

Translate this page into:
N-(substituted phenyl)-2-chloroacetamides: LSER and LFER study
⁎Corresponding author. Fax: +381 21 454065. borko.matijevic@dh.uns.ac.rs (Borko M. Matijević)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The UV absorption spectra of twelve N-(substituted phenyl)-2-chloroacetamides were recorded in eighteen solvents. The effect of specific and non-specific solvent–solute interactions on the absorption maxima shifts was evaluated by using the Kamlet–Taft solvent parameter set, i.e. applying linear solvation energy relationships (LSER) principles. Optimized geometries and experimental results were interpreted by using DFT (B3LYP/6-311+G(d,p) method) and time-dependent density functional (TD-DFT) method. Overall electron density in both ground and excited state was obtained by the use of Quantum Theory of Atoms in Molecules, i.e. Bader’s analysis. It was found that both solvent and substituents cause appropriate change of the extent of conjugation in the molecules that further affect their intra-molecular charge transfer (ICT) character. Linear free energy relationships (LFERs) were applied to the substituent-induced NMR chemical shifts (SCS) using single substituent parameter (SSP) and dual substituent parameter (DSP) model. Transmission mode of the electronic effects of substituent was discussed according to the results of theoretical calculations and results of LFER correlations. Comparative analysis of presented results with the ones published for structurally similar series of amide which contained cyano group, instead chlorine, provides additional information on the impact of present group to the properties of investigated compound.
Keywords
N-(substituted phenyl)-2-chloroacetamides
UV absorption spectra
Solvent effects
Substituent effects
Molecular geometry optimization
Bader’ analysis

1 Introduction
Since 1952, when Monsanto started to use chloroacetamides as herbicides (Hamm, 1974), these compounds have attracted great attention. Different chloroacetamides have been screened and some found commercial application such as substituted phenyl chloroacetamides, i.e. substituted acetanilides (alachlor, acetochlor, butachlor, metolachlor, propachlor). In addition chloroacetamides, were used for the synthesis of other biologically active compounds (Hernandez-Nunez et al., 2009; Liang et al., 2010 and Gincburg et al., 2010). Due to the importance of chloroacetamides they have been subject of many papers and patents as well as reviews. Some of the reviews on the synthesis of chloroacetamides were published by Jacobs and Heidelberger (1927). Reactivity and degradation of chloroacetamides were also reviewed (Jaworski et al., 1969 and Liu et al., 2011). The derivatives of acetamide are organic compounds of significant interest in medicinal chemistry. They manifest a number of pharmacological properties and biological activities: analgesic and antipyretic (Jawed et al., 2010 and Ozkay et al., 2011), antileishmanial (Pacheco et al., 2013), antitumor (Kaldrikyan et al., 2009), insecticidal (Hladik et al., 2006), antimicrobial, bactericidal and fungicidal (Ertan et al., 2007) and also have agricultural and toxicological applications. Some of the chloroacetamide derivatives are used for the treatment of diabetes (Hayakawa et al., 2010). Biological activity that a molecule may exhibit, in addition to the chemical structure, is highly dependent on the type and strength of interactions that it can achieve with its environment. Investigation of these interactions contributes significantly to the prediction of biological activity of newly synthesized molecules.
So far, our investigations in the chemistry of amides included synthesis and identification of new compounds including mass spectral study (Petrović et al., 1998; Mijin et al., 2004), UV absorption spectral study of N-(4-substituted phenyl)-2,3-diphenylpropanamides (Valentić et al., 2006) in various solvents, and investigation of mass fragmentation of N-alkyl and N-(substituted phenyl) cyanoacetamides (Ilić et al., 2010).
As in previous report (Ilić et al., 2010) in the first part of this work a series of twelve N-(substituted phenyl)-2-chloroacetamides were synthesized and characterized (Fig. 1). UV absorption spectra were recorded in the range from 220 to 350 nm in eighteen solvents of different polarity. Quantification of the solvent effects: dipolarity/polarizability and the hydrogen-bonding ability on the UV spectral shifts was interpreted by means of the Kamlet–Taft equation (Kamlet et al., 1981), i.e. by applying linear solvation energy relationship – LSER – in the form of Eq. (1):
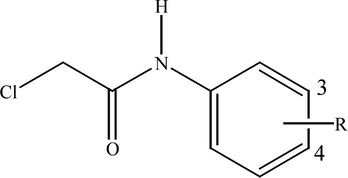
Structure of the investigated N-(substituted phenyl)-2-chloroacetamides, where R is: 4-H (1); 4-CH3 (2); 4-OCH3 (3); 4-Cl (4); 4-Br (5); 4-F (6); 4-I (7); 4-COCH3 (8); 4-OH (9); 4-CN (10); 3-CN (11); 3-Br (12).
In the second part of the work principles of linear free energy relationship, i.e. LFER analysis, were applied to the UV and SCS (substituent induced NMR chemical shift relative to parent compound) data in the studied compounds. For quantitative assessment of the substituent effects on the absorption frequencies, Hammett Eq. (2) was applied (Hammett, 1937):
The transmission of polar (field/inductive) and resonance effects from the substituent to the carbon atoms of interest was studied using Eq. (3):
In general, the UV and NMR data were analyzed by the use of LSER and LFER models, respectively, in order to evaluate solvent/solute interactions and substituent effects on solvatochromism and the extent of ICT in N-(substituted phenyl)-2-chloroacetamides. Geometries of compounds were optimized by DFT calculations (B3LYP/6-311+G(d,p) method). Time-dependent density functional theory (TD-DFT) was applied for estimation of the transition energy. TD-DFT was successfully applied for description of solvated organic molecules with a possibility of estimation of electronic density transfer (Antonov and Stoyanov, 1993), and the influence of molecular geometry on ICT (Barone and Polimeno, 2007). The solvatochromism and transmission of substituent effects were discussed in relation to the result of charge distribution analysis (Bader’s analysis).
2 Experimental
2.1 Synthesis of N-(substituted phenyl)-2-chloroacetamides
In a dry three-necked flask equipped with reflux condenser, thermometer, calcium chloride drying tube and addition funnel, were added 56 mmol of corresponding amine and 66 mmol of potassium carbonate in 50 ml dichloromethane at room temperature. Chloroacetyl chloride (33 mmol) was added dropwise in a course of half an hour and after that reaction mixture was heated at reflux for 4 h. After cooling to room temperature, reaction product was poured into 100 ml of cold water. Water solution was extracted with two portions of 100 ml CH2Cl2, and organic phase was dried with sodium sulfate. After filtration and evaporation of the solvent, crude residual material was crystallized from diethyl ether/petrol ether solvent mixture until constant melting point was obtained. Structure of the investigated N-(substituted phenyl)-2-chloroacetamides is shown in Fig. 1.
2.2 Characterization methods and spectral analysis
The chemical structure and purity of the synthesized compounds were verified by melting point, IR, 1H and 13C NMR spectra. Fourier-transform infrared (FTIR) spectra were recorded in transmission mode using a BOMEM (Hartmann & Braun) spectrometer. 1H and 13C NMR spectra were determined in an appropriate solvent on a Varian-Gemini 2000 MHz spectrometer using TMS as internal standard. The chemical shifts are expressed in ppm values referenced to TMS (δH = 0 ppm) in 1H NMR spectra, and to the residual solvent signal (δC = 39.5 ppm) in 13C NMR spectra. The chemical shifts were assigned by the complementary use of DEPT-, two dimensional 1H–13C correlation HETCOR- and by selective INEPT long-range experiments. Their full characterization is presented in Supplementary Material (Tables S1 and S2; NMR data are given on pages 4 and 5). UV absorption spectra were obtained with a Cintra 1010 spectrophotometer. The UV spectra were taken in spectro quality solvents (Fluka) at a fixed concentration of 4 × 10−5 mol dm−3. The absorption spectra were measured in λmax (nm) and the wave numbers (cm−1) were calculated by relation νmax = 1/λmax. All spectra were recorded at ambient temperature.
