

Translate this page into:
Theoretical study of new 3-(methylthio)-8-phenyl-8H-thieno[2,3-b]indole derivatives for application in DSSC: Solvent effect, adsorption process on the surface of TiO2
⁎Corresponding author. y.karzazi@ump.ac.ma (Yasser Karzazi)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
This investigation aims to elucidate the influence of chemical modifications of the π-bridge on the optoelectronic properties of a newly designed series of five dyes Di (i = 1–5). In this regard, the molecules are designed with 3-(methylthio)-8-phenyl-8H-thieno[2,3-b] indole as electron-donating fragments and cyanoacrylic acid as electron-accepting groups. To attain this goal, the optoelectronic properties of the Di dyes and the reference dye MKZ-39 were theoretically investigated in both the gas phase (GP) and Toluene solvent (TS) using Density Functional Theory (DFT). In addition, to predict the absorption spectrum and excited state properties of theoretical Di molecules, we have resorted to Time-dependent Density Functional theory (TD-DFT). Furthermore, the Monte Carlo approach was employed to theoretically evaluate the process of dye adsorption onto the surface of TiO2 (1 1 0) anatase to determine the most stable TiO2-dye-100 C7H8 complex. The results presented demonstrate that the selected dyes possess auspicious characteristics as potential promoters for solar cells, including a redshift of λMax that can reach 561 nm, a nearly complete Light-Harvesting Efficiency (LHE) of 99 %, and a higher open-circuit voltage (VOC) compared to the reference molecule. Our results could open new perspectives for further experimental research into the chemical-structural modifications of these sensitizers.
Keywords
Indole
DSSCs
DFT
Monte Carlo
TiO2 anatase

1 Introduction
The world is currently experiencing a global crisis in terms of energy of fossil origin (Solangi et al., 2011) and the search for new alternatives with renewable sources at lower cost and respectful of the environment is fundamental. Among the available options, solar energy is inexhaustible, clean, renewable and non-polluting. This type of technology is based on converting solar photons into energy through solar cells based on semiconductors. However, the manufacture of solar cells is still prohibitive in terms of energy cost. The development of a new type of solar cell based on dye-sensitized (DSSC), over the past three decades, has emerged as one of the most promising contenders for practical photovoltaic devices (Grätzel, 2001; O’Regan and Grätzel, 1991). This is attributed to their low manufacturing costs, high conversion efficiency, and ease of electronic properties control. This is why research in this area is very active around the world including our laboratory (Arbouch et al., 2020; Arbouch et al. (2017)). Ru-based organometallics complexes are often used as sensitizers due to their good stability and photoreception properties with a high molar extinction coefficient (Baviskar et al., 2014; Grätzel, 2003). However, the implementation of these types of semiconductors has also faced difficulties related to the complexity of the synthesis processes, manufacturing cost, and above all, the use of chemical agents that are toxic to the environment (Amao and Komori, 2004). To circumvent this challenge, important research work has been envisaged to synthesize new metal-free dyes. In this respect, metal-free organic dyes as potential photosensitizers for DSSCs have aroused a great deal of interest given their spectacular characteristics, among them, we can mention: great architecture flexibility, low manufacturing cost, optoelectronic properties that are easily adjustable, and significant molar extinction coefficients (Mishra et al., 2009a).
In general, organic dyes are formed from donor and acceptor fragments bridged either by a saturated conjugated chain or cyclic bridges (D-π–A) (Babu et al., 2015; Chiu et al., 2016; Malapaka et al., 2012; Shalini et al., 2016), this kind of dye is considered to be the most encouraging and successful model for improving the DSSC photovoltaic process because the D-π–A configuration can effectively increase the charge transfer (CT). The characteristics of the organic semiconductor can be modified by applying a few modifications to each part of the basic dye pattern, leading to a change in the photovoltaic properties (Gang et al., 2014; Wan et al., 2012). Various (D-π–A) sensitizers based on heteroaromatic systems such as indole (Ito et al., 2006; Santhini et al., 2019; Yuan et al., 2016), thiazole (Dessì et al., 2020; Menzel et al., 2012), tri- phenylamine (Hagberg et al., 2007; Hosseinzadeh et al., 2016; Xu et al., 2008) and coumarin (Hara et al., 2005; Hara et al., (2003a); Hara et al., (2003b); Hara et al., (2003c)), have been shown to behave as interesting dyes for DSSCs with good power conversion efficiencies (PCE), thanks to their electronic structures capable of acting as good electron donors. In fact, Indole-based dyes have shown an efficiency of up to 9.5 % (Ito et al., 2008), mainly due to their excellent ability to donate electrons and their exceptional ability to capture light. In parallel to the electron-donor part, the acceptor segment is also recognized for its importance in controlling the performance of the DSSC (Babu et al., 2015). In this aspect, various acceptor groups such as cyanoacetic acid have been found to be effective in most cases, mainly due to their electron retention characteristics and their ability to anchor to the TiO2 surface via an ester bond. Besides, we must not forget the crucial role of π-conjugation bridges such as thiophene derivatives, which have been demonstrating excellent CT efficiency (Mishra et al., 2009b). Their existence between the donor and acceptor groups not only reinforces the π-conjugation but also increases the overall stability of the molecules thus obtained. Moreover, these thiophene derivatives can also be used to reduce the bandgap and thus shift the maximum absorption peaks to higher wavelengths with an increased molar extinction coefficient (Wang et al., 2007).
In this study, we have focused our interest on the simulation and study of new dyes with a general D-π-A structure, in which the 3-(methylthio)-8-phenyl-8H-thieno[2,3-b]indole group behaves as an effective electron donor attached to a different π-spacer which is further linked to cyanoacetic acid as an acceptor/anchoring unit (as depicted in Fig. 1). MKZ-39 (R) was chosen as a reference because it has a simple structure where some groups can be changed to improve optical and electronic properties, and it has also a relatively high PCE value of up to 7.4 %.![Structure of the studied dyes based on 3-(methylthio)-8-phenyl-8H-thieno[2,3-b]indole and reference molecule R.](/content/184/2024/17/1/img/10.1016_j.arabjc.2023.105457-fig1.png)
Structure of the studied dyes based on 3-(methylthio)-8-phenyl-8H-thieno[2,3-b]indole and reference molecule R.
To describe the desired properties of the dyes, quantum chemistry modelling using DFT and TD-DFT has been successfully used to understand and predict optoelectronic characteristics of molecules, including molecular geometry, highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), band gap energy (Eeg), UV–vis absorption spectra, open-circuit voltage (VOC), reorganization energies ( ), electron injection driving force (ΔGinject), exciting state lifetime, electron density difference (EDD), transition density matrix analysis (TDM) and adsorption energy (Eads). Our work, informed by the results obtained from these parameters, serves as a valuable roadmap to guide the synthesis of designed molecules (D1-D6) with the overarching goal of discovering exceptionally efficient dye-sensitized solar cells (DSSCs). This knowledge paves the way for more efficient and targeted research in the field of photovoltaics.
