

Translate this page into:
Theoretical study for determining the type of interactions between a GG block of an alginate chain with metals Cu2+, Mn2+, Ca2+ and Mg2+
⁎Corresponding author. delgaradh@gmail.com (Carlos Soloaga Ardiles) csoloaga@academicos.uta.cl (Carlos Soloaga Ardiles)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
The alginate interaction with divalent cations has been the subject of extensive study since this biopolymer forms hydrogels in the presence of divalent cations (ionic crosslinking), which exhibit important properties with applications in fields such as medicine. It has been proposed that the properties of the hydrogel formed from the ionic crosslinking process depend, among several factors, on the type of interactions between the polymer and divalent metals. In this work, theoretical calculations based on density functional theory (DFT) were used to determine the chemical stability and the type of interaction between alginate and Cu2+, Mn2+, Ca2+ and Mg2+ cations. For this, the lowest energy geometries were determined at the B3LYP 6-31G (d, p) level of theory for the systems formed with Ca2+ and Mg2+, while those formed with Cu2+ and Mn2+ used effective central potential (ECP) with LanL2DZ bases. The following models were used to characterize the bonds between the oxygen atoms of the polymer chain with the different metals; theory of non-covalent interactions (NCI), natural bonding orbitals (NBO) method, and topological analysis of the electronic location function (ELF). Our results showed that the properties such as chemical stability of the systems would be correlated with the degree of covalence of the bonds and the delocalization phenomena charge, where the systems constituted with Cu2+ and Mn2+ formed bonds with highly polarized covalent characteristics, while Ca2+ and Mg2+ form bonds of the electrostatic type. Nevertheless, our results support the idea proposed by previous studies; regarding the lack of correlation between the alginate for divalent cations affinity with the strength of the interactions between these metals and the polymer.
Keywords
Alginate
Alginate affinity
Alginate metal interaction

1 Introduction
Alginate hydrogels, friendly to the environment, are very popular due to their wide use in the food industry, cosmetics, pharmacy, and others (Hasan et al., 2020a, 2020b, 2020c). Alginate is a biopolymer widely researched for the removal of metals from dilute aqueous solutions. One of its known applications is the adsorption of different metal ions such as Cu2+, Mn2+, Ca2+ and Mg2+, which may be present in aqueous tributaries (Chen et al., 2020). Another important application of this biopolymer is its stabilizing capacity for nanoparticles, used in the degradation of certain pollutant (Hasan et al., 2020a, 2020b, 2020c). However, they also attract particular interest in modern biomedicine and regenerative medicine for applications in drug delivery systems and tissue engineering (Henkel et al., 2013). Bone is the most frequently transplanted tissue, together with cartilage. These, followed by skin, have focused on scientific efforts to achieve their regeneration; however, the results have not been optimal (Bouhadir et al., 2000). Among the different materials used for this purpose, the use of biomaterials with high water content, such as alginate hydrogels, represents an interesting proposal. However, the main disadvantage is their complex degradation and the use of high molecular weight alginates to obtain mechanical properties similar to those of hard tissues (Xu and Lam, 2018). One of the methods aimed at improving the properties of hydrogels is ionic crosslinking. This method combines alginates with divalent cations (Bidarra et al., 2014). Alginates present different affinities towards different divalent ions, giving rise to gels with different stability, permeability and strength, depending on the metal (Mørch et al., 2012). Based on such studies, the following order of alginate affinity to different divalent ions has been established: Pb > Cu > Cd > Ba > Ba > Sr > Sr > Ca > Co Ni, Zn, Mn > Mg, which would determine the physical properties of ionically crosslinked alginate gels (Haug, 1961).
Alginate is the generic term used to refer to salts and derivatives of alginic acid. This polysaccharide occurs as a mixture of insoluble salts, mostly sodium and potassium in brown algae (Phaeophyceae). Alginic acid is a high molecular weight polysaccharide consisting of different proportions of D-mannuronic acid and L-guluronic acid. The variation in the proportions of both acids depends mainly on the species of algae used (Hasan, 2020). The algae synthesize the alginate initially as a polymer of mannuronic acid, which they subsequently modified by transforming units of mannuronic acid into guluronic acid through an enzymatic epimerization. The final product contains zones formed by mannuronic acid another formed by guluronic acid, and other zones formed by both alternating acids (Wang et al., 2019). The zones of mannuronic acid are almost flat, with a structure similar to a belt, while those of guluronic acid have a structure with recesses and protrusions, as shown in Fig. 1.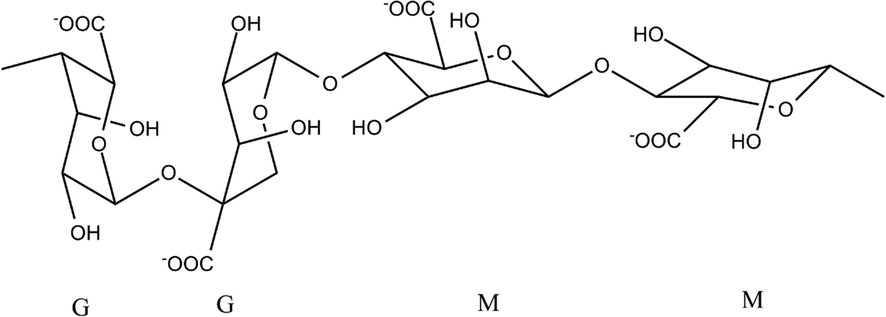
Structure of blocks G and M of alginate.
The divalent cations interact mostly with monomer G blocks, which would be determinant in the properties of the hydrogel, where the bonds would be with the carboxyl groups (–COO–) and hydroxyl groups (–OH) (Li et al., 2007). The mechanism consists of divalent ions with four carboxyl groups to form the egg crate model (Grant et al., 1973). This coordinating would influence the hydrogel's properties; for example, Zn2+ cations interact with alginate at different sites concerning Ca2+ ions, being less selective, which generates a more extensive crosslinking (Chan et al., 2002). On the other hand, attempts have been made to determine the ionic character of the metal–oxygen bonds in the formation of metal-alginate complexes, correlating the change of the asymmetric vibration of the carboxylic group (–COO–), concerning a reference element, which would allow determining the ionic percentage of the bond (PIB) (Filipiuk et al., 2005). Other authors, however, mention that this method may present erroneous results due to an erroneous assignment of the ν (COO–) peak by FTIR infrared spectroscopy (Papageorgiou et al., 2010).
However, results have been reported based on theoretical calculations of interaction energy, where it is concluded that metal–oxygen bonds with transition metals would be of the covalent type and electrostatic interactions for alkali earth elements (Agulhon et al., 2012). Both studies suggest that the affinity of alginate for different divalent cations would not correlate with the metal-polymer bond's strength. From these considerations, our main objective is to deepen the chemical nature of the interaction between alginate polymer with different divalent metals to provide information that contributes to the improvement in the methods of obtaining alginate hydrogels. In the present study, the problem is approached from a theoretical perspective, using the methods proposed by the density functional theory (DFT), non-covalent interaction (NCI) theory, natural orbital analysis (NBO), and electronic location function (ELF) analysis. For our purposes, the model used considers that the main interactions would occur with the oxygens of the functional groups derived from carboxylic acids and hydroxyl groups present in the polymer's G-blocks (Kohn, 1975). The model consists of a chain composed of two MM blocks linked to two GG blocks, which were made to interact with the following divalent metal ions; Cu2+, Mn2+, Ca2+ and Mg2+. The choice of these elements is since Cu2+ and Ca2+ cations have a high affinity for alginate, while Mn2+ and Mg2+ have the lowest affinity.
2 Theoretical calculations
2.1 Model
Our model consists of an alginate chain composed of two M−blocks linked to two G-blocks through a GM glycosidic bond (Fig. 1). The torsion angle of each glycosidic bond present in the molecule was modified to obtain the lowest energy structure. Each structure was optimized by density functional theory (DFT) in the gas phase, with the B3LYP hybrid at the 6-31G (d, p) level of theory. Then, this structure was re-optimized in each metal understudy, modifying the distances and orientations towards the oxygens of the carboxylic group and the hydroxyl group present in the GG block until the lowest energy structure was obtained. The ALG-Cu (ALG-M: ALG: alginate; M: Cu2+, Mn2+, Ca2+, Mg2+) and ALG-Mn systems were optimized by effective central potential (ECP) with the LanL2DZ bases (Yang, 2009), while ALG-Mg and ALG-Ca with the Gaussian 6-31G (d, P) bases (Bekri, 2018).
3 Computational method
The optimization of the systems' geometry understudy, molecular orbital energy calculation, and global reactivity indexes were performed using G09 software. The calculations of the NCI index, ELF, populations, variance, relative flux, bond polarization, and visualization of the ELF diagrams and NBO orbitals were performed using Multiwfn 3.7 software. The VMD package was used to visualize the NCI isosurfaces, while the Chimera X 1.0 software was used to visualize the attractors' location. The NBO calculations were obtained using an internal module of G09.
The DFT is a mechano-quantum model in which the main factor is the electronic probability density, from which it is possible to determine the properties of systems composed of atoms and molecules. Within this model, it is possible to represent energy and any other property of the system by the number of electrons and the external potential. From this have emerged various indices of global and local reactivity. These indices arise from information obtained from orbital borders such as HOMO and LUMO (Obot et al., 2015). The global reactivity indexes used in this work are the following:
Electrophilicity
: This index is a measure of the energy stabilization of the system when saturated with electrons that come from the environment.
Hardness
: This is a global property of the system and measures its resistance to the change in its electronic distribution. It is defined as:
3.1 Reduced density gradient (RDG) and NCI index
To understand non-covalent interactions (NCI), of great interest in the chemical and physical properties of molecules, the reduced density gradient (RDG) is used (Boto et al., 2020). In this theory, the density Laplacian decomposes into the sum of its contributions from the three eigenvalues of the Hessian matrix of the electron density at a given point, such that being . The local virial theorem indicates that the main contributions to the potential energy V(r) (attractive) and the kinetic energy G(r) (repulsive) of the system are related to the eigenvalues of the Hessian at each point in space. The NCI index is based on the reduced density gradient (RDG) given by Eq. (4). This index is a dimensionless quantity used to describe the deviations from a homogeneous electronic distribution.
