

Translate this page into:
Theoretical study of the catalytic effect of TM-C4H4 and TM-C5H5 (TM = Cr, Ti, V, Sc) on the activation of O2 at the cathode and CH3OH at the anode in “CH3OH-O2″ fuel cell via DFT computational method
⁎Corresponding author. m_aghaie@iau-tnb.ac.ir (Mehran Aghaie) marmin20042000@yahoo.com (Mehran Aghaie)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
The study of the interaction of oxygen molecule with organometallic compounds containing transition metals has always been an interesting subject for research, as the O2 activation by them is a measure of the efficiency of organometallic catalysts. This study examined the catalytic effect of some organometallic compounds containing transition metals with the general formula of TM-CmHm on increasing the reaction rate of “methanol-oxygen” fuel cell using the method of density functional theory (DFT) and PW91 method as well as the basis set of 6-31G(d) (TM represents each of Sc, Ti, V and Cr and m is equal to 4 or 5). The structures of the mentioned compounds were optimized by the natural bonding orbital calculations (NBO). Then, the diagrams of the density state (DOS) were plotted for them and afterward, gap energy (Eg), chemical hardness (η) ، chemical potential (µ) and electrophilicity (ω) were calculated in each case. The Eg and η of ScC5H5 were smaller than that of the others, indicating greater reactivity. Theoretical calculations showed that each of the TMs in TM-CmHm had a partial negative electric charge, which was higher in ScC5H5 than in others causing higher chemical activity. The adsorption energies of O2 and CH3OH were calculated and reported on each of the TM-CmHm. The adsorption energy of O2 and CH3OH on ScC5H5 was more negative than that of the other TM-CmHm used. This confirms that the compound had a greater efficiency and ability to play a catalytic role in the “oxygen - methanol” fuel cell. The results showed that the bond length of O=O increased by about 24% on average due to its adsorption on ScC5H5. The O-H bond length was significantly increased in response to the methanol adsorption process on ScC5H5. Increasing the bond length caused instability of the bond and raising its chemical activity and reactivity. The adsorption kinetics of each of O2 and CH3OH on ScC5H5 were also studied by the above-computational method. The potential energy variation of the “adsorbate/adsorbent” system in terms of reaction coordinate (bond length of O=O or O-H) in each case was evaluated with a one transition state observed in each case.
Keywords
Fuel cell
Catalyst
DFT
Adsorption

1 Introduction
The issue of energy is becoming increasingly important in today’s societies (Bakr et al., 2017). With the advancement of technology, the demand for energy is increasing rapidly (Holdren, 1991). On the other hand, fossil fuel resources, which are currently the main source of energy supply, are declining besides, their use is associated with serious environmental pollution. Thus, achieving clean and renewable energy is a basic and essential need of human societies (Kivrak et al., 2011; Ulas et al., 2018; Raza et al., 2016). There is significant potential for access to clean and renewable energy, for example, use of wind farms to generate electricity, application of solar cells to provide heat and electricity, usage of the tidal power of the seas to generate energy, the use of nuclear energy with all the necessary precautions, utilization of fuel cells and power plants consisting of them in the electricity generation and other fields etc. (Arshad et al., 2019; Vasseghian et al., 2020).
Fuel cells have several important advantages; first, their use in various fields is associated with the minimum damage and environmental pollution. Secondly, the conversion efficiency of chemical energy to electrical energy is high in them and may reach as large as 90%, while the mentioned efficiency in combustion power plants is often less than 30%. Further, continuous use of reactant and product recovery is possible in them. Also, fuel cells in multiple and interconnected form, called fuel pile, have a wide range of applications in various fields, such as their use in spacecraft, reconnaissance drones, reconnaissance submarines, hybrid automobiles, fuel cell power plants, etc. (Chen et al., 2019; Xu et al., 2007; Mohsin et al., 2019; Kulikovsky, 2014). Despite the above advantages, the main problem in the design and application of fuel cells is that the reaction rate of the cell under normal conditions is small and almost without functional usefulness. Thus, since the introduction of fuel cells, many studies have been done to find solutions to enhance the reaction rate in them. The use of various catalysts to increase the rate of anodic and cathodic half reactions of this species of cells is the result of such research (Hashmi and Hutchings, 2006; Omidvar, 2018). In this regard, many composite and nanocomposite electrodes with significant catalytic properties have been used to be applied in the anode and cathode of fuel cells. Indeed, the reaction of the cell on the surface of such composite and nanocomposite electrodes takes place much faster (Gong et al., 2012; Konkol and Okuda, 2008).
Platinum is widely used as a catalyst in the oxygen reduction reaction at the cathode, but it is deactivated in the presence of carbon monoxide. Platinum, on the other hand, is very expensive and has many limitations for commercialization. The search for cheap, platinum-free catalysts has recently become a hot topic (Chen et al., 2017; Nie et al., 2015). Considering the growing importance of fuel cells and their wide applications in various fields, many researchers in the field of theory and computation have focused on the theoretical study of fuel cells and their mechanism (Carrette et al., 2001; Griffith et al., 2000). Indeed, many theoretical studies and calculations based on classical mechanics and quantum mechanics have been done, especially using the DFT computational method (Winter and Brodd, 2004). Previously, the adsorption of O2 molecules on BN nanoclusters has been studied theoretically (Omidvar, 2017).