2.3 Molecular geometry optimization and theoretical absorption spectra calculation
The ground state geometries of compounds 1–12 were fully optimized with DFT method (more specifically the Becke’s three-parameter exchange functional (B3) and the Lee–Yang–Parr correlation functional (LYP)) with 6-311+G(d,p) basis set in gas phase, without symmetry constrain, and with default tight convergence criteria. For Iodine atom a specially designed basis set of 6-311G quality with added supplementary functions was used (Glukhovstev et al., 1995). Global minima were found for each molecule. Harmonic vibrational frequencies have been evaluated at the same level to confirm the nature of the stationary points found. The frontier molecular orbital energies: EHOMO for highest occupied molecular orbital (HOMO) and ELUMO for lowest unoccupied molecular orbital (LUMO) and HOMO–LUMO energy gaps (Egap) were also calculated with the same methods. Theoretical absorption spectra were calculated with TD-DFT method, more specifically with CAM-B3LYP functional (Yanai et al., 2004) and 6-311++G(d,p) basis set on gas phase optimized geometries. Solvents in the TD-DFT calculations were simulated with standard polarized continuum model (PCM) (Tomasi, 2004). The ground and excited state electron densities were calculated in gas phase with CAM-B3LYP/6-311++G(d,p) method on ground state optimized geometries. All DFT calculations were done with Gaussian09 software (Frisch et al., 2009). The Bader’s analysis was done on charge density grid with program “Bader” (Wiggins et al., 2009). Density difference maps were plotted as difference between electron densities of the excited state and the ground state, in program gOpenMol (Laaksonen, 1992).
2.4 Regression analysis
The correlation analysis was carried out using Origin 8.6 software that considers the 95% confidence level. The goodness of fit is discussed using the correlation coefficient (R), the standard error of the estimate (S), and Fisher’s significance test (F).
3 Results and discussion
In this work a series of twelve N-(substituted phenyl)-2-chloroacetamides with a substituent in meta- and para-position were synthesized and characterized. In order to get better insight into the transmission mode of substituent effects and electron density shift in investigated amides an LFER analysis of NMR data of N—H, C1 and C⚌O carbons was performed. Evaluation of the solvent/solute interactions of N-(substituted phenyl)-2-chloroacetamides in the solvent of different properties was performed by applying LSER analysis to the UV absorption maxima shifts.
The UV absorption maxima shifts (Table 1) can be attributed to different π → π∗ transitions involving the π-electronic system of the investigated compounds (Fig. 2). The π-delocalization originates mainly from the π-electron density shift from the substituted benzene unit (π1-unit), influenced by the electron-donor substituent, to the C⚌O group of the amide moiety. Opposite is true for electron-acceptor substituted compounds. Also discussion on the contribution of electronic effect of chlorine in acid residue on electron density shift was included. The presence of amide hydrogen contributes to the hydrogen bond donor and acceptor capabilities of N-(substituted phenyl)-2-chloroacetamides with respect to surrounding environment (Fig. 2).
1
2
3
4
5
6
7
8
9
10
11
12
1
Methanol
40.88
40.25
39.38
38.95
39.50
41.16
38.99
35.11
39.68
37.57
40.13
40.35
2
Ethanol
40.72
39.98
39.14
39.73
39.33
40.99
38.89
34.99
39.33
37.39
39.98
40.19
3
1-Propanol
40.66
39.94
39.13
39.63
39.28
40.96
38.82
34.87
39.23
37.26
39.90
40.18
4
2-Propanol
40.58
39.90
39.10
39.61
39.24
40.88
38.79
34.87
39.21
37.23
39.88
40.15
5
1-Butanol
40.54
39.88
39.03
39.53
39.23
40.72
38.79
34.80
39.13
37.19
39.85
40.09
6
2-Butanol
40.40
39.85
39.00
39.50
39.21
40.65
38.73
34.92
39.03
37.18
39.79
39.98
7
2-Methyl-2-propanol
40.62
39.80
38.96
39.43
39.12
40.57
38.82
34.80
38.99
37.13
39.68
39.92
8
Water
41.58
40.83
40.30
40.46
40.04
41.93
39.48
35.80
40.62
38.10
40.51
40.88
9
Dioxane
40.92
40.46
39.52
40.08
39.58
41.20
39.08
35.63
39.97
37.56
40.07
40.51
10
Acetonitrile
41.10
40.56
39.73
40.09
39.68
41.54
39.23
35.67
40.04
37.66
40.30
40.62
11
Chloroform
40.83
40.35
39.43
40.04
39.63
40.93
39.03
35.96
39.82
37.85
40.01
40.46
12
Hexane
40.15
39.73
38.81
39.30
38.83
40.05
38.65
35.47
–
37.16
39.39
39.87
13
Heptane
40.23
39.70
38.78
39.32
38.79
40.00
38.63
35.51
–
37.22
39.37
39.89
14
Methyl acetate
39.82
39.35
39.51
39.80
39.48
39.68
39.08
35.64
39.44
37.62
40.12
40.58
15
Ethyl acetate
39.89
39.28
38.36
39.71
39.38
39.52
38.99
35.53
39.41
37.50
40.03
40.45
16
Ethylene glycol
–
40.39
39.63
40.03
39.69
–
39.14
35.40
39.63
37.75
40.42
40.63
17
DMSOa
38.60
38.29
38.18
38.32
38.09
–
38.18
34.79
38.08
36.78
38.66
38.74
18
Acetic acid
–
39.98
39.40
39.87
39.47
–
39.01
35.23
39.70
37.76
40.03
–
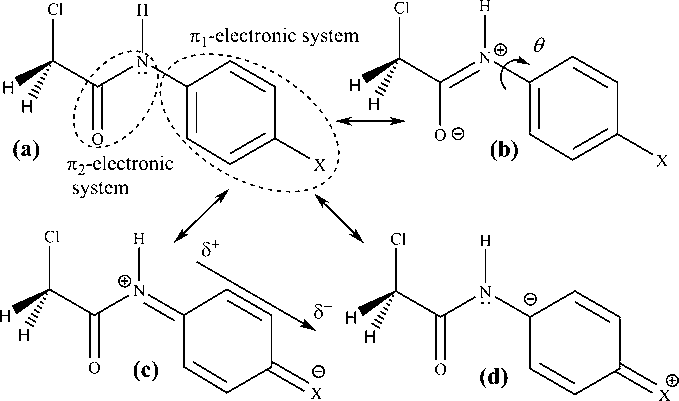
Resonance structures of N-(4-substituted phenyl)-2-chloroacetamides with defined π-electronic units (a), amide group resonance (b), as well as electron-acceptor (c), and electron-donor (d) resonance substituent effect participation in the overall electron density shift.
