

Translate this page into:
Prospecting native and analogous peptides with anti-SARS-CoV-2 potential derived from the trypsin inhibitor purified from tamarind seeds
⁎Corresponding author at: Biochemistry and Molecular Biology Postgraduate Program, Biosciences Center, Federal University of Rio Grande do Norte, Natal, RN 59078970, Brazil. ana.morais@ufrn.br (Ana Heloneida de Araújo Morais)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The study aimed to prospect in silico native and analogous peptides with anti-SARS-CoV-2 potential derived from the trypsin inhibitor purified from tamarind seeds (TTIp). From the most stable theoretical model of TTIp (TTIp 56/287), in silico cleavage was performed for the theoretical identification of native peptides and generation of analogous peptides. The anti-SARS-CoV-2 potential was investigated through molecular dynamics (MD) simulation between the peptides and binding sites of transmembrane serine protease 2 (TMPRSS2), responsible for the entry of SARS-CoV-2 into the host cell. Five native and analogous peptides were obtained and validated through chemical and physical parameters. The best interaction potential energy (IPE) occurred between TMPRSS2 and one of the native peptides obtained by cleavage with trypsin and its analogous peptide. Thus, both peptides showed many hydrophobic residues, a common physical–chemical property among the peptides that inhibit the entry of enveloped viruses, such as SARS-CoV-2, present in specific drugs to treat COVID-19.
Keywords
Antitrypsin activity
COVID-19
Protein
Plant bioactive compounds

Data availability
All data generated or analyzed during this study are included in this article. Further inquiries can be directed to the corresponding author.
1 Introduction
COVID-19, an infectious disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has spread rapidly since 2019, as it is transmitted through the airways, becoming a serious threat to health systems around the world. Its consequences range from self-limiting flu to fulminant pneumonia, culminating in respiratory failure and death (Del Rio and Malani, 2020). In December, the World Health Organization announced that this virus had already been responsible for 6.630.082 deaths worldwide (World Health Organization, 2022).
Recently it was observed that the SARS-CoV-2 genome encodes 3C-like protease (3CL pro), a cysteine-type protease (Pillaiyar et al., 2020), formally known as C30 endopeptidase, a chymotrypsin-like (Rota et al., 2003). This protease is considered a key component in the processing of viral polyproteins (Yu et al., 2020) that release non-structural proteins and structural proteins, such as spike (S), envelope (E), membrane (M) and nucleocapsid (N) glycoproteins (Forni et al., 2017), being important in viral ribonucleic acid (RNA) replication and transcription (Liu et al., 2020).
Hoffmann et al. (2020) observed that transmembrane serine protease 2 (TMPRSS2) is required to promote the entry of SARS-CoV-2 into host cells. Besides, the access of the virus into human cells occurs through glycoprotein S, which is found on the surface of the virus. Additionally, Iwata-Yoshikawa et al. (2022) state that TMPRSS2 is a critical virulence factor even for the Omicron variant.
Briefly, TMPRSS2-mediated protein S cleavage may trigger membrane fusion to release the viral genome into the cytoplasm (Belouzard et al., 2012). The SARS-CoV-2 infects cells via a critical protein–protein interaction (PPI) between the SARS-CoV S-protein receptor binding domain (RBD) with the protease domain (PD) of the human cell surface receptor angiotensin-converting enzyme 2 (ACE-2) (Morgan et al., 2021; Ramirez-Acosta et al., 2022).
Some cells do not express TMPRSS2, or expression is very low (Bartolomeo et al., 2022). In this case, the fusion process can occur by cathepsin L in endosomes after clathrin-dependent endocytosis (Chen et al., 2021). Thus, it is highlighted that several cell surface molecules are involved in the entry of SARS-CoV-2, including the main ACE-2 receptor, TMPRSS2, and other potential helper receptors or cofactors (Peng et al., 2021; Walls et al., 2020).
Once inside cells, SARS-CoV-2 hijacks the protein-making machinery through cross-talk with the autophagic machinery (Hui et al., 2021) and mitochondrial metabolism (Gatti et al., 2020) for its replication.
Because of this, molecules from different natural sources have been used as a source of new drug prototypes, responsible for most of the chemical products launched by the pharmaceutical industry to treat many diseases (Jackson et al., 2018). Researchers worldwide have tried identifying new therapeutic options to control COVID-19, such as monoclonal antibodies, anticoagulants, and the most diverse antivirals: remdesivir, molnupiravir, and nirmatrelvir (Drożdżal et al., 2021).
In silico prospecting of these new drugs has been carried out through numerous studies of computer simulations, either by docking or MD, interacting different substances with multiple targets, such as 3CLpro and TMPRSS2 proteases, which are related to the infection by SARS-CoV-2 (Jukič et al., 2020; Molavi et al., 2021).
New substances and drugs on the market have been investigated regarding their anti-SARS-CoV-2 potential. Therefore, the Food and Drug Administration (FDA) has authorized the use of the antivirals Veklury™ (remdesivir), Paxlovid™ (nirmatrelvir and ritonavir), and Lagevrio™ (molnupiravir) for the treatment of COVID-19 in adults. Paxlovid™ and Veklury™ were also allowed for pediatric patients. However, Lagevrio™ is not authorized for use in patients under the age of 18 years because it may affect bone and cartilage growth (Food and Drug Administration, 2022).
Among the natural products, protease inhibitors are widely studied, especially trypsin inhibitors, extracted from seeds, purified, characterized, and evaluated for diverse functionalities and bioactivities (Morais et al., 2021). Some examples are trypsin inhibitors that inhibit other serine proteases, such as chymotrypsin and human neutrophil elastase (Ascenzi et al., 2003; Lima et al., 2017). TMPRSS2 belongs to the same family of serine proteases, so these trypsin inhibitors are potential agents of TMPRSS2 inhibition.
Additionally, several works have been published with the use of antiviral peptides, protease inhibitors, antioxidant peptides, and anti-toxin peptides, among others, which are registered in Antimicrobial Peptide Database and coming from natural sources, in addition to containing less than 100 amino acid residues (Wang et al., 2016).
In this scenario, studies indicate that the bioactive compounds of tamarind seeds (Tamarindus indica L.) exhibit valuable bioactivities such as antioxidant, antidiarrheal and anti-inflammatory activity (Gupta and Gupta, 2016; Waqas et al., 2015). Recently, antimicrobial activity was observed in silico of a peptide derived from TTIp (Oliveira et al., 2023), which has been extensively studied by the group NutriSBioativoS from UFRN/Brazil.
Recent studies have shown that TTI acts on different mechanisms involving obesity control and its associated effects, such as the production of hormones related to satiety, affecting the central nervous system and the small intestine; reduced food consumption and weight gain; improvement of lipid profile; and reduction of the inflammatory process related to obesity, regardless of weight loss (Lima et al., 2019).
Medeiros et al. (2018) purified and characterized the trypsin inhibitor extracted from tamarind seeds, which showed a mass of 19 kDa by mass spectrometry. Through partial primary sequencing, 54 amino acid residues were found. Subsequently, the inhibitor, TTIp, had its sequencing completed, presenting 184 amino acid residues. The TTIp modeling was performed by homology, obtaining five three-dimensional models that were validated that allowed the development of in silico studies (Medeiros et al., 2021). The most stable theoretical model, number 56 and conformation 287 (TTIp 56/287), have been widely analyzed in silico through MD simulations regarding the interaction potential energy with several protein structures, including membrane receptors, bacterial lipid bilayer, and trypsin, a serine protease, as well as TMPRSS2 (Costa et al., 2022; Medeiros et al., 2021; Oliveira et al., 2023).
