

Translate this page into:
Identification of potential Indonesian marine invertebrate bioactive compounds as TMPRSS2 and SARS-CoV-2 Omicron spike protein inhibitors through computational screening
⁎Corresponding author. arry.yanuar@ui.ac.id (Arry Yanuar)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The coronavirus pandemic led to the announcement of a worldwide health emergency. The SARS-CoV-2 Omicron variant, which swiftly spread worldwide, has fueled existing challenges. Appropriate medication is necessary to avoid severe SARS-CoV-2 disease. The human TMPRSS2 and SARS-CoV-2 Omicron spike protein, which are required for viral entry into the host phase, were identified as the target proteins through computational screening. Structure-based virtual screening; molecular docking; absorption, distribution, metabolism, excretion, and toxicity (ADMET) analysis; and molecular dynamics simulation were the methods applied for TMPRSS2 and spike protein inhibitors. Bioactive marine invertebrates from Indonesia were employed as test ligands. Camostat and nafamostat (co-crystal) were utilized as reference ligands against TMPRSS2, whereas mefloquine was used as a reference ligand against spike protein. Following a molecular docking and dynamics simulation, we found that acanthomanzamine C has remarkable effectiveness against TMPRSS2 and spike protein. Compared to camostat (-8.25 kcal/mol), nafamostat (-6.52 kcal/mol), and mefloquine (-6.34 kcal/mol), acanthomanzamine C binds to TMPRSS2 and spike protein with binding energies of −9.75 kcal/mol and −9.19 kcal/mol, respectively. Furthermore, slight variances in the MD simulation demonstrated consistent binding to TMPRSS2 and spike protein after the initial 50 ns. These results are highly valuable in the search for a treatment for SARS-CoV-2 infection.
Keywords
ADMET
Marine invertebrates
Molecular docking
Molecular dynamics
SARS-CoV-2
TMPRSS2

1 Introduction
Coronavirus disease 2019 (COVID-19) is a worldwide medical health emergency caused by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). The COVID-19 pandemic began in December 2019 and is still ongoing (Riley et al. 2021). The introduction of novel SARS-CoV-2 mutations has also posed difficulties in the antiviral discovery process. The SARS-CoV-2 Omicron B.1.1.529 strain swiftly spread throughout the world (Behl et al. 2022). The Omicron variant of SARS-CoV-2 has 37 mutations spread across the trimeric protein domain. A total of 15 mutations are in the receptor binding domain of the spike protein, which can change the spread of infection and infectivity. The RBD of the omicron B.1.1.529 variant comprises the crucial mutations G446S, N501Y, G496S, Y505H, Q493R, E484A, T478K, S477N, K417N, G339D, S371L, S373P, S375F, N440K, and Q498R (Bharathi et al. 2022). Because of the high number of mutations in the Omicron variant, there is a risk that it will evade detection by the body’s antibodies following vaccination (Hoffmann et al. 2022).
According to preclinical trials, case reports, and clinical trials, current COVID-19 antivirals have limitations, such as the possibility of side effects and significant drug interactions (Brunetti et al. 2021; Hanai et al. 2022; Koseki et al. 2022; Touafchia et al. 2021; U.S. Food and Drug Administration 2022). Therefore, research on new anti-SARS-CoV-2 drugs as a better therapeutic option is essential.
Human receptors or enzymes are involved in the SARS-CoV-2 invasive process and have been identified as a potential new therapy method (Joshi, Joshi, and Degani 2020; Kim, Read, and Fauci 2020). The virus connects to the cell surface and degrades trimer stability before spike protein fusion when the SARS-CoV-2 spike protein binds to human angiotensin-converting enzyme 2. The cleavage of spike protein typically happens sequentially, with the furin enzyme in the S1/S2 subunit cleaving first, followed by transmembrane protease serine 2 (TMPRSS2) in the S2 subunit (Ma et al. 2021). To alter and activate spike proteins, SARS-CoV-2 employs a human TMPRSS2 (Hoffmann et al. 2020). TMPRSS2 is vital for the proteolytic activation of SARS-CoV-2 by cleaving the spike protein and beginning viral membrane fusion with human cellular membranes (Hu et al. 2021). As a result, inhibiting TMPRSS2 and the binding between TMPRSS2 and SARS-CoV-2 Omicron spike protein are prospective targets for the establishment of innovative COVID-19 medicines.
Finding a lead molecule is crucial in developing new medications, especially when looking for bioactive compounds found in nature. Frequently, natural substances are used to treat viral infections and boost the immune system. (Ma et al. 2021). Secondary metabolites of Indonesian marine invertebrates have been shown to exhibit antibacterial, antifungal, anticancer, and antiviral properties (Izzati et al. 2021; Nurrachma et al. 2021).
Several studies have been carried out on the antiviral action of marine invertebrate secondary metabolites. Bioactive compounds that have antiviral activity include mollamide B from the tunicate of Didemnum molle (Donia et al. 2008); C-nucleosides, spongouridine, and spongothymidine from the marine sponge Cryptothecea crypt (Datta, Nath Talapatra, and Swarnakar 2015); norbatzelladine L from the marine sponge species Monanchora; and norcembrane, a diterpenoid from the soft coral Sinularia gyrosa (Senan and Karthika 2021). However, none of these trials has focused on anti-SARS-CoV-2 antibodies.
Drug development in the laboratory using experimental approaches takes a long time and is relatively expensive (Horizny 2019). As a result, the computational technique is a valuable resource for reducing drug discovery time. Compounds with TMPRSS2 inhibitory activity have been the subject of computational analysis, including mozenavir (Mamidala et al. 2022); camostat mesylate analogs (Sharma et al. 2022); ZINC3830554 (Barge et al. 2021); gabexate (Hu et al. 2021); and capreomycin, aspoxicillin, and fosamprenavir (Hatmal et al. 2021). In addition, several computational studies on the molecular docking of the SARS-CoV-2 Alpha spike protein binding site with TMPRSS2 have been conducted (Hussain et al. 2020; Salleh and Deris 2022). Among the computational studies on the inhibition of the SARS-CoV-2 spike protein are ZINC02111387, ZINC02122196, SN00074072, and ZINC04090608 (Power et al. 2022); 28-demethyl-beta-amyrone, 24-Noroleana-3,12-diene, and stigmasterol (Paul et al. 2022); and dicaffeoylquinic acid and diacetylcurcumin (Singh et al. 2021).
So far, no in silico studies have examined the secondary metabolites of Indonesian marine invertebrates that block TMPRSS2 and SARS-CoV-2 Omicron spike protein. Therefore, this study aimed to identify secondary metabolites from Indonesian marine invertebrates that can suppress TMPRSS2 and the binding between TMPRSS2 and SARS-CoV-2 Omicron spike protein by combining structure-based virtual screening, molecular docking, ADMET analysis, and molecular dynamics simulation.
2 Materials and methods
2.1 Collection of macromolecules
The human TMPRSS2 crystal structure complex with nafamostat as an inhibitor (PDB ID: 7MEQ) and SARS-CoV-2 Omicron spike protein B.1.1.529 (PDB ID: 7QO7) three-dimensional structures were obtained from the Research Collaboratory for Structural Bioinformatics Protein Data Bank website (https://www.rcsb.org/). TMPRSS2 is a single-chain proenzyme (Fraser et al. 2022), while the SARS-CoV-2 Omicron spike protein structure is a trimer spike with monomers A, B, and C (Ni et al. 2021). Table 1 outlines the structural characteristics of TMPRSS2 and SARS-CoV-2 Omicron spike protein.