The RDG is determined as follows:
The regions where the electron density and have low values correspond to regions where non-covalent interactions occur.
Non-covalent interactions can be classified into different categories: electrostatics, π effects, van der Waals forces, and hydrophobic effects (Boto et al., 2020).
3.2 Natural bond orbital (NBO)
NBO analysis is a helpful method, widely described (Weinhold et al., 2016) and used to describe the nature of interactions between organic molecules with different metals (Lin et al., 2020) in terms of charge transfer from a filled orbital
(electron donor) to the valence of an anti-bonding orbital
(electron acceptor), resulting in so-called charge transfer or donor–acceptor interactions. This analysis method reveals important chemical information from the results obtained by transforming a wave function in the fundamental state, from which localized orbitals corresponding to the chemical notion of Lewis-type bonding are obtained. The interaction energy by charge transfer is called the second-order stabilization energy (
), which is estimated by the secondary perturbation theory, according to the following equation (Lu et al., 2019):
Additionally, it is possible to relate the magnitude of the Fock matrix elements to the bonding and anti-bonding orbitals' symmetry, which allows interpreting the energetic stabilization in terms of the generalized principle of maximum overlap between bonds and anti-bonds (Platts and Baker, 2020).
In this work, the concept of second-order stabilization energy ( ) and natural bond orbital overlap will be used to try to describe the effect of the oxygen-metal interaction in the systems under study.
3.3 Electron localization function (ELF)
In quantum chemistry, the electron localization function (ELF) estimates the probability of finding an electron in the neighboring space of a reference electron located at a certain point and with the same rotation. The electron pair density provides the probability of finding an electron of a particular spin at a point in space when the second electron of the opposite spin is at a certain distance (Liu et al., 2018). Within this context, the interpretation proposed by Savin et al. given by Eq. (6) indicates that the electron pair density can be generalized to any density-independent of spin (Fuentealba et al., 2007).
The first term on the right-hand side of Eq. (6) represents the kinetic energy density of the molecule under study, and the second term on the right-hand side is the von Weizsacker kinetic energy density which is exact for the hydrogen atom, exact for the helium atom in the HF approximation and exact for a boson system (Pilmé, 2017). Physically, this measures the extent of the spatial location of the reference electron and provides a method for mapping the probability of electron pairs in multielectronic systems. When applied to molecules, an ELF analysis shows a clear separation between the nucleus and the valence electron and shows covalent bonds and solitary pairs. As ELF is a scalar function, its gradient field's topological analysis determines local maxima (attractors) and the corresponding basins. The gradient vector field of ELF
allows dividing the Euclidean space into basins of attractors where electron pairs are located (Zhao et al., 2020). If the basin contains a nucleus (except a proton), it is a central basin (C). Otherwise, it is a valence basin (V). Invariably, a valence basin will be connected with at least one central basin. A vacancy representing a lone pair of electrons will be connected to a single central vacancy, and its attractor will be called monosynaptic. A basin representing a covalent bond will be connected with two central basins, and its attractor will be called disynaptic. Important properties of a chemical system can be examined by specific properties that basins exhibit, among those considered in this study are; the average population, the variance (σ2), and the relative flux
. The basin’s population is obtained by integrating the electron density over the volume of the basin, according to Eq. (7). At the same time, σ2 represents the fluctuation of the electron pair, it is given by Eq. (8), and finally, the relative flux indicates the electron charge fluctuations for a given basin is described by Eq. (9) (Chauvin et al., 2016). Within this same context, the polarity of a bond can be determined by the model proposed by Raud and Jansen, based on the combination of the complementary topological partitions of the electron density, obtained from the theory of atoms in molecules (AIM) and the electronic localization function (ELF) (Raub and Jansen, 2001).
4 Results and discussion
4.1 Reactivity descriptors
As mentioned above, the electronic chemical potential characterizes the ability of a system to exchange electrons with the medium, where the electronic transfer occurs from the highest to the lowest potential. Table 1 shows that ALG-Cu and ALG-Mn have the highest electronic chemical potential. These results show that the interaction of Cu+ 2 and Mn +2 ions with the polymer makes the formed systems less susceptible to electron exchange with the medium, while the higher values shown by ALG-Ca and ALG-Mg indicate that the electron density of these systems can vary spontaneously (Fekri et al., 2020), which makes them more reactive. The principle of maximum hardness (PMH) states that molecular systems in equilibrium tend to high hardness states. This principle is defined as a measure of resistance to change in electron density. Therefore, the molecule's stability and reactivity are related to the chemical hardness (η) (Pearson, 1993). The hardness values (Table 1) show that ALG-Cu is the system with the highest value for this reactivity index, followed by ALG-Mn.
ALG-Cu (eV)
ALG-Mn (eV)
ALG-Ca(eV)
ALG-Mg (eV)
−0.75
−0.77
−4.31
−4.25
1.53
1.42
0.59
0.92
0.18
0.21
52.49
9.77
Furthermore, the systems formed with the metals Ca +2 and Mg +2 presented lower values. In this sense, the Ca +2 and Mg +2 ions form the least stable systems, where ALG-Ca would be the most reactive (Chandrasekaran and Kumar, 2015). Finally, the electrophilicity values indicate that the ALG-Ca and ALG-Mg systems are the ones that would tend to stabilize to a larger extent when accepting electron density from their environment, according to the definition of this reactivity index (Chattaraj and Giri, 2009). These values confirm the higher reactivity of these systems.
4.2 NCI
For the characterization of the bonds in the ALG-M systems, the theory of non-covalent interactions (NCI) was used. Binding and nonbinding interactions can be identified by the sign of the second Hessian eigenvalue (
). The analysis results are shown in a graph of the reduced gradient of the density v/s the electronic density (
) multiplied by the sign
. The non-covalent/weak interactions are given by a low density; for example, Van der Walls interactions. The low density and low-density gradient that is
, indicates strong attractive interactions (blue surface isosurface), attractive weak interactions (green surface isosurface), and
indicates steric repulsion (red isosurface) (Contreras-García et al., 2016). The NCI isosurfaces for the ALG-Cu and ALG-Mn systems are shown in Figs. 2a and 2b. These results indicate that the strongest metal-alginate interactions occur with atoms of the chain's outermost oxygens, corresponding to carboxylic acid functional groups (–COO-) and hydroxyl groups (–OH). These oxygens are the closest and least sterically hindered; red and blue isosurfaces represent this in the form of rings on the metal–oxygen bond line.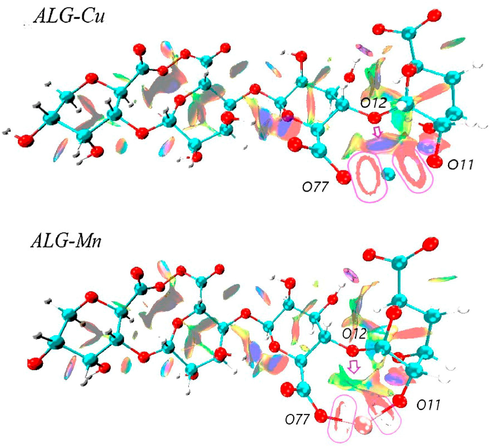
Isosurfaces of the reduced density gradient with s = 0.6 a.u. for ALG-Cu and ALG-Mn.
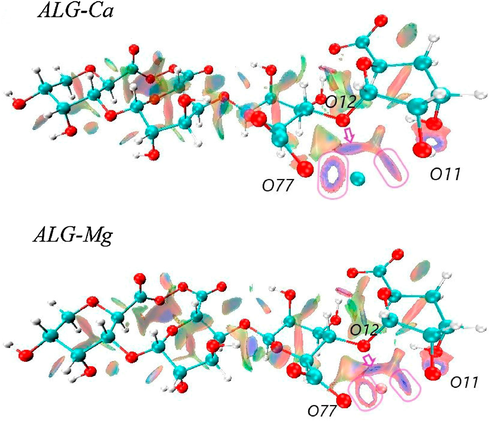
Isosurfaces of the reduced density gradient with s = 0.6 a.u. for ALG-Ca and ALG-Mg.
The formation of such rings indicates that the electron density higher than 0.05 could not be appreciated, and only the weak interaction area is observed, i.e., the central (empty) area of the ring represents a strong bond (Lefebvre et al., 2017). The isosurfaces were obtained for an index = 0.6, which accounts for an interesting difference. In the case of the ALG-Cu and ALG-Mn systems, these ring-shaped surfaces are fainter, concerning the systems with Ca and Mg. Moreover, the O77-Mn and O11-Mn interactions appear to be slightly stronger due to a lower electron density presence associated with weak bonds. Meanwhile, for the O77-Ca bond, the ring-shaped isosurface is more closed, suggesting that the weakest O77-M interaction occurs in the ALG-Ca system. It can also be seen that the O11-Metal interaction is only strong in the presence of transition metals. As for the interaction of ether oxygen (-O-), which is the most sterically hindered, the isosurface show slight differences between systems. For example, in ALG-Mn, it is broad and shows a faint blue color, which accounts for the weakest O12-M bonding of the systems under study.
Meanwhile, in ALG-Cu, the isosurface is of a more intense blue color and comprises a large area of the metal-binding region, which accounts for a stronger interaction. This description is very similar to that observed for the O12-Mg and O12-Ca bonds. Finally, intermolecular interactions are also observed, mainly by hydrogen bridges between the hydroxyl groups' hydrogens with neighboring oxygen atoms. According to the structural modifications that the polymer may undergo, these interactions vary in intensity due to their interaction with different metals. On the other hand, these results agree with those reported in previous experimental studies, where the strongest interaction between alginate and divalent metals occurs with the oxygen of the carboxyl group of the G-blocks (Lu et al., 2005). Also, our results show a strong interaction between the –OH groups with Cu2+ and Mn2+ ions.