Thus, short peptides of the entire protein are used to inhibit the larger protein (Krizsan et al., 2015). Considering that TTIp is a large molecule composed of 184 amino acid residues (Medeiros et al., 2021) and the possibility that it acts by inhibiting proteases such as TMPRSS2, the prospection of peptides derived from this protein is also a promising strategy, given the properties already demonstrated for it.
Therefore, our hypothesis to be investigated is that native peptides and their analogs from TTIp 56/287 may interact with TMPRSS2. Thus, this study aimed to prospect peptides from TTIp and evaluate their potential for interaction with this cell surface protease, which will contribute to understanding the numerous functionalities attributed to the TTIp.
2 Material and methods
The TTI with a defined sequence and three-dimensional modeling (TTIp 56/287), according to Medeiros et al. (2021), was subjected to a theoretical cleavage using the ExPASy server and the PeptideCutter analysis tool for the identification of native peptides (https://web.expasy.org/peptide_cutter). The proteinases used, individually, were pepsin, trypsin, and chymotrypsin, selected according to Kuba et al. (2005). The sequence and application of cleavage products from the theoretical model of TTIp 56/287 and its analogues with potential inhibitory activity on TMPRSS2 are patented processes under protocol number BR 10 2022 020330 0.
The length of the obtained peptides was evaluated, and those with at least ten amino acids in their primary structure were selected. The prediction of the three-dimensional structure of the selected peptides was obtained using the trRosetta server (https://yanglab.nankai.edu.cn/trRosetta/). The theoretical models were validated using the MolProbity server, with subsequent analysis of parameters, such as the Ramachandran plot, bond sizes, and angles, to optimize each theoretical structure obtained (https://molprobity.biochem.duke.edu/).
The search for similarity between selected native peptides derived from TTIp 56/287 and peptides present in other plant species was carried out by searching for peptides from protease inhibitors in the literature (Hellinger and Gruber, 2019; Schütz et al., 2020). Alignment of these sequences with native molecules was performed using the ClustalW2 server (https://www.ebi.ac.uk/Tools/msa/clustalw2/) to evaluate conserved residues.
Subsequently, rational planning was carried out to obtain analogous peptides,with modifications made to the native peptides to make them more nonpolar, maintaining their hydrophobic moment and making the net charge more negative. This choice, as well as the option of amino acids inserted in the analogous peptides, was based on the amino acids of the catalytic triad and the binding sites of the substrate of the TMPRSS2, responsible for the entry of the SARS-CoV-2 in the host cell (Vardhan and Sahoo, 2022). The number of residues present in the native sequences was maintained.
After the rational planning of the molecules, the analogous peptides with the same hydrophobic moment as the native peptides were selected. Then, the prediction of the peptide’s three-dimensional structure was obtained with the validation of the models, as previously mentioned. After that, the models were analyzed for stability using the Molecular Orbital PACkage (MOPAC) program, and those that showed better stability were chosen to be submitted to MD.
Subsequently, the analogous molecules were submitted to alignment with the primary sequences of the protease inhibitors. Then, the physicochemical characteristics (hydrophobicity, hydrophobic moment, and net charge) of all molecules - native and analogous - and the helical wheel projections were obtained using the Heliquest server ( https://heliquest.ipmc.cnrs.fr/cgi-bin/ComputParamsV2.py).
To build the TMPRSS2 model, a serine protease (PDB ID: 2OQ5) was used as a template, using the ExPASy server and the Swiss-Model analysis tool ( https://swissmodel.expasy.org/), and the model validation was performed using the MolProbity server.
The theoretical model of TMPRSS2 created in this study was attached to the lipid bilayer, built with the CHARMM GUI server ( https://www.charmm-gui.org/) to simulate a mammalian membrane together with the membrane protein TMPRSS2. 1:1 M ratio mixture of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS) was used to create this model. The hydrophobic amino acids 84–106 of TMPRSS2 were placed internally in the lipid bilayer.
All MD simulations were performed using the GROMACS software version 2018.4, implemented with the CHARMM36 force field. TIP3P water molecules (Jorgensen et al., 1983) were used to solvate the simulated systems, which were neutralized by adding counter-ions 256 sodium and 111 chloride ions.
The leap-frog algorithm was used to integrate the motion equations with a time interval of 2.0 fs. Long-range interactions were modeled using the particle-mesh Ewald sum (PME) method, with a cut-off radius of 1.2 nm. Van der Waals interactions were calculated using the same tool. Bonds involving hydrogen atoms were kept constant using the LINear Constraints Solver (LINCS) algorithm (Hess et al., 1997).
In all MD simulations, the Nosé–Hoover thermostat set the system temperature at 310 K. The pressure in value of 1.0 bar was controlled using the Parrinello–Rahman barostat in the number of particles, pressure, and temperature (NPT) ensemble simulations. The geometry of the systems was minimized using the steepest descent algorithm (Arfken et al., 2012) with 10.000 steps and a tolerance of 10 kJ.mol−1.nm−1 followed by the conjugate gradient algorithm (Hestenes and Stiefel, 1952) with 10.000 steps and tolerance of 10 kJ.mol−1.nm−1. Two 200 ps equilibrium dynamics were performed, the first using the number of particles, volume, and temperature (NVT) ensemble and the second using the NPT ensemble. Finally, 200 ns production MD simulations using the NVT ensemble were performed for each system to determine the adsorptions of peptides validated with TMPRSS2.
The interaction potential energy (IPE) calculation was used (Freitas et al., 2017) to analyze the energy between the peptides and TMPRSS2. IPE is the total interaction between two groups, in this case, between peptides and TMPRSS2. The IPE calculation includes the sum of contributions from van der Waals and Coulomb (electrostatic) energies, as seen in equation (1) below:
IPEi,j is the interaction energy between a group of atoms i and a group of atoms j, represented in this work by the group of atoms of the system (TMPRSS2 and each selected peptide); Ni and Nj are the total numbers of atoms in the groups i e j, respectively. VvdW (rij) are the Van der Waals contributions, and Velec (rij) are the electrostatic contributions. This method is commonly used to define the interaction energies between protein–ligand and protein–protein (Ribeiro et al., 2016).
3 Results
Individual cleavage with proteolytic enzymes generated 41 peptides by pepsin, 24 by trypsin, and 109 by chymotrypsin. The five longest peptides were used to obtain the three-dimensional structure: two by cleavage with pepsin, two by cleavage with trypsin, and one by cleavage with chymotrypsin (Fig. 1). The peptide obtained by cleavage with chymotrypsin had a length of less than ten amino acids, requiring the addition of glycine to enable the use of the trRosetta tool to obtain the three-dimensional structure of the peptide.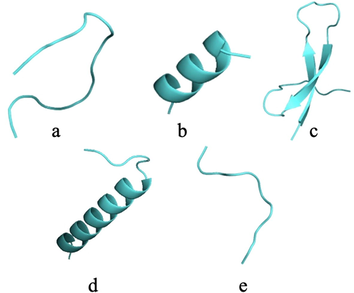
Three-dimensional structure of native peptides obtained by cleavage of TTIp 56/287. a: Pep1 - Peptide obtained by cleavage with pepsin. b: Pep2 - Peptide obtained by cleavage with pepsin. c: Tryp1 - Peptide obtained by cleavage with trypsin. d: Tryp2 - Peptide obtained by cleavage with trypsin. e: Chym1 - Peptide obtained by cleavage with chymotrypsin. TTIp 56/287: trypsin inhibitor purified from tamarind seeds (TTIp), model number 56, in conformation number 287.