Protein structure
PDB ID
Method
Resolution
Sequence length
Number of chains
TMPRSS2
7MEQ
X-RAY diffraction
1.95 Å
395
A
SARS-CoV-2 Omicron spike protein
7QO7
Electron microscopy
3.02 Å
1,285
A, B, C
2.2 Preparation and optimization of macromolecules
Unwanted ligands and TMPRSS2 and SARS-CoV-2 Omicron spike chain C structures were removed from water molecules. The macromolecules were visually examined using the Chimera tool to ensure that the empty amino acid residues were filled. Modeller 10.3 was applied to create all of the missing gaps and loops (Fiser et al., 2000). The ModLoop web server (https://modbase.compbio.ucsf.edu/modloop) was used to model and enhance the loops. Autodock Tools 1.5.6 was utilized to incorporate polar hydrogens and Kollman united atom charges in the 3D structures of the proteins (Trott and Olson 2009).
2.3 Collection and preparation of ligands
Overall, 137 secondary metabolites of Indonesian marine invertebrates were utilized as test ligands (Izzati et al. 2021; Nurrachma et al. 2021). Camostat was used as a reference inhibitor of TMPRSS2, and mefloquine was utilized as a reference inhibitor of the spike protein.
MarvinSketch 20.14 was employed to sketch the test ligand molecules, and the three-dimensional structures were created by adding polar hydrogen. The 3D structure of the ligand molecule was created by adding hydrogen atoms using BIOVIA Discovery Studio Visualizer 2021 software. As reference ligands, the 3D structures of camostat and mefloquine were retrieved from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/). Meanwhile, the nafamostat’s 3D structure was obtained by separating the nafamostat co-crystal bound to the TMPRSS2 protein.
2.4 Determination of the binding site of SARS-CoV-2 Omicron spike protein with TMPRSS2: Protein-Protein Interaction (PPI)
The structures of the SARS-CoV-2 Omicron spike protein and TMPRSS2 were docked by the HADDOCK 2.4 web server (https://wenmr.science.uu.nl/haddock2.4/) based on a previous study by Salleh and Deris (2022). The model with the best Z-HADDOCK score and the largest number of clusters was chosen to investigate the binding energy (Delta G) and dissociation constant (KD) utilizing the PRODIGY web server (https://wenmr.science.uu.nl/ prodigy/). Predictions of surface contact hydrogen bonds between proteins were carried out via the PDBePISA web server (https://www.ebi.ac.uk/pdbe/pisa/).
2.5 Virtual screening
Virtual screening of the test ligands to TMPRSS2 and SARS-CoV-2 Omicron spike protein was performed using PyRx-Autodock Vina 0.9.8 (Trott and Olson 2009). The coordinates of the TMPRSS2 active site used were × = -9.755 Å, y = -6.247 Å, and z = 20.307 Å, with a grid box size of 68 × 68 × 68 pts and a grid spacing of 0.375 Å. According to the protein–protein interaction (PPI) data, the coordinates of the SARS-CoV-2 Omicron spike protein were based on the amino acid residues of the spike protein that were in contact to create hydrogen bonds with the TMPRSS2 protein. The virtual screening binding affinity energy scores were sorted, and the top 20 test ligand complexes were examined using the molecular docking method.
2.6 Molecular docking
The top 20 ligand–protein complexes identified through virtual screening were confirmed using molecular docking with the AutoDock Tools program. Validation was acceptable if the RMSD was less than 2 Å (Allen and Rizzo 2014). The number of Genetic Algorithm runs was set to 50, the population size was 250, the maximum number of evals was 2,500,000 (medium parameter), and the maximum number of generations was 27,000 (Gurung et al. 2022). The 10 docking-validated test ligands with the best binding affinity energy values were examined for binding affinity energy (Delta G), inhibition constants (Ki), and ligand interactions with amino acid residues. The BIOVIA Discovery Studio Visualizer tool was used to visualize the complexes’ test ligand–protein and reference ligand–protein interactions of amino acids.
2.7 ADMET prediction analysis
The ADMETlab 2.0 web server (https://admetmesh.scbdd.com) was used to analyze drug similarities and estimate absorption, distribution, metabolism, excretion, and toxicity profiles (Sharma et al. 2022). Lipinski’s rules, which consider molecular weight, the log P partition coefficient value, the number of hydrogen bond donors, and the number of hydrogen bond acceptors, were used to see if the compounds were similar to drugs. In addition to Lipinski’s rules, the absorption, distribution, metabolism, excretion, and toxicity (ADMET) profiles of bioactive compounds must fulfil the following criteria: topological polar surface area (TPSA) ≤ 75 Å2, human intestinal absorption (HIA) ≤ 0.7, half-life (T1/2) ≤ 0.8, carcinogenicity ≤ 0.3, and AMES toxicity ≤ 0.3 (Lazniewski et al. 2022). Molecular dynamics simulation was examined on the best test ligand that met all of the ADMET analysis requirements.
2.8 Molecular dynamic simulation
After selecting a ligand molecule based on ADMET profile analysis, molecular dynamic simulation using AMBER MD software was performed in multiple stages. First, macromolecules and ligands were prepared, and then the topologies and coordinates of macromolecules, ligands, and macromolecule-ligand complexes were established in a vacuum and in aqueous solvents. The next phase was energy minimization and equilibration. During the equilibration step, the volume was held constant, and the temperature was increased to 310 K. Three factors needed to be verified before production began. Temperature, density, and potential energy must all be held constant, with a temperature of 310 K and a density of 1 g/mL. Simulations for 100 ns were performed for the production to be generated with a stable balanced system (Sharma et al. 2022). The Cpptraj subprograms were used to assess the findings of the molecular dynamics simulation. Root mean square deviation (RMSD) and root mean square fluctuation (RMSF) were the parameters studied.
3 Results and discussion
Structure-based drug design is a computer-assisted drug discovery technique that identifies potential active chemical substances focused on the three-dimensional structure of a target protein (Charoute et al. 2022). Computational screening of natural bioactive compounds can expedite the antiviral drug discovery process during the COVID-19 pandemic and help researchers identify the inhibitors with the most potential to specifically address the SARS-CoV-2 cell entrance paths.
Inhibiting human receptors or enzymes implicated in the SARS-CoV-2 invasive process has been proposed as a potential novel COVID-19 therapeutic development method (Joshi et al. 2020; Kim et al. 2020). The target macromolecules in this study were TMPRSS2 and SARS-CoV-2 Omicron spike protein. Full-length TMPRSS2 with PDB ID 7MEQ, which has a co-crystal native ligand nafamostat, and SARS-CoV-2 Omicron spike protein with PDB ID 7QO7, which has no co-crystal and for which only chain C is employed, were chosen. Visually, using the Chimera program, in TMPRSS2, four parts of the missing loop were identified—namely, the amino acid residues between SER163 and SER167, ASP202 and SER208, ALA216 and ILE221, and ASN249 and ILE256. The spike protein had five missing loop parts: the amino acid residues between SER146 and MET148, TYR245 and SER253, LYS441 and GLY444, THR673 and SER686, and LEU838 and ASP845. Fig. 1 depicts the macromolecules before and after all the missing amino acid residues were constructed and all the loops were finalized. The dotted line in the figure shows the missing loops, while the blue line indicates the modeled loop.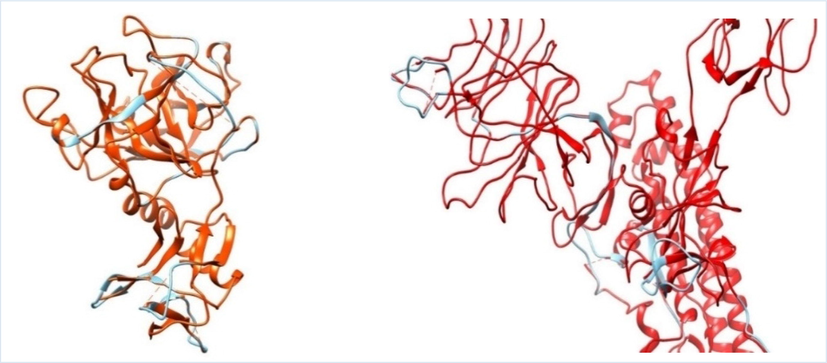
Superposition of macromolecules before and after modeling: A) TMPRSS2 loop before (orange dotted line) and after modeling (blue line) and B) SARS-CoV-2 Omicron variant spike protein chain C loop before (red dotted line) and after modeling (blue line).