DFT calculation is the novel addition to the field of quantum chemistry calculations. It is probably an approach understatement to state that DFT has strongly influenced the evolution of quantum chemistry during the last 15 years. DFT approach is based on the Hohenberg–Kohn paradigm, which states that the electron density and electronic Hamiltonian have a functional relationship, which allows the computation of all ground-state molecular properties without a wave function. This means that it is possible to obtain the properties of a molecule after determination of only three coordinates, regardless of molecular size. However, we do not know exactly what is the nature of this functional relationship. The only approach is to build trial exchange–correlation functionals and assess their relevance and accuracy. In its current Kohn–Sham formulation DFT is a method still very much under development, and although it is a long way away from its promise, the modern-day Kohn–Sham DFT has still massive computational advantages over ab initio methods and can be applied just as easily via implementations in modern-day commercial software packages. Current implementations are for example the functionals B3LYP, and BP86 which have been shown to have significant advantages over ab initio approaches since their performance is roughly equivalent to the electron correlation MP2 method at the cost of only a HF/SCF level calculation. Another way of utilizing this advantage is to use lower-level density functional equivalent in performance to HF/SCF approaches and trade off advantages in speed against increase in quality of for example size of basis sets to allow a more accurate description of molecular systems. With current compute power, in our experience full geometry optimizations of systems with around 30 heavy atoms are easily accessible with DFT methods like the functionals mentioned above (B3LYP), providing accurate and valuable information on molecular geometries and properties. In addition to the above-mentioned advantages in speed and performance of DFT over traditional ab initio methods, it is also differentiated by its ability to accurately describe the electronic properties of transition metals and their complexes.
Some useful kinetic researches have been done on the catalytic role of the composite of graphen and some transition metals that can be considered for promoting this work (Gao et al., 2018; Yang et al., 2020; Yang et al., 2020). In addition, a valuable computational study has been performed on the catalytic role of some special organometallic compounds containing some transition metals. The catalytic effect of these compounds in increasing the rate of cathodic half-reactions of fuel cells has also been considered computationally (Bazvand et al., 2019).
Organometallic compounds are very broad and familiar and have many applications, in each of which there is at least one carbon–metal bond. When alkaline metals or some alkaline earth metals are involved in the structure of organometallic compounds, the carbon–metal bond is ionic. In other cases, the bond may be covalent or coordinance (Negishi and Takahashi, 1988). In organometallics with transition metals, (n-1)d, np and ns valence bond orbitals are regularly used to form chemical bonds. The half-fullness of the mentioned orbitals in the transition metals allows these metals to act as both electron donors and electron acceptors, and in processes with the participation of ligands as σ-donor/ π-acceptor, where the 18-electron valence rule is observed. This rule states that when the organometallic compounds with the participation of transition metals are thermodynamically stable that, the sum of the d-valence electrons of the metal and the electrons supplied by the ligands reaches 18. Such an electron configuration is established in metallocene, including Ferrocene with the formula (C5H5)2Fe. Thus, this compound and similar compounds have significant thermodynamic stability at the same time, they have significant thermal stability (the melting and boiling points of ferrocene are 174 °C and 249 °C, respectively). (C5H5)2Ti, (C5H5)2V, (C5H5)2Co, etc. are also among metallocenes.
Organometallic compounds have been widely used in both research and industrial fields (Dutton and Ragogna, 2011; Jensen, 1982; Jezequel et al., 2001). A special group of them, which contain transition metals, have effectively exhibited catalytic properties. In addition, they have also been used in areas such as the preparation of polymers, reducing the viscosity of some industrial oils, preparation and transfer of drugs, petrochemical industries and refining of gasoline and gasoline, etc. (Collins et al., 2019).
Organometallic compounds in general are somewhat unstable against thermodynamic oxidation, but some of them with the general formula of C4H4M, (C4H4)2M, C5H5M, (C5H5)2M, etc. have good thermal stability. Note that despite their relative instability to thermodynamic oxidation, these compounds have high kinetic stability under normal conditions, whose oxidation rate is very low and can be neglected in these conditions. For this reason, they can be safely used as effective and inexpensive catalysts in catalytic reactions, especially in fuel cell reactions. These compounds are also stable in the presence of oxygen under normal conditions and can be used as a catalyst to destabilize oxygen and increase its reactivity in the cathode of fuel cells (Narváez-Pita et al., 2017; Singh et al., 2019).
In general, some organometallic compounds with the participation of transition metals can well play a significant catalytic role in increasing the reaction rate of “methanol-oxygen” fuel cell and other similar fuel cells. There are several methods to synthesize organometallic compounds (Jones and Gilman, 1954; Parkins et al., 1984; Spessard and Miessler, 2009) as metal metal interconversions, hydrogen metal interconversion, addition of organoalkali compounds to carbon- carbon double bonds, direct metalation of hydrocarbons, reaction of free radicals with metals, electrolysis of ketones to form RM compounds. One of the most used methods to synthesize organomettalic compounds is to react the pure metal with specific organic molecules for example metal is react with any alkyl or aryl- halides to form the reagent. In double-decomposition reactions, metal halides exchange with alkylating reagents or in hydrometalation a molecule with a metal- hydrogen bond reacts with a double bond in an organic compound.
2 Computational details
NBO calculations were performed based on DFT method for the studied species and then the negative charge accumulated on the TM atom in each optimized TM-CmHm was estimated. DOS diagrams were then plotted by Gauss sum software for the optimized structures of the studied organometallic compounds. Using the above diagrams, the highest occupied molecular orbital energy (EHOMO) and the lowest unoccupied molecular orbital energy (ELUMO) were calculated for each of them. Using these energies, Eg was estimated for each organometallic compound according to Eq. (1). In addition, the chemical hardness (η), chemical potential (µ), and electrophilicity (ω) were calculated for each of them according to Eqs. (2)–(4). Also, the adsorption energy (Eads) of each of O2 and CH3OH on each of the considered organometallic compound under the optimal conditions of the structures, was calculated and analyzed according to Eq. (5).
R/TM-CmHm indicates the organometallics to which a reactant (O2 or CH3OH) is adsorbed.
The adsorption kinetics of each of O2 and CH3OH on ScC5H5 were also evaluated by the previously introduced computational method. The diagram of the potential energy change of the “adsorbent/adsorbate” system was plotted in terms of the reaction coordinate (bond length O = O or O-H) in each case. Hence, the activation energies and the potential energy changes in each case were estimated.