2 Computational details
Throughout this paper, the DFT calculation will be employed as the most suitable method for investigating the electronic structure and photovoltaic properties of the dyes, providing a comprehensive understanding of their behaviour and performance in solar cells. The theoretical calculations of optoelectronics parameters were carried out with the GAUSSIAN 09 software package (Frisch et al., 2009), and GAUSS VIEW 6.0 interface was used for results visualization (Dennington et al., 2019). The geometrical ground state structures of studied compounds have been optimized with the XC function Lee–Yang–Parr gradient-corrected correlation (B3LYP) (Becke, 1993). This functional is considered a common choice for DFT calculations due to its good balance between accuracy and computational cost, making it appropriate for the study of both ground and excited states of molecules (Bouakline et al., 2023; Bouzineb et al., 2020; Britel et al., 2023; Britel et al. (2022)). Additionally, frequency calculations are needed to confirm that the optimized dye structures are stable when imaginary frequencies are missing.
The accuracy and computation time of calculations strongly depend on the chosen basis set. Therefore, in our study, we used the split-valence (double-ζ) basis sets 6–31(d,p) with polarization functions on both heavy atoms and hydrogen. This choice of basis set allowed us to accurately describe molecular electronic properties (Hachi et al., 2021; Zhao et al., 2020). Pastore et al. demonstrated that introducing diffuse functions to this basis set did not significantly affect the precision of theoretical predictions (Pastore et al., 2010). Besides, the solvation effect in the context of TD-DFT was implicitly incorporated by using the polarizable continuum model (PCM) (Becke, 1993) and toluene has been chosen as a solvent in order to ensure comparability of our theoretical results with those of the experiment for the reference R in toluene (Zhang et al., 2010).
The calculations at the TD-DFT level for the free dyes have been carried out using different functionals: WB97XD (Chai and Head-Gordon, 2008), PBEPBE(Doust Mohammadi and Abdullah, 2021), B3LYP, PW91PW91(Wang et al., 2020), M06-2X and CAM-B3LYP) to determine the optical properties such as the oscillator strengths (f), the vertical excitation energies (Eex) and the absorption absorption maximum wavelength . Notably, the CAM-B3LYP functional exhibited strong agreement with experimental results, as demonstrated in Supplementary Material (SM) Table S1. B3LYP and CAMB3LYP functionals have been largely used in the literature and have been shown to provide accurate results for similar types of our molecules (Britel et al. (2022); Pedone, 2013; Yang et al., 2015). Nevertheless, B3LYP gives good results for structural and energetic studies but fails when dealing with excitations and CT at the TD-DFT theory level. Conversely, CAMB3LYP takes into account the long-range correction to correctly describe the excited states and transition energies (Yanai et al., 2004). Similarly, we have varied the complexity of the basis set, starting with 6-31G and ending with 6-31G(d,p)++, keeping the same functional in order to observe the effect of the basis set on the shift of the UV–Vis spectrum. (See Table S2 for additional data in SM).
The adsorption dye on the TiO2 anatase crystal was performed by Molecular dynamics (MD) simulations using the adsorption locator module based on the Metropolis Monte Carlo (Allalen, 2006) approach incorporated in Biovia Materials Studio v8.0 (BIOVIA (2017)). The model used here to represent the TiO2 anatase surface (1 1 0) is constructed by a slab thickness of 5.0 Å, a vacuum of 30 Å along the Z axis and a supercell (6x8). Thus, a simulation box (56x42x38 Å) with periodic boundary conditions was used to evaluate the interaction between a single dye molecule Di, 100 molecules of Toluene and the surface of the TiO2 (1 1 0) anatase. The atomic parameters used in the geometry optimization are those given by the COMPASS force field (Amrani et al., 2023; Bunte and Sun, 2000; Sun, 1998).
3 Results and discussion
3.1 Geometrical stability
The goal of the ground-state geometric optimization is to predict the most stable structure, which is achieved by determining parameters such as bond lengths, bond angles and dihedrals, see Table 1 and Table 2. The connection between the electron-donor moiety and the π-spacer (r1) is in the range of 1.443 to 1.446 Å and 1.442 to 1.44 Å for the gas and solvent phases, respectively, hence the parameter r1 is hardly affected when passing from GP to TS. At the same time, the connection between the electron acceptor and the conjugated π-bridge (r2) is in the range of 1.422 to 1.443 Å and 1.418 Å to 1.440 Å for GP and TS respectively, so the parameter r2 becomes shorter (more stabilized (Bourass et al., 2017)) when going from GP to TS. Furthermore, the growth orders of r1 and r2 in the solvent are D5 < D3 < D4 < D1 < D2 and D3 < D2 < D1 < D4 < D5, respectively. In DSSCs, the distance between the donor group and the π-bridge is crucial, the CT is more efficient when the binding distance is shorter between these two moieties (Ninis et al., 2013). Besides, the dihedral angle plays a critical role in determining the stability of a molecule. Specifically, when the dihedral angle is small, the atoms tend to arrange themselves in a relatively planar configuration, which in turn leads to increased stability of the molecule. Therefore, the coplanar molecular structure favours intramolecular charge transfer (ICT) between the electron donor and acceptor groups (Ninis et al., 2013). However, as the dihedral angle increases, the atoms can come into closer proximity to each other, resulting in steric interference and increased energy (Wu et al., 2019). In summary, the values of the dihedral angles presented in Table 2, show that the molecule D4 has the most coplanar structure.
DYE
Gas
Toluene
r1 (Å)
r2 (Å)
r1 (Å)
r2 (Å)
D1
1.446
1.423
1.444
1.421
D2
1.446
1.423
1.445
1.420
D3
1.444
1.422
1.442
1.418
D4
1.445
1.427
1.443
1.424
D5
1.443
1.443
1.441
1.440
DYE
Gas
Toluene
Dihedral angle
Dihedral angle
D1
156.68−178.97
153.48−179.07
–
–
D2
−147.50−179.68
−146.32−179.79
165.16
166.38
D3
−167.12178.60
−164.78178.38
–
–
D4
−178.45179.03
−176.56179.62
−177.02
−179.20
D5
−153.53−177.75
−152.76179.96
175.60
174.60
3.2 Frontier molecular orbital (FMO) energy
To improve dye efficiency, it is crucial to make appropriate selections for both the donor group and the π-bridge. The HOMO and LUMO energy levels, as well as the energy gap (Egap), for the ground-state configurations of the dyes, were calculated at the B3LYP/6-31G(d,p) level, as summarized in Table S3. It's worth highlighting that optimizing the efficiency of the DSSC device involves choosing dyes with HOMO energy levels (EHOMO) that closely match the electrolyte redox potential
at −4.8 eV and the LUMO energy level must be positioned higher than the conduction band (CB) of the TiO2 (-4.0 eV) (Barea et al., 2010), as illustrated in Fig. 2. It can be observed that all dyes exhibit larger promising electron injection potential than the R molecule. This is attributed to the most potent electron donor group with the highest EHOMOs, which are in a relatively narrow range, from −2.63 to −2.89 eV in the GP and −2.71 to −2.90 eV in the TS. Notably, these values are higher than the energy level of the TiO2 conduction band.