The electronic absorption spectra of the investigated compounds were recorded in eighteen different solvents in the range from 220 to 350 nm, and showed one main absorption maximum. As an example, Fig. 3 shows absorption spectra of all investigated derivates of N-(substituted phenyl)-2-chloroacetamides in ethanol. In general, the presence of both electron-donating and electron-accepting substituents in the molecule causes bathochromic shift of the absorption maximum relative to unsubstituted molecule 1, except in the case of compounds 6 and 12 (with F and 3-Br substituents, respectively) in some solvents. The lowest bathochromic shift is noticed in the presence of weak electron-donating CH3 groups and meta-substituted compounds, and the largest shift was recorded in the presence of strong electron-attracting acetyl and cyano groups. This phenomenon has been observed in the N-(4-substituted phenyl)benzamides and interpreted by the compensation of influence of electron-acceptor groups with the effect of the local polarization of the amide groups. As a consequence bathochromic shifts were observed in the spectra of corresponding amides (Ušćumlić and Petrović, 2002; Valentić et al., 2006). The deviation of the fluorine, comparing to the other halogen derivatives, has been noted and discussed earlier (Ušćumlić and Petrović, 2002).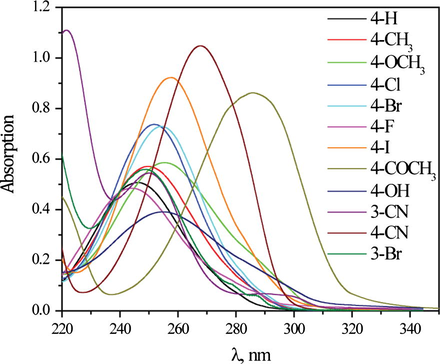
The absorption spectra of N-(substituted phenyl)-2-chloroacetamides in ethanol.
From the presented UV–Vis spectra (Fig. 3) it could be noticed diversity of the spectra, but all of them show appropriate shifts which depends on substituent present in investigated molecules and solvent used. Data from Table 1 show small shift of the absorption maximum in protic solvents for N-phenyl-2-chloroacetamide. Significantly larger bathochromic shifts were observed in aprotic solvents, and largest shifts were found in DMSO for N-(substituted phenyl)-2-chloroacetamides. This result is similar to previous finding for N-(substituted phenyl)-2-cyanoacetamides in DMSO and DMF (Matijević et al., 2014). Observation that most of compounds show bathochromic shift in solvent used is a strong indication that π → π∗ transition is a main process that contributes to electronic transition during molecule excitation. The difference in stabilization of the ground and excited states indicates that the intensity of solute/solvent interactions changes during the excitation. It also indicates that extended resonance interaction through π-electronic systems of the molecule (Fig. 2), significantly affects contribution of the appropriate electronic transitions involved during excitation.
The difference in stabilization of the ground and excited states of the studied compounds was observed as a small bathochromic shift in all solvents, with the exception of the larger bathochromic shift noticed for all compounds in DMSO. It is characteristic of molecules of low dipolarity in the ground-state, in which processes of the electronic transitions are associated with intra-molecular charge transfer (Reichardt, 2003). These kinds of compounds include acetamide derivatives where π-electron delocalization within amide group is operative, i.e. lone pair of amide nitrogen interaction with π-electrons of carbonyl group (Fig. 2(b)). Unlike other used solvents, DMSO posses significantly pronounced polar character, high value of relative permittivity (46.7), and also very high value of dipole moment (3.96 D). Higher bathochromic shifts of the investigated compounds in aprotic dipolar solvent DMSO can be explained by the fact that, at high relative permittivity of the surrounding medium, the energy necessary to bring about charge separation in the excited state is relatively small. When these molecules are in the excited state, the dipole may occur as a result of electron delocalization between the donor and acceptor atoms/groups present in investigated compounds. If the solvent can stabilize the induced dipole in the excited state, the electronic transition will require less energy, and due to this absorption bands will be shifted bathochromically.
The difference in the absorption spectra of compounds with the same substituent in different positions, e.g. compounds 5 and 12, and 10 and 11 (R = Br and CN) in positions 4 and 3, respectively, was also observed. In both cases it was noticed that the substituent in position 4 causes a larger bathochromic shift comparing to the compounds with the substituent in position 3, as it was previously found (Mijin et al., 2006). Substituent from para-position contributes to increased direct conjugational electronic density shift to the rest of molecule thus enabling effective extended π-electron delocalization. As a consequence, such effect supports larger extent of n,π-resonance in amide group, in that way formation of local dipolar structure in the amide part of the molecule, i.e. π2-unit, was noticed (Fig. 2(d)). Effective π-electron transfer from substituted phenyl ring contributes to formation of such molecular orbital structure of frontier orbital region which enables energetically favorable electronic excitations observed as bathochromic shift in corresponding spectra. All meta-substituent showed moderate influences on absorption maxima shifts due to lower extent of direct π-electron delocalization. In these compounds transmission of the electronic effect was achieved by adjustment of the orientation of the induced dipole at substituted phenyl ring achieving, through the space, electrostatic stabilization/destabilization of surrounding/neighboring charges in π2-unit.
3.1 Solvent effects on UV absorption spectra
Recording the UV spectra of the investigated derivatives in solvents of different properties allowed evaluation of solute/solvent interactions by applying LSER and quantum chemical analysis. As an example the absorption spectra of the compound 1 in protic (Fig. 4(a)) and aprotic solvents (Fig. 4(b)) were presented. According to the data in Fig. 4 and Table 1 a low variation of νmax shift (∼8 to 19 nm) was noticed. Generally, the observed changes in the spectral behavior were more pronounced in aprotic solvent than in protic one. As it was previously explained this phenomenon probably occurs due to the solvent induced effective intramolecular charge transfer participation of the electron density shift in the course of electron excitation (Reichardt, 2003). The contribution of specific and nonspecific solvent–solute interactions was quantitatively evaluated by the use of LSER Eq. (1). The LSER concepts developed by Kamlet and Taft are widely and successfully used for quantitative treatments of solvation effects. This treatment assumes attractive/repulsive solvent/solute interactions and enables an estimation of the ability of the solvated molecule to create different interaction with surrounding solvent molecule. Correlation results obtained by the use of Kamlet–Taft (Eq. (1)) are given in Table 2. Evaluation of the solvent influence on the νmax, from Table 1, was correlated with Kamlet–Taft solvents parameters π∗, α and β (Table S3), taken from the literature (Marcus, 1993). Correlation analysis was conducted by using multiple regression analysis.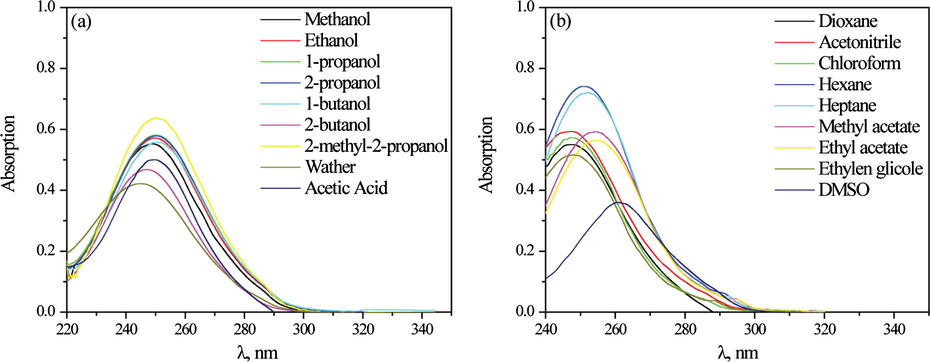
Absorption spectra of N-(4-methyl phenyl)-2-chloroacetamide in protic (a) and aprotic (b) solvents.