3 Results and discussion
The structure of each of the following compounds including ScC5H5, ScC4H4, TiC5H5, TiC4H4, VC5H5, VC4H4, CrC5H5, and CrC4H4 was optimized via Gaussian 09 software (Fig. 1). DFT calculations predicted bond lengths of TM-C in TM-CmHm compounds ranging from 1.990 in (CrC5H5) to 2.465 Å in (ScC5H5). Then, the change in the bond length in the presence of O2 and CH3OH molecules was also investigated.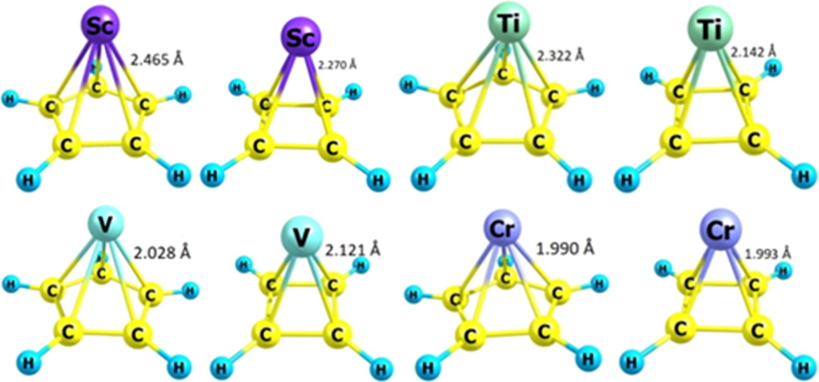
Optimized structure of each of the organometallic compounds TMCmHm (TM = Sc, Ti, V, Cr and m = 4 or 5). The TM-C bond length has been also mentioned in each optimized structure.
DOS diagram was plotted for each of the optimized structures such as TM-CmHm to allow EHOMO and ELUMO of each structure to be estimated. For example, two of them, related to ScC4H4 and ScC5H5 respectively, have been shown in Fig. 2.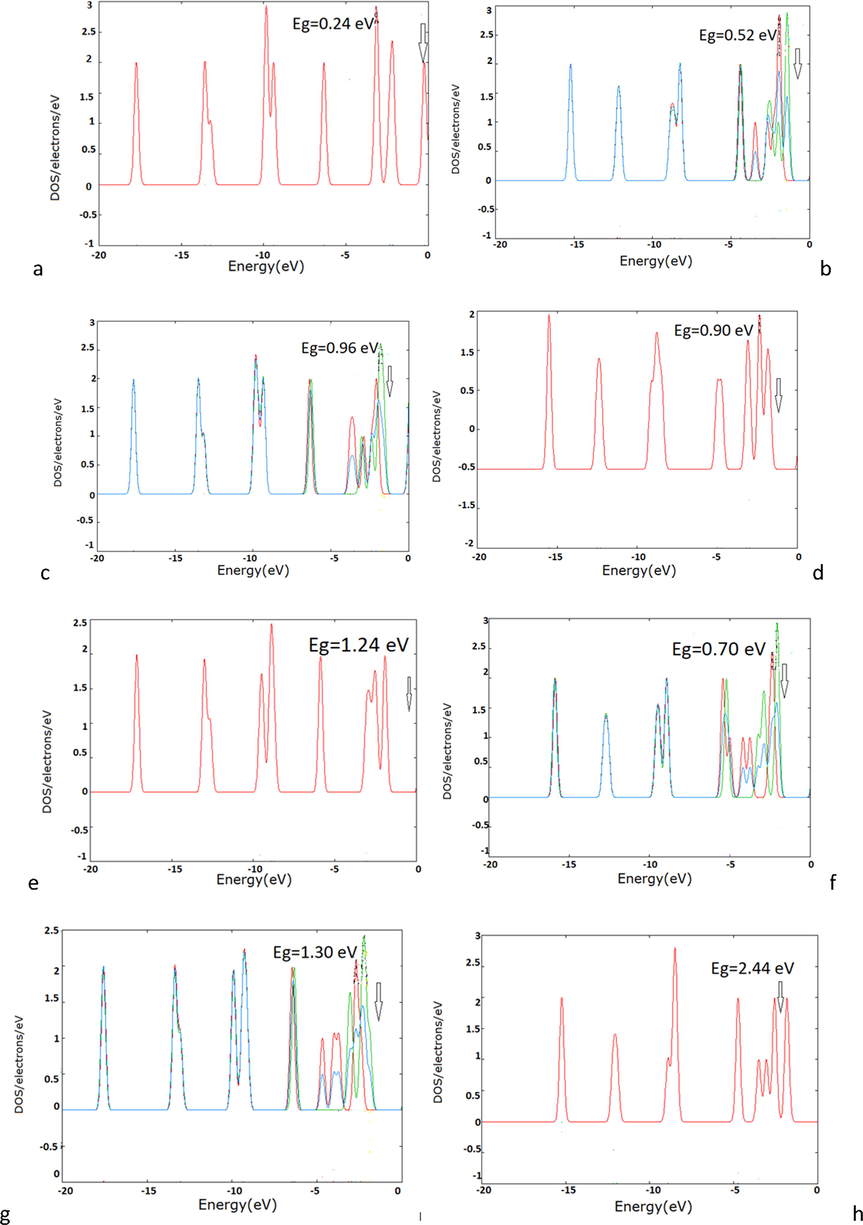
Density of state (DOS) for a) ScC5H5 b) ScC4H4 c) TiC5H5 d) TiC4H4 e) VC5H5 f)VC4H4 g) CrC5H5 h) CrC4H4.
The calculated values of Eg, η, µ and ω of each of the optimized TMCmHm have been collected in Table 1. The smaller Eg leads to higher chemical activity and reactivity. Thus, looking at the Eg values collected in Table 1, it is concluded that ScC5H5 with the lowest Eg = 0.24 eV has the highest chemical affinity and reactivity compared to other studied organometallics. Thus, this compound is a good candidate for playing the catalytic role in the reaction of “CH3OH-O2″ fuel cell and the similar cells.