4.3 NBO
4.3.1 NBO ALG-Cu
In Table 2, the NBO results for the ALG-Cu system are shown. This analysis predicts a σ (O77-Cu) bond. The anti-bonding orbital σ*(O77 -Cu), formed by the
orbital of oxygen with the
orbital of copper, participates in different interactions that bring high stability to the structure (Zülfikaroğlu et al., 2018). The highest value of
was for the interaction of LP 2 (O77) (donor) with the anti-bonding orbital σ*(O77 -Cu) (acceptor), where the latter presents a high occupancy, which would give instability to the O77-Cu bond (Lu et al., 2019). In Fig. 3A, the NBO orbitals for the anti-bonding orbital σ*(O77 -Cu) interactions with O11, O12, and O77 are shown. For the interaction between LP 2 (O77) and σ*(O77 -Cu), the decrease of the donor density (green-yellow (positive) and violet (negative) region) and an increase of this in the anti-bonding orbital (green (positive) and blue (negative) region) are appreciated, generating a broad interaction region located around O77, indicating a strong polarization of the bond (Lee et al., 2013). This interaction allowed reducing the energy of the system by 35.24 Kcal mol−1. Another important interaction was LP 2 (O77), with the anti-bonding orbital σ*(O77 -Cu). This interaction presented the second-highest value of
. The σ*(O77 -Cu) acts as an acceptor of LP 2 (O11) (donor), of low occupancy, a product of electronic donation to the anti-bonding orbital, thus decreasing its instability due to its high occupancy. NBO orbitals represent this interaction in Fig. 3B. Here, it is observed how the donor's positive density regions (LP 2 (O11)) and the acceptor generate a region of interaction smaller than that visualized with O77, which is expressed in a lower value of
. For O12, the perturbation energy values were lower. In Fig. 3C, the NBO orbitals of the interaction between LP 2 (O12) and σ*(O77 -Cu) can be observed. The overlap of the orbital of the lone pair of O12 with the σ* of the O77-Cu bond is low. The sum of the perturbation energies was 63.95 Kcal mol−1 for O77-Cu, for O11-Cu 59.44 Kcal mol−1 and for O12-Cu 15.57 Kcal mol−1. The above is related to that indicated by the NCI isosurfaces, which show a slightly stronger interaction with O77.
Donor (i)
Occupancy
Acceptor (j)
Occupancy
E(2) Kcal mol−1
σ (C6-O11)
1.99219
LP* 7 (Cu)
0.05719
2.91
σ (O11-H42)
1.98884
LP* 6 (Cu)
0.09353
3.16
σ (O11-H42)
LP* 7 (Cu)
0.05719
2.89
σ (C21-O77)
1.99510
LP* 7 (Cu)
2.86
σ (C21-O77)
σ*(O77 –Cu)
0.16663
3.26
σ* (C21-O76)
0.29437
σ*(O77 –Cu)
3.45
LP 1 (O11)
1.96733
LP* 7 (Cu)
0.05719
4.39
LP 1 (O11)
LP* 8 (Cu)
0.03830
2.13
LP 2 (O11)
1.86336
LP* 6 (Cu)
0.09353
18.25
LP 2 (O11)
LP* 7 (Cu)
0.05719
10.31
LP 2 (O11)
σ*(O77-Cu)
0.16663
24.36
LP 1 (O12)
1.95326
LP* 6 (Cu)
0.09353
2.14
LP 1 (O12)
LP* 8 (Cu)
0.03830
3.10
LP 2 (O12)
1.89306
LP* 6 (Cu)
0.09353
2.34
LP 2 (O12)
LP* 8 (Cu)
0.03830
4.79
LP 2 (O12)
σ*(O77 –Cu)
0.16663
3.20
LP 1 (O77)
1.93078
LP* 7 (Cu)
0.05719
9.19
LP 2 (O77)
1.80574
LP* 6 (Cu)
0.09353
2.74
LP 2 (O77)
LP* 7 (Cu)
0.05719
16.78
LP 2 (O77)
σ*(O77 –Cu)
0.16663
35.24
LP 5 (Cu)
1.98263
σ*(O77 –Cu)
3.12
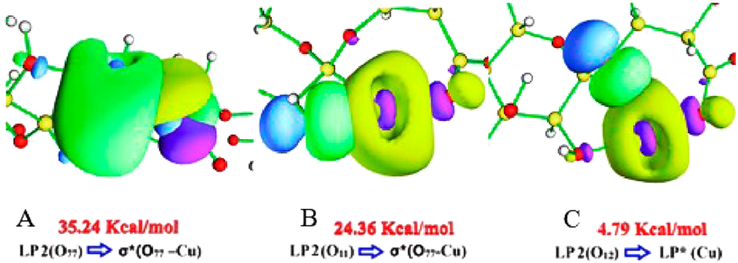
ABC NBO orbitals for the most important interactions, in terms of
, in ALG-Cu.
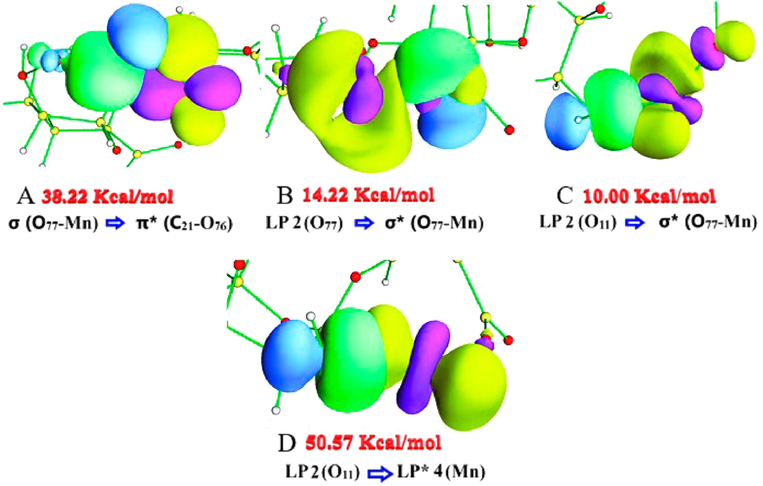
4.3.2 NBO ALG-Mn
Table 3. Presents the NBO results for the ALG-Mn system. The NBO analysis predicts the O77-Mn bond formed by an
orbital of manganese to the
orbital of oxygen. The σ-bonding orbital (O77-Mn) acts as a donor, and the anti-bonding π* (C21-O76) as an acceptor, which generated a high structural stabilization, with one of the highest values
(38.22 Kcal mol−1). The donor orbital occupancy is low, while that of the acceptor is high, revealing a strong charge transfer. The fact that the occupancy in the anti-bonding orbital is high decreases the stability of the C21-O76 bond (Raju et al., 2020), of which O77 is a part. Furthermore, the anti-bonding orbital σ* (O77-Mn) acts as an acceptor in several stabilizing system interactions. Among these were those of LP 2 (O77) and LP 2 (O11), which transfer charge to the anti-bonding orbital, generating moderate values of
. Another interaction to consider was the anti-bonding orbital π* (C21-O76) with σ* (O77-Mn). The former presented a high occupancy, as already mentioned. The charge transfer performed by π* (C21-O76) would allow it to decrease the bond instability of the carboxylic group of O77. In Fig. 4, the NBO orbitals are shown for the interactions that presented high values of
. Among these are those involving σ (O77-Mn) and π* (C21-O76). In the σ → π* donation, a broad region of interaction is observed, where there is a decrease in density in the donor (green and blue colored surfaces) and an increase in density in the acceptor, indicating charge transfer (Fig. 4A). In the interactions in which σ* acts as an acceptor for LP 2 (O11) and LP 2 (O77) (Fig. 4B and 4C), the overlap decreases, leading to a drop in the perturbation energy values. Meanwhile, the interaction LP 2 (O11) (donor) and
LP* 4 (Mn) (acceptor), where the
orbital of Mn and the hybrid
orbital of the sp3 hybridization oxygen (75% character-p), present a high overlap, generating the highest value of
(50.57 Kcal mol−1) (Fig. 4D). Meanwhile, the interaction between LP 2 (O77) (donor) and LP* 5 (Mn) (acceptor) presented the third highest value of
due to a strong charge transfer from donor to acceptor. For O12-Mn, lower values of , were obtained where LP 1 (O12) and LP 1 (O12) transfer charge to LP* (Mn). The latter presented the lowest occupancy value concerning the other anti-bonding orbitals presented in Table 3. The latter suggests little charge transfer resulting in a very weak interaction between O12 and the metal. The sum of
for O77-Mn was 74.99 Kcal mol−1, for O11-Mn 80.22 Kcal mol−1, and for O12-Mn was 2.72 Kcal mol−1. Finally, the perturbation energy values for the O77-Mn and O11-Mn bond presented higher values than in ALG-Cu, which agrees with the NCI isosurfaces, suggesting that the O77-Mn and O11-Mn interaction would be slightly stronger than O11-Cu and O77-Cu, while with O12, copper presents a stronger interaction.