Findings in the literature made it possible to align the primary sequences of native peptides from TTIp 56/287 and its analogues with sequences of peptides from plant protease inhibitors and the polypeptide present in bovine lung, thus identifying conserved residues. Among these plant protease inhibitors, the most remarkable similarities occurred between the native peptides derived from TTIp 56/287 and peptides from Arabidopsis thaliana and Cucurbita maxima, which strongly inhibit serine proteases (Hellinger and Gruber, 2019), as well as the trypsin inhibitor extracted from tamarind seeds (Tamarindus indica L.) (Carvalho et al., 2020).
The alignment between the primary structure of the validated native TTIp 56/287 peptides and peptide sequences from plant and bovine lung protease inhibitors can be observed (Supplementary Fig. S1).
There are also conserved residues between the native peptides Pep1 (LTVSQTPIDIPIGL), Tryp1 (GGGLGLSNDDDGNCPLTVSQTPIDIPIGLPVR), Tryp2 (ISHITTALSLNIEFTIAPACAPKPAR), Chym1 (TPIDIPIGLG), and the bovine lung aprotinin polypeptide, which is a natural inhibitor of broad-ranging serine proteases, able to inhibit TMPRSS2 (Schütz et al., 2020). However, no conserved residues were found between Pep2 (a peptide that did not interact with TMPRSS2) and aprotinin. Therefore, the conserved amino acids between Tryp1 (best interaction potential energy with TMPRSS2) and aprotinin (composed of Glycine and Aspartic Acid) may be strongly related to the interaction with TMPRSS2 and, consequently, to the potential of these peptides to inhibit this protease.
Regarding the peptides analogues to the native peptides of TTIp 56/287, the rational planning of the molecules led to the development of 2.658 new analogous peptides. We selected 32 that had the same hydrophobic moment as the native peptides. But, we chose the five most stable peptides (Fig. 2) to perform the MD.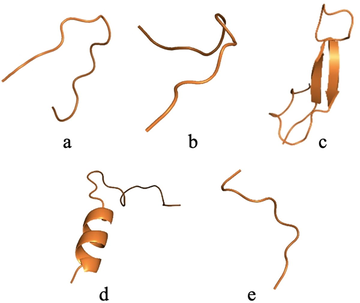
Three-dimensional structure of analogous peptides obtained by cleavage of TTIp 56/287. a: ID 32 – Peptide analogue to Pep1. b: ID 76 - Peptide analogue to Pep2. c: ID 63 - Peptide analogue to Tryp1. d: ID 336 - Peptide analogue to Tryp2. e: ID 101 - Peptide analogue to Chym1. TTIp 56/287: trypsin inhibitor purified from tamarind seeds (TTIp), model number 56, in conformation number 287.
We observed the alignment between the primary structures of the peptides analogues to the validated TTIp 56/287 peptides and the sequences of peptides derived from plant and bovine lung protease inhibitors (Supplementary Fig. S2).
The greatest similarities occurred between analogues peptides with the Arabidopsis thaliana peptides. In general, except for ID 76 (SSRARISGLTTA), all peptides showed similarity with peptides from Cucurbita maxima, Ecballium elaterium, and with Kalata B1 from the Oldenlandia affinis plant.
It should be noted that there were conserved amino acid residues between the peptides ID 63 (Aspartic Acid and Cysteine), ID 76 (Glycine and Leucine), and ID 336 (Phenylalanine and Cysteine) and amino acid residues present in bovine lung aprotinin.
Below are comparative data of the predicted physicochemical properties in silico for the native sequences of the TTIp 56/287 peptides and the analogous peptides (Table 1). By observing the hydrophobicity of the peptides prospected in this study, we found Tryp1 (GGGLGLSNDDDGNCPLTVSQTPIDIPIGLPVR) and ID 63 (GGGLGLSNDDDGNCPLTVSQTPIDLPIGLPVR), which exhibited better interaction with TMPRSS2, containing a majority of hydrophobic residues, especially Leucine, Isoleucine, Cysteine, and Valine.
ID
Hydrophobicity
Hydrophobic moment
Surface charge
Pep1
0.782
0.235
−1
ID 32
0.775
0.235
−1
Pep2
0.228
0.186
2
ID 76
0.208
0.185
2
Tryp1
0.437
0.106
−3
ID 63
0.434
0.106
−3
Tryp2
0.586
0.067
1
ID 336
0.621
0.067
1
Chym1
0.803
0.303
−1
ID 101
0.802
0.303
−1
ID: identification; Pep1 – Peptide obtained by cleavage with pepsin; Pep2 – Peptide obtained by cleavage with pepsin; Tryp1 – Peptide obtained by cleavage with trypsin; Tryp2 – Peptide obtained by cleavage with trypsin; Chym1 – Peptide obtained by cleavage with chymotrypsin; ID 32 – Peptide analogue to Pep1; ID 76 – Peptide analogue to Pep2; ID 63 – Peptide analogue to Tryp1; ID 336 – Peptide analogue to Tryp2; ID 101 – Peptide analogue to Chym1; TTIp 56/287: trypsin inhibitor purified from tamarind seeds (TTIp), model number 56, in conformation number 287.
Helical wheel projections are obtained from native and analogous peptides (Fig. 3 and Fig. 4).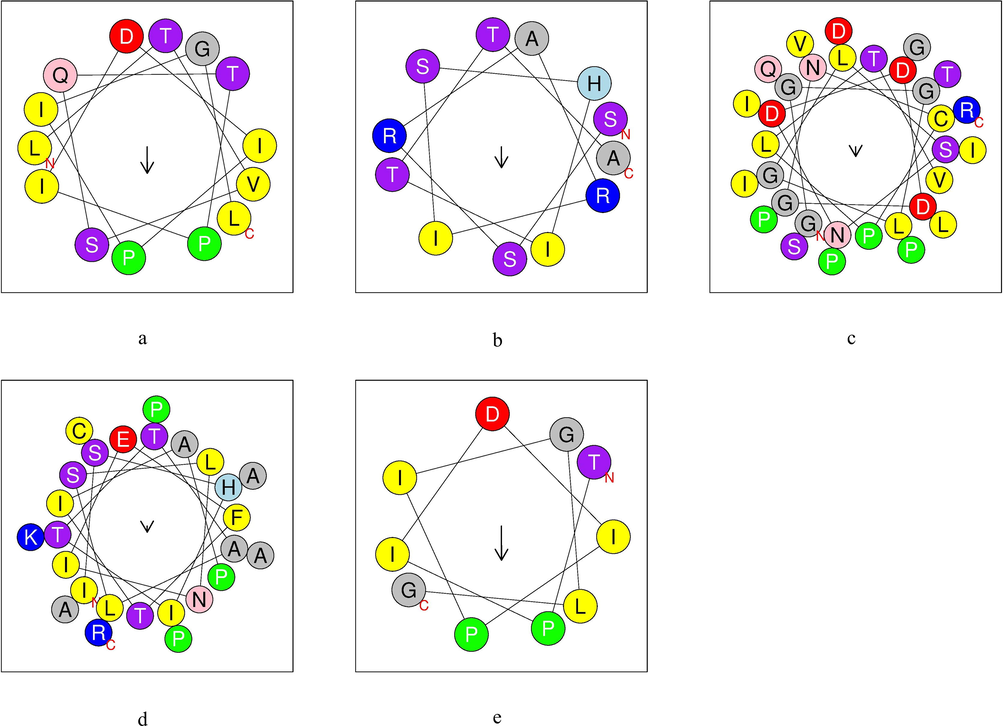
α-helix structure of native peptides obtained by cleavage of TTIp 56/287. a: Pep1 – Peptide obtained by cleavage with pepsin; b: Pep2 – Peptide obtained by cleavage with pepsin; c: Tryp1 – Peptide obtained by cleavage with trypsin; d: Tryp2 – Peptide obtained by cleavage with trypsin; e: Chym1 – Peptide obtained by cleavage with chymotrypsin; Yellow – hydrophobic residues; purple – serine and threonine residues; dark blue – basic residues; red – acid residues; pink – asparagine and glutamine residues; gray – alanine and glycine residues; light blue – histidine residues; green – proline residues. TTIp 56/287: trypsin inhibitor purified from tamarind seeds (TTIp), model number 56, in conformation number 287.