Structure
Charge
Eg
η
µ
ω
ScC5H5
−0.879
0.24
0.12
−2.81
32.90
ScC4H4
−0.835
0.52
0.26
−3.08
18.24
TiC5H5
−0.766
0.96
0.48
−2.96
9.13
TiC4H4
−0.772
0.90
0.45
−3.09
10.61
VC5H5
−0.453
1.24
o.62
−3.57
10.28
VC4H4
−0.527
0.70
0.35
- 3.50
17.50
CrC5H5
-0.485
1.30
0.65
-2.85
6.25
CrC4H4
−0.483
2.44
1.22
−2.82
3.26
Chemical hardness (η) is a measure of a molecule's resistance against breaking and participating in chemical reactions. The role and effect of η are similar to Eg. The smaller Eg causes smaller η. For example, both Eg and η of ScC5H5 are less than the other studied organometallic compounds.
Chemical potential (µ) is a measure of chemical stability. In some cases the more negative the µ value of a molecule, the more stable it is. µ, associated with ScC5H5, is algebraically more positive than others. Thus, its chemical stability is lower than that of other studied organometallics, which is consistent with previous conclusions. Interestingly, the variation trends of each Eg, η and µ have good consistency with the predictions derived from them. For example, all three point out that the chemical activity and reactivity of ScC5H5 is higher than that of the other organometallics used in this study and, thus has a more pronounced catalytic property. As such, this compound with participation in the anode and cathode structure of “methanol-oxygen” fuel cell can significantly increase its anodic and cathodic half-reactions and replace expensive catalysts such as platinum, rhodium, iridium as well as their composite as an abundant and inexpensive catalyst.
Using NBO calculations and analysis, it was found that the TM atom in TM-CmHm has a small amount of negative electric charge. The amount of charge in TMC4H4 varies from 0.483 basic units of charge or electron charge (au) to 0.835au and in TMC5H5 varies from 0.453 to 0.879au (Table 1). It can be observed that Sc in ScC5H5 has the highest negative electric charge and therefore ScC5H5 has the highest chemical activity among the organometallics in this study (Table 1). In Table 1, in addition to the electrical charge accumulated on the TMs, the gap energy (Eg), chemical hardness (η), and chemical potential (µ) of each of the organometallic compounds used in this study have been also listed.
Thus, when O2 and CH3OH are separately in contact with TM-CmHm, some negative electric charge from TM-CmHm flows to the π* orbitals of O=O and O-H in CH3OH, augments the length of these bonds, and increases their chemical activity, which in turn causes to elevated the rate of cathodic and anodic half-reactions in the “methanol-oxygen” fuel cell, respectively. Thus, ScC5H5 can be considered as an inexpensive and effective catalyst to increase the rate of “methanol-oxygen” fuel cell reaction. Definitely, other studied organometallics also have some of these catalytic properties, but to a lesser extent.
A very important point that should be considered about the data in Table 1 is that the variation trends of charge, Eg, η and µ in the studied organometallics are satisfactorily consistent, which is a good sign of the scientific validity of the calculations. To further investigate this issue, a plot of each of the electric charges collected on the TMs, η and µ in terms of Eg was drawn (Fig. 3). According to the diagrams, the trend of variations in each of them in terms of Eg is descending or ascending, which is fairly a good sign that the results of the calculations are compatible with each other.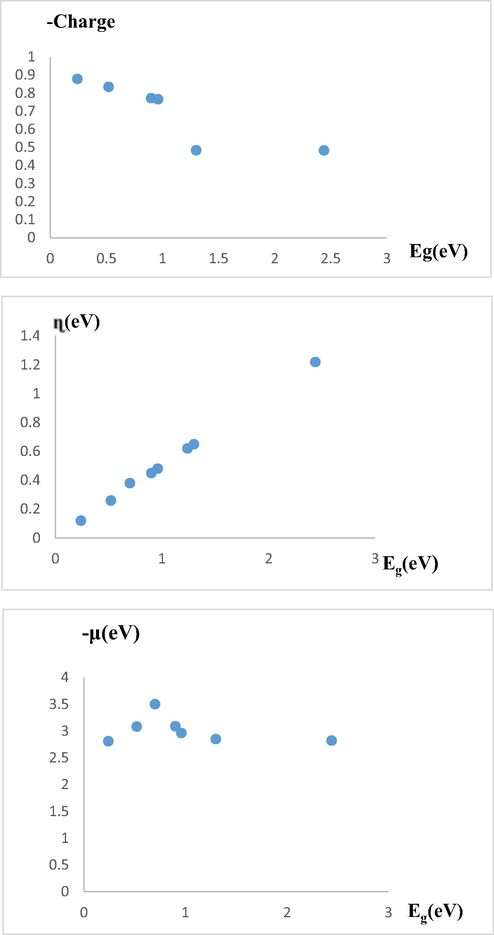
Diagram of change (a) charge in terms of Eg, (b) η in terms of Eg, and µ in terms of Eg.
Considering the relation between η and Eg (η = 1/2 (ELUMO – EHOMO) = 1/2Eg,), one can easily found that there is a linear relationship between them.