Donor (i)
Occupancy
Acceptor (j)
Occupancy
E(2) Kcal mol−1
σ (C6-O11)
1.99154
LP* 4 (Mn)
0.16684
3.18
σ (O11-H42)
1.98906
LP* 4 (Mn)
3.99
σ (O11-H42)
LP* 6 (Mn)
0.04227
2.55
σ (C21-O77)
1.99362
σ* (O77-Mn)
0.07984
2.77
σ (O77-Mn)
1.80640
π* (C21-O76)
0.25870
38.22
π* (C21-O76)
0.25870
σ* (O77-Mn)
0.07984
8.44
LP 1 (O11)
1.94344
LP* 6 (Mn)
0.04227
3.34
LP 1 (O11)
σ* (O77-Mn)
0.07984
4.62
LP 2 (O11)
1.84993
LP* 4 (Mn)
0.16684
50.57
LP 2 (O11)
LP* 5 (Mn)
0.13032
4.00
LP 2 (O11)
LP* 6 (Mn)
0.04227
7.69
LP 2 (O11)
σ* (O77-Mn)
0.07984
10.00
LP 1 (O12)
1.95808
LP* 7 (Mn)
0.02868
1.37
LP 2 (O12)
1.89984
LP* 7 (Mn)
1.35
CR 1 (O77)
1.99970
RY* 2 (Mn)
0.01064
2.73
LP 1 (O77)
1.88007
LP* 4 (Mn)
0.16684
10.51
LP 1 (O77)
LP* 5 (Mn)
0.13032
2.57
LP 1 (O77)
LP* 6 (Mn)
0.04227
10.73
LP 2 (O77)
1.80832
LP* 5 (Mn)
0.13032
30.68
LP 2 (O77)
LP* 6 (Mn)
0.04227
3.55
LP 2 (O77)
σ* (O77-Mn)
0.07984
14.22
ABC NBO orbitals for the most important interactions, in terms of
, in ALG-Mn.
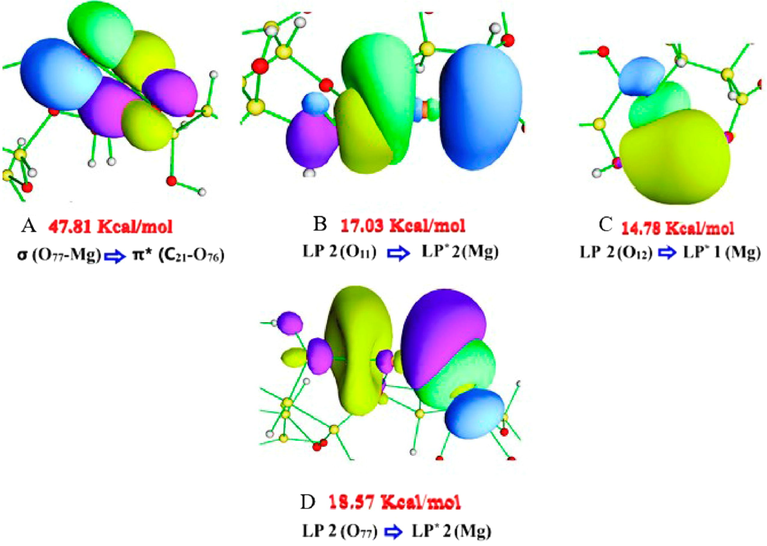
4.3.3 NBO ALG-Mg
Table 4 shows the NBO results for the ALG-Mg system. This analysis predicted an O77-Mg bond. The highest value of
was for the interaction between σ (O77-Mg) and π* (C21-O76) (47.81 Kcal mol-1), where, according to the occupancy values of the donor and acceptor, there is a strong charge transfer, which would weaken the C21-O76 bond, due to the high occupancy of the π* orbital. This bond would decrease its instability by charge transfer to LP* 2 (Mg). This interaction's high stability is due to the delocalization of the electronic density in the carboxylic group. In Fig. 5A, the NBO orbitals for the σ → π* interaction are shown. A high overlap is observed, giving rise to a π interaction, where there is a decrease in the donor density (green–blue surfaces) and even an increase in that of the acceptor (green-yellow-violet surfaces), indicating the strong charge transfer already mentioned. The interaction of LP 2 (O11) with LP* 2 (Mg) generates the highest value
for the interaction between O11 and Mg (17.03 Kcal mol-1), but lower than those obtained in the ALG-Cu and ALG-Mn systems, where there is charge transfer from an n to an n* orbital. The latter showed a moderate occupancy value. This interaction is visualized through NBO orbitals in Fig. 5B. In this, an overlap σ, between the hybrid orbital
of LP 2 (O11), of hybridization
with 73% -
character, with the pz orbital of LP* 2 (Mg) of hybridization
and 99.35% p-character, is seen. The predicted interactions for O12 differ from the previous systems analyzed in that these generate higher stability with the metal. The highest value of
, obtained for this interaction, was 14.78 Kcal mol-1, where the donor occupancy is low (LP 2 (O12)), and the acceptor occupancy is high (LP* 1 (Mg)), accounting for the strong charge transfer. Fig. 5C shows the NBO orbitals for LP 2 (O12) interaction with LP* 1 (Mg). The
hybrid orbital of the oxygen lone pair, of hybridization sp3.75 with 79% -
character overlaps with the
orbital of the n*, which presents an increase in the density region, the product of the charge transfer. Then, O77 presents the highest value of
for the interaction between LP 2 (O77) and LP* 2 (Mg) (18.57 Kcal mol−1), where the donor and acceptor occupancy values suggest a low charge transfer. It is highlighted that O77 generates a higher amount of stabilizing interactions, as shown in Table 4. Nevertheless, given the perturbation energy values, the O77-Mg interactions are smaller than those observed with Cu2+ and Mn2+. NBO orbitals represent this interaction in Fig. 5D. An overlapping σ is appreciated between the hybrid py orbital of LP 2 (O77) of hybridization
with 78% -
character and the
orbital of LP* (Mg), with 99.35% p-character. Finally, an LP* 1 (Mg) interaction with σ* (O77-Mg) generates a low charge transfer to the anti-bonding sigma orbital. Therefore, the O77-Mg bond stability is less affected; the O12-Mg interaction results in higher stability than that generated with Cu2+ and Mn2+, due to the ionic radius of Mg2+ (the smallest of the metals under study) and its low electron density. The sum of
for the O77-Mg interaction was 57.88 Kcal mol−1, for O11-Mg 33.37 Kcal mol−1, and for O12-Mg 43.98 Kcal mol−1. These results agree with those given by the NCI index, which indicates that Mg interacts more weakly with O77 and O11, concerning Cu2+ and Mn2+, while the binding with O12 is stronger for Mg2+. On the other hand, the most stabilizing interaction, which corresponds to the charge transfer from the σ bond (O77-Mg) to π* (C21-O76), suggests that the electronic delocalization produced in the carboxylic group would be the one that most stabilizes the O77-Mg interaction.
Donor (i)
Occupancy
Acceptor (j)
Occupancy
E(2) Kcal mol−1
σ (C6-O11)
1.99123
LP* 2 (Mg)
0.08039
2.43
σ (O11-H42)
1.98855
LP* 2 (Mg)
3.56
σ (O12-H13)
1.98812
LP* 1 (Mg)
0.05227
2.07
σ (C21-O77)
1.99316
LP* 2 (Mg)
0.08039
2.96
σ (O77-Mg)
1.80464
π* (C21-O76)
0.22042
47.81
CR 1 (O11)
1.99984
LP* 2 (Mg)
0.08039
2.26
LP 1 (O11)
1.96682
LP* 3 (Mg)
0.05227
2.69
LP 2 (O11)
1.90001
LP* 1 (Mg)
0.13797
6.80
LP 2 (O11)
LP* 2 (Mg)
0.08039
17.03
LP 2 (O11)
LP* 3 (Mg)
0.05227
6.85
LP 1 (O12)
1.94652
LP* 1 (Mg)
0.13797
5.59
LP 1 (O12)
LP* 2 (Mg)
0.08039
2.66
LP 2 (O12)
1.89639
LP* 1 (Mg)
0.13797
14.78
LP 2 (O12)
LP* 2 (Mg)
0.08039
3.01
LP 2 (O12)
LP* 3 (Mg)
0.05227
8.03
LP 2 (O12)
σ* (O77-Mg)
0.08384
9.91
CR 1 (O77)
1.99979
LP* 2 (Mg)
0.08039
2.81
LP 1 (O77)
1.93189
LP* 2 (Mg)
0.08039
10.47
LP 2 (O77)
1.85541
LP* 1 (Mg)
0.13797
15.89
LP 2 (O77)
LP* 2 (Mg)
0.08039
18.57
LP 2 (O77)
σ* (O77-Mg)
0.08384
10.14
LP* 1 (Mg)
0.13797
σ* (O77-Mg)
2.73
ABC NBO orbitals for the most important interactions, in terms of
, in ALG-Mg.
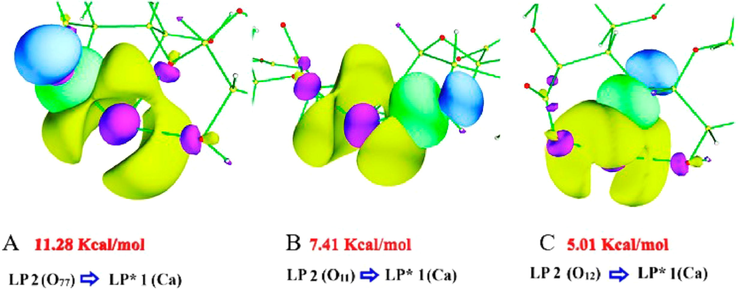
4.3.4 NBO ALG-Ca
Table 5 presents the NBO results for ALG-Ca. The NBO analysis did not predict any O-Ca bonds. Calcium interactions with oxygens occur through charge transfer from LP 2 (O11), LP 2 (O12), and LP 2 (O77) lone pairs to LP* 1 (Ca). The values
for the O11 interaction with Ca are low and lower than the systems already analyzed. The highest value obtained was for LP 2 (O11) and LP* 1 (Ca) (7.41 Kcal mol−1), where the occupancy of this LP* 1 (Ca) is the highest and it is the one that participates as an acceptor in the most stabilizing interactions with the three oxygens under study. Fig. 6A shows the NBO orbitals for the interactions, as mentioned earlier. In these, it can be observed how the localized orbitals of LP 2 (O11), LP 2 (O12), and LP 2 (O77) interact with LP* 1 (Ca), which presents an increase in its density region as a result of the charge transfer of the mentioned oxygens (Fig. 6A). The interaction LP 2 (O77) and LP* 1 (Ca) produced the highest value of hyperconjugative interaction energy
(11.28 Kcal mol−1). In Fig. 6B, the frontal overlap between the NBO orbitals involved in this interaction is observed. In the same image, the NBO orbitals for the interaction that generated the highest value between O12 and the metal (5.01 Kcal mol−1) can be noticed (Fig. 6C). In this case, there is a sigma overlap between the n orbital of LP 2 (O12) and the aforementioned n* orbital of calcium. The interactions where calcium acts as a donor were not considered in the table because they have low
values. The sum of the values
for O77-Ca was 41.96 Kcal mol−1, O11-Ca 23.65 Kcal mol−1, and O12-Ca 16.85 Kcal mol−1. This analysis reveals that the O-metal interactions for ALG-Ca, considered mainly as O → Mg charge transfer, generate little stability to the structure and lower electronic delocalization concerning ALG-Cu and ALG-Mn.