Camostat mesylate is an established oral serine protease inhibitor of TMPRSS2, the human transmembrane surface protease, and a promising COVID-19 antiviral drug (Breining et al. 2021). Camostat is the reference ligand that inhibits TMPRSS2, while mefloquine inhibits the SARS-CoV-2 spike protein. According to a previous study, camostat binds strongly with the catalytic triad of TMPRSS2 residues HIS296 and SER441, and the protein–ligand combination is stable in molecular dynamics simulations (Sonawane et al. 2021). In this study, the results of camostat molecular docking with TMPRSS2 revealed similar outcomes, as interactions formed hydrogen bonds with amino acids HIS296, SER441, GLY464, ASP435, and SER436 (Fig. 2A). Furthermore, Camostat mesylate exhibited significant antiviral activity in vitro and in vivo against SARS-CoV-2 (Hoffmann et al. 2020; Zhou et al. 2015).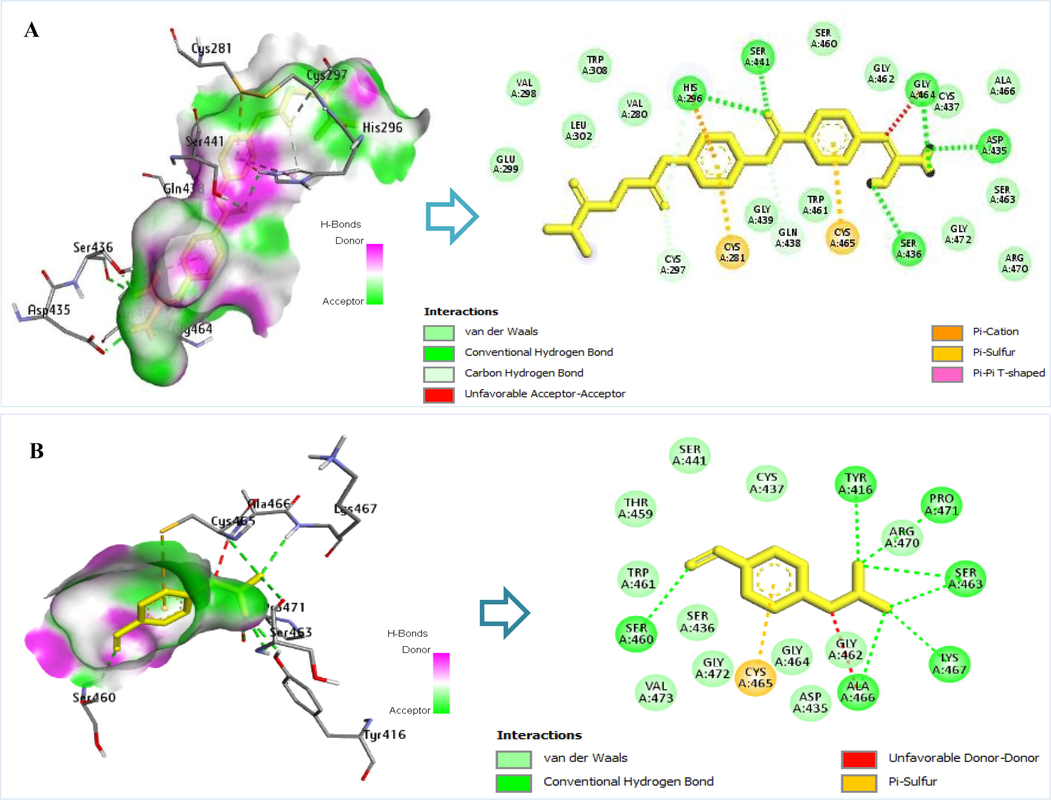
Interaction of amino acid residues TMPRSS2 with reference ligands: A) Camostat’s interaction with TMPRSS2 amino acid residues and B) nafamostat’s interaction with TMPRSS2 amino acid residues.
This study also investigated nafamostat, a co-crystal TMPRSS2 macromolecule. Nafamostat, a clinically available serine protease inhibitor, has been recognized as the primary and most powerful inhibitor for the epithelial cell entry of Coronavirus during the Middle East respiratory syndrome epidemic (Yamamoto et al. 2016). To this end, it has been demonstrated that nafamostat mesylate inhibits SARS-COV-2 entry into human epithelial cells when the clinical evaluation threshold is 10 nM (Yamamoto et al. 2016). In this study, nafamostat’s molecular docking results with TMPRSS2 were comparable to those of another study (Vardhan and Sahoo 2022). It interacts with SER463 amino acids to form hydrogen bonds and with ASP435, CYS437, and SER441 amino acids to form hydrophobic bonds (Fig. 2B).
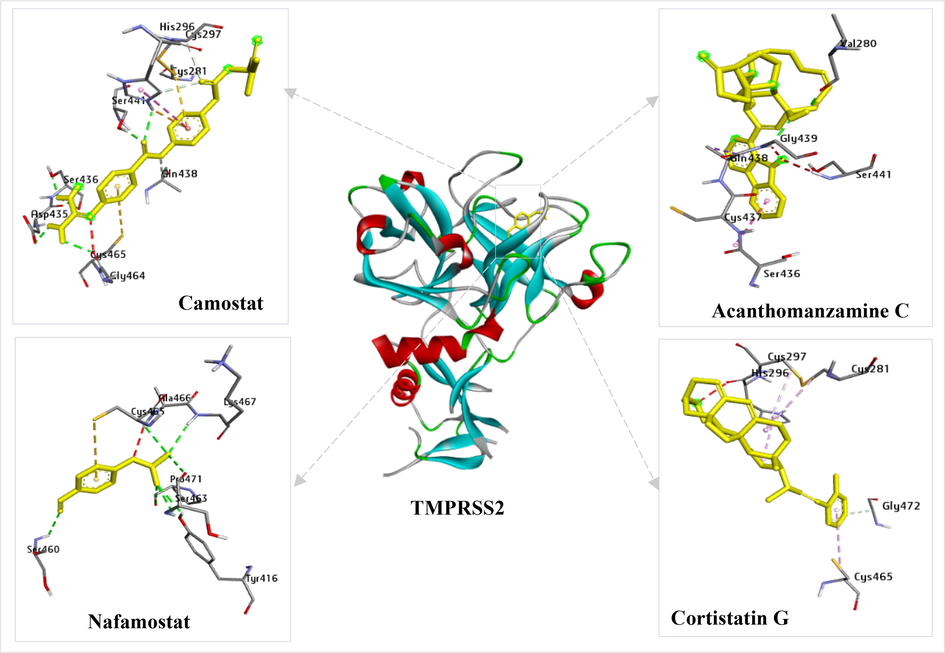
Mefloquine, which is approved for the treatment of malaria, exhibits antiviral activity against both MERS-CoV and SARS-CoV (Dyall et al. 2014). Computationally, mefloquine inhibits SARS-CoV-2 spike protein activity with a binding affinity of −6.7 kcal/mol (Prashantha et al. 2021), whereas the binding affinity in our docking investigation was −6.34 kcal/mol. In vitro, mefloquine, a derivative of hydroxychloroquine, exhibits greater anti-SARS-CoV-2 activity in TMPRSS2 gene overexpressed VeroE6 cells than hydroxychloroquine, with an IC50 of 1.28 M, indicating that it might prevent virus entry when the virus attaches to the target host cell receptor (Shionoya et al. 2021). Although the structures of mefloquine and chloroquine have similarities, there is a change in the quinoline core in mefloquine, which results in a change in pK and log P values. A high pK can encourage lysosomal trapping of mefloquine in lung tissue. In addition, due to its higher lipophilicity, mefloquine has a high volume of lung distribution (Sacramento et al. 2022).