To evaluate the adsorption advancement of O2 and CH3OH onto the studied organometallics, each of the optimized O2 and CH3OH was separately placed in parallel at a suitable distance from each of the TM-CmHm according to Fig. 4. Then, their optimal structure and the structure-dependent energy of each optimized adsorbent/adsorbate pairs were evaluated. The optimal pair structures in relation to O2/TM-CmHm have been shown in Fig. 4 and in relation to CH3OH/TM-CmHm in Fig. 5. The adsorption energy (Eads) in each case was calculated according to Eq. (5) as reported in Table 3. According to research conducted (Narváez-Pita et al., 2017), the more negative the adsorption energy of the pair, such as O2/TM-CmHm, the higher its catalytic efficiency. Thus, according to the data presented in Tables 2 and 3, the catalytic efficiency of ScC5H5 in destabilizing and activating O2 and CH3OH in the “ methanol- oxygen” fuel cell is higher. At the same time, other organometallic compounds studied in this study have also significant catalytic properties but less than ScC5H5 (see Tables 2 and 3).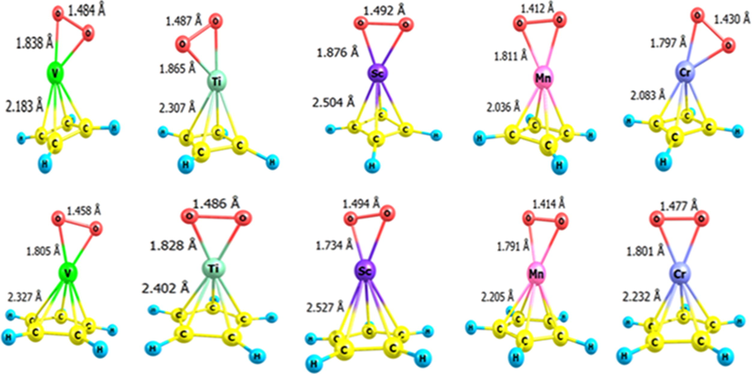
Optimized structure of each of the O2/TM-CmHm pairs (TM = Sc, Ti, V, Cr and m = 4.5). The required bond lengths on each structure have been presented.
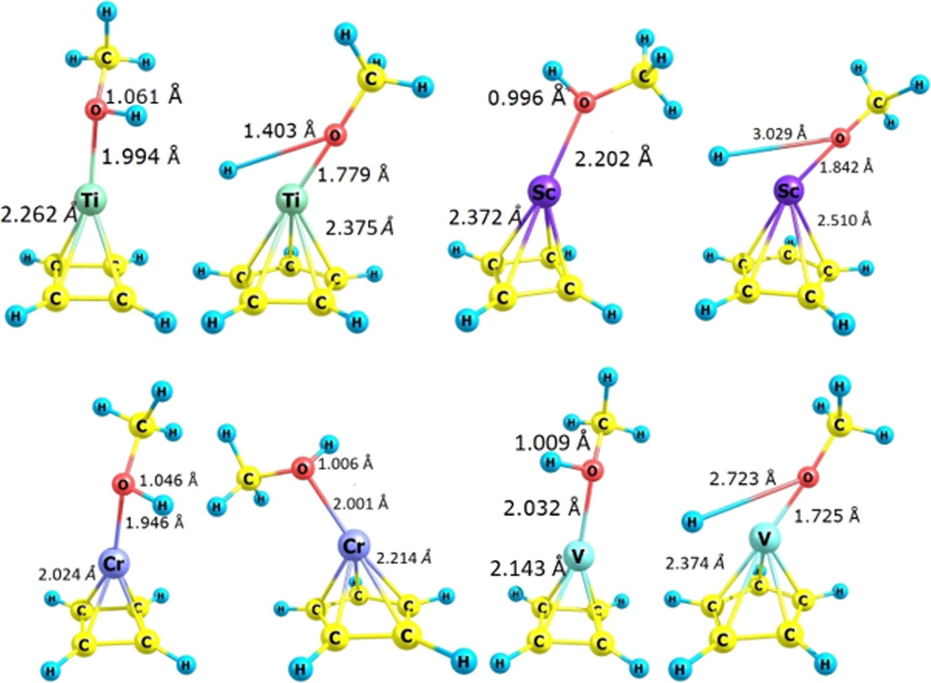
Optimized structures of each pair of TMCmHm/R (TM = Sc, Ti, V, Cr and m = 4 or 5 and R = CH3OH. Other required bond lengths have been given on each structure in the figure.
Structure
−Charge
Eads
r (O2)
O2/ScC5H5
0.899
−7.492
1.494
O2/ScC4H4
0.879
−7.394
1.492
O2/TiC5H5
0.697
−7.245
1.486
O2/TiC4H4
0.733
−7.349
1.486
O2/VC5H5
0.540
−6.112
1.477
O2/VC4H4
0.629
−6.716
1.430
O2/CrC5H5
0.584
−6.412
1.477
O2/CrC4H4
0.539
−6.050
1.430
structure
-Charge
Eads
r(OH)
CH3OH /ScC5H5
0.501
−4.169
3.029
CH3OH /ScC4H4
0.493
−1.061
0.996
CH3OH /TiC5H5
0.094
−4.108
1.403
CH3OH /TiC4H4
0.480
−1.442
1.061
CH3OH /VC5H5
0.043
−4.132
2.023
CH3OH /VC4H4
0.315
−1.523
1.000
CH3OH /CrC5H5
0.170
−1.360
1.006
CH3OH /CrC4H4
0.143
−1.714
1.046
When O2 is adsorbed on the desired organometallic compound, a small amount of negative electric charge is transferred from the organometallic compound to the O = O bond and increases the bond length, causing O2 instability. These changes, in turn, increase O2 reactivity. The bond length of O2 in the free state according to the experimental results is 1.210 Å (Singh et al., 2019); after the adsorption onto ScC5H5, ScC4H4, TiC4H4, TiC5H5, VC5H5, VC4H4, CrC5H5 and CrC5H5, it becomes 1. 494, 1 492, 1.486, 1.486, 1.458, 1.484, 1.430, and 1.477 Å respectively. It has shown the highest elongation in O2/ScC5H5 pair compared to other organometallics used and indicates that O2/ScC5H5 has a more effective catalytic role. The bond length of TM-Cs also increased after the adsorption of O2 molecules on them, with the largest increase related to Sc-C, which rose from 2.464 to 2.492 Å. This also indicates the greater catalytic role of ScC5H5.
