 by microalloying with Ag. Scr. Metall.et materialia. 1992;26(7):1031-1036.
- [Google Scholar]
- Site preference of refractory elements in Ni-based single-crystal Superalloys alloying with Ru: from first principles. Advanced Materials Research: Trans Tech Publ; 2012. p. :3-12.
- Structural intermetallics, Warrendale, PA (United States). Minerals: Metals and Materials Society; 1993.
- Site occupancy of alloying elements in γ′ phase of nickel-base single crystal superalloys. Intermetallics. 2020;121:106772
- [Google Scholar]
- Analysis of γ′/γ equilibrium in Ni−Al−X alloys by the. Metall. Trans. A. 1989;20(4):649-664.
- [Google Scholar]
- High-temperature site preference and atomic short-range ordering characteristics of ternary alloying elements in γ′-Ni3Al intermetallics. Phil. Mag.. 2017;97(29):2615-2631.
- [Google Scholar]
- The Site Preferences of Transition Elements and Their Synergistic Effects on the Bonding Strengthening and Structural Stability of γ′-Ni3 Al Precipitates in Ni-Based Superalloys: A First-Principles Investigation. Metallurgical and Materials Transactions A. 2021;52(6):2298-2313.
- [Google Scholar]
- Elastic constants of hexagonal transition metals: Theory. Phys. Rev. B. 1995;51(24):17431.
- [Google Scholar]
- Investigation on precipitation phenomena of Ni–22Cr–12Co–9Mo alloy aged and crept at high temperature. Int. J. Press. Vessels Pip.. 2008;85(1–2):63-71.
- [Google Scholar]
- Effects of Ir or Ta alloying addition on interdiffusion of L12–Ni3Al. Intermetallics. 2008;16(9):1095-1103.
- [Google Scholar]
- Site preference and alloying effect of platinum group metals in γ′ -Ni3Al. Acta Mater.. 2004;52(18):5427-5433.
- [Google Scholar]
- Effects of platinum on the interdiffusion and oxidation behavior of Ni-Al-based alloys. Materials Science Forum: Trans Tech Publ; 2004. p. :213-222.
- Structure and yield strength of directionally solidified Ni3Al intermetallic premelted with MoSi2 phase. Intermetallics. 1999;7(1):109-114.
- [Google Scholar]
- Strengthening effects of alloying elements W and Re on Ni3Al: A first-principles study. Comput. Mater. Sci.. 2018;144:23-31.
- [Google Scholar]
- Determination of site preference of Cu and Ge in Ni3Al. Surf. Sci.. 1992;266(1–3):358-363.
- [Google Scholar]
- Elastic and thermodynamic properties of the Ti2AlNb orthorhombic phase from first-principles calculations, physica status solidi (b). 2017;254(6):1600634.
- The effects of refractory elements on Ni-excesses and Ni-depletions at γ (fcc)/ γ′ (L12) interfaces in model Ni-based superalloys: Atom-probe tomographic experiments and first-principles calculations. Acta Mater.. 2016;121:288-298.
- [Google Scholar]
- Phys. Lett. 1983;42:672.
- Toughening mechanism of Ni3Al alloys by B-doping. J. Mater. Sci. Lett.. 1998;17(23):1967-1969.
- [Google Scholar]
- Synthesis of nickel aluminides by mechanical alloying. Mater. Lett.. 1988;7(1–2):51-54.
- [Google Scholar]
- Computational predictions of energy materials using density functional theory. Nat. Rev. Mater.. 2016;1(1):1-13.
- [Google Scholar]
- Site preference of ternary alloying elements in Ni3Al: A first-principles study. Acta Mater.. 2006;54(4):1147-1154.
- [Google Scholar]
- Site preference of alloying additions in intermetallic compounds. J. Phys.: Condens. Matter. 1993;5(36):6653.
- [Google Scholar]
- The elastic constants of Ni3Al at 0 and 23.5 ℃, Physica status solidi (a). 1981;64(1):335-342.
- Effects of alloying elements on elastic properties of Ni3Al by first-principles calculations. Intermetallics. 2010;18(6):1163-1171.
- [Google Scholar]
- Effects of alloying elements on thermal expansions of γ-Ni and γ′-Ni3Al by first-principles calculations. Acta Mater.. 2012;60(4):1846-1856.
- [Google Scholar]
- Microtwinning and other shearing mechanisms at intermediate temperatures in Ni-based superalloys. Prog. Mater Sci.. 2009;54(6):839-873.
- [Google Scholar]
- Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B. 1996;54(16):11169.
- [Google Scholar]
- From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B. 1999;59(3):1758.
- [Google Scholar]
- An ab initio investigation of the effect of alloying elements on the elastic properties and magnetic behavior of Ni3Al. Comput. Mater. Sci.. 2015;101:39-46.
- [Google Scholar]
- Elastic properties of metals and alloys, I. Iron, nickel, and iron-nickel alloys, Journal of Physical and Chemical Reference Data. 1973;2(3):531-618.
- [Google Scholar]
- Reproducibility in density functional theory calculations of solids. Science. 2016;351(6280):aad3000.