This study investigated 137 compounds of secondary metabolites isolated from Indonesian marine invertebrates as test ligands. Alkaloids, terpenoids, peptides, polyketides, macrolides, steroids, saponins, benzoyl derivatives, and cembranoids were identified among the secondary metabolites derived from marine sponges, marine tunicates, and soft corals. These secondary metabolites have been linked to a variety of biological functions, including antiviral activity (Izzati et al. 2021; Nurrachma et al. 2021). Most of the 137 secondary metabolite compounds found in Indonesian marine invertebrates were alkaloids, which made up about 60% of all compounds. After alkaloids, the next most common compounds were terpenoids (18%) and peptides (10%). Other secondary metabolite compounds were found at rates below 6% (Supplementary Table 1).
3.1 Protein-Protein interaction: determination binding site of SARS-CoV-2 Omicron spike protein
The SARS-CoV-2 Omicron spike protein was docked with TMPRSS2 to find where the spike protein binds. This information was then used in virtual screening with test ligands. When the SARS-CoV-2 spike protein binds to TMPRSS2, cleavage occurs in the S2 subunit (Ma et al. 2021), which activates the membrane fusion of the SARS-CoV-2 spike protein into the host cell (Evans and Liu 2021; Simmons et al. 2013; Zhu et al. 2021). The test ligand can prevent protease cleavage and thereby prevent SARS-CoV-2 from entering cells via endocytosis by preventing the interaction of the SARS-CoV-2 spike protein with TMPRSS2.
Protein-protein docking yielded 10 clusters with a total of 161 structures, accounting for 80% of the water-refined HADDOCK model. Cluster 1 had the best HADDOCK score of −69.3 ± 4.0, the largest cluster size of 38, and a Z-score of −0.8 (Supplementary File).acknow
The Z-score indicates the number of standard deviations from the cluster mean; the lower the value, the better. The cluster 1 model was then used to calculate binding energy (Delta G) and dissociation constant (KD) utilizing the PRODIGY web server (https://wenmr.science.uu.nl/prodigy/). The investigation yielded binding energies (Delta G) of −9.5 kcal/mol and dissociation constants (KD) of 1.1 × 10–7 M. The PDBePISA web server was used to predict surface contact hydrogen bonding between proteins. The surface contact hydrogen bonds formed between the SARS-CoV-2 Omicron spike protein and TMPRSS2 can be seen in Table 2.
Residue of amino acids in the SARS-CoV-2 Omicron spike protein
Distance of hydrogen bonds (Å)
Residue of amino acid in the TMPRSS2
ALA681
3.67
GLY462
ALA685
1.70
ARG490
GLN604
1.81
LYS390
GLY636
3.44
GLY391
SER683
3.04
GLY462
SER634
3.77
LYS390
SER686
3.85
GLY462
SER686
3.76
GLY464
THR635
3.50
GLY391
THR635
2.28
GLN438
TYR633
1.77
LYS390
The BIOVIA Discovery Studio Visualizer program was used to depict the cluster 1 model (Fig. 3). TMPRSS2 bonded only to a single binding site in the active residues 685 and 686 of the 10 clusters of the SARS-CoV-2 Omicron spike protein. The bonds between TMPRSS2 and spike protein were enhanced by the formation of many h-bonds, one of which was the bond produced by GLY 462 from TMPRSS2 and SER 686 from the spike proteins’ cleavage site 1. Although the cleavage site of the spike protein did not directly contact all TMPRSS2 binding sites, the amino acid residues of the spike protein engaged with the residues of amino acids of TMPRSS2 clamped to the TMPRSS2 active site. Once attached, this flexible loop can alter conformation, bringing cleavage sites 685 and 686 of the spike protein nearer to the catalytic active site of TMPRSS2. In the two-step mechanism, this results in the enzymatic cleavage of S1/S2 and S’ and the fusion of the host cell membrane (Saxena et al. 2020).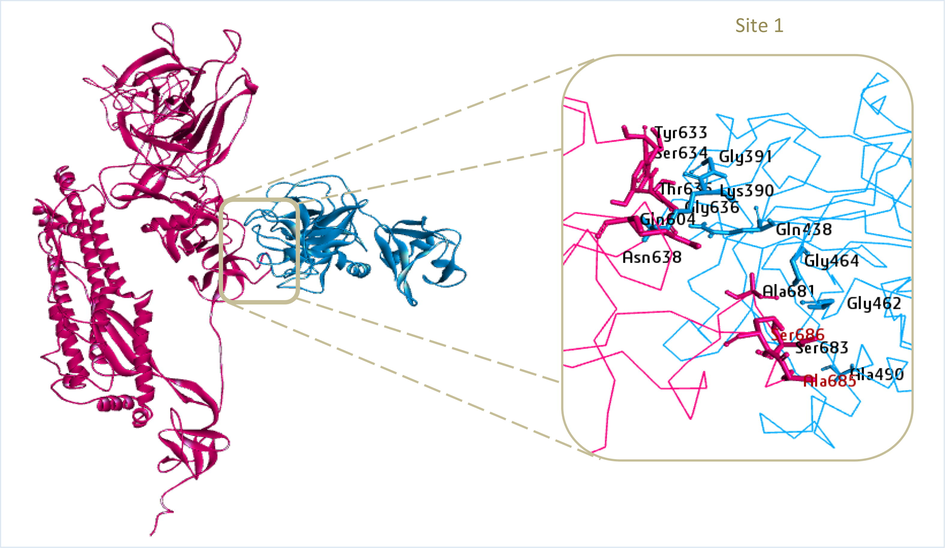
Result of TMPRSS2 protein docking (PDB ID: 7MEQ) with Omicron variant SARS-CoV-2 spike protein (PDB ID: 7QO7) at cleavage site 1 active residue 686 and 685 (red residues). The SARS-CoV-2 Omicron variant spike protein is represented by magenta, while TMPRSS2 is represented by blue.
3.2 Docking validation of TMPRSS2 protein and SARS-CoV-2 Omicron spike protein
Docking method validation is acceptable if the RMSD value less than 2 Å (Allen and Rizzo 2014). The deviation distance from the co-crystal or native ligand bond location with the protein after redocking to the actual co-crystal bond position is defined as the RMSD. The TMPRSS2 protein (7MEQ) and the SARS-CoV-2 Omicron spike protein (7QO7) were validated by redocking the reference ligands.