The comparison of the magnitude of the absorption energy in O2/ TM-CmHm pairs was as follows
CrC4H4 < VC5H5 < CrC5H5 < VC4H4 < TiC5H5 < TiC4H4 < ScC4H4 < ScC5H5. These energies from ScC5H5 to CrC4H4 were −7.492, −7. 394, −7. 245, −7. 349, −6.112, −6.716, −6.412 and −6.050 eV respectively.
The comparison of the magnitude of the charge transfer process from TMs to adsorbed O2 molecule was as follows:
O2/ScC5H5(0.899au) > O2/ScC4H4(0.879au) > O2/TiC4H4(0.733au) > O2/TiC5H5(0.697au) O2/VC4H4(0.629au) > O2/CrC5H5(0.584au) > O2/VC5H5(0.540au) > O2/CrC4H4(0.539au)
Generally, the highest charge transfer and the highest adsorption advancement belonged to O2 adsorption onto ScC5H5 compound. Thus, this compound is considered as the most reactive organometallic compound; thus, its catalytic ability is higher than the others.
In the case of adsorbed CH3OH, a small amount of negative electric charge is transferred from the organometallic compound to O-H in CH3OH, increasing its length, resulting in the instability of O-H in CH3OH. These changes in turn boost the reactivity of CH3OH in response to the adsorption process. Thus, whenever the desired organometallic compound in the anode of the “ methanol -oxygen” fuel cell participates, the anodic half-reaction rate increases as a result of the catalytic role of the organometallic compound used, which is very economically significant.
The O-H bond length in methanol in the free state is 0.986 Å, but after its adsorption onto the desired organometallic compounds and the formation of CH3OH/ScC5H5, CH3OH/ScC4H4, CH3OH/TiC4H4, CH3OH/TiC5H5, CH3OH/VC5H5, CH3OH/VC4H4, CH3OH/Cr3C5H5 and CH3OH/Cr3C5H5 pairs, it reaches the values of 3.020, 0.996, 1.403, 1.061, 2.723, 1.009, 1.006, 1.046 Å respectively. The highest value is related to CH3OH/ScC5H5, which confirms that ScC5H5 has a more effective catalytic role in the fuel cell. The bond length of TM-Cs increased after adsorption of CH3OH molecule on them with the largest increase related to Sc-C, which rose from 2.464 to 2.510 Å. As such, the methanol molecule becomes more active to participate in the anodic half reaction.
Thus, in general, it can be concluded that both O2 and CH3OH become significantly unstable in response to adsorption onto the organometallic compounds used in this study. As a result of the elongation of the O = O and O-H bonds, they participate in the “ methanol- oxygen ” fuel cell half-reactions with greater chemical activity, thus increasing the reaction rate of the cell.
The comparison of the magnitude of the charge transfer trend from TM in TMCmHm to O-H (methanol) is as follows:
CH3OH/ ScC5H5 > CH3OH/ScC4H4) > CH3OH/VC4H4) > CH3OH/TiC4H4 > CH3OH/CrC5H5 > CH3OH/VC5H5
Hence, the charge transfer in the CH3OH/ScC5H5 pair is higher than in the others; thus, elongation of the O-H bond (in methanol) is also higher.
As previously noted, further elongation of the O = O or O-H bonds during the adsorption process is associated with a further rise in their reactivity. Thus, the catalytic property of ScC5H5 is greater in accelerating the anodic and cathodic half-reactions of “methanol-oxygen” cell than other organometallics used.
The adsorption energy in turn is more negative in the CH3OH/ScC5H5 pair than in other similar pairs; the more negative the absorption energy, the higher the negative electric charge transfer from the organometallic compound to the adsorbed species, making it more unstable and increasing its reactivity. Thus, ScC5H5 has more catalytic properties destabilizing the adsorbed CH3OH.
To study the kinetics of the adsorption process, the potential energy change of the adsorption process of each of O2 and CH3OH onto the organometallic compound ScC5H5 according to the 6-31G(d) approach was separately evaluated in terms of reaction coordinate (bond length of O = O) in the O2/ScC5H5 pair and O-H bond length in methanol in the CH3OH/ScC5H5 pair. The results have been shown in Figs. 6 and 7. In each case, the desired transition state (Ts) and its optimal energy were determined as displayed in the figure. The initial optimal potential energy of the adsorption process and its final state were also estimated, whose values are presented in Figs. 6 and 7. Based on the mentioned energies, it was possible to estimate the forward and reverse activation energy and the potential energy change associated with the O2 and CH3OH adsorption onto ScC5H5, which the estimated value of each of them is presented in Figs. 6 and 7. According to the mentioned values, performing the absorption process in the forward direction in each case is more favorable than the reverse direction, as it is associated with less activation energy. In addition, in each of the mentioned absorptions, only one transition state was observed. Each of the pairs O2/ScC5H5 and CH3OH/ScC5H5 was associated with a reduction in potential energy in the transition from the initial state to the final state, which is considered as the driving factor for the spontaneous advancement of each of the mentioned pairs from the initial state to the final state.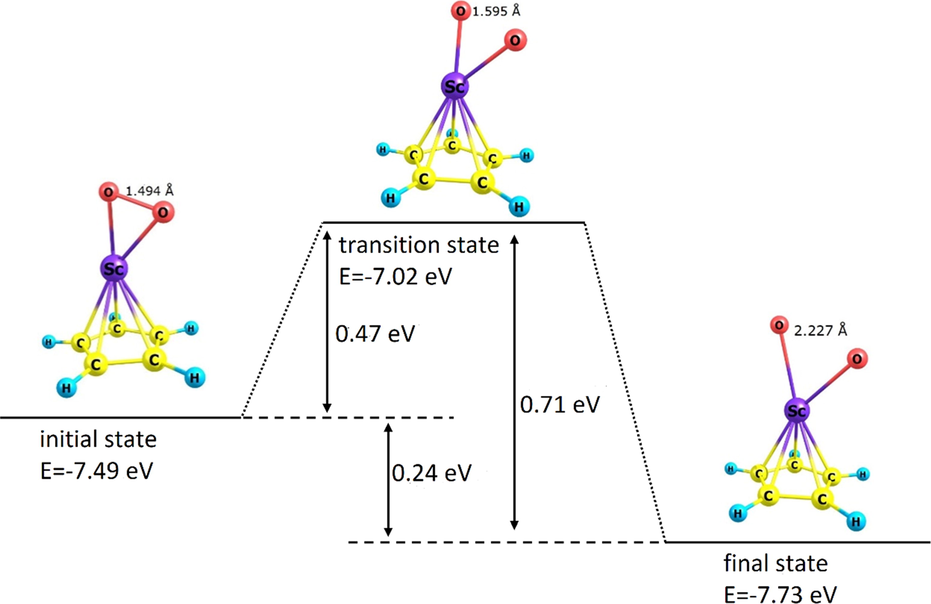
Diagram of potential energy change of the O2/ScC5H5 pair in terms of the reaction coordinate (bond length of absorbed O = O) in the activation of O2 by the organometallic compound ScC5H5. Potential energies, activation energies, and potential energy change have been presented on the figure.