- [Google Scholar]
- Atomic site location by channelling enhanced microanalysis (ALCHEMI) in γ′-strengthened Ni-and Pt-base alloys. Acta Mater.. 2008;56(16):4267-4276.
- [Google Scholar]
- Investigation of the elemental partitioning behaviour and site preference in ternary model nickel-based superalloys by atom probe tomography and first-principles calculations. Phil. Mag.. 2016;96(21):2204-2218.
- [Google Scholar]
- Thermal behavior during combustion synthesis on intermetallic compound of Ni–Al system. J. Mater. Sci. Lett.. 1999;18(5):395-398.
- [Google Scholar]
- Atomic ordering characteristics of Ni3Al intermetallics with substitutional ternary additions. Acta Mater.. 1997;45(3):1077-1083.
- [Google Scholar]
- APFIM characterization of single-crystal PWA 1480 nickel-base superalloy. Appl. Surf. Sci.. 1994;76:172-176.
- [Google Scholar]
- 104 The physical and mechanical properties of NiAl. Acta Metall. Mater.. 1993;41(3):649-684.
- [Google Scholar]
- Microstructural characterization of Udimet 720: A nickel-base alloy. J. Phys. Coll.. 1988;49(C6) C6-391–C6-396
- [Google Scholar]
- The location of atoms in Re-and V-containing multicomponent nickel-base single-crystal superalloys. Appl. Surf. Sci.. 1994;76:177-183.
- [Google Scholar]
- Physical and mechanical properties of the B2 compound NiAl. Int. Mater. Rev.. 1993;38(4):193-232.
- [Google Scholar]
- Physical properties of crystals: their representation by tensors and matrices. Oxford University Press; 1985.
- Site-preferences and local spin-polarization of transition metal solute atoms in B2 type Ni–Al alloys. Intermetallics. 2009;17(6):441-444.
- [Google Scholar]
- Order–disorder transition in nanocrystalline Ni3Al prepared by a chemical route. Physica E. 2006;31(2):224-227.
- [Google Scholar]
- Influence of N-vacancy on the electronic and optical properties of bulk GaN from first-principles investigations. Int. J. Energy Res.. 2021;45(10):15512-15520.
- [Google Scholar]
- First-principles investigation of structural stability, electronic and optical properties of suboxide (Zr3O) Mater. Sci. Eng., B. 2022;281:115746
- [Google Scholar]
- Exploring the novel structure, transportable capacity and thermodynamic properties of TiH2 hydrogen storage material. Int. J. Energy Res.. 2020;44(6):4997-5007.
- [Google Scholar]
- Influence of alloying elements on the mechanical and thermodynamic properties of ZrB2 boride. Vacuum. 2022;198:110898
- [Google Scholar]
- Generalized gradient approximation made simple. Phys. Rev. Lett.. 1996;77(18):3865.
- [Google Scholar]
- Theoretical predictions of structure and related properties of intermetallics. Mater. Sci. Technol.. 1992;8(4):345-349.
- [Google Scholar]
- Nickel-based superalloys for advanced turbine engines: chemistry, microstructure and properties. J. Propul. Power. 2006;22(2):361-374.
- [Google Scholar]
- Temperature and composition dependence of the elastic constants of Ni3Al. Metallurgical and Materials Transactions A. 1999;30(9):2403-2408.
- [Google Scholar]
- First-principles investigation of solution mechanism of C in TM-Si-C matrix as the potential high-temperature ceramics. J. Am. Ceram. Soc.. 2022;105(4):2858-2868.
- [Google Scholar]
- New insight into the structural stability, ductility and melting point of Mo5SiB2 under high-pressure environment. Vacuum. 2022;196:110727
- [Google Scholar]
- First-principles investigation of oxidation mechanism of Al-doped Mo5Si3 silicide. Ceram. Int.. 2022;48(8):11518-11526.
- [Google Scholar]
- First-principles investigation of equilibrium phase, mechanical and thermodynamic properties of the Nowotny TM5Si3C ternary phase. Ceram. Int.. 2022;48(14):20438-20445.
- [Google Scholar]
- First-principles prediction of structure and mechanical properties of TM5SiC2 ternary silicides. Vacuum. 2022;199:110981
- [Google Scholar]
- Relations between the elastic moduli and the plastic properties of polycrystalline pure metals, The London, Edinburgh, and Dublin Philosophical Magazine and Journal of. Science. 1954;45(367):823-843.
- [Google Scholar]
- A study of ternary element site substitution in Ni 3Al using pseudopotential orbital radii based structure maps. Scr. Mater.. 1996;34(11)
- [Google Scholar]
- The alloying behaviour and mechanical properties of polycrystalline Ni3Al (γ′ phase) with ternary additions. J. Mater. Sci.. 1975;10(3):505-514.
- [Google Scholar]
- The Superalloys. CBO9780511541285: Fundamentals and Applications Cambridge Cambridge University Press 10.1017; 2006.
- Calculated site substitution in ternary γ′ IH-Ni3Al: Temperature and composition effects. Phys. Rev. B. 1997;55(2):856.