Substituent
ν0 10−3 cm−1
S
a
b
Ra
Sb
Fc
Remarkd
H
40.237 (±0.055)
1.297 (±0.112)
−e
−0.200 (±0.085)
0.959
0.088
70.1
14, 15, 17
CH3
39.794 (±0.054)
1.268 (±0.102)
−0.345 (±0089)
−0.259 (±0.078)
0.950
0.086
69.1
14,15, 17
OCH3
38.859 (±0.055)
1.362 (±0.096)
−0.133 (±0.072)
−0.376 (±0.078)
0.955
0.090
91.4
17
Cl
39.404 (±0062)
1.045 (±0.105)
–
−0.352 (±0.087)
0.917
0.100
48.1
17
4-Br
39.910 (±0.040)
1.129 (±0.068)
−0.060 (±0.051)
−0.183 (±0.056)
0.965
0.065
119.1
17
F
40.107 (±0.064)
1.930 (±0.130)
−0.251 (±0.119)
–
0.972
0.102
104.5
14, 15
I
38.679 (±0.031)
0.795 (±0.053)
−0.086 (±0.039)
−0.200 (±0.043)
0.957
0.050
97.6
17
COCH3
35.564 (±0.041)
0.689 (±0.070)
−0.318 (±0.052)
−0.884 (±0.058)
0.975
0.067
169.8
17
OH
38.890 (±0.314)
1.707 (±0.408)
–
−0.597 (±0.235)
0.911
0.156
34.2
16, 17
3-CN
39.4443 (±0.034)
1.148 (±0.058)
−0.101 (±0.044)
–
0.973
0.056
157.9
17
4-CN
37.277 (±0.043)
0.821 (±0.073)
0.073 (±0.054)
−0.543 (±0.060)
0.953
0.069
87.3
17
3-Br
39.946 (±0.033)
1.127 (±0.056)
−0.222 (±0.046)
−0.239 (±0.048)
0.976
0.053
163.4
17
The results of multivariate data fitting by the use of Kamlet–Taft model show good correlation (R = 0.911 − 0.976) between the absorption frequencies of investigated compounds and solvent parameters. The obtained linear correlation (Fig. 5) between experimental absorption frequencies, νexp, and those that are calculated by using Eq. (2), νcalc, confirms that the chosen solvatochromic model is adequate.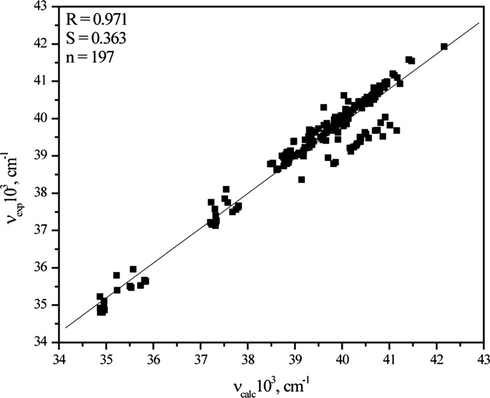
Linear correlation of experimental, νexp, versus calculated, νcalc, values obtained by using Eq. (1).
The results presented in Table 2 show that in all investigated derivatives of N-(4-substituted phenyl)-2-chloroacetamides (except for comp. 8), the absolute value of s is greater than sum of coefficients a and b, which means that the solvent dipolarity/polarizability plays a major role to the solvatochromism of these compounds in comparison with contribution of hydrogen-bonding interactions.
For all investigated compounds, as can be seen from the data in Table 2, the positive value of the coefficient s was obtained. This is an indication that hypsochromic shift (blue shift) is expected with increasing polarity of the solvent, which is in agreement with the theoretical prediction (Antić-Jovanović, 2006). Negative values of coefficients a and b, only exception is coefficient a found for comp. 10, indicate that an increase in the solvent acidity or basicity leads to bathochromic shift of the absorption maximum of investigated compounds.
Assessment of influence of the solvent on the absorption spectra of investigated chloroacetamide derivatives can be achieved by calculating percentage contribution of appropriate solvatochromic parameters (Table S4). In this way a quantitative evaluation of solvent effect on the solvatochromic behavior is obtained for the investigated compounds. As it can be observed from the presented results, for most compounds the dipolarity/polarizability solvent effect has the dominant influence on the solvatochromic behavior of studied compounds (57.13–91.91%). The lowest value, 36.44%, of the percentage contribution of solvent dipolarity/polarizability was found for compound 8 (Table S4). It means that contribution of solvent dipolarity/polarizability in both ground and excited states is fairly similar, and this effect prevails in ground state. Hydrogen-bonding solvent basicity or acidity has significantly smaller influence on the spectral shift of investigated compounds, except in the case of comp. 8 (COCH3 substituent), where the HBA solvent effect has the largest contribution. Specific interactions achieved through hydrogen bonding, expressed by solvent acidity and basicity, can be attributed mainly to the carbonyl moiety and NH hydrogen, and they are moderately affected by the substituent. Presented results, obtained by the use of Kamlet–Taft model, indicate that the solvent effects on UV absorption maximum spectra of the investigated compounds are very complex due to diversity of the contribution of both solvent and substituent effect. This also indicated that the participation of lone electron pair at amide nitrogen is different between derivatives with electron-donating and electron-accepting substituents.
3.2 Substituent effects on UV absorption spectra: LFER analysis
A comprehensive analysis of substituent effects on absorption spectra of the investigated molecules, by using LFER principles in the form of the Hammett equation, i.e. Eq. (2), was performed. The data from Table 1 confirm that the positions of the ultraviolet absorption frequencies depend on the nature of the substituent on the benzene ring. In an attempt to assess substituent effects on ICT of the investigated molecules, the principles of liner free energy relationships (LFER) were applied to the UV spectral data. The plot of the νmax versus σm/p Hammett substituent constant (Table S5) in ethanol is shown in Fig. 6. In Fig. 6, two separate linear correlation lines can be noticed. First correlation includes electron donor substituent and fluor, and the second includes only halogen substituted compounds. Strong electron-acceptors exhibit deviations from this trend showing a parabolic correlation. Different behavior of halogen substituents than other electron acceptor substituents may be explained by their specific properties. All halogen substituents are considered “modified” due to positive resonance effect in contrast to negative effect of the “real” electron-acceptor (COCH3 and CN) substituent (Valentić et al., 2006). Similar correlation was obtained for other used solvents. The correlation results are given in Table 3.
The plot of νmax versus σm/p of investigated compounds in ethanol.