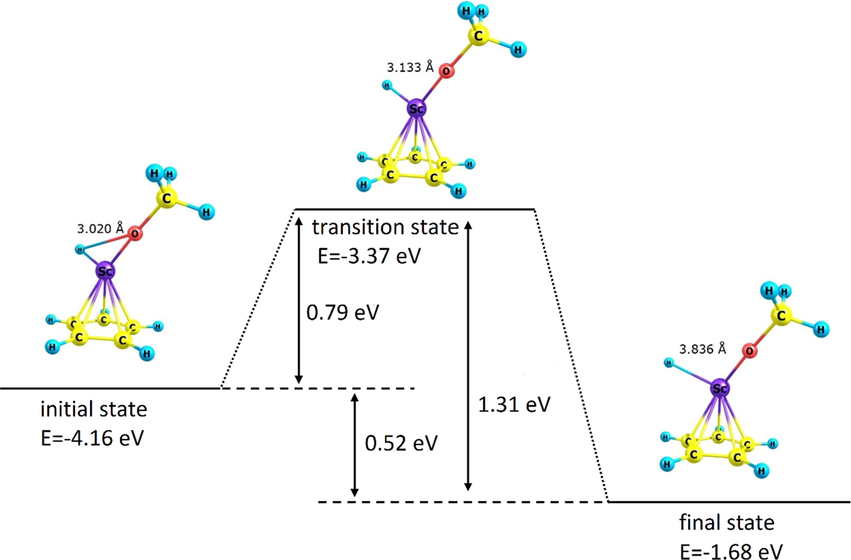
Diagram of potential energy change of CH3OH/ScC5H5 pair in terms of the reaction coordinate (bond length of O-H in CH3OH) in CH3OH activation by ScC5H5 organometallic compound. Potential energies, activation energies, and potential energy change have been presented in the figure.
4 Conclusion
Using density function theory and PW91 method, the natural bonding orbitals were evaluted for each of the organometallics of this study with plotting the optimal structure for each of them. Then, the diagram of the density of states in each case was plotted, which made it possible to estimate Eg, η, µ, ω and the electric charge collected on the TM in each TMCmHm. Eg and η were lower for ScC5H5 than for the others. This proves that the ScC5H5 is less resistant to breakage and has higher reactivity. Thus, this organometallic compound can be considered as a suitable catalyst in increasing the reaction rate of “methanol-oxygen” fuel cell. Comparison of the variation trend of each Eg, electric charge, η and µ in TMCmHm showed good agreement with each other, which can be concluded as a measure of computational validity. The results of the calculations revealed that the amount of negative charge accumulated on Sc in ScC5H5 was also higher than other organometallics used, which in turn suggests that ScC5H5 has higher tendency for sending some negative electric charge to spices in contact with it. Hence, when each of O2 and CH3OH is in contact with each of the TMCmHm, they will gain an additional amount of negative charge, causing the desired bonds to stretch and become unstable. It was observed that ScC5H5, which has a higher negative charge on Sc, emits more negative charge to O2 and O-H in methanol in contact with it making them more unstable and more reactive for participating in the considered half reaction of “CH3OH-O2″ fuel cell. The other studied organometallic compounds exhibited a lower catalytic effect; thus, the reaction rate of the ”CH3OH-O2″ fuel cell with ScC5H5 in the anode and cathode structure in the role of catalyst was significantly increased. Comparison of the adsorption energies of O2 and CH3OH on each TMCmHm showed that the adsorption energies of each of O2 and CH3OH on ScC5H5 were more negative than the other organometallics used, which also confirms that the compound ScC5H5 has a more pronounced catalytic effect in increasing the reaction rate of the “methanol-oxygen” fuel cell. Then, the kinetics of the adsorption of each of O2 and CH3OH onto the ScC5H5 compound were studied computationally. Also, the potential energy levels of the initial adsorption state, transition state, and final adsorption state were calculated. In this way, it became possible to estimate the forward and reverse activation energies of the absorption process and the potential energy change associated with it. It was found that the adsorption process in each case in the forward direction had a better chance for occurrence and was associated with a significant reduction in potential energy. This is a favorable factor for the adsorption advancement in thermodynamics point of view.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Energy and exergy analysis of fuel cells: A review. Therm. Sci. Eng. Prog.. 2019;9:308-321.
- [CrossRef] [Google Scholar]
- A mini review on health and environmental risks of oil and gas industry undesired products: Hydrogen sulfide and Carbon monoxide. Int. J. Ecosyst. Ecol. Sci.. 2017;7:883-894.
- [Google Scholar]
- Organometallic compound as an efficient catalyst toward oxygen reduction reaction. Inorg. Chem. Commun.. 2019;108:107520
- [CrossRef] [Google Scholar]
- Electrocatalytic O 2 reduction on Pt: multiple roles of oxygenated adsorbates, nature of active sites, and origin of overpotential. J. Phys. Chem. C. 2017;121:6209-6217.