The TMPRSS2 protein worked by redocking nafamostat, a native ligand for TMPRSS2. The parameter of the grid box was 68 × 68 × 68 pts, the grid spacing was 0.375 Å, and the coordinates corresponding to the bond site were × = -9.755 Å, y = -6.247 Å, and z = 20.307 Å. In the same way, mefloquine, a reference ligand, was docked to validate the SARS-CoV-2 Omicron spike protein. The parameter of the grid box was 76 × 76 × 76 pts, grid spacing was 0.375 Å, and the coordinates were × = 233.664 Å, y = 171.257 Å, and z = 197.310 Å. This grid box was set up based on the amino acid residues on the spike protein that interact with TMPRSS2 to form hydrogen bonds. The PyMOL tool was used to align nafamostat and mefloquine ligands before and after redocking, and the findings indicate that the RMSD values were 0.424 Å and 0.057 Å, respectively (Fig. 4A, B). Based on these findings, the docking method’s validation produces good results, is acceptable, and could be utilized for virtual ligand screening tests.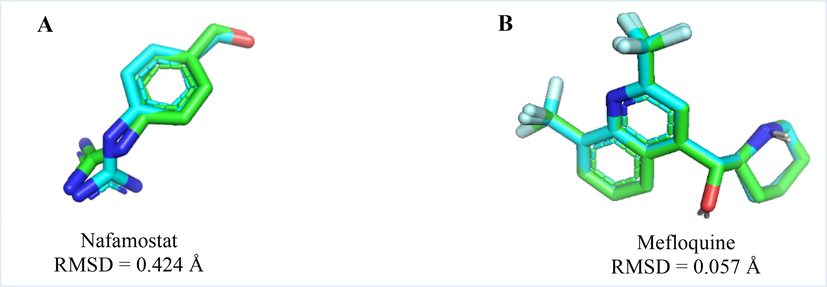
Alignment of nafamostat (A) and mefloquine (B) for validation of the TMPRSS2 protein and the SARS-CoV-2 Omicron spike protein, respectively. The light blue color indicates the position of the ligand before redocking, while the light green color shows its position after redocking.
3.3 Molecular docking-based virtual screening and ADMET analysis
According to the virtual screening and docking validation results, each of the 10 compounds exhibited the highest binding affinity energy for the TMPRSS2 protein and the SARS-CoV-2 Omicron spike protein, as shown in Tables 3 and 4. Compounds with affinity energies ranging from −10.43 to −8.63 kcal/mol were discovered during the screening of test ligands with TMPRSS2 (Table 3). The bond affinity energy of the test ligand exceeds that of the nafamostat co-crystal, which is −6.52 kcal/mol. Camostat has a lower comparable ligand bonding affinity energy (-8.25 kcal/mol) than nafamostat. In contrast, the findings of screening test ligands with SARS-CoV-2 Omicron spike protein (Table 4) revealed that compounds had an affinity energy range of −9.68 to −8.70 kcal/mol. The binding affinity energy of the tested ligands was higher than the binding affinity energy of the reference ligand mefloquine, which was −6.34 kcal/mol. Molecular docking-based virtual screening revealed that the test ligands derived from the secondary metabolites of Indonesian marine invertebrates were more successful than the reference ligands at inhibiting TMPRSS2 and SARS-CoV-2 Omicron spike protein. However, more research must be done to ensure that the hit compounds work against COVID-19 in vitro and in vivo.
No.
Compound
Bioactive class
Species
Mol. weight (g/mol)
Docking results
Binding affinity (kcal/mol)
Ki
RMSD (Å)
H-bond interaction
Hydrophobic interaction
Reference Ligands
1
Camostat
–
–
398.40
−8.25
894.50 nM
0.040
HIS296, SER441, GLY464, ASP435, SER436
GLU299, VAL298, LEU302, TRP308, VAL280, SER460, GLY462, CYS437, ALA466, SER463, GLY472, ARG470, TRP461, GLY439
2
Nafamostat (co-crystal)
–
–
347.37
−6.52
16.49 µM
0.134
SER460, TYR416, PRO471, SER463, LYS467, ALA466
TRP461, THR459, SER441, CYS437, ARG470, GLY462, GLY464, ASP435, GLY472, SER436, VAL473
Test Ligands
1
Acanthomanzamine A
Alkaloid
Acanthostrongylophora ingens
594.32
−9.67
81.71 nM
1.643
SER460, LYS342, LYS340
GLN438, CYS297, LEU302, VAL280, CYS437, GLY464, GLY439, CYS465, SER441, GLY462, SER339, THR341
2
Acanthomanzamine C
Alkaloid
Acanthostrongylophora ingens
562.33
−9.75
70.93 nM
0.234
GLY439, VAL280
THR393, TRP461, THR459, CYS437, ASP440, CYS297, HIS296, CYS281, HIS279
3
Acanthomanzamine D
Alkaloid
Acanthostrongylophora ingens
562.37
−9.30
151.19 nM
0.178
GLY462
LYS342, GLN438, SER463, HIS296, SER441, GLY439, SER460, THR459, VAL473, GLY472, SER436, CYS437, GLY464, GLU389
4
Acanthomanzamine E
Alkaloid
Acanthostrongylophora ingens
576.38
−9.93
52.91 nM
0.033
GLY462
THR341, SER460, SER441, GLN438, CYS437, GLY464, SER463
5
Cortistatin G
Alkaloid
Corticium complex
458.33
−9.39
131.53 nM
0.825
–
GLU299, LEU302, VAL 280, GLY439, SER441, GLN438, SER460, THR459, TRP461, SER436, VAL473, GLY462, GLY464, CYS437
6
Cortistatin J
Alkaloid
Corticium complex
438.27
−8.83
338.08 nM
0.078
GLN438
SER441, TRP461, VAL473, THR459, GLY472, GLY462, CYS437, GLY464, GLU389, THR393, LYS392
7
Cortistatin L
Alkaloid
Corticium complex
456.28
−9.29
154.76 nM
0.199
GLY462
LYS340, SER441, GLY439, CYS437, SER436, GLY472, CYS465, SER460, SER463
8
Jaspamide Q
Peptide
Jaspis splendens
630.34
−8.63
470.44 nM
0.296
SER436, ASP 435, SER460
SER463, GLN438, GLU389, GLY464, THR459, GLY462, CYS437, GLY462, HIS296, LYS340, LEU419
9
Jaspamide R
Peptide
Jaspis splendens
786.16
−9.60
91.44 nM
0.392
SER441, GLY464, GLY391
LYS342, THR459, TRP461, VAL473, SER436, GLY472, CYS465, LYS392, GLU389
10
Pre-neo-kauluamine
Alkaloid
Acanthostrongylophora ingens
580.34
−10.43
22.66 nM
0.406
VAL473, SER436, ASP435, GLY462
GLY472, CYS437, THR459, SER441, SER460, GLN438, GLY439, LYS390, LYS342, GLY464, SER463, TRP461, CYS465
No.