Calculated energy level diagram of the dyes at the B3LYP/6-31G(d,p) level.
Generally, a smaller Egap, which represents the energy difference between the ground state and the excited state, makes the electronic transition easier. This means that dye with a smaller Egap (D5) is more efficient at absorbing light of longer wavelengths, particularly in the infrared range. This enhanced absorption of longer-wavelength light is beneficial for the overall performance of DSSCs because it allows the system to harness a broader spectrum of solar energy. As a result, these devices can achieve a higher short-circuit current density (JSC) and increased overall conversion efficiency (Nazeeruddin et al., 1993). It's important to note that in the present study, the solvation step has been found to lower both the ELUMO and the Egap for the six dyes under investigation. This adjustment has the potential to further optimize the performance of these dyes.
Examining the isosurfaces of the HOMO and LUMO orbitals associated with MKZ-39 and Di dyes is essential to gain insights into the charge separation process, as illustrated in Fig. 3. A remarkable feature is the broader charge distribution in all dyes compared to photosensitizer R. HOMO electrons are widely dispersed in these molecules, with their highest electron density residing in the donor groups and their concentration relatively low in the acceptor group. For LUMOs, the main localization is around the cyanoacrylic acid units, with D1-5 displaying the highest electron density in the π-spacer groups. Consequently, when the HOMO-LUMO state is excited through irradiation, it triggers electron migration from the donor group, passing through the π-spacer, and ultimately reaching the acceptor group. This electron migration process leads to charge transfer (CT) and eventually to the TiO2 surface.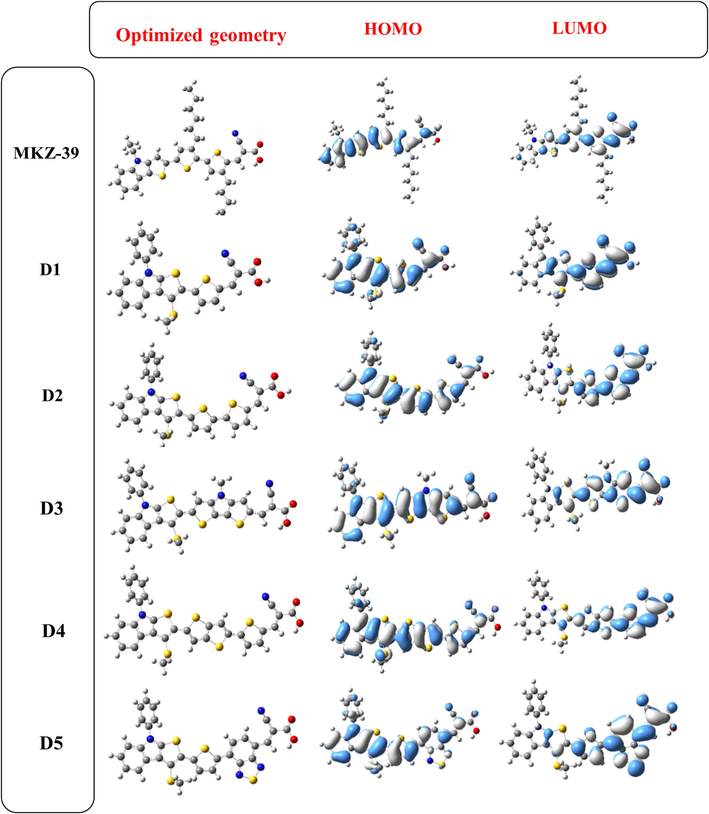
Structural optimization and electronic distribution.
3.3 Optical characteristics of the investigated dyes
The optical spectra of the dyes were calculated in the gaseous phase and in Toluene by the TD-DFT method using the solvent model C-PCM at the CAM-B3LYP/6–31(d,p) level of theory. The ability to harvest the light in the active layer is directly related to the absorption wavelength range and absorptivity. Theoretical UV–vis absorption profiles of dyes in GP and TS are shown in Fig. 4. The theoretical max reported in the following corresponds to the first singlet excited states with an electronic transition from HOMO to LUMO. All dyes present two prominent absorption bands; the first one in the UV region observed at (300–370 nm) is due to a π - π* electronic transition of the dye fragments and the second occurs in the visible region (394–561 nm) is attributed to ICT between the donor and the anchor/acceptor unit that could lead to the formation of a charge-separated excited state. All the dyes revealed a red shift of the maximum absorption wavelength (λmax) in the ICT band when moving from the GP to the TS. λmax follows the same trend D5 > MKZ-39 > D3 > D4 > D2 > D1 in GP and TS. Among these dyes, D5 stands out as the most promising candidate due to its substantial redshift in λmax, signifying its potential for efficient ICT and enhanced performance in relevant applications.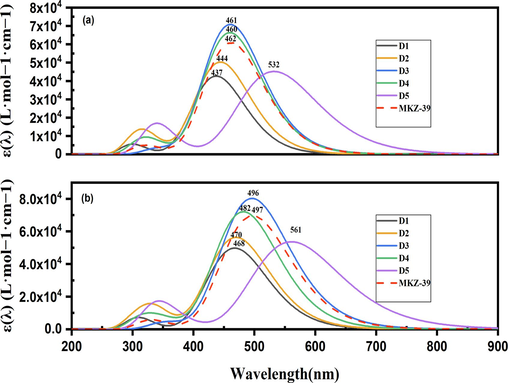
Calculated absorption spectra of the dyes at the CAM-B3LYP/6-31G(d,p) level. (a) in GP (b): in TS.
The excitation process parameters, as outlined in Table 3, reveal a distinct trend in the values of the parameter “ f.” Specifically, the order of values is consistently D3 > MKZ-39 > D4 > D2 > D5 > D1 in both the GP and TS. Moving from the GP to the TS, there is a uniform increase in the oscillator strength value for MKZ-39 and D5. However, it is noteworthy that among all the dyes examined, the most significant increase in f, indicative of the ICT process, is observed in the case of D3. This implies that D3 exhibits the most pronounced change in the excitation process when moving from the ground state to the transition state. This increase in efficiency is due to the more effective ICT upon excitation of electrons between the indole donor and the acceptor, facilitated by the π-conjugation spacer. Furthermore, both experimental and theoretical absorption wavelengths for semiconductors are highly consistent (99.1 %), indicating that the theoretical level proposed in our study is suitable for this comparison.