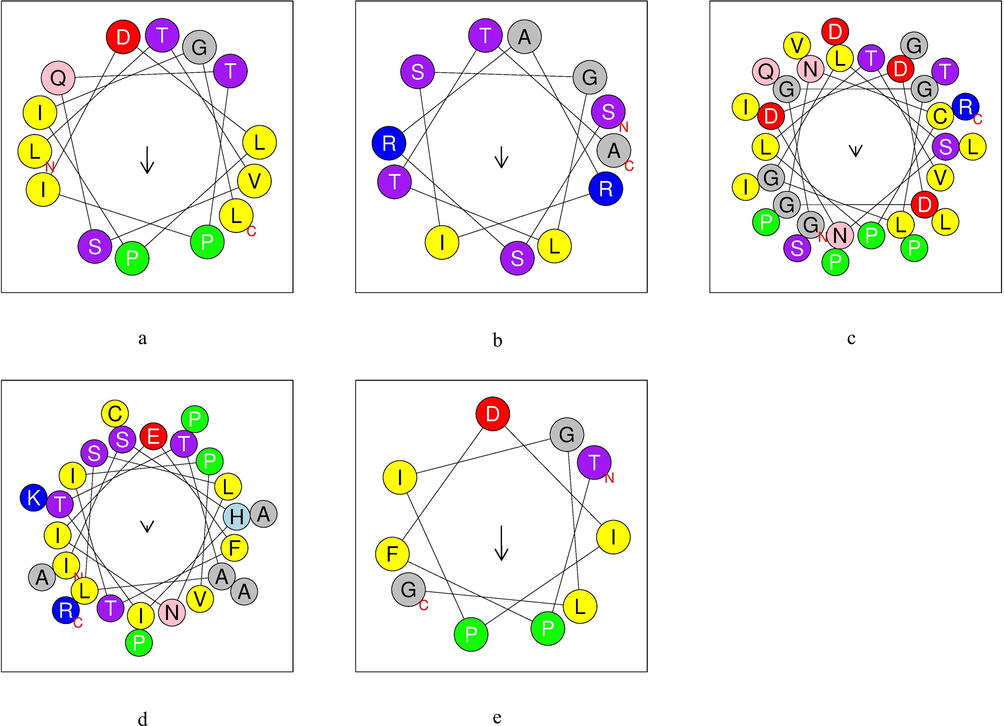
α-helix structure of analogous peptides obtained by cleavage of TTIp 56/287. a: ID 32 – Peptide analogue to Pep1; b: ID 76 – Peptide analogue to Pep2; c: ID 63 – Peptide analogue to Tryp1; d: ID 336 – Peptide analogue to Tryp2; e: ID 101 – Peptide analogue to Chym1. Yellow – hydrophobic residues; purple – serine and threonine residues; dark blue – basic residues; red – acid residues; pink – asparagine and glutamine residues; gray – alanine and glycine residues; light blue – histidine residues; green – proline residues. TTIp 56/287: trypsin inhibitor purified from tamarind seeds (TTIp), model number 56, in conformation number 287.
The theoretical model of TMPRSS2 created in this study can be seen below (Supplementary Fig. S3).
The final structural analysis of the interaction between theoretical models of native TTIp 56/287 peptides and their analogues after 200 ns simulation with TMPRSS2 is shown below (Fig. 5 and Fig. 6).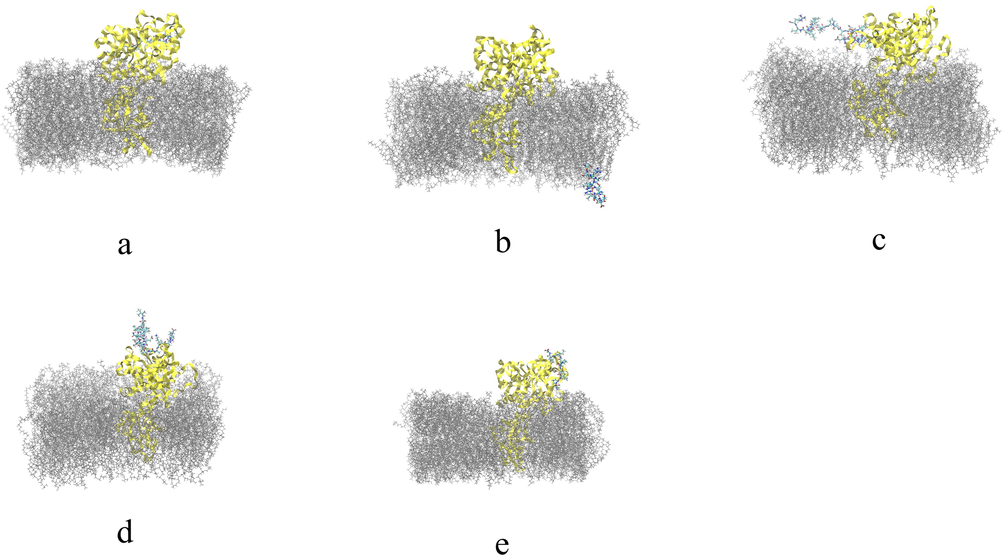
Final structural analysis of the interaction between theoretical models of native peptides obtained by cleavage of TTIp 56/287 with TMPRSS2. a: Pep1 – Peptide obtained by cleavage with pepsin; b: Pep2 – Peptide obtained by cleavage with pepsin; c: Tryp1 – Peptide obtained by cleavage with trypsin; d: Tryp2 – Peptide obtained by cleavage with trypsin; e: Chym1 – Peptide obtained by cleavage with chymotrypsin. TTIp 56/287: trypsin inhibitor purified from tamarind seeds (TTIp), model number 56, in conformation number 287. TMPRSS2: transmembrane serine protease 2.