- [Google Scholar]
- The Monte Carlo simulation of ordering kinetics in Ni-base superalloys. Mater. Sci. Eng., A. 1997;223(1–2):1-9.
- [Google Scholar]
- Schreiber, E., Anderson, O.L., Soga, N., Bell, J.F., 1975. Elastic constants and their measurement.
- First-principles elastic constants of α-and θ-Al2O3. Appl. Phys. Lett.. 2007;90(10):101909
- [Google Scholar]
- Site determination of Fe, Co and Cr atoms added in Ni3Al by electron channelling enhanced microanalysis. Trans. Jpn. Inst. Met.. 1988;29(12):956-961.
- [Google Scholar]
- First principles study of site substitution of ternary elements in NiAl. Acta Mater.. 2001;49(9):1647-1654.
- [Google Scholar]
- First-Principles Study of Structural, Mechanical, and Thermodynamic Properties of Refractory Metals (Rh, Ir, W, Ta, Nb, Mo, Re, and Os). Materials Science Forum: Trans Tech Publ; 2020. p. :1017-1030.
- The temperature dependence of the strength of pseudo-binary platinum-based L12 alloys with B-subgroup elements. J. Mater. Sci.. 1981;16(10):2737-2744.
- [Google Scholar]
- First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Phys. Rev. B. 2008;78(13):134106
- [Google Scholar]
- Phase-partitioning and site-substitution patterns of molybdenum in a model Ni-Al-Mo superalloy: An atom-probe tomographic and first-principles study. Appl. Phys. Lett.. 2012;101(12):121910
- [Google Scholar]
- Site preference and lattice relaxation around 4d and 5d refractory elements in Ni3Al. Journal of synchrotron radiation. 2016;23(1):286-292.
- [Google Scholar]
- Energetics and electronic structure of Re and Ta in the γ′ phase of Ni-based superalloys. Phys. Rev. B. 2001;65(3):035101
- [Google Scholar]
- Influence of the alloying element Re on the ideal tensile and shear strength of γ′ -Ni3Al. Scr. Mater.. 2009;61(2):197-200.
- [Google Scholar]
- Pressure-induced structure, electronic, thermodynamic and mechanical properties of Ti2AlNb orthorhombic phase by first-principles calculations. Rare Met.. 2021;40(10):1-11.
- [Google Scholar]
- First principle investigation of crystal lattice structure, thermodynamics and mechanical properties in ZnZrAl2 intermetallic compound. Solid State Commun.. 2016;247:82-87.
- [Google Scholar]
- The influence of Mn atom location on the electronic structure of Ni3Al1− xMnx alloys: LMTO calculation and X-ray spectroscopy. J. Alloy. Compd.. 2004;362(1–2):189-197.
- [Google Scholar]
- Alloying element additions to Ni3Al: Site preferences and effects on elastic properties from first-principles calculations. Comput. Mater. Sci.. 2012;53(1):436-443.
- [Google Scholar]
- Prediction of the ordering behaviours of the orthorhombic phase based on Ti2AlNb alloys by combining thermodynamic model with ab initio calculation. Intermetallics. 2008;16(1):42-51.
- [Google Scholar]
- Prediction of the site ordering behaviours of elements in C15 NbCr2-based intermetallics by combining thermodynamic model with ab-initio calculation. Intermetallics. 2013;35:104-109.
- [Google Scholar]
- A reasonable approach to describe the atom distributions and configurational entropy in high entropy alloys based on site preference. Intermetallics. 2022;144:107489
- [Google Scholar]
- Understanding pop-in phenomena in FeNi3 nanoindentation. Intermetallics. 2015;67:111-120.
- [Google Scholar]
- Thermodynamic, structural and elastic properties of Co3X (X= Ti, Ta, W, V, Al) compounds from first-principles calculations. Intermetallics. 2013;32:303-311.
- [Google Scholar]
- Accelerating exploitation of Co-Al-based superalloys from theoretical study. Mater. Des.. 2018;142:139-148.
- [Google Scholar]
- Influence of noble metals on the electronic and optical properties of LiH hydride: First-principles calculations. Int. J. Hydrogen Energy. 2021;46(71):35342-35350.
- [Google Scholar]
- The first-principles study on the doping effect of Re in Ni3Al. Prog. Nat. Sci.. 2008;18(7):861-866.
- [Google Scholar]
- Synergistic effect of co-alloying elements on site preferences and elastic properties of Ni3Al: A first-principles study. Intermetallics. 2015;65:75-80.
- [Google Scholar]
- Prediction of the site occupations of the ThMn12-type intermetallics YFe12−xMox by combining thermodynamic model with ab-initio calculations. Intermetallics. 2010;18(8):1465-1469.
- [Google Scholar]
- The partitioning and site preference of rhenium or ruthenium in model nickel-based superalloys: An atom-probe tomographic and first-principles study. Appl. Phys. Lett.. 2008;93(17):171905
- [Google Scholar]
- First-principles study of Ni/Ni3Al interface doped with Re, Ta and W. Comput. Mater. Sci.. 2020;175:109586
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2022.104278.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1
Supplementary data 2
Supplementary data 2