Solvents
Electron donating substituenta
Halogen substituentb
νo 103 (cm−1)
ρ 103 (cm−1)
Rc
Sd
Fe
νo 103 (cm−1)
ρ 103 (cm−1)
Rc
Sd
Fe
Methanol
40,812 (±0.317)
3.773 (±1.297)
0.899
0.354
8.46
38.209 (±0.504)
5.684 (±1.873)
0.906
0.297
9.21
Ethanol
40.648 (±0.285)
4.224 (±1.165)
0.932
0.318
13.14
38.128 (±0.505)
5.466 (±1.874)
0.900
0.297
8.51
1-propanol
40.606 (±0.247)
4.277 (±1.010)
0.948
0.276
17.92
37.986 (±0.460)
5.794 (±1.709)
0.923
0.271
11.50
2-propanol
40.531 (±0.245)
4.115 (±1.002)
0.945
0.274
16.85
37.956 (±0.471)
5.794 (±1.750)
0.920
0.277
10.96
1-butanol
40.504 (±0.250)
4.240 (±1.025)
0.946
0.280
17.11
37.974 (±0.406)
5.575 (±1.509)
0.934
0.239
13.65
2-butanol
40.400 (±0.227)
4.099 (±0.930)
0.952
0.254
19.44
38.083 (±0.342)
4.815 (±1.270)
0.937
0.201
10.08
2-methyl-2-propanol
40.564 (±0.238)
4.800 (±0.973)
0.961
0.266
24.31
38.083 (±0.342)
4.815 (±1.270)
0.937
0.201
14.38
Water
41.445 (±0.300)
3.011 (±1.229)
0.866
0.336
6.01
38.745 (±0.608)
5.709 (±2.259)
0.873
0.358
4.10
Chloroform
40.780 (±0.345)
3.322 (±1.413)
0.857
0.386
5.52
38.299 (±0.629)
5.791 (±2.337)
0.869
0.370
6.14
Dioxane
40.895 (±0.322)
3.533 (±1.318)
0.884
0.360
4.52
38.290 (±0.628)
5.911 (±2.331)
0.873
0.369
6.43
Acetonitrile
41.042 (±0.305)
3.381 (±1.250)
0.886
0.341
7.31
38.384 (±0.505)
5.908 (±1.875)
0.912
0.297
9.92
Hexane
40.255 (±0.216)
4.729 (±0.886)
0.967
0.242
28.52
37.735 (±0.424)
5.536 (±1.573)
0.928
0.249
12.39
Heptane
40.317 (±0.196)
5.073 (±0.801)
0.976
0.219
40.08
37.683 (±0.472)
5.726 (±1.753)
0.918
0.277
10.66
Methyl acetate
–
–
–
–
–
38.010 (±0.356)
6.700 (±1.320)
0.963
0.209
25.74
Ethyl acetate
–
–
–
–
–
37.959 (±0.365)
6.500 (±1.356)
0.959
0.215
22.95
Ethylene glycol
40.909 (±0.619)
3.800 (±2.194)
0.866
0.310
3.00
38.252 (±0.503)
6.294 (±1.868)
0.922
0.296
11.34
DMSO
38.573 (±0.038)
1.411 (±0.154)
0.988
0.042
83.09
37.537 (±0.210)
3.003 (±0.780)
0.939
0.123
14.84
Acetic acid
–
–
–
–
–
36.634 (±1.487)
13.200 (±6.928)
0.885
0.282
3.63
The correlation results (Table 3), classified in the two groups (columns) of substituent-dependent correlation results, reflect the complexity of solvent and substituent electronic effects on absorption maxima shifts. The linear correlations of absorption frequency, νmax, and substituent constants, σm/p, gave a wide range of proportionality constants in the range from 1.411 to 6.700. The correlation results (Table 3), obtained separately for the electron-donor and halogen substituted chloroacetamides, reflect different transmission modes of electronic substituent effects through differently oriented π-electronic units (Fig. 2).
The introductions of an electron-donor substituent contribute to the positive solvatochromism and lower sensitivity of the absorption maxima shifts to substituent effect. Slightly higher values of proportionality constant obtained for halogen substituted derivatives showed significant influences of the electronic substituent effects on the π-electronic density shifts through resonance structure presented in Fig. 2. This indicates that balanced contribution of the effect of halogen substituent and amide group resonance, i.e. n,π-conjugation, causes higher sensitivity of electronic transition energy to substituent effect in the solvent of different properties. Exceptionally low ρ value in DMSO found for first series and low in second series could be explained by high relative permittivity of surrounding medium by which the energy necessary to bring about charge separation in the excited state is relatively small. Such condition gives rise to a lower susceptibility to electronic substituent effects.
3.3 Substituent effect on NMR data: LFER analysis
An extensive analysis of 1H and 13C NMR chemical shifts has been done in order to get better insight into the influence of their geometry on transmission of particular substituent effect. In order to analyze substituent influences on SCS of atoms of interest, LFER in the form of SSP equation with σ, σ− or σ+ substituent constants (Hansch et al., 1995; Kubinyi, 1993) was applied for N-(substituted phenyl)-2-chloroacetamides, and the best correlation results are presented in Table 4.
Atom
Scale
ρ
h
Ra
Sb
Fc
nd
N—H
σm/p
0.562 (±0.031)
0.001 (±0.011)
0.987
0.030
338
11e
C⚌O
σm/p
1.276 (±0.078)
-0.145 (±0.030)
0.982
0.090
267
12
C⚌O
σ+/σ−
0.755 (±0.047)
−0.012 (±0.027)
0.981
0.091
254
12
C1
σm/p
7.029 (±0.838)
−1.436 (±0.384)
0.948
1.098
70
10f
The results of SSP correlations for all carbons with σ are of good precision. Good correlation between SCS and σ indicates that electronic effects control the substituent-induced changes of electron density at C1 and C⚌O carbon. Electron-withdrawing substituents cause a downfield shift, indicating deshielding at the N—H proton, and C1 and C⚌O carbons.
According to the observed ρ values for carbon atoms and N—H proton, it is apparent that SCS of C1 show an increased susceptibility to substituent effects, and normal substituent effect is observed for all atoms of interest. Good correlation obtained for C⚌O carbon with combination of σ+ and σ– constant (Kubinyi, 1993), so-called electrophilic and nucleophilic substituent constants, respectively, is given in Table 4. It means that extended resonance interaction within π1-unit could be effectively transmitted to C⚌O group by resonance and field transmitted resonance to π2-unit, and their contribution depends on the electronic effect of substituent present. Correlation results (Table 4) for C1 and C⚌O carbons also indicate appropriate contribution of nitrogen lone pair participation in resonance interaction within amide group (Fig. 2(b)), i.e. n,π-conjugation, to overall electronic density shift. Planarity of electron-accepting carbonyl group enables higher extent of electronic interaction with phenyl ring with electron-donating substituent. In that way, a higher extent of resonance interaction is operative within π2-resonance unit causing enhancement of electron-withdrawing ability of carbonyl group.
Although SSP analysis uses an additive blend of inductive and resonance parameters of substituents, it presented a satisfactory description of substituent electronic effects in correlations using Eq. (2). Evaluation of the separate contributions of inductive and resonance effects of substituent (X), the regression according to Eq. (3), DSP analysis, using σI and σR substituent constants (Hansch et al., 1995) was carried out, and the results are given in Table 5.
Atom
scale
ρI
ρR
h
Ra
Sb
Fc
λd
n
N—H
σR
0.649 (±0.064)
0.668 (±0.063)
0.005 (±0.026)
0.979
0.040
93
1.03
11e
C⚌O
σR
1.337 (±0.215)
1.729 (±0.194)
–0.094 (±0.088)
0.961
0.138
54
1.29
12
C1
σR
5.331 (±1.723)
15.886 (±1.923)
–0.152 (±0.698)
0.956
1.050
39
2.97
10f
Generally, both polar (inductive/filed) and resonance substituent effects have different contribution at all atoms (Table 5). Results of DSP fits are similar or slightly better than SSP correlations, indicating that short distance transmission of substituent effect could be adequately described by substituent σ values. The observed ρI and ρR values for N—H atom indicate similar contribution of the field and resonance effect. Resonance effect is somewhat higher value at carbonyl and C1 atoms, and for all atoms it shows a negligible alternation of its contribution regarding particular atom position in molecular structure of investigated compounds.