Compound
Bioactive class
Species
Mol. weight (g/mol)
Docking results
Binding affinity (kcal/mol)
Ki
RMSD (Å)
H-bond interaction
Hydrophobic interaction
Reference Ligand
1
Mefloquine
–
–
378.31
−6.34
22.68 µM
0.465
VAL605, VAL632, ARG631, TYR633
GLY649, GLN604, VAL607, ASP291, TRP630
Test Ligands
1
Acantholactam
Alkaloid
Acanthostrongylophora ingens
594.32
−8.93
283.47 nM
0.76
ARG631, VAL605
LEU290, ASN603, ASP291, GLN604, ILE689, GLY649, VAL632, PRO292, LEU293, SER688, SER637
2
Acanthomanzamine A
Alkaloid
Acanthostrongylophora ingens
594.32
−8.91
294.52 nM
0.733
VAL605, VAL632, THR629, LEU290
ALA606, VAL607, ARG631, ASP291, TRP630, LEU293, GLN604, ASN603
3
Acanthomanzamine B
Alkaloid
Acanthostrongylophora ingens
545.36
−8.70
420.11 nM
0.230
VAL605, ASP291
VAL632, PRO292, ARG631, ASN603, SER294, LEU293, SER637, GLN604, GLY649, ALA606
4
Acanthomanzamine C
Alkaloid
Acanthostrongylophora ingens
562.33
−9.19
183.69 nM
0.090
ARG631, VAL632, VAL605
TRP630, THR629, PRO292, LEU290, ASP291, ASN603, GLY649, SER688, ALA650
5
Acanthomanzamine D
Alkaloid
Acanthostrongylophora ingens
562.37
−9.14
198 nM
0.068
VAL605
ALA606, VAL607, THR629, ARG631, GLN604, ASP291, ASN603, LEU293
6
Acanthomanzamine E
Alkaloid
Acanthostrongylophora ingens
576.38
−8.98
262.83 nM
1.412
VAL605
ARG631, VAL632, ASP291, ASN603, GLY636, SER637, GLN604, GLY649, ILE689
7
Cortistatin J
Alkaloid
Corticium complex
438.27
−8.97
264.46 nM
0.018
SER688
ILE689, GLY649, GLN674, ASN638, SER680, SER682, GLY636
8
Jaspamide R
Peptida
Jaspis splendens
786.16
−9.33
144.30 nM
0.251
VAL632, VAL605, ASP291
TRP630, ARG631, VAL607, ALA606, GLY649, SER637, SER688
9
Pre-neo-kauluamine
Alkaloid
Acanthostrongylophora ingens
580.34
−9.68
79.62 nM
0.078
GLY649, ILE689, GLN604
VAL605, LEU293, PRO292, GLY636, SER634, ASP291, ALA650, SER688
10
Psammaplysin L
Alkaloid
Aplysinella strongylata
770.81
−8.80
353.78 nM
1.422
VAL605, GLN604, ALA681
LEU293, PRO292, ILE689, GLU651, ASN638, SER680, SER637, SER682, GLY636, SER688, GLY649, TYR633
A novel drug’s pharmacokinetics and toxicity must be known before it can be developed. The results show that acanthomanzamine C, cortistatin G, cortistatin J, and cortistatin L met the ADMET analysis requirements (Table 5) and indicated feasible oral administration. TPSA (Topological Polar Surface Area) represents a molecule's total surface of polar atoms, while HIA (Human Intestinal Absorption) shows how well the drug is absorbed in the gastrointestinal tract. TPSA and HIA parameters are useful indicators to describe the bioaccessibility and bioavailability of a compound. Parameter T1/2 (half-life) is used to set the interval of drug administration. Carcinogenicity and AMES toxicity are important to know whether a compound has carcinogenic or mutagenic effects. The BIOVIA Discovery Studio Visualizer tool was used to depict the top two test ligands and their reference ligands with the greatest binding affinity energy to each TMPRSS2 (Fig. 5) and SARS-CoV-2 Omicron spike protein (Fig. 6). The details of the H-bond interactions and those with hydrophobic interactions can be seen in Tables 3 and 4.
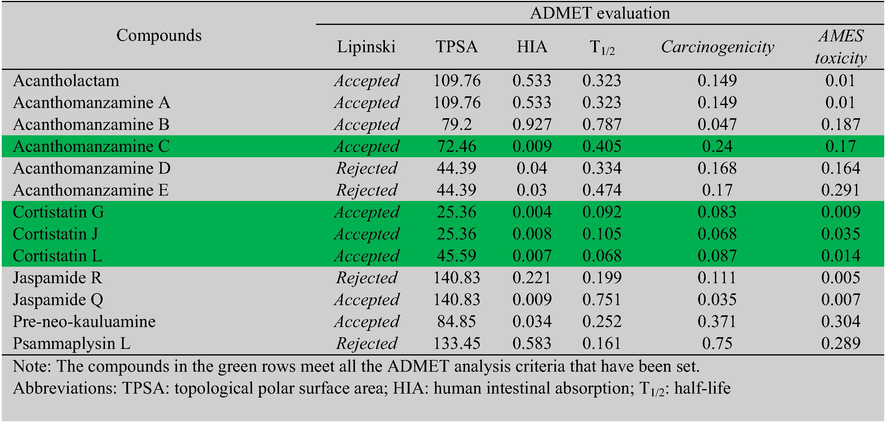
Interactions of the reference ligands (camostat and nafamostat) and the test ligands (acanthomanzamine C and cortistatin G) with TMPRSS2 amino acid residues.
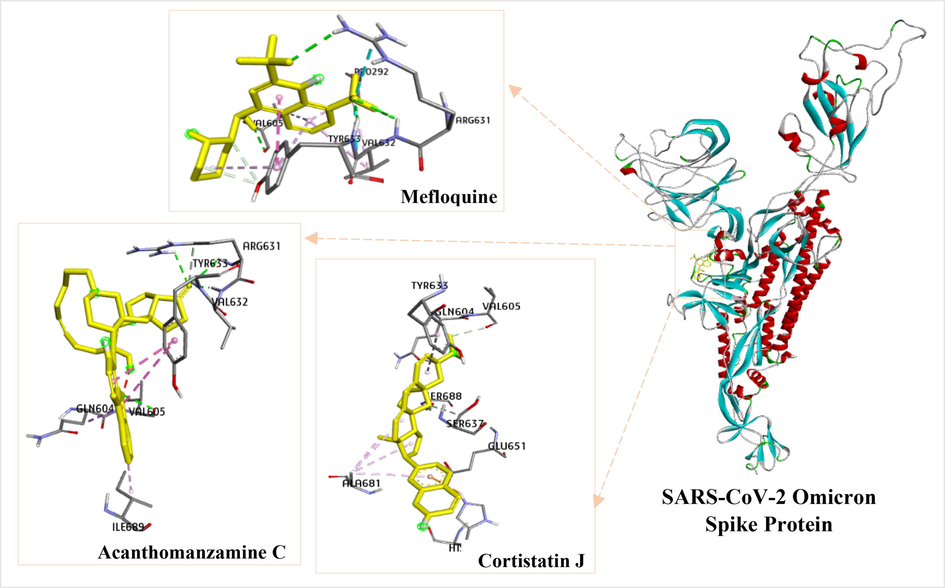
Interactions of the reference ligand (mefloquine) and the test ligands (acanthomanzamine C and cortistatin J) with SARS-CoV-2 Omicron spike protein amino acid residues.
3.4 Molecular dynamics simulation studies
Acanthomanzamine C, the primary molecule in the complex with TMPRSS2 and SARS-CoV-2 Omicron spike protein, was subjected to molecular dynamics simulations to determine the extent of this compound’s interactions with TMPRSS2 and SARS-CoV-2 Omicron spike protein. The most important ways to evaluate protein dynamics are RMSD and RMSF analyses.