DYE
Gas
Toluene
(eV)
f
Transition
(eV)
f
Transition
D1
437.45
2.83
1.05
467.78
2.65
1.23
315.51
3.93
0.02
H-1
322.45
3.84
0.03
H-1
298.96
4.14
0.12
H-2
308.13
4.02
0.16
H-2
D2
444.49
2.79
1.24
H-1
470.05
2.63
1.4
322.93
3.84
0.24
H-2
335.91
3.69
0.32
H-2
L
300.46
4.13
0.14
H-1
L + 1
302.25
4.10
0.15
H-1
L + 1
D3
460.64
2.69
1.75
H-1
496.22
2.49
1.98
345.25
3.59
0.08
H-3
358.07
3.46
0.10
H-3
L
329.98
3.75
0.01
H-2
L
343.44
3.61
0.02
H-2
L
D4
432.90
2.86
1.30
465.73
2.66
1.43
319.26
3.88
0.01
H
L + 1
332.98
3.72
0.05
H-2
L
296.71
4.18
0.11
H
L + 1
307.18
4.03
0.12
H
L + 1
D5
532.32
2.33
1.11
561.03
2.21
1.32
363.58
3.41
0.07
H-2
L
371.48
3.34
0.12
H-1
L
337.14
3.67
0.37
H
L + 2
337.82
3.67
0.38
H
L + 2
MKZ-39 (Zhang et al., 2010)
461.80
2.68
1.50
497.2(502a)
2.49
1.71
338.86
3.65
0.01
H-2
L
353.56
3.50
0.02
H-1
L + 1
320.50
3.86
0.11
H-2
L
329.46
3.76
0.13
H-2
L
3.4 Mechanism of electron-hole charge transfer
We have specifically calculated the 2D Transition Density Matrix (TDM) plots and Electron Density Difference Maps (EDDM) of our designed molecules using multiwfn 3.8 software(Lu and Chen, 2012) at the CAM-B3LYP/6-31G** level of theory. In order to depict the ability of the exciton dissociation into free charges and the Intramolecular charge transfer process, it is informative to examine the EDDM between the excited (S1) and ground states (S0). The EDDM as shown in Fig. 5, demonstrate that a charge transfer has occurred between the excited and ground states for all the dyes under investigation. The blue isosurface corresponds to the region of electron concentration, while the green isosurface indicates the regions where holes remain after electron departure. It is evident that there is a reduction in electron density localized around the primary electron-donating indole group and the π-bridge segments. Furthermore, there is an increase in electron density observed on the π-linker and the cyanoacrylic acid units.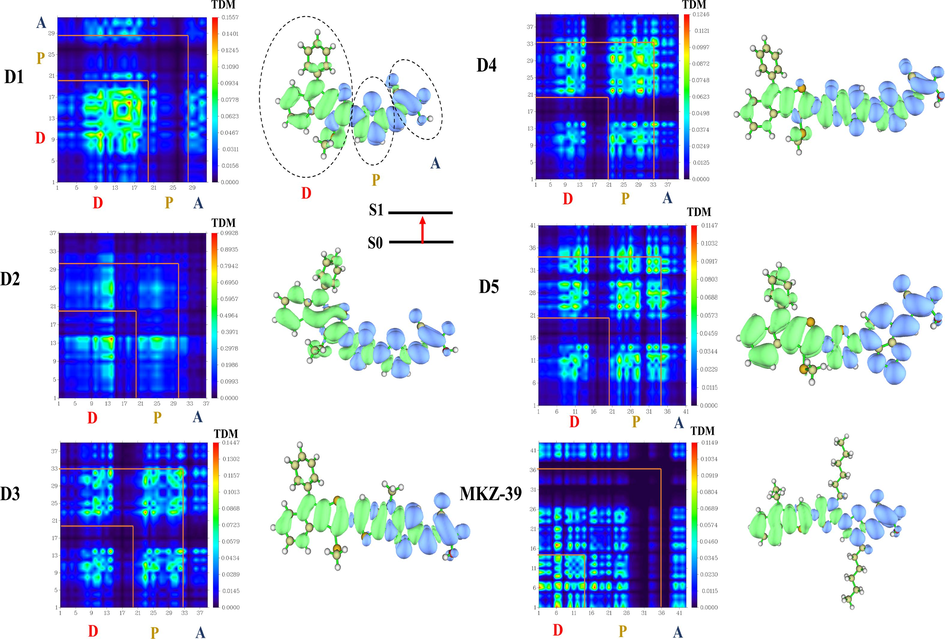
Simulated TDM and EDD for the investigated dyes Di (i = 1, 5) and the reference molecule in toluene at S1 (the influence of hydrogen atoms has been disregarded due to their minimal impact on electronic transitions), Donor (D), Bridge (P) and Acceptor (A).
Transition Density Matrix (TDM) is also an important metric to visually represent ICT during electronic transitions in compounds D1 to D5. TDM maps are widely recognized as a valuable tool for studying the probability of exciton escape from the influence of coulombien attraction(Li et al., 2015; Li et al. (2011)). As revealed by Fig. 5, the electron-hole coherences within the first excited states in all molecules have a large magnitude off-diagonal boxes (D-D, P-P, A-A), suggesting a strong electronic coupling between the donor and acceptor moieties, facilitating efficient charge transfer. These findings are further supported by the results obtained from EDDMs. D1, D3, D4, D5 and MKZ-39 exhibit a pronounced concentration within the diagonal box (D-D, P-P, A-A) during the process of photoexcitation. This concentration serves as a compelling demonstration of the successful charge transfer from the donor to the acceptor unit and the dissociation of excitons in these molecules is much easier, which is consistent with the situation presented by EDDM. Notably, the TDMs transition demonstrates an effective contribution to the bridge unit, highlighting its role in facilitating and mediating the electron-hole dynamics in the system. As Fig. 5 illustrates, a distinct relationship becomes evident between the extent of dissociation and the degree of conjugation within the bridge. Conspicuously, among the compounds under consideration, MKZ-39 and D5 stand out as they exhibit a significant intra-molecular charge transfer, accompanied by a smaller degree of electron-hole coherence. This characteristic makes these dyes particularly conducive to the efficient dissociation of excitations. Furthermore, there is the possibility of electron transfer observed in the off-diagonal portion of the analysis, which could further enhance the charge transfer behaviour of these molecules, contributing to their overall performance. It is clear that the solvation of dyes does not affect the overall shape of TDM, this is due to the property of toluene, which is a relatively non-polar solvent (see Fig. S1 in SM).