Donor (i)
Occupancy
Acceptor (j)
Occupancy
E(2) Kcal mol−1
LP 2 (O11)
1.94508
LP* 3 (Ca)
0.04333
3.78
LP 2 (O11)
LP* 4 (Ca)
0.02629
4.34
LP 2 (O11)
LP* 1 (Ca)
0.10299
7.41
LP 2 (O11)
LP* 3 (Ca)
0.04333
3.78
LP 2 (O11)
LP* 4 (Ca)
0.02629
4.34
LP 1 (O12)
1.95197
LP* 1 (Ca)
0.10299
4.05
LP 1 (O12)
LP* 3 (Ca)
0.04333
2.68
LP 1 (O12)
LP* 4 (Ca)
0.02629
2.26
LP 2 (O12)
1.92360
LP* 1 (Ca)
0.10299
5.01
LP 2 (O12)
LP* 4 (Ca)
0.02629
2.85
LP 1 (O77)
1.94007
LP* 1 (Ca)
0.10299
9.41
LP 1 (O77)
LP* 2 (Ca)
0.04468
2.15
LP 1 (O77)
LP* 3 (Ca)
8.45
LP 2 (O77)
1.89529
LP* 1 (Ca)
0.10299
11.28
LP 2 (O77)
LP* 3 (Ca)
0.04333
4.60
LP 3 (O77)
1.73465
LP* 2 (Ca)
0.04468
2.92
LP 3 (O77)
LP* 5 (Ca)
0.02029
3.15
ABC NBO orbitals for the most important interactions, in terms of
, in ALG-Ca.
4.4 ELF topological analysis
4.4.1 ALG-Cu
We will try to deepen the nature of bonding by employing ELF topological analysis and attractor analysis. Fig. 7A shows the diagrams of the electronic localization function and the monosynaptic attractors, which correspond to the nonbonding electrons of the oxygen atoms interacting with the metal and their respective populations for the different systems under study. Table 6 shows these populations, variance, and relative flux, where, also, attractors of other oxygens present in the system are considered. High ELF values (approximately 0.8–1.0) are red; intermediate ELF values are represented by orange, green, and blue regions. The lower end of the scale is represented by white. The analysis of the attractors tells us that if the position of the attractor tends to be between the centers (bond line), the covalence increases until a completely symmetrical topology is achieved (pure covalent bond) (Savin et al., 1997). The copper atom presents the core's degenerate attractors with a total electronic population of 27.75e, lower than the expected value of 28e (isolated atom); this would result from the 3d contribution to the valence region (Fuster et al., 2004). There are also delocalized valence attractors in the outermost region of the copper atom (V (Cu), V' (Cu), and V'' (Cu)), with low electronic populations. This is related to the low delocalized electron density between O11-Cu-O77, represented in the ELF diagram (Fig. 7B), by the blue-colored region surrounding O77, Cu, and O11, to a lesser extent O12 (Fig. 7C). The ELF value is 0.40, lower than the homogeneous gas reference value, indicating high electronic delocalization in the mentioned region (Jerabek et al., 2018). It is observed as the attractors of the lone pairs V (O11), V (O12), and V (O77) are oriented towards the metal, and their electronic populations are low if compared to the respective attractors present in this system Table 6. On the other hand, the attractors representing the other lone pair of these oxygens (V' (O11), V' (O12), and V' (O77)) have higher populations if the same comparison is made. The former indicates electronic donation from the oxygens to the metal, and the latter is explained by an asymmetric redistribution of the electron density generated by charge transfer from the metal (Durlak et al., 2009). The monosynaptic basin V (O11) (highlighted in Fig. 7) is located at the junction line between O11 and Cu (binding distance of 1.95 A0), with a low electron population of 2.06e (the average value was 2.32e) (Table 6). The total population for V (O11) and V'(O11) was 4.72e, which is below the average for other basins of lone pairs of oxygens of the same type in this system (4.75e), suggesting electron density donation O11 → Cu (Chaudret et al., 2011). According to Silvi & Savin, the position of V (O11) can be interpreted, as a dative covalent bond type, where the pair of electrons forming this bond are contributed by oxygen; this is also supported by the high value of
, which indicates a high electronic delocalization in the metal-binding region (Savin et al., 1996). On the other hand, the ELF diagram (Fig. 7B) shows us that the non-bonding pair of O77 and O11 interacting with copper are oriented towards the metal and present high delocalization (orange region), mainly O77. The monosynaptic basin V (O12) is oriented towards copper, close to the binding line, with a population of 2.33e (lower value than the average of 2.46e), indicating charge transfer O12 → Cu. The total population for V (O12) and V' (O12) was 5.00e (higher than average), suggesting Cu → O12 charge transfer. The high electron flux, given by the value of
, suggests that a high electron density (concerning the total population of this attractor) is delocalized, but not in the direction of binding to Cu, as suggested by its localization; this can be observed in the ELF diagrams, where the Cu-O12 interaction region presents an ELF value close to zero. Considering also that the binding distance was 2.48 A0, it is suggested that the O12-Cu interaction is mainly electrostatic. The position of the monosynaptic basin V (O77) is similar to that of V (O11), with a population of 2.46e (low value to the average of 2.73e), while V' (O77) 3.47e (high value), and between them, they add up to the highest population (5.93e), concerning the average (
= 5.45e). The O77-Cu bond distance was 1.84
. The attractor's location and populations and the short binding distance suggest that electron density donation and back-donation phenomena between O77 and copper give a covalent character; this is also reflected in the variance (
) value, indicating a considerable increase of electronic flux in the bonding region.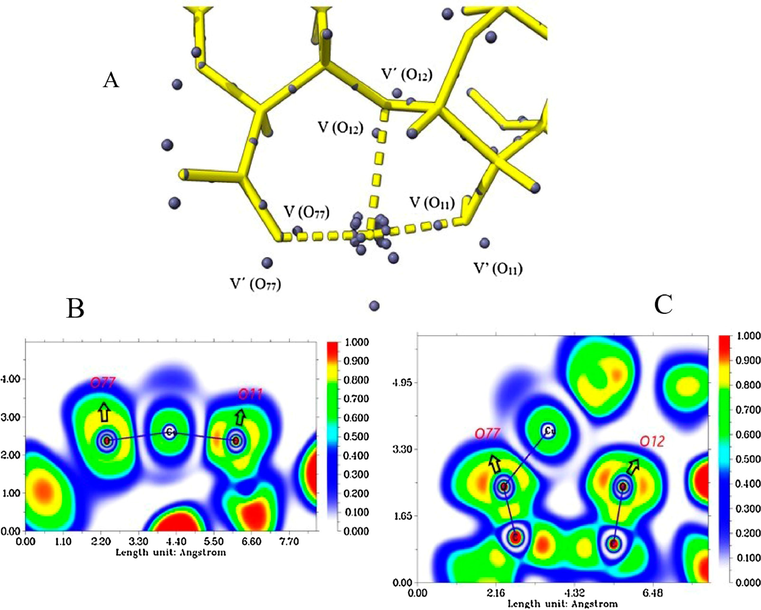
Attractor populations of the lone pairs of O11, O12, O77 and valence attractors and the copper core (A). ELF diagrams for the Cu-(O77, O11) interaction (B), and for the Cu-(O12, O77) interaction (C).
Attractor ALG-Cu
Populations
-OH/, –CO/, -O-
–OH/, –CO/, -O-
(–OH/, –CO/, -O-)
(O)
2.32e
2.65e
2.43e
0.28
0.66
0.86
0.22
0.43
0.36
́(O)
2.43e
2.80e
2.49e
0.47
1.23
1.17
0.27
0.27
0.47
V (O)
2.06e
2.46e
2.33e
1.13
1.27
1.18
0.55
0.52
0.50
V́(O)
2.67e
3.47e
2.67e
1.19
1.42
1.23
0.45
0.41
0.46
Meanwhile, the high electron population in V' (O77) suggests that this lone pair contributes to the π C21 = O77 bond. Finally, the analysis of the relative basin fluctuation (Table 6) shows us a high electronic delocalization in O11, O12, O77, and Cu. These high values suggest that this interaction could be represented by a system where the charge supported by each atom involved in the interaction varies continuously in time, in the manner of a resonant structure (Poater et al., 2005). This is in agreement with the hyperconjugative energy values of the NBO analysis. It is suggested that this system's chemical stability would be related to the electronic delocalization produced in the oxygen-metal interaction. Finally, the atomic contributions to the V (O) localization basins are analyzed below. The V (O11) contribution is given by 95.60% contributed by the oxygen lone pair and 4.4% by Cu. That is, of the total 2.06e, 1.96e are contributed by oxygen and only 0.1e by copper. For V (O77), the contributions are given by 94.51% of the lone pair of O77 and 5.50% of Cu; therefore, 2.32e are contributed by O77 and 0.14 by copper. According to several authors, the above agrees with the interpretation of an O → Cu dative bond (Mierzwa et al., 2020). These results indicate that these bonds are strongly polarized. The high populations of V (O77) and V' (O77) could be explained by the high electronic delocalization of the carboxylic group, as shown by NBO results and ELF attractor analysis. The V (O12) contributions are given by 98.90% O12 and 1.1% Cu in terms of populations indicating that O12 and only 0.03e by Cu contribute 2.3e; this is related to the lower interaction between O12 and the metal, which corresponds with the idea of electrostatic interaction.