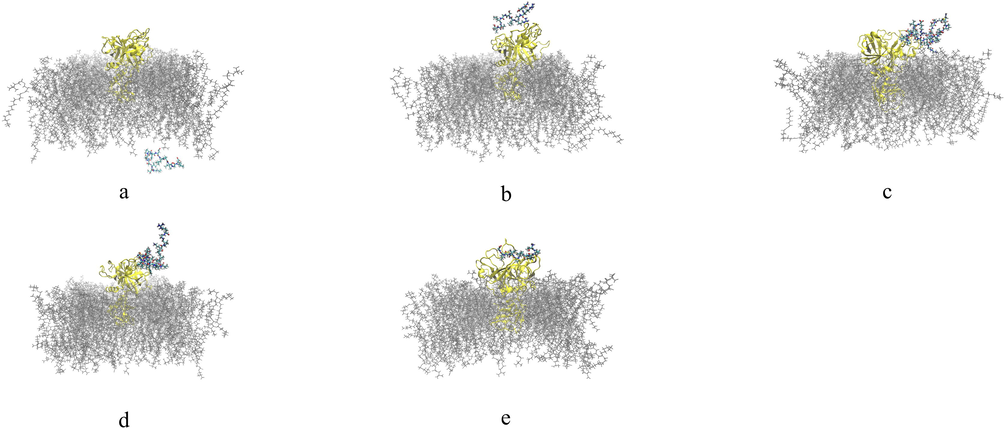
Final structural analysis of the interaction between theoretical models of analogous peptides obtained by cleavage of TTIp 56/287 with TMPRSS2. a: ID 32 – Peptide analogue to Pep1; b: ID 76 – Peptide analogue to Pep2; c: ID 63 – Peptide analogue to Tryp1; d: ID 336 – Peptide analogue to Tryp2; e: ID 101 – Peptide analogue to Chym1. TTIp 56/287: trypsin inhibitor purified from tamarind seeds (TTIp), model number 56, in conformation number 287. TMPRSS2: transmembrane serine protease 2.
The IPE values of the 200 ns simulation of native peptides with TMPRSS2 (Fig. 7) show the best interaction capacity of Tryp1 (peptide obtained by cleavage with trypsin (IPE = −287.48 kJ.mol−1), followed by Tryp2 (peptide also obtained by cleavage with trypsin. IPE = −267.61 kJ.mol−1), Chym1 (peptide obtained by cleavage with chymotrypsin. IPE = −195.51 kJ.mol−1), and Pep1 (peptide obtained by cleavage with pepsin. IPE = −171.71 kJ.mol−1). Pep2 (peptide obtained by cleavage with pepsin) showed no interaction. IPE = 0.00 kJ.mol−1. Among the analogues, the peptide ID 63, with a sequence analogue to the peptide Tryp1, showed the best interaction capacity (IPE = −293.49 kJ.mol−1), followed by ID 101, analogue to the peptide Chym1 (IPE = −270.71 kJ.mol−1); ID 76, analogue to Pep2 (IPE = −248.22 kJ.mol−1); ID 336, analogue to Tryp2 (IPE = −158.58 kJ.mol−1); and ID 32, analogue to Pep1 (IPE = −42.15 kJ.mol−1).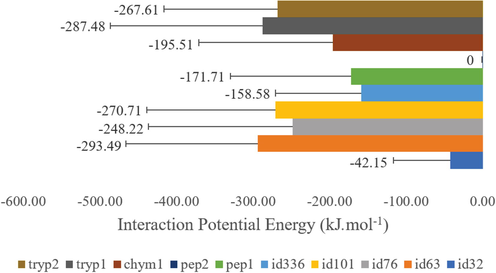
Interaction potential energy of native and analogous peptides obtained by cleavage of TTIp 56/287. Pep1 – Peptide obtained by cleavage with pepsin; Pep2 – Peptide obtained by cleavage with pepsin; Tryp1 – Peptide obtained by cleavage with trypsin; Tryp2 – Peptide obtained by cleavage with trypsin; Chym1 – Peptide obtained by cleavage with; ID 32 – Peptide analogue to Pep1; ID 76 – Peptide analogue to Pep2; ID 63 – Peptide analogue to Tryp1; ID 336 – Peptide analogue to Tryp2; ID 101 – Peptide analogue to Chym1. TTIp 56/287: trypsin inhibitor purified from tamarind seeds (TTIp), model number 56, in conformation number 287.
The root-mean-square deviation (RMSD) analysis was performed based on C-α as a reference during the whole trajectory, utilizing the structure initial of protein as starting point.
The mean RMSD values for the TMPRSS2-Pep1, TMPRSS2-Pep2, TMPRSS2-Chym1, TMPRSS2-Tryp1, and TMPRSS2-Tryp2 complexes were, respectively, 1.23 nm, 1.90 nm, 0.94 nm, 1.21 nm, and 0.99 nm. In addition, these complexes reached equilibrium at 80–200 ns. On the other hand, the mean values of RMSD for the TMPRSS2-ID32, TMPRSS2-ID63, TMPRSS2-ID76, TMPRSS2-ID101, and TMPRSS2-ID336 complexes were, respectively, 1.54 nm, 0.85 nm, 0.46 nm, 0.45 nm, and 0.68 nm. Regarding the equilibrium time, the TMPRSS2-ID32, TMPRSS2-ID101, and TMPRSS2-ID336 complexes reached 120–200 ns, 100–200 ns, and 10–100 ns, respectively. The TMPRSS2-ID63 and TMPRSS2-ID76 complexes reached equilibrium time in 25–200 ns (Fig. 8).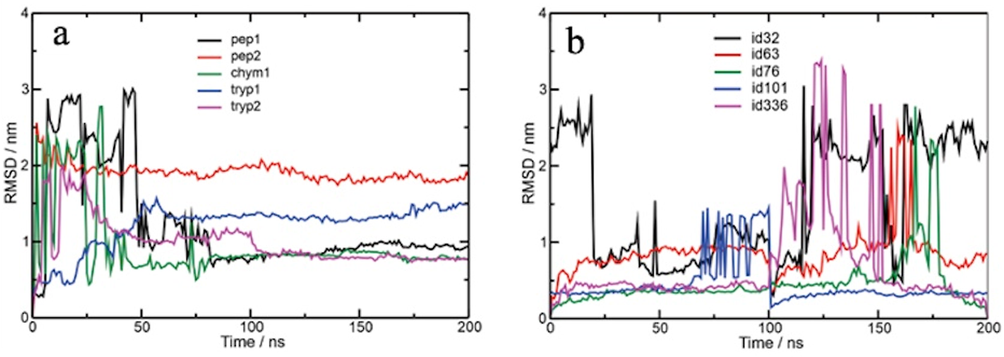
Root-mean-square deviation (RMSD) of molecular dynamics simulations of native and analogous peptides obtained by cleavage of TTIp 56/287 with TMPRSS2. a: RMSD of the native peptides; the black line represents Pep1 obtained by cleavage of TTIp 56/287 with pepsin; the red line represents Pep2 obtained by cleavage of TTIp 56/287 with pepsin; the green line represents Chym1 obtained by cleavage of TTIp 56/287 with chymotrypsin; the blue line represents Tryp1 obtained by cleavage of TTIp 56/287 with trypsin, and the purple line represents Tryp2 obtained by cleavage of TTIp 56/287 with trypsin. b: RMSD of analogous peptides; the black line represents the analogue to Pep1 (ID 32); the red line represents the analogue to Tryp1 (ID 63); the green line represents the analogue to Pep2 (ID 76); the blue line represents the analogue to Chym1 (ID 101), and the purple line represents the analogue to Tryp2 (ID 336). nm: nanometers. ns: nanoseconds. TTIp 56/287: trypsin inhibitor purified from tamarind seeds (TTIp), model number 56, in conformation number 287. TMPRSS2: transmembrane serine protease 2.