3.5 Light-harvesting efficiency (LHE) and open-circuit voltage (Voc)
The primary determinant of the active layer's performance is the LHE parameter. It provides a comprehensive insight into the photon absorption and the efficiency of injecting excited electrons at the TiO2 conduction band (CB). This parameter can be calculated using Eq. (S4). All the systems Di have high LHE values in the range of 0.91 to 0.98 in GP and 0.94 to 0.99 in TS, see Table 4. These values are comparable with the corresponding reference dye values of 0.96 and 0.98, respectively. Additionally, Voc is also a key parameter as it is directly proportional to the conversion rate. Our findings reveal significant variations in Voc values, with values ranging from 0.67 to 1.37 eV in the gas phase and from 0.66 to 1.29 eV in Toluene. Generally, the higher the ELUMO value, the higher the open-circuit voltage value. In fact, the higher values of ELUMO (-2.63 eV in GP and −2.71 eV in TS) and Voc (1.37 V in GP and 1.29 eV in Toluene) are encountered for sensitizer D1. We emphasize that Voc can be calculated according to Eq. (S2).
Dye
(eV)
(eV)
LHE
Voc(V)
ΔGinje(eV)
(eV)
(ps)
Gas
D1
5.36
2.53
0.91
1.37
−1.47
0.56
274
D2
5.19
2.40
0.94
1.29
−1.60
0.39
238
D3
5.12
2.43
0.98
1.31
−1.57
0.32
181
D4
5.21
2.35
0.95
1.11
−1.65
0.41
217
D5
5.32
2.99
0.92
0.67
−1.01
0.52
382
MKZ-39
5.18
2.50
0.96
1.25
−1.36
0.38
214
Toluene
D1
5.39
2.74
0.94
1.29
−1.26
0.59
266
D2
5.23
2.44
0.96
1.27
−1.56
0.43
237
D3
5.10
2.41
0.99
1.27
−1.59
0.30
187
D4
5.19
2.33
0.96
1.10
−1.67
0.39
228
D5
5.27
3.06
0.95
0.66
−0.94
0.47
357
MKZ-39
5.18
2.69
0.98
1.20
−1.31
0.38
217
3.6 Free energy of electron injection ΔGinje
Referring to Table 4, it is evident that the ΔGinje values for the dyes are consistently negative. This indicates that the energy levels of the excited states in these dyes are consistently situated above the conduction band (CB) of TiO2, facilitating the spontaneous injection of charges into the titanium oxide. The driving force for injection, as determined by Eq. (S6), falls within the range of −1.65 to −1.01 eV in one case and −1.67 to −0.94 eV in the other. Interestingly, the order of these values is as follows: D4 < D2 < D3 < D1 < MKZ-39 < D5 and D4 < D3 < D2 < D1 < MKZ-39 < D5 in the other, for both the gas and solvent phases, respectively. For the dyes in TS, ΔGinje have known a remarkable decrease since the solvent has increased the energy of , except for dye D4 which underwent the EHOMO higher. Among all the dyes, the most negative value of ΔGinject is calculated for D4 (-1.67 eV) and by the way we expect this dye to have a higher value of JSC.
3.7 Dye regeneration energy
The efficiency of the devices can be enhanced by selecting dyes with an EHOMO more closely to the ERP( / ), but a sufficient driving force must be delivered to provide an effective regeneration of the dye by the redox agent. The minimum driving force necessary to ensure the quasi-quantitative recombination of the electron with the oxidized sensitizer by the oxidation of iodide to triiodide is about 0.2 to 0.3 eV (GrÃtzel (2009)). Generally, if the energy induced by the thermodynamic forces necessary for the regeneration of the pigments is too great, it can be dispersed in the form of heat involved in the regeneration process and this contributes to a considerable loss of efficiency of the DSSCs (Ning et al., 2008). Table 4 show that the values of the studied dyes are below the ERP ( / ) and are sufficient for the dye regeneration process by electron-hole pairs, wich it can be obtained from Eq. (S7). Besides, we can classify the values of regeneration energy as follows: D3 < MKZ-39 < D2 < D4 < D5 < D1 in GP, and D3 < MKZ-39 < D4 < D2 < D5 < D1 in TS. This observation underscores that exhibits an ascending trend in correlation with the electron donor potential of the donor group.
3.8 Excitation lifetime
The lifetime of the first excited state plays a very important role in the effectiveness of CT. In fact, sensitizers that have longer electron lifetimes can contribute more to CT and are able to maintain their cationic form for longer periods. By increasing the lifetime of the excited states, the rearrangement process of the electron-hole pair is retarded and the efficiency of the photovoltaic cell is improved. The lifetimes of the excited states of the dyes can be calculated as follows where f is the oscillator strength and the excitation energy (cm−1). Comparing the lifetimes in the gas phase and toluene, we can observe variations in τ for different dyes and the reference molecule (see Table 4). As a general trend, the excited state lifetimes are typically longer in the gas phase for most dyes. D5 has the longest excited state lifetime in both conditions, indicating that it retains its excited state for a longer time. This property could be advantageous in applications where a longer excited state is needed such as our case.
3.9 Monte Carlo simulations
To understand the interaction of indole derivatives with the titanium dioxide surface, a Monte Carlo simulation was used to construct the adsorption structures of the compound. Subsequently, the most appropriate configuration for a TiO2 (1 1 0)-Dye-100 C7H8 system can be reasonably predicted through the evaluation of interaction energies. As determined by the adsorption locator module, the adsorption energy distributions for systems composed of TiO2 (1 1 0)-Dye-100 C7H8 are represented in Fig. 6.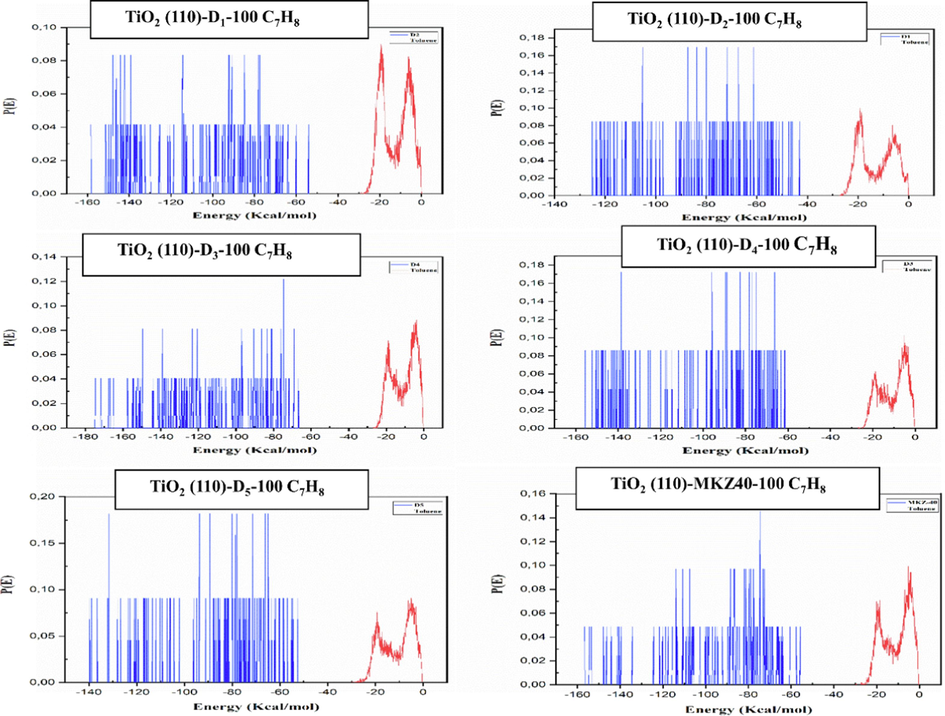
Distribution of the adsorption energy chart for the TiO2 (1 1 0)-Dye-100 C7H8 system during the energy optimization process.