4.4.2 ALG-Mn
Fig. 8 shows the attractors and ELF diagrams for the interaction between O11, O12, and O77 with Mn. In the ELF diagrams (Fig. 8A and 8B), the nonbonding electron pairs of the oxygens, the core electrons, and the Mn atom's valence electrons can be observed. Also, a region with delocalized electron density can be observed with ELF values close to 0.4, distributed between Mn-O77 and to a lesser extent with O11, similar to that described in the ALG-Cu system. On the other hand, Table 7 shows the average values of populations, variance (
), and relative fluctuation
for the monosynaptic oxygen attractors (lone pairs); O11, O12, and O77. The total population for the core attractors was 20.27e, well below the formal value of 23e; this would indicate the contribution of 3d electrons to the valence region (Mierzwa et al., 2018). For the lone pair attractors of O11 and O77, the same trend seen in the ALG-Cu system is observed in terms of decreasing the metal-oriented lone pairs populations and the asymmetric electron density distribution. V (O11) presents the lowest electron population (2.14e for an average of 2.32e). The total population for V (O11) and V' (O11) was 4.69e, a low value compared to the average (4.74e).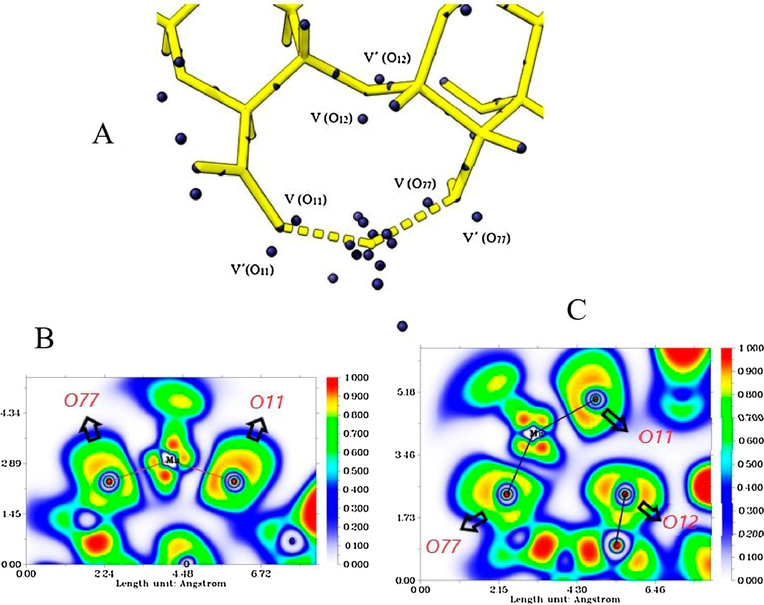
Attractor populations of the lone pairs of O11, O12, O77 and valence, and core attractors of manganese (A). ELF diagrams for the Mn-(O77, O11) interaction (B) and for the Mn-(O11, O12, O77) interaction (C).
Atractor ALG-Mn
Poblaciones
–OH/, –CO/, -O-
–OH/, –CO/, -O-
(–OH/, –CO/, -O-)
(O)
2.32e
2.60e
2.43e
1.11
1.17
1.22
0.48
0.46
0.48
́(O)
2.43e
2.78e
2.50e
1.12
1.21
1.18
0.46
0.43
0.39
V (O)
2.14e
2.48e
2.41e
1.14
1.24
1.18
0.53
0.50
0.49
V́(O)
2.55e
3.40e
2.56e
1.18
1.43
1.21
0.46
0.42
0.47
Additionally, the bond length was 1.97 A0. The above and the localization of V (O11) suggest that this interaction presents some covalent character, where the electron density donation from O11 to Mn would be the dominant process; this agrees with the interpretation of Silvi et al. regarding the nature of the dative bond concerning the attractor's position. The idea of the degree of covalence of this bond is also supported by the high electronic flux given by (a higher value for this type of oxygens in the ALG-Mn system, considering its low population), which would be evidencing the high delocalization, product of the electronic exchange between these nuclei. The V attractor (O77) is oriented towards the metal and located near the bond line. It presents a low population of 2.48e for an average of 2.69e, indicating a donation from O77 to the metal. The sum of the populations of V (O77) and V' (O77) was 5.88e, higher than the average (5.38e).
Meanwhile, the bond length was 1.81 A0. The short binding distance, the orientation of V (O77), and the population analysis suggest some covalent nature via both donating and back-donating processes. The ELF diagrams (Fig. 8A and 8B) show the lone pairs of O77 highly delocalized, and an interaction region encompassed by the aforementioned non-bonding electron density is appreciated. On the other hand, the high value of shows a high electronic flux due to the exchange of electrons between both elements. On the other hand, the high population of V' (O77) (3.40e) suggests that it contributes to the π C21 = O77 bond, as described in ALG-Cu. V' (O12) is oriented toward Mn but not in the bond line, which has a length of 3.03 A0. The populations of V (O12) and V' (O12) are closer to the average values (Table 7), indicating a lower interaction between O12 and Mn.
On the other hand, the ELF diagram shows that the bonding region between O12 and Mn presents an ELF value close to zero, and no delocalized electron density shared with the other atoms understudy is observed. This value is also reflected in the variance, which presented a low value (Table 7), i.e., the pair of nonbonding electrons that interact with Mn are largely located in O12; the values of the relative fluctuation confirmed this, showing a high electronic delocalization between O11-Mn-O77. Meanwhile, the value of obtained for O12 is within the average, confirming that the charge would be more localized on the O12 atom, which reaffirms the description of weak electrostatic bonding. The analysis of contributions to the V (O) basin yielded the following results: V (O11) = 98.08% (2.10e) O11; Mn = 1.92% (0.04e), for V (O77) = 97.78% (2.43e) and 2.22% (0.05e). These results have the same interpretation given for these attractors in the ALG-Cu system. That is, bonds of dative character, highly polarized between copper with O11 and O77. For V (O12), a contribution of 100% O12 was obtained, which confirms the scarce interaction between this oxygen and manganese.
4.4.3 ALG-Mg
In Fig. 9, the ELF diagrams, the attractors, and their electronic populations in the ALG-Mg system are presented. In Fig. 9B and 9C, the different electronic shells of Mg can be seen, with almost spherical symmetry, very similar to that of the isolated atom, which is indicative of a mainly electrostatic bonding (Zou et al., 2018). In Fig. 9A, the core attractors on the Mg atom are seen to be highly localized. The population of these degenerate attractors was 7.88e, a value well below that expected for 12e Mg, suggesting electron density contribution from the inner shells to the valence regions and strong charge transfer. The V attractor (O11), with a population of 2.34e (slightly higher than the average for this type of attractor (2.30e), is oriented towards the metal and is located at the bond line.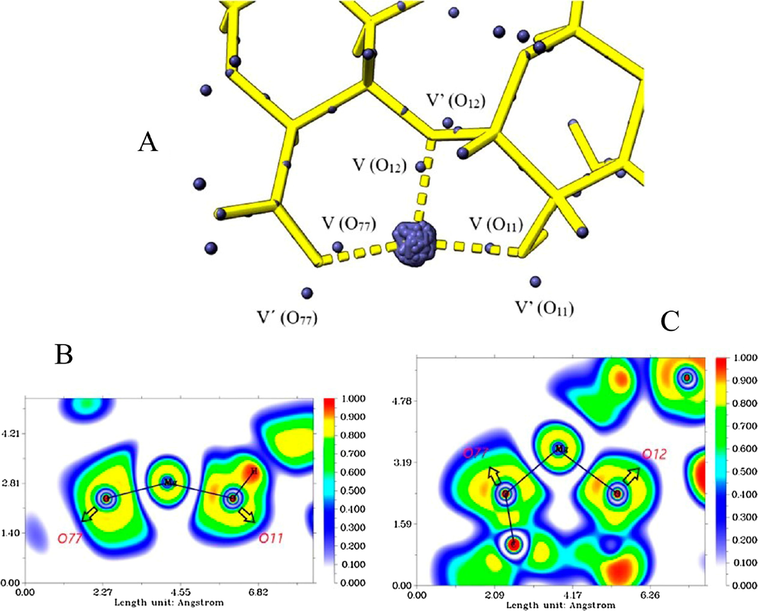
Attractor populations of the lone pairs of O11, O12, O77, magnesium core, and valence attractors (A). ELF diagrams for the Mg-(O77, O11) interaction (B) and for the Mg-(O11, O12, and O77) interaction (C).
The O11-Mg bond distance was 1.97 A0, and the total population for V (O11) and V' (O11) was 4.87e; this is a higher value than the average of –OH-type oxygens in the ALG-Mg (4.73e), suggesting that this bond is formed mainly by Mg → O11 charge transfer. The electronic localization function diagram shows the Mg-O11 interaction, where the binding region presents an ELF value close to zero, which accounts for the electrostatic character of this interaction. The variance value supports the above, which was slightly higher than the average; however, considering the populations, the electronic flux does not vary appreciably, showing that the electronic density is located mainly in the oxygen atom. Meanwhile, the monosynaptic basin V (O77) presents an electronic population of 2.67e, a value higher than the average for this type of attractor (2.58e), and is oriented towards the metal, close to the bond line.
The total population for O77 non-bonding attractors was 6.15e, a high value compared to the average (5.34e), indicating a strong charge transfer from Mg to O77. The O77-Mg bond length was 1.84 A0. The ELF diagram analysis (Fig. 9A and 9B) shows that the interaction region between O77 and Mg has an ELF value close to zero, which would indicate a mainly electrostatic binding type. Meanwhile, the value is higher than the average value, which suggests an increase in the electronic flux. However, this attractor's population tells us that this increase is very slight, showing that the electron density is located mainly in O77, which allows us to suggest a strong electrostatic interaction between O77 and Mg.