4 Discussion
MD studies between TTIp and trypsin, carried out by our research group, indicate conformation number 287 presented the lowest value of interaction potential energy, favoring the bioprospecting of this protein in the pharmaceutical industry (Medeiros et al., 2021). Furthermore, in vitro data obtained by our group demonstrate high antitrypsin activity of TTI in its isolated, purified, or nanoencapsulated form (Costa et al., 2022; Queiroz et al., 2018).
Considering the functional diversity of the TTI and knowing that the bioactivity of peptides is determined by their composition and an amino acid sequence, prospecting TTI peptides to verify which present more potent anti-SARS-CoV-2 activity is an excellent strategy (Du and Li, 2022). Furthermore, it is also highlighted that peptides have high specificity for target tissues, resulting in little or no toxicity, and undergo little or no accumulation in the body (Sarmadi and Ismail, 2010).
Enzymatic hydrolysis is considered one of the most used strategies in the peptide industrial isolation and prospection. Enzymes such as trypsin, pepsin, flavourzyme, and pancreatin have become substantially used in these processes (Kehinde and Sharma, 2020). Additionally, peptides have been obtained from hydrolysis using the enzyme chymotrypsin (Dang et al., 2019).
In this context, proteinases such as pepsin, chymotrypsin, and trypsin have been frequently used in protein hydrolysis to obtain angiotensin-converting enzyme (ACE) inhibitor peptides (Kuba et al., 2005). Studies have shown that the TTI acts on different mechanisms that involve the control of obesity and its associated effects, such as the production of hormones related to satiety, affecting the central nervous system and the small intestine; reduced food consumption and weight gain; improvement of the lipid profile; and reduction of the inflammatory process related to obesity, regardless of weight loss (Lima et al., 2019). Thus, considering that TTI is a promising molecule concerning the mechanisms associated with the inhibition of ACE-2 (Morais et al., 2021), was chosen to use the enzymes pepsin, trypsin, and chymotrypsin in this study to obtain the peptides from the theoretical TTIp (TTIp 56/287) by enzymatic hydrolysis in silico.
Regarding physicochemical properties, amino acids 84–106 of TMPRSS2 are known to be hydrophobic and probably compose the transmembrane domain (Paoloni-Giacobino et al., 1997). This information was necessary for the analogous peptides prospection from the native peptides derived from TTIp 56/287 and for the assembly of the MD simulation system between TMPRSS2 and each peptide obtained from the TTIp 56/287 cleavage and their analogues since the main objective of this study was to evaluate the anti-SARS-CoV-2 activity of these peptides. Thus, the MD simulation was planned considering all the physicochemical characteristics of TMPRSS2.
It is relevant to highlight the mixture of POPC and POPS used to build the TMPRSS2 model served to ensure structural stability during MD simulations (Aliper et al., 2022) due to the presence of hydrophobic tails in these phospholipids (Kumar and Sastry, 2021). Therefore, suggesting that hydrophobic interactions mediate structural stability.
In this study, each analogous peptide presented values close to its native peptides for hydrophobicity and hydrophobic moment values. Considering the substrate binding site has a neutral charge (Vardhan and Sahoo, 2022), it was necessary to change amino acid residues that are also neutral (nonpolar) to obtain the analogous peptides. For this, the reduction of the hydrophobic moment was avoided, thus making the replacement by aromatic amino acids. Consequently, those with the same hydrophobic moment as the native peptides were chosen to select analogous peptides. In addition, the net charges of the peptides were mostly negative, which is relevant, considering that the catalytic site of TMPRSS2 is positive (Vardhan and Sahoo, 2022).
According to Badani et al. (2014), the peptides that inhibit the entry of enveloped viruses have a common physicochemical property: hydrophobicity and/or amphipacity. These data indicated that such peptides are excellent candidates against SARS-CoV-2 (an enveloped virus) (Hu et al., 2021). As mentioned above, cysteine is also present in aprotinin, which can inhibit TMPRSS2 (Schütz et al., 2020), reinforcing this potential of the peptides prospected here.
We emphasize that all TMPRSS2-peptides complexes showed stability in interactions, especially the TMPRSS2-Chym1, TMPRSS2-Tryp2, TMPRSS2-ID63, TMPRSS2-ID76, TMPRSS2-ID101, and TMPRSS2-ID336 complexes, due to the low RMSD values observed. Therefore, the prospection of native peptides of TTIp 56/287 and its analogues is an excellent alternative for inhibiting of TMPRSS2. Peptides are preferred over whole proteins as they generally have a low molecular weight, are easily digestible structures and are more bioavailable than proteins for a variety of biological activities in the human body, and commonly exhibit greater bioactivity than the parent protein (Chalamaiah et al., 2018; Reyes-Díaz et al., 2019).
Peptides and natural compounds from plants, and their analogues, have shown potent anti-SARS-CoV-2 and anti-protease activities, being considered attractive molecules in drug discovery against COVID-19 (Dinda et al., 2022). Relative to existing pharmacological treatments, camostat, nafamostat, and gabexate - clinically approved in Japan and initially intended for treating pancreatitis - may also suppress TMPRSS2 activity (Yamaya et al., 2020). However, to date, information on specific and safe antiviral drugs for treating infectious disease caused by SARS-CoV-2 is scarce, and current clinical treatments of COVID-19 with antiviral, antimalarial, and immunomodulatory drugs repurposed from cocktail therapy are controversial. Only remdesivir, developed initially against Ebola infection, was found to be effective in inhibiting SARS-CoV-2 infection in vitro, and it has been approved by the FDA to treat patients with severe COVID-19 (Dinda et al., 2022).
Recently, a docking and MD study using peptides from the Data Repository of Antimicrobial Peptides (DRAMP) found that among the four peptides with the highest binding affinities to the main protease of SARS-CoV-2 (Mpro), DRAMP00877 (from Chassalia parviflora) showed better interaction strength, indicating the potential to inhibit this protease (Mahmud et al., 2021). This information reinforces the potential of the peptides prospected in our study as anti-SARS-CoV-2 agents based on the IPE presented for TMPRSS2.
Furthermore, Chen et al. (2021) suggested that homoharringtonine, an alkaloid isolated from the plant Cephalotaxus harringtonii, prevented SARS-CoV-2 infection in human lung cancer Calu-3 cells by inhibiting viral entry by suppressing the expression and activity of TMPRSS2, corroborating that the interaction of molecules with this protease can enable its inhibition. Our study, the first in which TTIp focused on SARS-CoV-2 infection, demonstrated that the peptides interacted with TMPRSS2 in silico. This significant result points to a new perspective on the TTIp activity. This finding made the patent registration possible (BR 10 2022 020330 0).
Therefore, the peptides from TTIp 56/287 and its analogues evaluated, which showed good interaction with TMPRSS2, are excellent candidates as inhibitors of SARS-CoV-2 infection since compounds and/or molecules targeting the TMPRSS2 inhibition exhibit highly efficient antiviral effects regardless of SARS-CoV-2 gender and variant (Hashimoto et al., 2021). However, this is still a preliminary study, as the peptides prospected here need to be synthesized to deeply investigate the effects in the in silico analyses. Additionally, secondary studies need to be developed to verify other bioactive activities of the prospected peptides since these molecules, when cleaved, lose their three-dimensional structure, which results in loss of function. Thus, there is no guarantee that the peptides obtained in this study have the same functions previously demonstrated for the TTIp.