The x-axis represents energy in kilojoules per mole (kJ/mol), and the y-axis shows the probability of finding a molecule with a specific energy level P(E). Notably, the chart highlights that dye molecules exhibit a broader range of adsorption energies when compared to toluene molecules. This is primarily due to the larger and more complex nature of dye molecules, allowing them to interact with the TiO2 surface in a wider variety of ways. Moreover, the chart demonstrates that the most probable adsorption energy for dye molecules is considerably more negative than that for toluene molecules. This suggests that dye molecules form stronger bonds with the TiO2 surface, indicating more robust adsorption.
During Monte Carlo simulations, the positions of the atoms or molecules are randomly perturbed, and the energy of the system is subsequently computed at each step. Through repeated iterations of this process, each bound structure was energetically optimized by adjusting the entire system. The resulting energies for the TiO2 (1 1 0)-Dye-100 C7H8 systems in TS are presented in Fig. 7.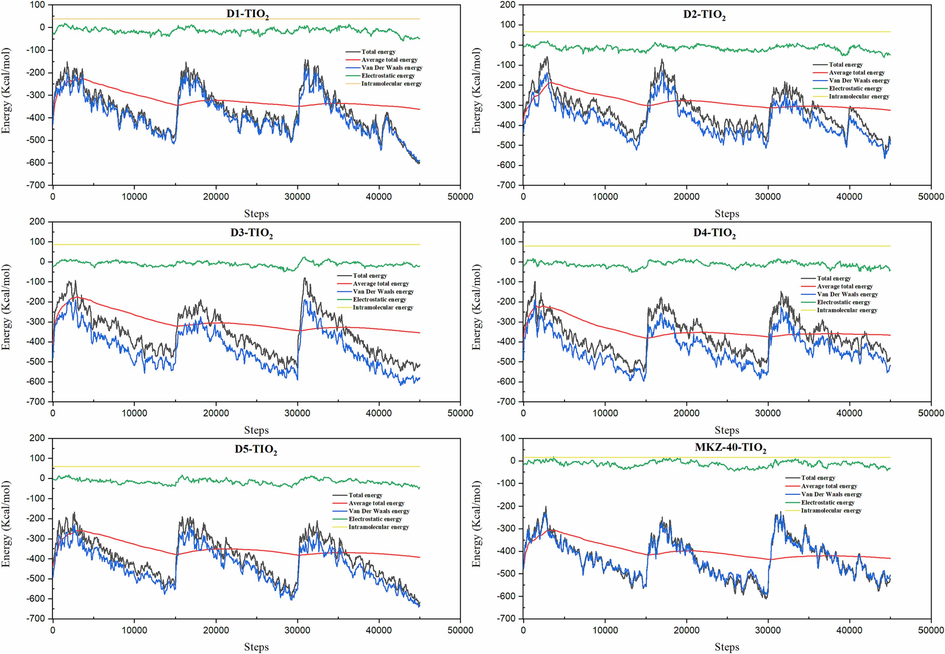
Total energy distribution graphic of the TiO2 (1 1 0)-Dye-100 C7H8 system.
The top and side views of the most stable adsorption forms of the dyes on the TiO2 (1 1 0) surface are presented in Fig. 8. These images depict the most effective adsorption modes for the TiO2 (1 1 0)-Dye-100 C7H8 systems, as determined by the adsorption locator module. The active centres in the interface between the dye molecules (D1, D2, D3 and D4) and the oxide surface are clearly the oxygen (-O-) and hydrogen (H-) atoms, as evidenced by Fig. 8. The calculated contact angles between the dyes D2 and D4 and the TiO2 (1 1 0) surface are close to 90°, indicating the successful action of the anchoring group, which facilitates the transfer of charges from the dye to the TiO2 CB. Moreover, for D1, D3, D5, and MKZ-39, the contact angle is close to 180° demonstrating an increase of the adsorbate-adsorbent contact surface since these dyes are adsorbed in an almost planar form on the TiO2 (1 1 0) surface.
Top and side views of the equilibrium adsorption configurations for TiO2 (1 1 0)-Dye-100 C7H8complexes.
Table 5 shows the results and description of the lowest adsorption configurations of TiO2 (1 1 0)-Dye-100 C7H8 systems using the adsorption locator module (all values are in kcal/mol). The energy required when the relaxed adsorbed components Di are adsorbed onto the substrate (TiO2 (1 1 0) surface) is reported by the adsorption energy (Eads). Overall, adsorption is widely recognized as the main interaction process that allows the guaranteed injection of electrons onto the TiO2 (1 1 0) surface.
Structures TiO2-Dye
Total energy (Kcal/mol)
Adsorption Energy (Kcal/mol)
Rigid adsorption Energy (Kcal/mol)
Deformation Energy (Kcal/mol)
Dye: dEad/dMi
Toluene: dEad/dMi
TiO2-D1
−1338 0.652
−1376.921
−1351.487
−25.433
−89.962
−0.414
TiO2-D2
−1195.223
−1260.839
−1219.379
−41.459
−73.860
−3.174
TiO2-D3
−1089.598
−1176.619
−1143.521
–33.097
−142.491
−0.603
TiO2-D4
−1120.604
−1200.142
−1161.267
−38.873
−135.822
−0.809
TiO2-D5
−933.325
−1203.086
−1102.626
−100.459
−221.358
−0,848
TiO2-MZK-39
−1117.033
−1132.111
−1094.582
−37.528
−79.586
−0.402
In Table 5, one of the listed variables is the total energy of the substrate/adsorbate system, which represents the sum of all constituent energies. The adsorption energy reflects the energy change that occurs when the relaxed adsorbate component Di is adsorbed onto the TiO2 (1 1 0) substrate, and this is often determined using Eq. (S9). Besides, the rigid adsorption energy measures the energy released (or required) when the unrelaxed Di dye is adsorbed on the TiO2 (1 1 0) surface in the presence of 100 molecules of Toluene before the geometry optimization phase. The deformation energy represents the energy released when the adsorbate Di is relaxed by the geometry optimisation step on the TiO2 (1 1 0) surface. Similarly, the rigid adsorption energy and the deformation energy for the components of the adsorbate are combined to generate the adsorption energy. While, the ratio (dEads/dNi) also shows the desorption energy of the Di molecule, where one of the adsorbed molecules has been removed.