Meanwhile, the monosynaptic basin V (O12) is oriented towards the metal but is not located in the O12-Mg bond line. It presents a population of 2.51e, which is above the average value in the system for this type of attractors in ALG-Mg (2.38e). The sum of V (O12) and V' (O12) populations was 5.17e. This value is higher than the average (4.87e). Fig. 9C shows that the interaction region between O12 and Mg presents an ELF value close to zero, which indicates a type of electrostatic bond. On the other hand, the variance value is slightly higher than the average value, indicating no significant increase in the electron density flux. However, the short O12-Mg bond distance was 1.97 A0 (the smallest for O12-metal interactions), indicating that this O12-Mg interaction is the strongest in which O12 participates, concerning the other systems under study. Finally, the analysis of the relative fluctuation (
) revealed that the electron density tends to be little delocalized; this is consistent with electrostatic bonds between localized negative charges, the oxygens, and the positively charged metal. An important factor to consider is that the absence of 3d electrons would prevent further stabilization by non-bonding electron density delocalization. The basin contribution values of the attractors of the non-bonding pairs of O11, O12 and O77 were as follows: 97.67% (2.29e) O11 and 2.33% (0.05e) Mg; 98.29% (2.47e) O12 and 1.71% (0.04e); 97.15% (2.59e) O77 and 2.85% (0.07e). As can be seen, the metal contributions are low, and the above analyses show that the charge is localized on the oxygens; all this reaffirms the electrostatic description of these bonds (see Table 8).
Atractor ALG-Mg
Poblaciones
–OH/, –CO/, -O-
–OH/, –CO/, -O-
(–OH/, –CO/, -O-)
(O)
2.30
2.59
2.38
1.12
1.20
1.20
0.48
0.46
0.49
́(O)
2.43
2.76
2.49
1.13
1.14
1.15
0.46
0.43
0.47
V (O)
2.34
2.67
2.51
1.15
1.27
1.23
0.49
0.48
0.49
V́(O)
2.53
3.48
2.66
1.15
1.39
1.24
0.45
0.40
0.47
4.4.4 ALG-Ca
The attractors' populations are presented in Fig. 10A, 10B, and 10C, the ELF diagrams for the ALG-Ca system are shown. In this, the 3 oxygens analyzed are the ones that presented the largest electronic populations (Table 9). The degenerate attractors of the calcium atom core have a population of 15.92e, lower value compared to the expected formal value of 20e. Furthermore, the ELF diagrams (Fig. 10B and 10C) reveal the different electronic layers of the calcium atom, which shows symmetry similar to that of the isolated atom, suggesting a type of electrostatic interaction.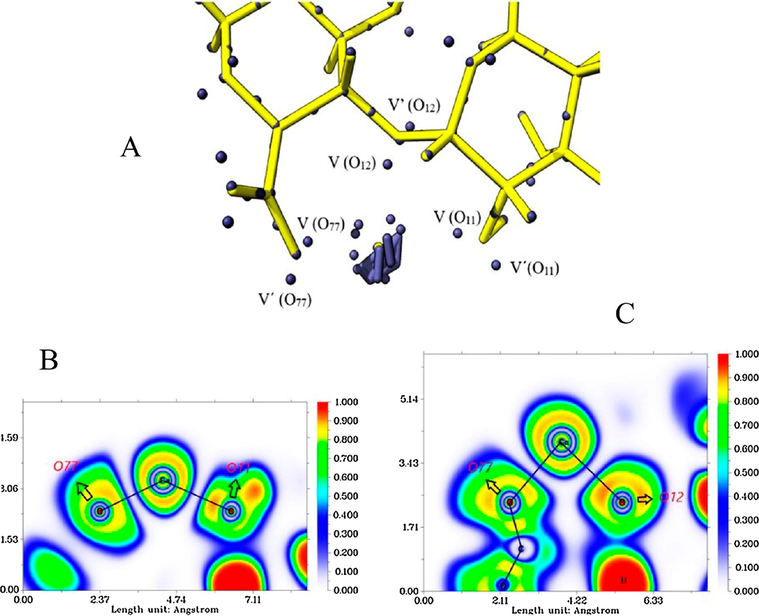
Attractor populations of the lone pairs of O11, O12, O77, valence, and calcium core attractors (A). ELF diagrams for the Ca-(O77, O11) interaction (B) and for the Ca-(O12, O77) interaction (C).
Attractor ALG-Ca
Populations
–OH/, –CO/, -O-
–OH/, –CO/, -O-
(–OH/, –CO/, -O-)
(O)
2.43
2.60
2.43
1.10
1.20
1.12
0.45
0.46
0.46
́(O)
2.34
2.81
2.46
1.13
1.18
1.17
0.48
0.42
0.47
V (O)
2.32
2.79
2.45
1.15
1.30
1.20
0.49
0.49
0.49
V́(O)
2.55
3.25
2.69
1.16
1.35
1.24
0.45
0.42
0.47
The V (O12) population was 2.45e which is slightly above average for this type of attractors (2.43e), suggesting no O12 → Ca donation. The total population for the V (O12) and V'(O12) attractors was 5.14e, which is above average (4.89e) for this type of attractors in the ALG-Ca system, suggesting electronic Ca → O12 donation. The ELF value for the O12-Ca interaction region presents a value close to zero, indicating binding of electrostatic character. Additionally, the high bond distance (3.37 A0) reveals the weakest oxygen-metal interaction of the systems under study. The analysis of tells us that the electronic flux increases due to charge transfer from the metal, while the relative flux is slightly higher than the average value (Table 9). The above would confirm the description of a predominantly electrostatic bonding, where the electron density would be located at O12. The V (O77) presented a population of 2.79e, a value higher than the average (2.60e), which indicates electronic transfer Ca → O77. The sum of both attractors was 6.04e, a value that exceeds the average for attractors of the same type of oxygens (5.41e). The charge transfer would explain this value from the metal to this oxygen and, as seen in previous systems, by V'(O77) contribution in the π C21 = O77 bond. The O77-Ca bond distance was 2.52 A0, the highest value obtained for the O77 interaction with the different metals. The ELF diagrams suggest a strong electrostatic attraction due to the near-zero value of the electronic localization function. The analysis of variance shows an increase in the electronic flux caused by the charge transfer from the metal, which would suggest a strong electronic delocalization; however, the value of the relative flux indicates that such delocalization is slightly higher than the average values of for this system, which supports the idea of electrostatic interaction.
The V(O11) attractor had a population of 2.32e, a value lower than the average of 2.43e, suggesting an electronic donation to the metal. Also, the total population for V(O11) and V'(O11) was 4.87e, a value higher than the average for –OH-oxygen attractors (4.77e). The above indicates Ca → O11 electron transfer. The analysis of variance and electronic flux shows a situation similar to that of O77, i.e., an increase in electronic flux, in this case, due to donation and retro-donation processes, which generate a delocalization only slightly higher than the average values presented in Table 9; this implies a mainly electrostatic bonding. The ELF diagrams Fig. 10B and 10C) also support this electrostatic bonding, which shows the same situation of ELF values close to zero in the interaction region.
Finally, as in ALG-Mg, the oxygen-metal interaction can be represented by negative charges located on the oxygens and positive charges located on the metal. Finally, the contributions to the attractors V (O11), V (O12) and V (O77), were as follows: 98.66% (2.29e) O11 and 1.34% (0.05e) Ca; 98.86% (2.42e) O12 and 1.14% (0.02e) Ca; 97.15% (2.59e) O77 and 2.85% (0.04e) Ca. The above is related to the ALG-Mg system's values and supports the electrostatic description of the bonds.
According to the NBO, ELF, and NCI index results, for the different systems under study, the strongest interaction with O77 follows the following order: Cu Mn > Mg > Ca, where Mn and Cu present polar covalent bonding, while Ca and Mg electrostatic bonds. For O11: Cu Mn ≫ Mg > Ca, where Mn and Cu present polar covalent bonds, while Ca and Mg electrostatic bonds and for O12: Mg > Cu Ca ≫ Mn, all these interactions are electrostatic. Finally, these results correlate with the analysis of the overall reactivity indices. According to our results, the stability order is as follows: ALG-Cu > ALG-Mn > ALG-Mg > ALG-Ca.
Our results contradict some previous studies regarding the type of interactions between alginate and divalent cations. It has been mentioned, for example, that cations such as Mn2+ would bind weakly to the polymer, mainly by electrostatic interactions (Emmerichs et al., 2004). It has also been reported that Mg2+ binds more weakly than calcium because the former shows little or no tendency to form hydrogels (Topuz et al., 2012). Furthermore, as already mentioned, other studies indicate that the degree of affinity is not correlated with the bond's strength (see Refs. 12 and 13), whose results, concerning the type of interactions, agree with those presented in this work. Finally, our study supports the idea of the lack of correlation between bond strength and affinity.
5 Conclusions
The overall reactivity indices showed higher stability for the systems under study, given the following order: ALG-Cu > ALG-Mn > ALG-Mg > ALG-Ca, which seems to be correlated with the degree of electronic delocalization and the degree of covalency of the bonds. As for the nature of the bonds in the ALG-Cu and ALG-Mn systems, these were between the carboxylic acid and alcohol groups' oxygens and proved to be of polar covalent, whereas with the ether oxygen, it is predominantly electrostatic. For the latter, Mn presented a weak interaction, and Mg the strongest. Whereas, in ALG-Ca and ALG-Mg, all the oxygen-metal bonds studied were of electrostatic nature.
Acknowledgements
The authors would like to thank Dr. María Camarada, Director of the Center for Applied Nanotechnology, Faculty of Sciences Universidad Mayor, for providing the means to carry out this study. Cristian Castro thanks the regular FONDECYT project 1130676.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Structure of alginate gels: interaction of diuronate units with divalent cations from density functional calculations. Biomacromolecules. 2012;13:1899-1907.
- [CrossRef] [Google Scholar]
- Bekri, L., Zouaoui-Rabah, M., Springborg, M., & Rahal, M.S.J.J.o.m.m., 2018. A structural DFT study of MM, GG, MG, and GM alginic acid disaccharides and reactivity of the MG metallic complexes, Journal of molecular modeling, 24, 1-13.