Finally, it was impossible to perform in vitro or in vivo analyses that confirmed that TTIp inhibits the activity of TMPRSS2 or influences the expression of this protease. These are also essential aspects for future investigations, including biochemical assays. Thus, TTIp 56/287 and its peptides should be further explored in future studies to make their use as specific drugs in successfully treating COVID-19 and other diseases. Furthermore, it is noteworthy that the model of TTIp 56/287 was generated from homology modeling, being a limiting factor, which reinforces the need to carry out other experiments with the molecule and with its peptides.
5 Conclusion
The peptides from TTIp 56/287 obtained by cleavage with trypsin and its analogous peptide showed good interaction with TMPRSS2 and the best IPE. Therefore, are excellent candidates as inhibitors of SARS-CoV-2 infection targeting the TMPRSS2 inhibition. The interaction occurred between TMPRSS2 and this native peptide obtained by cleavage with trypsin and its analogous peptide, also showed many hydrophobic residues, a common physical–chemical property among the peptides that inhibit the entry of enveloped viruses, such as SARS-CoV-2, present in specific drugs to treat COVID-19.
CRediT authorship contribution statement
Anna Beatriz Santana Luz: Formal analysis, Data curation, Investigation, Methodology, Validation, Writing – original draft. Amanda Fernandes de Medeiros: Methodology, Writing – review & editing. Lucas Lima Bezerra: Formal analysis, Investigation, Methodology, Writing – review & editing. Mayara Santa Rosa Lima: Investigation, Methodology, Writing – review & editing. Annemberg Salvino Pereira: Investigation, Methodology, Writing – review & editing, Methodology, Writing – review & editing. Thais Souza Passos: Investigation, Methodology, Writing – review & editing. Norberto de Kássio Vieira Monteiro: Formal analysis, Investigation, Methodology, Writing – review & editing. Emilly Guedes Oliveira e Silva: Methodology, Writing – review & editing. Ana Heloneida de Araújo Morais: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Supervision, Writing – review & editing.
Acknowledgments
The authors thank the Federal University of Rio Grande do Norte (UFRN), especially the Pro-Rectory of Postgraduate and the Pro-Rectory of Research, for all efforts dedicated to supporting the research in our institution.
Funding
This work was supported for scholarship and payment of fees by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior [Finance Code 001 – CAPES].
Ethics Committee
Considering that these in silico studies and, therefore, studies with humans and/or animals were not carried out, the free and informed consent term (FICF). Thus, approving the Research Ethics Committee with Humans and/or Animals (CEP and CEUA, respectively) was unnecessary.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- A Uniquely Stable Trimeric Model of SARS-CoV-2 Spike Transmembrane Domain. Int. J. Mol. Sci.. 2022;23(16):9221.
- [CrossRef] [Google Scholar]
- Arfken, G.B., Harris, F.E., Weber, H.J., 2012. Mathematical Methods for Physicists: A Comprehensive Guide, seven ed. Academic Press. https://doi.org/10.1016/C2009-0-30629-7.
- The bovine basic pancreatic trypsin inhibitor (Kunitz inhibitor): a milestone protein. Curr. Protein Pept. Sci.. 2003;4(3):231-251.
- [CrossRef] [Google Scholar]
- Peptide entry inhibitors of enveloped viruses: the importance of interfacial hydrophobicity. Biochim. Biophys. Acta. 2014;1838(9):2180-2197.
- [CrossRef] [Google Scholar]
- SARS-CoV-2 infection and replication kinetics in different human cell types: The role of autophagy, cellular metabolism and ACE2 expression. Life Sci.. 2022;308:1-13.
- [CrossRef] [Google Scholar]
- Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses. 2012;4(6):1011-1033.
- [CrossRef] [Google Scholar]
- Tamarind Enzymatic Inhibitors: Activities and Health Application Perspectives. Food Rev. Int. 2020
- [CrossRef] [Google Scholar]
- Immunomodulatory and anticancer protein hydrolysates (peptides) from food proteins: A review. Food Chem.. 2018;245:205-222.
- [CrossRef] [Google Scholar]
- SARS-CoV-2 cell entry and targeted antiviral development. Acta Pharm. Sin. B.. 2021;11(12):3879-3888.
- [CrossRef] [Google Scholar]
- A high-throughput screen for TMPRSS2 expression identifies FDA-approved compounds that can limit SARS-CoV-2 entry. Nat. Commun.. 2021;12(1):3907.
- [CrossRef] [Google Scholar]
- An Insulin Receptor-Binding Multifunctional Protein from Tamarindus indica L. Presents a Hypoglycemic Effect in a Diet-Induced Type 2 Diabetes-Preclinical Study. Foods. 2022;11(15):2207.
- [CrossRef] [Google Scholar]
- In Vitro and in Vivo Studies on the Angiotensin-Converting Enzyme Inhibitory Activity Peptides Isolated from Broccoli Protein Hydrolysate. J. Agric. Food Chem.. 2019;67(24):6757-6764.
- [CrossRef] [Google Scholar]
- COVID-19-New Insights on a Rapidly Changing Epidemic. JAMA. 2020;323(14):1339-1340.
- [CrossRef] [Google Scholar]
- Some natural compounds and their analogues having potent anti-SARS-CoV-2 and anti-proteases activities as lead molecules in drug discovery for COVID-19. Eur. J. Med. Chem.. 2022;6:100079
- [CrossRef] [Google Scholar]
- An update on drugs with therapeutic potential for SARS-CoV-2 (COVID-19) treatment. Drug Resist. Updat.. 2021;59:100794
- [CrossRef] [Google Scholar]
- Review and perspective on bioactive peptides: A roadmap for research, development, and future opportunities. J. Agric. Food Res.. 2022;9:1-13.
- [CrossRef] [Google Scholar]
- Food and Drug Administration (FDA), 2022. FDA approves first treatment for COVID-19. https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-covid-19 (accessed 10 November 2022).
- Molecular Evolution of Human Coronavirus Genomes. Trends in Microbiol.. 2017;25(1):35-48.
- [CrossRef] [Google Scholar]
- Theoretical and Experimental Investigation of Acidity of the Glutamate Receptor Antagonist 6,7-Dinitro-1,4-dihydroquinoxaline-2,3-dione and Its Possible Implication in GluA2 Binding. J. Phys. Chem.. 2017;121(39):7414-7423.
- [CrossRef] [Google Scholar]
- Mitochondria Targeted Viral Replication and Survival Strategies-Prospective on SARS-CoV-2. Front. Pharmacol.. 2020;11:1-9.
- [CrossRef] [Google Scholar]
- Investigation of antidiarrhoeal activity of ethanolic extract of Tamarindus indica L. seeds in albino wistar rats. Asian J. Pharm.. 2016;10(4):1-5.
- [CrossRef] [Google Scholar]
- Dual inhibition of TMPRSS2 and Cathepsin B prevents SARS-CoV-2 infection in iPS cells. Mol. Ther. Nucleic Acids. 2021;26:1107-1114.
- [CrossRef] [Google Scholar]
- Peptide-based protease inhibitors from plants. Drug Discov. Today. 2019;24(9):1877-1889.
- [CrossRef] [Google Scholar]
- Hess, B., Bekker, H., Berendsen, H.J.C., Fraaije J.G.E.M., 1997. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem., 18, 1463–1472; https://doi.org/10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H.
- Methods of Conjugate Gradients for Solving Linear Systems. J. Res. Natl. Bur. Stand.. 1952;49(6):409-436.