The dynamics simulation of the ligands nafamostat (co-crystal), camostat (reference ligand), and acanthomanzamine C in association with TMPRSS2 protein was examined (Fig. 7A). The combination of nafamostat and TMPRSS2 was shown to be stable within the 100-ns period with RMSD less than 3 Å. In comparison to the nafamostat, the camostat and acanthomanzamine C exhibited an increase in RMSD after 40 ns, after which the graph stabilized up to 100 ns at RMSD less than 4 Å. Fig. 7B depicts the RMSD mefloquine (reference ligand) and acanthomanzamine C complexed with the SARS-CoV-2 Omicron spike protein. Mefloquine and acanthomanzamine C showed slight fluctuations but stabilized within the first 50 ns, with small variations found in the remaining duration at RMSD less than 4 Å. The RMSF figure (Fig. 8A, B) shows a high degree of variation in the amino acid residues implicated in the loop region of TMPRSS2 and SARS-CoV-2 Omicron spike protein, with maximum degrees of fluctuation reaching 6 Å and 4 Å, respectively. A careful examination reveals that the residues implicated in the ligand’s binding have low RMSF values.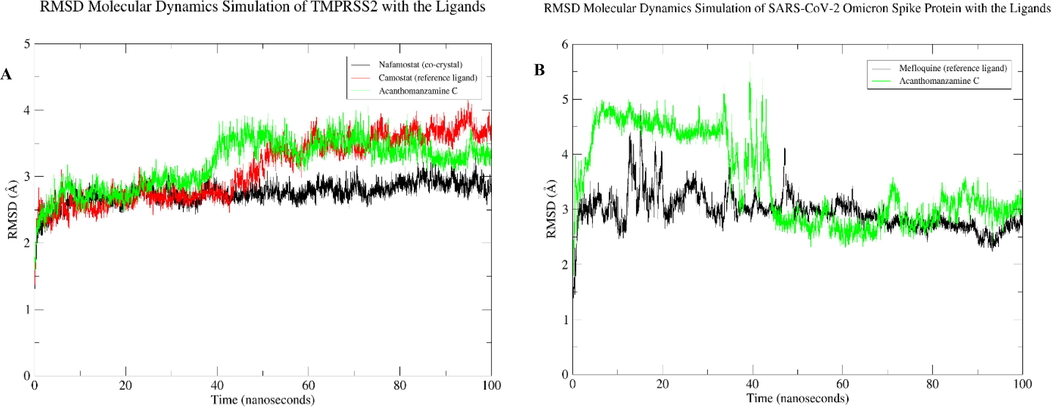
RMSD results for 100 ns: A) ligands in a complex with the TMPRSS2 and B) ligands in a complex with the SARS-CoV-2 Omicron spike protein.
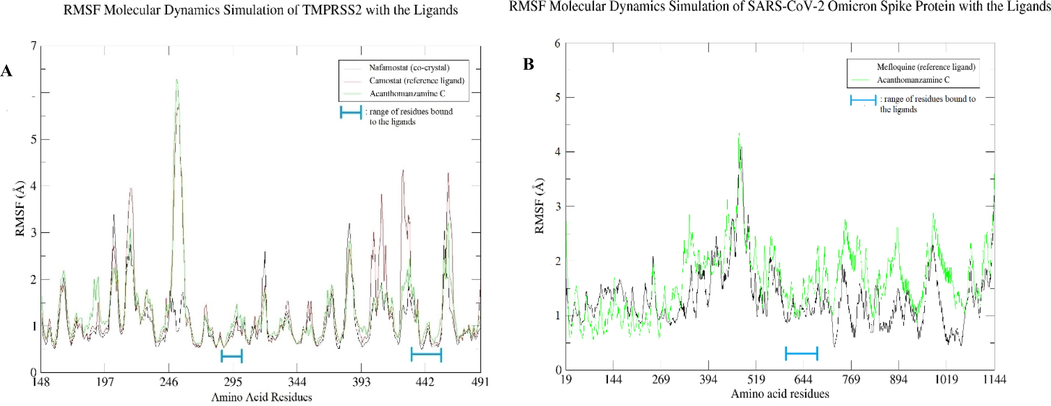
RMSF results for 100 ns: A) amino acid residues of the TMPRSS2 and B) amino acid residues of the SARS-CoV-2 Omicron spike protein.
4 Conclusions
Based on the results of molecular docking-based virtual screening and molecular dynamics simulations, the best compound, acanthomanzamine C, was chosen as a prospective inhibitor of TMPRSS2 and SARS-CoV-2 Omicron spike protein. Acanthomanzamine C demonstrated higher binding potential than nafamostat and camostat as reference ligands for TMPRSS2, as well as higher binding affinity energy than mefloquine as a spike protein reference ligand. Furthermore, in molecular dynamics simulation tests, acanthomanzamine C demonstrated persistent binding to TMPRSS2 and SARS-CoV-2 Omicron spike protein after the initial 50 ns, with minor variations. Therefore, we suggest continued studies on this hit molecule to identify a long-lasting cure for the COVID-19 pandemic.
Funding.
There was no external funding for this research.
Acknowledgement
During this research, the molecular dynamics simulation for SARS-CoV-2 Omicron spike protein with chosen ligands was performed at MAHAMERU BRIN HPC, National Research and Innovation Agency of Indonesia (BRIN).
Declaration of Competing Interests
The authors declare that they have no known competing financial interests or personal relationships that could have influenced the work reported in this paper.
References
- Implementation of the Hungarian algorithm to account for ligand symmetry and similarity in structure-based design. J. Chem. Inf. Model.. 2014;54(2):518-529.
- [CrossRef] [Google Scholar]
- In-Silico Screening for Identification of Potential Inhibitors against SARS-CoV-2 Transmembrane Serine Protease 2 (TMPRSS2) Eur. J. Pharm. Sci.. 2021;162(March):105820
- [CrossRef] [Google Scholar]
- There Is Nothing Exempt from the Peril of Mutation – The Omicron Spike. Biomed. Pharmacother.. 2022;148(February):112756
- [CrossRef] [Google Scholar]
- In Silico Screening of Bioactive Compounds of Representative Seaweeds to Inhibit SARS-CoV-2 ACE2-Bound Omicron B.1.1.529 Spike Protein Trimer. Mar. Drugs. 2022;20(2):148.
- [CrossRef] [Google Scholar]
- Camostat Mesylate against SARS-CoV-2 and COVID-19—Rationale, Dosing and Safety. Basic Clin. Paharmacol. Toxicol.. 2021;128(2):204-212.
- [CrossRef] [Google Scholar]
- Safety and Heart Rate Changes in Covid-19 Patients Treated with Remdesivir. Int. J. Infect. Dis.. 2021;112:254-327.
- [CrossRef] [Google Scholar]
- Computational Screening of Potential Drugs against COVID-19 Disease: The Neuropilin-1 Receptor as Molecular Target. VirusDisease. 2022;33(1):23-31.
- [CrossRef] [Google Scholar]
- Bioactive Compounds from Marine Invertebrates for Potential Medicines – An Overview. Int. Lett. Nat. Sci.. 2015;34:42-61.
- [CrossRef] [Google Scholar]
- Mollamides B and C, Cyclic Hexapeptides from the Indonesian Tunicate Didemnum Molle. J. Nat. Prod.. 2008;71(6):941-995.
- [CrossRef] [Google Scholar]
- Repurposing of Clinically Developed Drugs for Treatment of Middle East Respiratory Syndrome Coronavirus Infection. Antimicrob. Agents Chemother.. 2014;58(8):4885-4893.
- [CrossRef] [Google Scholar]
- Role of Host Factors in SARS-CoV-2 Entry. J. Biol. Chem.. 2021;297(1):100847
- [CrossRef] [Google Scholar]
- Modeling of Loops in Protein Structures. Protein Sci.. 2000;9:1753-1773.
- [CrossRef] [Google Scholar]
- Structure and Activity of Human TMPRSS2 Protease Implicated in SARS-CoV-2 Activation. Nat. Chem. Biol.. 2022;18(9):963-971.
- [CrossRef] [Google Scholar]
- Exploring the Phytochemicals of Platycodon Grandiflorus for TMPRSS2 Inhibition in the Search for SARS-CoV-2 Entry Inhibitors. J. King Saud Univ. – Sci.. 2022;34(6):102155
- [CrossRef] [Google Scholar]
- Evaluation of Risk Factors for Uric Acid Elevation in COVID-19 Patients Treated with Favipiravir. Diagn. Microbiol. Infect. Dis.. 2022;000:115640
- [CrossRef] [Google Scholar]
- SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181(2):271-280.e8.