- Injectable alginate hydrogels for cell delivery in tissue engineering. Acta Biomater.. 2014;10:1646-1662.
- [CrossRef] [Google Scholar]
- NCIPLOT4: Fast, Robust, and Quantitative Analysis of Noncovalent Interactions. J. Chem. Theory Comput.. 2020;16:4150-4158.
- [CrossRef] [Google Scholar]
- Sustained and controlled release of daunomycin from cross-linked poly (aldehyde guluronate) hydrogels. J. Pharm. Sci.. 2000;89:910-919.
- [CrossRef] [Google Scholar]
- Cross-linking mechanisms of calcium and zinc in production of alginate microspheres. Int. J. Pharm.. 2002;242:255-258.
- [CrossRef] [Google Scholar]
- Structural, spectral, thermodynamical, NLO, HOMO, LUMO and NBO analysis of fluconazole. Spectrochim. Acta Part A Mol. Biomol. Spectrosc.. 2015;150:974-991.
- [CrossRef] [Google Scholar]
- Electrophilicity index within a conceptual DFT framework. Ann. Rep. Sect. C (Physical Chemistry). 2009;105:13-39.
- [CrossRef] [Google Scholar]
- Correlation between electron localization and metal ion mutagenicity in DNA synthesis from QM/MM calculations. PCCP. 2011;13:11239-11247.
- [CrossRef] [Google Scholar]
- Applications of Topological Methods in Molecular Chemistry. 2016;Vol. 22:1-586.
- Molecular insights into the impacts of iron (III) ions on membrane fouling by alginate. Chemosphere. 2020;242:125232
- [CrossRef] [Google Scholar]
- A benchmark for the non-covalent interaction (NCI) index or… is it really all in the geometry? Theor. Chem. Acc.. 2016;135:1-14.
- [CrossRef] [Google Scholar]
- A Car-Parrinello and path integral molecular dynamics study of the intramolecular lithium bond in the lithium 2-pyridyl-N-oxide acetate. J. Chem. Phys.. 2009;131:024308
- [CrossRef] [Google Scholar]
- Interaction between alginates and manganese cations: identification of preferred cation binding sites. Int. J. Biol. Macromol.. 2004;34:73-79.
- [CrossRef] [Google Scholar]
- Adsorption of Metronidazole drug on the surface of nano fullerene C60 doped with Si, B and Al: A DFT study. Int. J. Nano Dimens.. 2020;11:346-354.
- [Google Scholar]
- Transition metal complexes with uronic acids. J. Mol. Struct.. 2005;744:705-709.
- [CrossRef] [Google Scholar]
- Understanding and using the electron localization function. Theor. Comput. Chem.. 2007;19:57-85.
- [CrossRef] [Google Scholar]
- A theoretical study of the bonding in NO,(NO) 2,(NO) 2− and (NO) 2 2− using a topological analysis of the electron localization function. PCCP. 2004;6:3228-3234.
- [CrossRef] [Google Scholar]
- Biological interactions between polysaccharides and divalent cations: the egg-box model. FEBS Lett.. 1973;32:195-198.
- [CrossRef] [Google Scholar]
- Synthesis, characterization and photo-catalytic activity of guar-gum-g-aliginate@ silver bionanocomposite material. RSC Adv.. 2020;10:7898-7911.
- [CrossRef] [Google Scholar]
- A highly efficient Ag nanoparticle-immobilized alginate-g-polyacrylonitrile hybrid photocatalyst for the degradation of nitrophenols. Polymers. 2020;12:3049.
- [CrossRef] [Google Scholar]
- Ecofriendly Green Synthesis of the ZnO-Doped CuO@ Alg Bionanocomposite for Efficient Oxidative Degradation of p-Nitrophenol. ACS Omega. 2020;5:32011-32022.
- [CrossRef] [Google Scholar]
- Affinity of some divalent metals to different types of alginates. Acta Chem. Scand.. 1961;15:1794-2000.
- [Google Scholar]
- Bone regeneration based on tissue engineering conceptions—a 21st century perspective. Bone Res.. 2013;1:216-248.
- [CrossRef] [Google Scholar]
- Electron and Nucleon Localization Functions in Superheavy Elements. Phys. Rev. Lett. 2018;120:053001
- [CrossRef] [Google Scholar]
- Kohn, R., 1975. Ion Binding On Polyuronats—Alginate Pectin. In: Khalaf, Moayad N. (Eds.) Green Polymers and Environmental Pollution Control, Apple Academic Press Inc, pp. 239–263.
- Natural bond orbital analysis in the ONETEP code: applications to large protein systems. J. Comput. Chem.. 2013;34:429-444.
- [CrossRef] [Google Scholar]
- Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. PCCP. 2017;19:17928-17936.
- [CrossRef] [Google Scholar]
- Reexamining the egg-box model in calcium− alginate gels with X-ray diffraction. Biomacromolecules. 2007;8:464-468.
- [CrossRef] [Google Scholar]
- A theoretical perspective of the agostic effect in early transition metal compounds. Coord. Chem. Rev.. 2020;419:213401
- [CrossRef] [Google Scholar]
- Identifying strong covalent interactions with Pauli energy. J. Phys. Chem. A. 2018;122:3087-3095.
- [CrossRef] [Google Scholar]
- Difference in concentration dependence of relaxation critical exponent n for alginate solutions at sol− gel transition induced by calcium cations. Biomacromolecules. 2005;6:2150-2156.
- [CrossRef] [Google Scholar]
- Analysis of Density Functional Tight Binding with Natural Bonding Orbitals. J. Phys. Chem. A. 2019;123:7439-7453.
- [CrossRef] [Google Scholar]
- The nature of the triple BB, double, BB, single, B-B, and one-electron, BB boron-boron bonds from the topological analysis of electron localisation function (ELF) perspective. J. Mol. Struct.. 2020;1221:128530
- [CrossRef] [Google Scholar]
- On the nature of the boron–copper interaction. Topological study of the electron localisation function (ELF) New J. Chem.. 2018;42:17096-17114.
- [CrossRef] [Google Scholar]
- Binding and leakage of barium in alginate microbeads. J. Biomed. Mater. Res. Part A. 2012;100:2939-2947.
- [CrossRef] [Google Scholar]
- Density functional theory (DFT) as a powerful tool for designing new organic corrosion inhibitors. Part 1: an overview. Corros. Sci.. 2015;99:1-30.
- [CrossRef] [Google Scholar]
- Metal–carboxylate interactions in metal–alginate complexes studied with FTIR spectroscopy. Carbohydr. Res.. 2010;345:469-473.
- [CrossRef] [Google Scholar]
- Electron localization function from density components. J. Comput. Chem.. 2017;38:204-210.
- [CrossRef] [Google Scholar]
- A computational investigation of orbital overlap versus energy degeneracy covalency in [UE 2] 2+(E= O, S, Se, Te) complexes. Dalton Trans.. 2020;49:1077-1088.
- [CrossRef] [Google Scholar]
- Theoretical evaluation of electron delocalization in aromatic molecules by means of atoms in molecules (AIM) and electron localization function (ELF) topological approaches. Chem. Rev.. 2005;105:3911-3947.
- [CrossRef] [Google Scholar]
- Metallophilic interactions: observations of the shortest metallophilicinteractions between closed shell (d 10⋯ d 10, d 10⋯ d 8, d 8⋯ d 8) metal ions [M⋯ M′ M= Hg (ii) and Pd (ii) and M′= Cu (i), Ag (i), Au (i), and Pd (ii)] Dalton Trans.. 2020;49:9099-9117.
- [CrossRef] [Google Scholar]
- A quantitative measure of bond polarity from the electron localization function and the theory of atoms in molecules. Theor. Chem. Acc.. 2001;106:223-232.
- [CrossRef] [Google Scholar]
- ELF: The electron localization function. Angew. Chem., Int. Ed. Engl.. 1997;36:1808-1832.
- [CrossRef] [Google Scholar]
- Topological analysis of the electron localization function applied to delocalized bonds. Can. J. Chem.. 1996;74:1088-1096.
- [CrossRef] [Google Scholar]
- Magnesium ions and alginate do form hydrogels: a rheological study. Soft Matter. 2012;8:4877-4881.
- [CrossRef] [Google Scholar]
- Alginate-based composites for environmental applications: a critical review. Crit. Rev. Environ. Sci. Technol.. 2019;49:318-356.
- [CrossRef] [Google Scholar]
- What is NBO analysis and how is it useful? Int. Rev. Phys. Chem.. 2016;35:399-440.
- [CrossRef] [Google Scholar]
- Xu, Z., Lam, M.T., 2018. Alginate application for heart and cardiovascular diseases. In: Alginates and Their Biomedical Applications, Springer, pp. 185–212. https://doi.org/10.1007/978-981-10-6910-9.
- Yang, Y., Weaver, M.N., & Merz Jr, K.M.J.T.J.o.P.C.A. (2009). Assessment of the “6-31+ G**+ LANL2DZ” mixed basis set coupled with density functional theory methods and the effective core potential: prediction of heats of formation and ionization potentials for first-row-transition-metal complexes. The Journal of Physical Chemistry A, 113(36), 9843-9851.
- Partitioning a Molecule into the Atomic Basins and the Resultant Atomic Charges from Quantum Chemical Topology Analysis of the Kohn-Sham Potential. J. Phys. Chem. A. 2020;124:5023-5032.
- [CrossRef] [Google Scholar]
- Rb3VO (O2) 2CO3: A Four-in-One Carbonatoperoxovanadate Exhibiting an Extremely Strong Second-Harmonic Generation Response. Angew. Chem. Int. Ed.. 2018;57:8619-8622.
- [CrossRef] [Google Scholar]
- A theoretical and experimental study on isonitrosoacetophenone nicotinoyl hydrazone: Crystal structure, spectroscopic properties, NBO, NPA and NLMO analyses and the investigation of interaction with some transition metals. J. Mol. Struct.. 2018;1162:125-139.
- [CrossRef] [Google Scholar]