Substituents exert relatively small influences on SCS of N—H proton. Spatial position of N—H atom with respect to the benzene ring indicates that anisotropic effect could exist, which generally has relatively small contribution to NMR chemical shifts change. The anisotropy effects depend on spatial arrangement, but it is independent of the nuclei being observed (Klod and Kleinpeter, 2001). Except this, steric effect also could be operative, and it includes all those phenomena which result in structural changes at measured sites, such as bond lengths and angles, effects due to size of the atom close enough to cause geometrical adaptation. Since optimized geometry of investigated compounds counts to lowest steric interactions, it could be assumed that the field effect (through the space induced π-polarization) has a highest contribution causing shielding at C1, N—H and C⚌O atoms. Contribution of diamagnetic anisotropy effect of the electron-donor substituted phenyl ring causes upfield shifts to N—H proton. Electron-donor substituents increase electron density at benzene ring and, hence, an increase in the shielding of N—H proton could be observed. Opposite is true for electron-acceptor.
Considering resonance structure (c) (Fig. 2), for electron-acceptor substituted N-(substituted phenyl)-2-chloroacetamides, a dipole on X (or near the C—X bond) is induced, and interaction of this dipole through molecular cavity results in the polarization of whole π1-unit. Resonance interaction within amide group (n,π-conjugation), presented by resonance structure in Fig. 2(b), could be of large significance and oppositely oriented with respect to interaction within π1-unit Fig. 2(c). The net result is that the electron-acceptor substituents decrease the electron density about the C1 and C⚌O carbons and, hence, decrease the shielding.
Electron-donor substituents via their +R effect, transmitted through the π1-electronic unit increase the electronic density at the C1 carbon, Fig. 2(d), shifting the corresponding signal at C1 carbon toward higher magnetic field. This considerably increases the extent of n,π-conjugation of nitrogen lone pair with π-electron of carbonyl group (Fig. 2(b)). The increased electron density on the C1 atom favors the delocalization of the free electron pair from amide nitrogen atom toward the C⚌O carbon on that way electron density at the C⚌O atom increases as the electron-donating power of substituent increase. Namely, this conjugation, which involves the amide nitrogen lone pair and π-electrons from the C⚌O carbonyl group, is possible only if the appropriate geometrical adjustment could provide overlap of the nitrogen lone pair and C⚌O π-electron (Fig. 2(d)). Such requirement is possible for trans-configuration of C—N amide bond. Significant contribution of resonance effect of substituent at C1 is in accord with the corresponding ρR values of 15.886 versus ρI of 5.331, which indicates that substituent effect on the electron density shift to the C1 atom could be presented as in Fig. 1(d). Somewhat different results were obtained for N-(substituted phenyl)-2-cyanoacetamides, and the high contribution of the resonance effect at C1, in accordance with the corresponding ρR values of 14.282 and λ = 1.39 (ρR/ρI) (Marinković et al., 2013), indicates similar transmission mode of substituent effect. These results indicate that, irrespective of chloro or cyano substituent, similar effect was found for C1 carbon. The transmission of substituent electronic effects through π-resonance units of investigated compounds takes place by contribution of both defined π-electronic unit and overall conjugated system. Ratio of their contribution depends on substitution pattern, as well as on solvent under consideration.
3.4 DFT, TD-DFT and Bader’s analysis. Evaluation of electronic transition and charge density change
An additional analysis of substituent effects on absorption frequencies and conformational changes of the studied compounds, necessitated quantum-chemical calculations, i.e. geometry optimization and charge density analysis. Geometry optimization of the investigated molecules was performed by the use of B3LYP functional with 6-311+G(d,p) basis set, and results of QM calculations are given in Table S6 and Fig. S1. As expected the higher stability of trans-isomer was obtained by around 8 kcal/mol (data not shown). The calculation of optimal geometry gives valuable results required for better understanding of the transmission of substituent effects, i.e. electron density distribution. Higher planarity molecule, i.e. molecule with low torsional angle θ (Fig. 2), induces red shift in the absorption spectra. In the investigated molecules these values are fairly similar (close to 0) which indicate significance of extended resonance interaction in electron-donor substituted compounds. In electron-acceptor substituted compounds appropriate contribution of n,π-conjugation (nitrogen lone pair participation) to overall electronic density shift, and opposite direction of interaction with π1-unit cause perturbation of overall π-electron density.
From the results presented in Table S6 a low influence of electronic substituent effects could be noticed. Some bond distance values’ changes are in the range of statistical errors and thus they are not suitable for evaluation purpose. An electron-donating substituent supports electron density shift from the π1-unit to the carbonyl group (Fig. 2(d)) supporting planarization of the molecule as a whole, and a slight decrease of the NH—(C⚌O) bond length (Table S6) is a consequence. This result is an additional support for the fact that the extended conjugation operative in the π1- and π2-units, i.e., the net π-electron density shifts toward carbonyl group causes increase in both n,π-conjugation in the π2-unit and carbonyl bond lengths increase (Table S6). Due to this the λmax in UV spectra of electron-donor substituted derivatives appeared at longer wavelengths.
The results are fairly opposite for the electron-acceptor substituted compounds. An increase in the NH—(C⚌O) bond length indicates that two opposite electron-accepting effects operate: electron accepting character of both carbonyl group in the π2-unit and substituted phenyl ring. The normal polarization operative in the π2-unit is suppressed and has opposite effect to the polarization in the extended conjugative system of the π1-unit. Nevertheless, the longer λmax are obtained in comparison with compound 1.
In order to check significance of HOMO and LUMO orbitals in electronic excitations detailed TD-DFT studies of all 12 compounds in different solvents (water, ethanol, chloroform and DMSO) were conducted using CAM-B3LYP functional. This functional was able to properly predict relative positions of absorption maxima for all the compounds (Fig. S2), with errors in absolute values of around 10 nm. A more detailed data of TD-DFT calculations for all the compounds in water are presented in Table S7. For compounds 1–6, 8, 9, 11 and 12 the second excited state is the excited state with highest oscillator strength, for compound 7 it is third excited state and for compound 10 it is the first excited state. For all the excited states with high oscillator strength values, HOMO–LUMO single particle excitation has the highest contribution to actual transition (Table S7) indicating that mechanism of electronic excitations and changes in the electron density distribution in excited states of the investigated molecules can be studied by calculation of the HOMO/LUMO energies (EHOMO/ELUMO) and orbital shapes, as well as energy gaps values, Egap, between the frontier orbitals. The results of these calculations are presented in Fig. 7.
The spatial distribution of HOMO and LUMO molecular orbitals and corresponding Egap in investigated compounds.
The results of calculation of HOMOs and LUMOs molecular orbitals and energy gaps are in accordance with shift of corresponding UV spectra and results of LSER correlations (Table 3). Compound 1 has the largest energy gap with 5.550 eV, while somewhat lower energy gaps for electron-donor substituted compounds, in the range 5.198–5.409, were found. This is consistent with red shift of their absorption maxima relative to comp. 1. Methoxy substituted compounds show lower Egap values with respect to compound 9 (OH subst.) probably due to large HBD value of OH group. A similar range of Egap values were observed for all electron-acceptor substituted compounds, and the lowest one, 4.738 eV, was found for compound 8. It means that electron-accepting phenyl group (π1-electronic system) and amido group (π2-electronic system) cause formation favorable molecular orbital structure as a response to electronic demand of these two electron-attracting systems (Fig. 2; structures (b) and (c)). The normal carbonyl groups polarization is suppressed and causes a slight bond length decrease of carbonyl groups. Nevertheless, the shifts to higher λmax are obtained in comparison with compound 1. Furthermore, no ICT processes can be clearly observed during the orbital transition process from HOMO to LUMO, given in Fig. 7.