- [CrossRef] [Google Scholar]
- SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181(2):271-280.e8.
- [CrossRef] [Google Scholar]
- Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol.. 2021;19(3):141-154.
- [CrossRef] [Google Scholar]
- SARS-CoV-2 promote autophagy to suppress type I interferon response. Signal Transduct. Target. Ther.. 2021;6(1):1-3.
- [CrossRef] [Google Scholar]
- Essential role of TMPRSS2 in SARS-CoV-2 infection in murine airways. Nat. Commun.. 2022;13(6100):1-11.
- [CrossRef] [Google Scholar]
- Discovery and development of new antibacterial drugs: learning from experience? J. Antimicrob. Chemother.. 2018;73(6):1452-1459.
- [CrossRef] [Google Scholar]
- Comparison of simple potential functions for simulating liquid water. J. Chem. Phys.. 1983;79(2):926-935.
- [CrossRef] [Google Scholar]
- Ensemble Docking Coupled to Linear Interaction Energy Calculations for Identification of Coronavirus Main Protease (3CLpro) Non-Covalent Small-Molecule Inhibitors. Molecules. 2020;25(24):1-11.
- [CrossRef] [Google Scholar]
- Recently isolated antidiabetic hydrolysates and peptides from multiple food sources: A review. Crit. Rev. Food Sci. Nutr.. 2020;60(2):322-340.
- [CrossRef] [Google Scholar]
- Short Proline-Rich Antimicrobial Peptides Inhibit Either the Bacterial 70S Ribosome or the Assembly of its Large 50S Subunit. Chembiochem. 2015;16(16):2304-2308.
- [CrossRef] [Google Scholar]
- Production of angiotensin I-converting enzyme inhibitory peptides from soybean protein with Monascus purpureus acid proteinase. Process. Bochem.. 2005;40:2191-2196.
- [CrossRef] [Google Scholar]
- Study of lipid heterogeneity on bilayer membranes using molecular dynamics simulations. J. Mol. Graph. Model.. 2021;108:108000
- [CrossRef] [Google Scholar]
- Gastroprotective and antielastase effects of protein inhibitors from Erythrina velutina seeds in an experimental ulcer model. Biochem. Cell Biol.. 2017;95(2):243-250.
- [CrossRef] [Google Scholar]
- Trypsin inhibitors: promising candidate satietogenic proteins as complementary treatment for obesity and metabolic disorders? J. Enzyme Inhib. Med. Chem.. 2019;34(1):405-419.
- [CrossRef] [Google Scholar]
- The development of Coronavirus 3C-Like protease (3CLpro) inhibitors from 2010 to 2020. Eur. J. Med. Chem.. 2020;206:1-18.
- [CrossRef] [Google Scholar]
- Antiviral peptides against the main protease of SARS-CoV-2: A molecular docking and dynamics study. Arab. J. Chem.. 2021;14(9):103315
- [CrossRef] [Google Scholar]
- Biochemical characterisation of a Kunitz-type inhibitor from Tamarindus indica L. seeds and its efficacy in reducing plasma leptin in an experimental model of obesity. J. Enzyme Inhib. Med. Chem.. 2018;33(1):334-348.
- [CrossRef] [Google Scholar]
- Structural insights and molecular dynamics into the inhibitory mechanism of a Kunitz-type trypsin inhibitor from Tamarindus indica L. J. Enzyme Inhib. Med. Chem.. 2021;36(1):480-490.
- [CrossRef] [Google Scholar]
- Identification of FDA approved drugs against SARS-CoV-2 RNA dependent RNA polymerase (RdRp) and 3-chymotrypsin-like protease (3CLpro), drug repurposing approach. Biomed. Pharmacother.. 2021;138:111544
- [CrossRef] [Google Scholar]
- Tamarind (Tamarindus indica L.) Seed a Candidate Protein Source with Potential for Combating SARS-CoV-2 Infection in Obesity. Drug Target Insights. 2021;15:5-12.
- [CrossRef] [Google Scholar]
- Stapled ACE2 peptidomimetics designed to target the SARS-CoV-2 spike protein do not prevent virus internalization. Peptide Sci.. 2021;113:1-8.
- [CrossRef] [Google Scholar]
- Prospecting in silico antibacterial activity of a peptide from trypsin inhibitor isolated from tamarind seed. J. Enzyme Inhib. Med. Chem.. 2023;38(1):67-83.
- [CrossRef] [Google Scholar]
- Cloning of the TMPRSS2 gene, which encodes a novel serine protease with transmembrane, LDLRA, and SRCR domains and maps to 21q22.3. Genomics. 1997;44(3):309-320.
- [CrossRef] [Google Scholar]
- Recent discovery and development of inhibitors targeting coronaviruses. Drug Discov. Today. 2020;25(4):1-21.
- [CrossRef] [Google Scholar]
- Chitosan-whey protein nanoparticles improve encapsulation efficiency and stability of a trypsin inhibitor isolated from Tamarindus indica L. Food Hydrocoll.. 2018;84:247-256.
- [CrossRef] [Google Scholar]
- Design and selection of peptides to block the SARS-CoV-2 receptor binding domain by molecular docking. J. Nanotechnol.. 2022;13:699-711.
- [CrossRef] [Google Scholar]
- Legume proteins as a promising source of anti-inflammatory peptides. Curr. Protein Pept. Sci.. 2019;20(12):1204-1217.
- [CrossRef] [Google Scholar]
- A xylose-stimulated xylanase-xylose binding protein chimera created by random nonhomologous recombination. Biotechnol. Biofuels. 2016;9:119.
- [CrossRef] [Google Scholar]
- Characterization of a Novel Coronavirus Associated with Severe Acute Respiratory Syndrome. Science. 2003;300(5624):1394-1399.
- [CrossRef] [Google Scholar]
- Antioxidative peptides from food proteins: a review. Peptides. 2010;31(10):1949-1956.
- [CrossRef] [Google Scholar]
- Peptide and peptide-based inhibitors of SARS-CoV-2 entry. Adv. Drug Deliv. Rev.. 2020;167:47-65.
- [CrossRef] [Google Scholar]
- Virtual screening by targeting proteolytic sites of furin and TMPRSS2 to propose potential compounds obstructing the entry of SARS-CoV-2 virus into human host cells. J. Tradit. Complement. Med.. 2022;12(1):6-15.
- [CrossRef] [Google Scholar]
- Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020;181(2):281-292.
- [CrossRef] [Google Scholar]
- APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res.. 2016;44(D1):D1087-D1093.
- [CrossRef] [Google Scholar]
- Skin Lightening and Sebum Control Efficacy of a Cosmetic Emulsion Containing Extract of Tamarind Seeds on Asian Skin Type. Lat. Am. J. Pharm.. 2015;34(3):570-575.
- [Google Scholar]
- World Health Organization (WHO), 2022. WHO Coronavirus (COVID-19) Dashboard. https://covid19.who.int/ (accessed 12 December 2022).
- Protease Inhibitors: Candidate Drugs to Inhibit Severe Acute Respiratory Syndrome Coronavirus 2 Replication. Tohoku J. Exp. Med.. 2020;251(1):27-30.
- [CrossRef] [Google Scholar]
- Computational screening of antagonists against the SARS-CoV-2 (COVID-19) coronavirus by molecular docking. Int. J. Antimicrob. Agents. 2020;56(2):1-6.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.104886.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