The fact that all of the adsorption energy values in Table 5 are negative indicates the possibility of spontaneous adsorption. Thus, the trend of the calculated adsorption energy values is: TiO2-MKZ-39 > TiO2-D3 > TiO2-D4 > TiO2-D5 > TiO2-D2 > TiO2-D1. Hence, the TiO2 (1 1 0)-D1 complex (-1376.921 Kcal/mol) is the most stable system and witnessed the strongest adsorption.
4 Conclusions
To simulate the ground state geometry and electronic structure of indole-based photosensitizers, DFT and TD-DFT methods were employed. Our results on the dyes Di were found to be comparable to previously reported experimental data, indicating the sturdiness of B3LYP and CAM-B3LYP functionals with a double-ζ basis set in computing electronic structure characteristics and determining the optical properties of dyes, respectively. Besides, the UV–vis spectra showed that the designed molecules Di have a λMax range of (465–561 nm) in the GP and (429–537 nm) in Toluene. Furthermore, the results also indicate that all the dyes exhibit higher oscillator strength, LHE, and a negative charge injection-free energy, which thermodynamically proves that CT to the CB of TiO2 is favoured, with a maximum value of −1.65 eV and −1.67 eV in both GP and TS, respectively. The adsorption process of the dyes on the TiO2 (1 1 0) surface was simulated using the Monte Carlo method, which revealed a negative value of the adsorption energy of the TiO2 (1 1 0)-Dye-100 C7H8 complex, confirming the spontaneous interaction between the Di dyes and the TiO2 surface. Upon careful consideration of all relevant parameters, it can be concluded that all the studied dyes, exhibit qualities that make them suitable candidates for DSSC.
CRediT authorship contribution statement
Mohammed Elkabous: Software, Formal analysis, Investigation, Writing – original draft. Yasser Karzazi: Conceptualization, Resources, Writing – review & editing, Supervision.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- M. Allalen Molecular Dynamics and Monte-Carlo Simulations of CoPt alloys. 2006.
- Bio-photovoltaic conversion device using chlorine-e6 derived from chlorophyll from Spirulina adsorbed on a nanocrystalline TiO2 film electrode. Biosens Bioelectron. 2004;19:843-847.
- [CrossRef] [Google Scholar]
- Ceratonia siliqua L seeds extract: Experimental analysis and simulation study. Mater Today Proc. 2023;72:3705-3711.
- [CrossRef] [Google Scholar]
- Influence of the nature of the anchoring group on electron injection processes at dye–titania interfaces. PCCP. 2017;19:29389-29401.
- [CrossRef] [Google Scholar]
- Influence of the nature of the anchoring group on the interfacial energy level alignment in dye-sensitized solar cells: A theoretical perspective. Phys Rev Mater. 2020;4:115401.
- [CrossRef] [Google Scholar]
- New D-Ï€-A type indole based chromogens for DSSC: Design, synthesis and performance studies. Dyes Pigm.. 2015;112:183-191.
- [CrossRef] [Google Scholar]
- Quantification of the effects of recombination and injection in the performance of dye-sensitized solar cells based on N-substituted carbazole dyes. J. Phys. Chem. C. 2010;114:19840-19848.
- [CrossRef] [Google Scholar]
- Cactus architecture of ZnO nanoparticles network through simple wet chemistry: Efficient dye sensitized solar cells. Mater Lett. 2014;116:91-93.
- [CrossRef] [Google Scholar]
- A new mixing of Hartree-Fock and local density-functional theories. J Chem Phys. 1993;98:1372-1377.
- [CrossRef] [Google Scholar]
- Materials Studio package, BIOVIA, Dassault Systèmes, San Diego, USA, 2017, n.d.
- Antioxidative activity of Pistacia lentiscus leaf extract main components: Experimental and theoretical study. Mater Today Proc. 2023;72:3275-3279.
- [CrossRef] [Google Scholar]
- The optoelectronic properties of organic material based on triphenylamine relevant to organic solar photovoltaic cells. New J. Chem.. 2017;41
- [CrossRef] [Google Scholar]
- Theoretical study of organic sensitizers based on 2, 6-diphenyl-4H-pyranylidene/1, 3, 4-oxadiazole for dye-sensitized solar cells. J Mol Model. 2020;26:1-12.
- [CrossRef] [Google Scholar]
- Theoretical investigation of the influence of π-spacer on photovoltaic performances in carbazole-based dyes for dye-sensitized solar cells applications. J Photochem Photobiol A Chem. 2022;428:113870.
- [CrossRef] [Google Scholar]
- Carbazole based D-πi-π-A dyes for DSSC applications: DFT/TDDFT study of the influence of πi-spacers on the photovoltaic performance. Chem Phys. 2023;565
- [CrossRef] [Google Scholar]
- Molecular modeling of energetic materials: The parameterization and validation of nitrate esters in the COMPASS force field. J. Phys. Chem. B. 2000;104:2477-2489.
- [CrossRef] [Google Scholar]
- Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. PCCP. 2008;10:6615-6620.
- [CrossRef] [Google Scholar]
- DPP containing D–π–A organic dyes toward highly efficient dye-sensitized solar cells. Dyes Pigm.. 2016;125:27-35.
- [CrossRef] [Google Scholar]
- R. Dennington T.A. Keith J.M. Millam 2019. GaussView Version 6.
- Thiazolo[5,4-d]thiazole-based organic sensitizers with improved spectral properties for application in greenhouse-integrated dye-sensitized solar cells. Sustainable Energy Fuels. 2020;4:2309-2321.
- [CrossRef] [Google Scholar]
- The adsorption of bromochlorodifluoromethane on pristine, Al, Ga, P, and As-doped boron nitride nanotubes: A study involving PBC-DFT, NBO analysis, and QTAIM. Comput Theor Chem. 2021;1193:113047.
- [CrossRef] [Google Scholar]
- GAUSSIAN09. Wallingford, CT, USA: Gaussian Inc.; 2009.
- Series of D-π-A system based on isoindigo dyes for DSSC: Synthesis, electrochemical and photovoltaic properties. Synth Met. 2014;187:17-23.
- [Google Scholar]
- Recent advances in sensitized mesoscopic solar cells. Acc Chem Res. 2009;42:1788-1798.
- [CrossRef] [Google Scholar]
- Dye-sensitized solar cells. J Photochem Photobiol C: Photochem Rev. 2003;4:145-153.
- [CrossRef] [Google Scholar]
- Theoretical design and characterization of D-A1-A based organic dyes for efficient DSSC by altering promising acceptor (A1) moiety. J Photochem Photobiol A Chem. 2021;407:113048.