- [CrossRef] [Google Scholar]
- DFT study of the two dimensional metal–organic frameworks X3(HITP)2 as the cathode electrocatalysts for fuel cell. Appl. Surf. Sci.. 2019;471:256-262.
- [CrossRef] [Google Scholar]
- Collins, R., Durrant, J.P., He, M., Layfield, R.A., 2019. Electronic structure and magnetic properties of rare-earth organometallic sandwich compounds, 1st ed, Handbook on the Physics and Chemistry of Rare Earths. Elsevier B.V. https://doi.org/10.1016/bs.hpcre.2019.05.002
- Dutton, J.L., Ragogna, P.J., 2011. Recent developments in the synthesis and isolation of p-block centered polycations. Coord. Chem. Rev. https://doi.org/10.1016/j.ccr.2011.01.028
- Support effects on adsorption and catalytic activation of O 2 in single atom iron catalysts with graphene-based substrates. Phys. Chem. Chem. Phys.. 2018;20:7333-7341.
- [CrossRef] [Google Scholar]
- Copper (II) removal by pectin-iron oxide magnetic nanocomposite adsorbent. Chem. Eng. J.. 2012;185–186:100-107.
- [CrossRef] [Google Scholar]
- Griffith, R., Patent, 2000. Electric Power Generation System Including Fuel Cells. USOO6110614A.
- Lewis acid-base interactions and adhesion theory. In: Rubber Chemistry and Technology. Allen Press; 1982. p. :881-901.
- [CrossRef] [Google Scholar]
- Supported metallocene catalysts by surface organometallic chemistry. synthesis, characterization, and reactivity in ethylene polymerization of oxide-supported mono- and biscyclopentadienyl zirconium alkyl complexes: establishment of structure/reactivity R. J. Am. Chem. Soc.. 2001;123:3520-3540.
- [CrossRef] [Google Scholar]
- Jones, R.G., Gilman, H., 1954. Methods of preparation of organometallic compounds, Chem. Rev. 54(5), 835–890 https://doi.org/10.1021/cr60171a004
- Carbon nanotube structures as support for ethanol electro-oxidation catalysis. Int. J. Chem. React. Eng.. 2011;9
- [CrossRef] [Google Scholar]
- Konkol, M., Okuda, J., 2008. Non-metallocene hydride complexes of the rare-earth metals. Coord. Chem. Rev. https://doi.org/10.1016/j.ccr.2007.09.019
- Kulikovsky, A.A., 2014 Physically-Based Analytical Polarization Curve of a PEM Fuel Cell. J.Electrochem.Soc. 161, F263–F270. doi:10.1149/2.028403jes.
- Mohsin, M., Raza, R., Mohsin-ul-Mulk, M., Yousaf, A., Hacker, V., 2019. Electrochemical characterization of polymer electrolyte membrane fuel cells and polarization curve analysis. Int. J. Hydrogen Energy. https://doi.org/10.1016/j.ijhydene.2019.08.246
- Ferrocene-steroid conjugates: Synthesis, structure and biological activity. J. Organomet. Chem.. 2017;846:113-120.
- [CrossRef] [Google Scholar]
- Organozirconium compounds in organic synthesis. Synthesis (Stuttg). 1988;1988:1-19.
- [CrossRef] [Google Scholar]
- Recent advancements in Pt and Pt-free catalysts for oxygen reduction reaction. Chem. Soc. Rev.. 2015;44:2168-2201.
- [CrossRef] [Google Scholar]
- Catalytic role of transition metals supported on niobium oxide in O2 activation. Appl. Surf. Sci.. 2018;434:1239-1247.
- [CrossRef] [Google Scholar]
- Catalytic activation of O 2 molecule by transition metal atoms deposited on the outer surface of BN nanocluster. J. Mol. Graph. Model.. 2017;77:218-224.
- [CrossRef] [Google Scholar]
- Parkins, A.W., Poller, R.C., 1984. An introduction to organometallic chemistry, Palgrave, London, 11-41 https://doi.org/10.1007/978-1-349-18198-8-2
- Raza, R., Akram, N., Javed, M.S., Rafique, A., Ullah, K., Ali, A., Saleem, M., Ahmed, R., 2016. Fuel cell technology for sustainable development in Pakistan - An over-view. Renew. Sustain. Energy Rev. https://doi.org/10.1016/j.rser.2015.08.049
- Ferrocene-appended pharmacophores: An exciting approach for modulating the biological potential of organic scaffolds. Dalt. Trans.. 2019;48:2840-2860.
- [CrossRef] [Google Scholar]
- Spessard, G.O., Miessler, G.L., 2009. Organometallic Chemistry, 2nd Ed Oxford University Press, 978-0-19-533099-1.
- Composition dependent activity of PdAgNi alloy catalysts for formic acid electrooxidation. J. Colloid Interface Sci.. 2018;532:47-57.
- [CrossRef] [Google Scholar]
- Pollutants degradation and power generation by photocatalytic fuel cells: A comprehensive review. Arab. J. Chem.. 2020;12:8458-8480.
- [CrossRef] [Google Scholar]
- What are batteries, fuel cells, and supercapacitors? Chem. Rev.. 2004;104:4245-4270.
- [CrossRef] [Google Scholar]
- Analysis of proton exchange membrane fuel cell polarization losses at elevated temperature 120°C and reduced relative humidity. Electrochim. Acta. 2007;52:3525-3533.
- [CrossRef] [Google Scholar]
- Theoretical study on double-atom catalysts supported with graphene for electroreduction of nitrogen into ammonia. Electrochim. Acta. 2020;335:135667.
- [CrossRef] [Google Scholar]
- Screening the activity of single-atom catalysts for the catalytic oxidation of sulfur dioxide with a kinetic activity model. Chem. Commun.. 2020;56:11657-11660.
- [CrossRef] [Google Scholar]