In order to obtain data on overall electronic density distribution, the Bader’s charge analysis for both ground state and highest oscillator strength excited state was performed. Bader’s theory of atoms in molecules is useful to define the charge enclosed within the Bader volume as a good approximation of the total electronic charge of an atom. Atom and ring numbering used in Bader’s analysis are given in Fig. S3. Difference of atomic charges in the excited and in the ground state (Δcharge) for appropriate atoms is given in Tables S8–S19 and electron density difference between excited state with highest oscillator strength and ground-state is represented in Fig. 8. Variation of substituent patterns indicates that contributions of donor–acceptor character of π-electronic systems are involved in the ICT mechanism of the investigated molecules.
Presentation of the electron density difference between excited and ground state (blue, electron density increase upon transition; red, electron density decrease upon transition).
According to the results shown in Tables S8–S19 and Fig. 8, it could be observed that acetyl substituted compound 8 exhibits largest extent of ICT. According to the character of the ICT processes, molecules can be formally divided into three groups: the first one includes compounds 1–6 and 9, the second compounds 7, 8 and 10 and the third compounds 11 and 12.
In the first group the studied molecules showed no observable ICT process, and electron density was mainly concentrated on phenyl and amide moieties in both ground and excited states: low to moderate Δcharge for the C2 and O4 atoms vary from −0.0007 to −0.0497, and from 0.0298 to 0.0545, respectively (Tables S8–S13 and S16). It designates electron density shift from carbonyl oxygen, and highest one was found for compound 6. Along with this, amide nitrogen gained a larger amount of charge (Δcharge): from 0.0963 to 0.1403, and again highest value was found for compound 6. The compound 9 showed negligible change of electron density at C2 carbon, −0.0007, which indicates that electron-accepting character of carbonyl oxygen is compensated by high electron-donating capability of hydroxyl group. This is reflected in higher electron density increases at O4, 0.0472, confirming electron density transfer to this carbon. Extent of observed charge transfer can be explained by different contribution of resonance substituent effect present in compounds 2, 3 and 9: low electro-donating methyl, moderate methoxy- and strong electron-donating hydroxy-substituent, respectively. It should be noted that Δcharge in hydroxy-substituted compound is similar to compound 3, which means that balance of higher proton-donating capabilities of hydroxy group and polarization in carbonyl groups is reflected in similar polarizability of π-electron density of compounds 3 and 9.
In the second group, which formally includes compounds 7, 8 and 10 with iodo and strong electron-accepting group, ICT process is different and significant, and highest extent was found in compound 8. It can be noticed that the overall electron density in compound 8 is separately populated on the 4-acetylphenyl ring in ground and it is largely shifted to amido group in excited state, confirming presence of significant ICT during excitation. Although, in compounds 8 and 10 with similar electronic properties of present substituent a different sensitivity of electron density was found, the calculation showed that different negative values of electron density change were found for C2: −0.0354 for comp. 8 and −0.152 for comp. 10. Large and important differences could be also observed for amide nitrogen (Tables S15 and S17): negligible Δcharge of 0.005 for N3 in comp. 8 and large value for comp. 10 of 0.1963 were found. These results indicate that complex influences of both substituent effect and carbonyl group polarization on electron density change at N3 were operative. Opposite behavior was found for O4: losses of electron, Δcharge is −0.0133, were noticed for comp. 8 and 0.0401 for comp. 10. Along with this, C13 phenyl carbon reflects direct substituent influences on electron density change at this carbon: −0.1245 for comp. 8 and −0.0551 for comp. 10. Obviously, the ICT process is the most feasible in compounds 8 and 10. For both compounds, the reason is high planarity which enables that the bond connecting the electron-acceptor group can decouple the orbitals of these two systems providing means by which significant charge transfer can occur. The highly polarizable ground and excited states of compound 8 can be stabilized by solvent/solute interactions with respect to the ground state. Due to this, the excited and ground states are closer and more rapid internal conversion is permitted.
In the third group, compounds 11 and 12 show moderate ICT which occurs from amido to cyano group in former compound, and oppositely from bromo to amido group in comp. 12. The calculated values Δcharge for C2 and N3 are of similar values: −0.0152 and −0.0992 and 0.1764 and 0.0748 for compounds 11 and 12, respectively. Higher and positive Δcharge value of 0.0852 for C13 carbon was found for comp. 11, while oppositely lower and negative value, −0.0326, was obtained for compound 12, which reflect different conjugational capabilities of the substituent in meta-position. Observed charge transfer can be explained by different extent of electron-accepting properties of cyano and bromo substituents. Balance of electron-accepting effect of both substituent, chloro atom in compound 11, and amido group contribute to electron density shift to the substituent in the course of excitation. Opposite process was found for compound 12 reflected in a modified behavior of bromo substituent, i.e. electron-donating effect of bromo atom was caused by stronger electron-accepting effect of amido group.
4 Conclusions
Experimental and theoretical data of investigated compounds were considered. Solvent effects on the absorption maxima shifts of the investigated compounds were successfully evaluated by using the Kamlet–Taft solvent parameter set. Solvent dipolarity/polarizability effect is the principal factor which influences the blue shift of absorption maxima indicating higher polarity/polarizability of the molecule in the ground state. The solvent hydrogen bonding acidity and basicity are less important effects which contribute to higher stabilization of the molecules in excited state. In general, according to presented results the extent of conjugational ability of the π-electron densities through localized or delocalized π-electronic systems of N-(substituted phenyl)-2-chloroacetamides determines their solvatochromic behavior.
The LFER analysis applied on νmax implies that solvent effects have significant influence on the transmission mode of substituent effects. Positive solvatochromism was found for all solvents. The LFER analysis of SCS data shows normal substituent effect at all carbons, and highest susceptibility to substituent effects was found for C1 carbon.
Quantum chemical calculations of the optimal geometries were obtained by the use of B3LYP functional with 6-311+G(d,p) basis set. Theoretical calculations and experimental results gave an insight into the influence of the molecular conformation on the transmission mode of substituent effects, and help in the analysis of the solvent–solute interactions. In addition, Bader’s analysis showed that similar processes of electronic excitation are operative in most of investigated compounds with most distinctive ICT observed in para-substituted acetyl- and cyano-derivatives, moderate process in meta-substituted compounds, while in halogen substituted compounds negligible ICT was noticed.
Acknowledgments
This work was supported by the Ministry of Education, Science and Technological development of the Republic of Serbia (Project 172013).
References
- Antić-Jovanović, A., 2006. Molekulska spektroskopija-spektrohemijski aspekti, Fakultet za fizičku hemiju, Beograd.
- Regarding a generalized scale of solvent polarities. J. Am. Chem. Soc.. 1977;99:8325-8327.
- [Google Scholar]
- Analysis of the overlapping bands in UV–Vis absorption spectroscopy. Appl. Spectrosc.. 1993;47:1030-1035.
- [Google Scholar]
- Integrated computational strategies for UV/vis spectra of large molecules in solution. Chem. Soc. Rev.. 2007;36:1724-1731.
- [Google Scholar]
- Synthesis and biological evaluation of new N-(2-hydroxy-4(or 5) nitro/aminophenyl) benzamides and phenylacetamides as antimicrobial agents. Bioorg. Med. Chem.. 2007;15:2032-2044.