- [CrossRef] [Google Scholar]
- The Omicron Variant Is Highly Resistant against Antibody-Mediated Neutralization: Implications for Control of the COVID-19 Pandemic. Cell. 2022;185(3):447-456.e11.
- [CrossRef] [Google Scholar]
- Horizny, C., 2019. “The Drug Discovery Process.” Taconic Models for Life. Retrieved February 11, 2022 (https://www.taconic.com/taconic-insights/quality/drug-development-process.html).
- Discovery of TMPRSS2 Inhibitors from Virtual Screening as a Potential Treatment of COVID-19. ACS Pharmacol. Translational Sci.. 2021;4(3):1124-1135.
- [CrossRef] [Google Scholar]
- Molecular Docking between Human Tmprss2 and Sars-Cov-2 Spike Protein: Conformation and Intermolecular Interactions. AIMS Microbiol.. 2020;6(3):350-360.
- [CrossRef] [Google Scholar]
- Chemical Diversity and Biological Activity of Secondary Metabolites Isolated from Indonesian Marine Invertebrates. Molecules. 2021;26(7)
- [CrossRef] [Google Scholar]
- Tackling SARS-CoV-2: Proposed Targets and Repurposed Drugs. Future Med. Chem.. 2020;12(17):1579-1601.
- [CrossRef] [Google Scholar]
- Baseline Uric Acid Levels and Steady-State Favipiravir Concentrations Are Associated with Occurrence of Hyperuricemia among COVID-19 Patients. Int. J. Infect. Dis.. 2022;115:218-223.
- [CrossRef] [Google Scholar]
- Drug Repurposing for Identification of Potential Spike Inhibitors for SARS-CoV-2 Using Molecular Docking and Molecular Dynamics Simulations. Methods 2022
- [CrossRef] [Google Scholar]
- Screening S Protein – ACE2 Blockers from Natural Products: Strategies and Advances in the Discovery of Potential Inhibitors of COVID-19. Eur. J. Medicinal Chem.. 2021;226
- [CrossRef] [Google Scholar]
- Docking-Generated Multiple Ligand Poses for Bootstrapping Bioactivity Classifying Machine Learning: Repurposing Covalent Inhibitors for COVID-19-Related TMPRSS2 as Case Study. Comput. Struct. Biotechnol. J.. 2021;19:4790-4824.
- [CrossRef] [Google Scholar]
- In Silico Prediction of Mozenavir as a Potential Drug for SARS-CoV-2 Infection via Binding Multiple Drug Targets. Saudi J. Biol. Sci.. 2022;29(2):840-887.
- [CrossRef] [Google Scholar]
- Ni, D., Lau, K., Turelli, P., Raclot, C., Beckert, B., Nazarov, S., Pojer, F., Myasnikov, A., Stahlberg, H. and Trono, D., 2021. Structural Analysis of the Spike of the Omicron SARS-COV-2 Variant by Cryo-EM and Implications for Immune Evasion. BioRxiv 2021.12.27.474250. doi: https://doi.org/10.1101/2021.12.27.474250.
- Cembranoids of soft corals: recent updates and their biological activities. Natural Products and Bioprospecting. 2021;11(3):243-306.
- [CrossRef] [Google Scholar]
- Computational Screening and Biochemical Analysis of Pistacia Integerrima and Pandanus Odorifer Plants to Find Effective Inhibitors against Receptor-Binding Domain (RBD) of the Spike Protein of SARS-Cov-2. Arab. J. Chem.. 2022;15(2):103600
- [CrossRef] [Google Scholar]
- Virtual Screening and in Vitro Validation of Natural Compound Inhibitors against SARS-CoV-2 Spike Protein. Bioorganic Chem.. 2022;119(November 2021)
- [CrossRef] [Google Scholar]
- Molecular Screening of Antimalarial, Antiviral, Anti-Inflammatory and HIV Protease Inhibitors against Spike Glycoprotein of Coronavirus. J. Mol. Graphics Modell.. 2021;102
- [CrossRef] [Google Scholar]
- Enhancing the prefusion conformational stability of SARS-CoV-2 spike protein through structure-guided design. Front. Immunol.. 2021;12(April):1-13.
- [CrossRef] [Google Scholar]
- Unlike Chloroquine, Mefloquine Inhibits SARS-CoV-2 Infection in Physiologically Relevant Cells. Viruses. 2022;14(2):1-18.
- [CrossRef] [Google Scholar]
- In Silico Molecular Characterization of Human TMPRSS2 Protease Polymorphic Variants and Associated SARS-CoV-2 Susceptibility. Life. 2022;12(2)
- [CrossRef] [Google Scholar]
- Chasing COVID-19 through SARS-CoV-2 Spike Glycoprotein. VirusDisease. 2020;31(4):399-407.
- [CrossRef] [Google Scholar]
- Bioactive Compounds from Marine Invertebrates and Their. Pharm. Potential. 2021;13(2):739-746.
- [CrossRef] [Google Scholar]
- Computational Screening of Camostat and Related Compounds against Human TMPRSS2: A Potential Treatment of COVID-19. Saudi Pharm. J. 2022:7.
- [CrossRef] [Google Scholar]
- Mefloquine, a Potent Anti-Severe Acute Respiratory Syndrome-Related Coronavirus 2 (SARS-CoV-2) Drug as an Entry Inhibitor in Vitro. Front. Microbiol.. 2021;12(April)
- [CrossRef] [Google Scholar]
- Proteolytic Activation of the SARS-Coronavirus Spike Protein: Cutting Enzymes at the Cutting Edge of Antiviral Research. Antiviral Res.. 2013;100(3):605-614.
- [CrossRef] [Google Scholar]
- Identification of Potential Plant Bioactive as SARS-CoV-2 Spike Protein and Human ACE2 Fusion Inhibitors. Comput. Biol. Med.. 2021;136(April):104631
- [CrossRef] [Google Scholar]
- Structural Insights and Inhibition Mechanism of TMPRSS2 by Experimentally Known Inhibitors Camostat Mesylate, Nafamostat and Bromhexine Hydrochloride to Control SARS-Coronavirus-2: A Molecular Modeling Approach. Inf. Med. Unlocked. 2021;24(February):100597
- [CrossRef] [Google Scholar]
- Serious Bradycardia and Remdesivir for Coronavirus 2019 (COVID-19): A New Safety Concerns. Clin. Microbiol. Infect.. 2021;27(5):791.e5-791.e8.
- [CrossRef] [Google Scholar]
- AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem.. 2009;31(2)
- [CrossRef] [Google Scholar]
- U.S. Food and Drug Administration, 2022. Fact Sheet for Healthcare Providers: Emergency Use Authorization for Molnupiravir.
- Virtual Screening by Targeting Proteolytic Sites of Furin and TMPRSS2 to Propose Potential Compounds Obstructing the Entry of SARS-CoV-2 Virus into Human Host Cells. J. Tradit. Complement. Med.. 2022;12(1):6-15.
- [CrossRef] [Google Scholar]
- Identification of Nafamostat as a Potent Inhibitor of Middle East Respiratory Syndrome Coronavirus s Protein-Mediated Membrane Fusion Using the Split-Protein-Based Cell-Cell Fusion Assay. Antimicrob. Agents Chemother.. 2016;60(11):6532-6659.
- [CrossRef] [Google Scholar]
- Protease Inhibitors Targeting Coronavirus and Filovirus Entry. Antiviral Res.. 2015;116:76-84.
- [CrossRef] [Google Scholar]
- Spontaneous Binding of Potential COVID-19 Drugs (Camostat and Nafamostat) to Human Serine Protease TMPRSS2. Comput. Struct. Biotechnol. J.. 2021;19:467-476.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.104984.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1
Supplementary data 2
Supplementary data 2