- [CrossRef] [Google Scholar]
- Tuning the HOMO and LUMO energy levels of organic chromophores for dye sensitized solar cells. J Org Chem. 2007;72:9550-9556.
- [CrossRef] [Google Scholar]
- Design of new coumarin dyes having thiophene moieties for highly efficient organic-dye-sensitized solar cells. New J. Chem.. 2003;27:783-785.
- [CrossRef] [Google Scholar]
- Molecular design of coumarin dyes for efficient dye-sensitized solar cells. J Phys Chem B. 2003;107:597-606.
- [CrossRef] [Google Scholar]
- Dye-sensitized nanocrystalline TiO2 solar cells based on novel coumarin dyes. Sol. Energy Mater. Sol. Cells. 2003;77:89-103.
- [CrossRef] [Google Scholar]
- Oligothiophene-containing coumarin dyes for efficient dye-sensitized solar cells. J Phys Chem B. 2005;109:15476-15482.
- [CrossRef] [Google Scholar]
- Novel D–π–A type triphenylamine based chromogens for DSSC: Design, synthesis and performance studies. New J. Chem.. 2016;40:8371-8381.
- [CrossRef] [Google Scholar]
- High-efficiency organic-dye- sensitized solar cells controlled by nanocrystalline-TiO2 electrode thickness. Adv. Mater.. 2006;18:1202-1205.
- [CrossRef] [Google Scholar]
- High-conversion-efficiency organic dye-sensitized solar cells with a novel indoline dye. Chem. Commun. 2008:5194-5196.
- [CrossRef] [Google Scholar]
- Theoretical design and characterization of pyridalthiadiazole-based chromophores with fast charge transfer at donor/acceptor interface toward small molecule organic photovoltaics. RSC Adv. 2015;5:29401-29411.
- [CrossRef] [Google Scholar]
- Theoretical characterization of the PC60BM:PDDTT model for an organic solar cell. J. Phys. Chem. C. 2011;115:21865-21873.
- [CrossRef] [Google Scholar]
- Multiwfn: A multifunctional wavefunction analyzer. J Comput Chem. 2012;33:580-592.
- [CrossRef] [Google Scholar]
- Simple metal-free organic D-π-A dyes with alkoxy- or fluorine substitutions: Application in dye sensitized solar cells. J Nanosci Nanotechnol. 2012;12:4489-4494.
- [CrossRef] [Google Scholar]
- 4-Methoxy-1,3-thiazole based donor-acceptor dyes: Characterization, X-ray structure, DFT calculations and test as sensitizers for DSSC. Dyes Pigm.. 2012;94:512-524.
- [CrossRef] [Google Scholar]
- Metal-free organic dyes for dye-sensitized solar cells: From structure: Property relationships to design rules. Angew. Chem. Int. Ed.. 2009;48:2474-2499.
- [Google Scholar]
- Conversion of light to electricity by cis-X2bis(2,2’-bipyridyl-4,4’-dicarboxylate)ruthenium(II) charge-transfer sensitizers (X = Cl-, Br-, I-, CN-, and SCN-) on nanocrystalline titanium dioxide electrodes. J Am Chem Soc. 1993;115:6382-6390.
- [CrossRef] [Google Scholar]
- Starburst triarylamine based dyes for efficient dye-sensitized solar cells. J Org Chem. 2008;73:3791-3797.
- [CrossRef] [Google Scholar]
- Investigations on optoelectronic properties of new low gap compounds based on pyrrole as solar cells materials. Journal of Applied Chemical Research. 2013;7:19-32.
- [Google Scholar]
- A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature. 1991;353:737-740.
- [CrossRef] [Google Scholar]
- A computational investigation of organic dyes for dye-sensitized solar cells: Benchmark, strategies, and open issues. J. Phys. Chem. C. 2010;114:7205-7212.
- [CrossRef] [Google Scholar]
- Role of solvent on charge transfer in 7-aminocoumarin dyes: New hints from TD-CAM-B3LYP and state specific PCM calculations. J Chem Theory Comput. 2013;9:4087-4096.
- [CrossRef] [Google Scholar]
- Indolo[3,2-b]indole donor-based D–π–A dyes for DSCs: Investigating the role of π-spacers towards recombination. New J. Chem.. 2019;43:862-873.
- [CrossRef] [Google Scholar]
- Status and outlook of sensitizers/dyes used in dye sensitized solar cells (DSSC): A review: Sensitizers for DSSC. Int J Energy Res. 2016;40
- [CrossRef] [Google Scholar]
- A review on global solar energy policy. Renew. Sustain. Energy Rev.. 2011;15:2149-2163.
- [CrossRef] [Google Scholar]
- COMPASS: An ab initio force-field optimized for condensed-phase applicationsoverview with details on alkane and benzene compounds. J. Phys. Chem. B. 1998;102:7338-7364.
- [CrossRef] [Google Scholar]
- Triphenylamine-based starburst dyes with carbazole and phenothiazine antennas for dye-sensitized solar cells. J Power Sources. 2012;199:426-431.
- [CrossRef] [Google Scholar]
- A high-light-harvesting-efficiency coumarin dye for stable dye-sensitized solar cells. Adv. Mater.. 2007;19:1138-1141.
- [CrossRef] [Google Scholar]
- Highly efficient dye-sensitized solar cells with composited food dyes. Ind Eng Chem Res. 2020;59:10457-10463.
- [CrossRef] [Google Scholar]
- Steric switching for thermally activated delayed fluorescence by controlling the dihedral angles between donor and acceptor in organoboron emitters. ACS Appl Mater Interfaces. 2019;11:10768-10776.
- [CrossRef] [Google Scholar]
- New triphenylamine-based dyes for dye-sensitized solar cells. J. Phys. Chem. C. 2008;112:874-880.
- [CrossRef] [Google Scholar]
- A new hybrid exchange–Correlation functional using the Coulomb-attenuating method (CAM-B3LYP) Chem Phys Lett. 2004;393:51-57.
- [CrossRef] [Google Scholar]
- Z. Yang C. Liu C. Shao C. Lin Y.L.-T.J. Physical of undefined, 2015. First-principles screening and design of novel triphenylamine-based D− π–A organic dyes for highly efficient dye-sensitized solar cells ACS Publications 119 2015 21852 21859 10.1021/acs.jpcc.5b05745.
- New D-D-π-A-type indol-triarylamine sensitizers for efficient dye-sensitized solar cells. Synth Met. 2016;215:21-27.
- [CrossRef] [Google Scholar]
- Organic dyes containing thieno[3,2-b ]indole donor for efficient dye-sensitized solar cells. J. Phys. Chem. C. 2010;114:18283-18290.
- [CrossRef] [Google Scholar]
- Enhanced photoelectric and photocatalysis performances of quinacridone derivatives by forming D-π-A-A structure. Sol. Energy. 2020;201:872-883.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.105457.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