- [Google Scholar]
- Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G.A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H.P., Izmaylov, A.F., Bloino, J., Zheng, G., Sonnenberg, J.L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J.A., Jr., Peralta, J.E., Ogliaro, F., Bearpark, M., Heyd, J.J., Brothers, E., Kudin, K.N., Staroverov, V.N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., Rega, N., Millam, J.M., Klene, M., Knox, J.E., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Martin, R.L., Morokuma, K., Zakrzewski, V.G., Voth, G.A., Salvador, P., Dannenberg, J.J., Dapprich, S., Daniels, A.D., Farkas, Ö., Foresman, J.B., Ortiz, J.V., Cioslowski, J., Fox, D.J., 2009. Gaussian 09, Revision D.01, Gaussian Inc., Wallingford, CT.
- Gincburg, A.L., Zigangirova, N.A., Tokarskaya, E.A., Zorina, V.V., Tartakovskaya, D.I., Krayushkin, M.M., Yarovenko, V.N., Zayakin, E.S., 2010. Preparation of bioactive oxamic acid thiohydrazide hydrazone derivatives for suppression of pathogenic bacteria and process for their production. WO 2010036147.
- Extension of Gaussian-2 (G2) theory to bromine-and iodine-containing molecules: use of effective core potentials. J. Chem. Phys.. 1995;103:1878-1885.
- [Google Scholar]
- Discovery, development, and current status of the chloroacetamide herbicides. Weed Sci.. 1974;22:541-545.
- [Google Scholar]
- The effect of structure upon the reactions of organic compounds. Benzene derivatives. J. Am. Chem. Soc.. 1937;59:96-103.
- [Google Scholar]
- Exploring QSAR, ACS Professional Reference Book. Washington: American Chemical Society; 1995.
- Hayakawa, M., Kido, Y., Nigawara, T., Okumura, M., Kanai, A., Maki, K., Amino, N., 2010. Phenylacetamide derivative. U.S. Patent 20100286171 A1.
- Synthesis and in vitro trichomonicidal, giardicidal and amebicidal activity of N-acetamide(sulfonamide)-2-methyl-4-nitro-1H-imidazoles. Eur. J. Med. Chem.. 2009;44:2975-2984.
- [Google Scholar]
- Hladik, M.L., Lynn Roberts, A., Bouwer, E.J., 2006. Chloroacetamide herbicides and their transformation products in drinking water. Awwa Research Foundation and U.S. Environmental Protection Agency, Washington
- EI/MS/MS spectra of N-monosubstituted cyanoacetamides. Chem. Ind. Chem. Eng. Q.. 2010;16:387-397.
- [Google Scholar]
- Chloroacetamide Organic Syntheses. Vol vol. 7. Weinheim: Wiley-VCH; 1927. p. :16-17.
- N-(2-hydroxy phenyl) acetamide inhibits inflammation-related cytokines and ROS in adjuvant-induced arthritic (AIA) rats. Int. Immunopharmacol.. 2010;10:900-905.
- [Google Scholar]
- Degradation Herbicides. New York: Marcel Dekker Inc; 1969.
- Synthesis and antitumor activity of some benzofuryl-substituted 1,2,4-triazoles. Pharm. Chem. J.. 2009;43:242-244.
- [Google Scholar]
- The solvatochromic comparison method. I. The β-scale of solvent hydrogen-bond acceptor (HBA) basicities. J. Am. Chem. Soc.. 1976;98:377-383.
- [Google Scholar]
- Linear solvation energy relationships. Part 3. Some reinterpretations of solvent effects based on correlations with solvent π and α values. J. Chem. Soc. Perk. T. II 1979:349-356.
- [Google Scholar]
- An examination of linear solvation energy relationships. Prog. Phys. Org. Chem.. 1981;13:485-630.
- [Google Scholar]
- Ab initio calculation of the anisotropy effect of multiple bonds and the ring current effect of arenes—application in conformational and configurational analysis. J. Chem. Soc. Perkin Trans. II 2001:1893-1899.
- [Google Scholar]
- Mannhold R., Krogsgaard-Larsen P., Timmerman H., eds. QSAR: Hansch Analysis and Related Approaches. Weinheim: Wiley-VCH; 1993.
- A graphics program for the analysis and display of molecular dynamics trajectories. J. Mol. Graph.. 1992;10:33-34.
- [Google Scholar]
- Synthesis and biological activity of some 1,3-dihydro-2H-3-benzazepin-2-ones with a piperazine moiety as bradycardic agents. Arch. Pharm.. 2010;343:114-119.
- [Google Scholar]
- Degradation of pesticides by UV and UV-based advanced oxidation processes: state-of-the-art. Acta Sci. Circums.. 2011;31:225-235.
- [Google Scholar]
- The properties of organic liquids that are relevant to their use as solvating solvents. Chem. Soc. Rev.. 1993;22:409-416.
- [Google Scholar]
- Substituent effect on IR, 1H- and 13C NMR spectral data in N-(substituted phenyl)-2-cyanoacetamides: a correlation study. Chem. Ind. Chem. Eng. Q.. 2013;19:67-78.
- [Google Scholar]
- Solvent and structural effects on the UV absorption spectra of N-(substituted phenyl)-2-cyanoacetamides. Spectrochim. Acta A. 2014;117:568-575.
- [Google Scholar]
- Alkylation of N-substituted 2-phenylacetamides. J. Serb. Chem. Soc.. 2004;69:711-736.
- [Google Scholar]
- Substituent and solvent effects on the UV/vis absorption spectra of 5-(3- and 4-substituted arylazo)-4,6-dimethyl-3-cyano-2-pyridones. Chem. Phys. Lett.. 2006;418:223-229.
- [Google Scholar]
- Synthesis and analgesic effects of 2-(2-carboxyphenylsulfanyl)-N-(4-substitutedphenyl)acetamide derivatives. Med. Chem. Res.. 2011;20:152-157.
- [Google Scholar]
- N-(4-((E)-3-arylacryloyl)phenyl)acetamide derivatives and their antileishmanial activity. J. Brazil. Chem. Soc.. 2013;24:1685-1690.
- [Google Scholar]
- Mass spectral study of N-substituted 2-phenylacetamides. Hem. Ind.. 1998;52:490-496.
- [Google Scholar]
- Solvent and Solvent Effects in Organic Chemistry (third ed.). Weinheim: Wiley-VCH; 2003. Updated and enlarged
- The general applicability of a fixed scale of inductive effects. II. Inductive effects of dipolar substituents in the reactivities of m- and p-substituted derivatives of benzene1,2. J. Am. Chem. Soc.. 1958;80:2436-2443.
- [Google Scholar]
- Thirty years of continuum solvation chemistry: a review, and prospects for the near future. Theor. Chem. Acc.. 2004;112:184-203.
- [Google Scholar]
- Reversal of substituent effect on electronic absorption spectra of N-(4-substituted phenyl)-benzamides in different solvents. Indian J. Chem. B. 2002;41:206-210.
- [Google Scholar]
- Solvent and substituent effect on electronic spectra of N-(4-substituted phenyl)-2,3-diphenylpropanamides. Arkivoc 2006:80-89.
- [Google Scholar]
- Excited state surfaces in density functional theory: a new twist on an old problem. J. Chem. Phys.. 2009;131:091101/1-091101/4.
- [Google Scholar]
- A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP) Chem. Phys. Lett.. 2004;393:51-57.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.arabjc.2015.09.008.
Appendix A
Supplementary material
Supplementary data 1
Supplementary data 1




