

Translate this page into:
Novel benzimidazole derivatives as anti-cervical cancer agents of potential multi-targeting kinase inhibitory activity
⁎Corresponding author. ea.mabrouk@nrc.sci.eg (Eman A. Abd El-Meguid)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
New functionalized benzimidazole derivatives were designed, synthesized and evaluated for their in vitro anti-cervical cancer activity. The most promising derivatives were evaluated in vitro as multi-targeting kinase inhibitors against EGFR, HER2, PDGFR-β and VEGFR-2. Compound 13 exhibited significant apoptotic activity and cell cycle arrest at G2/M phase. A molecular docking study was carried out to represent the binding modes of the promising compounds in the active pocket of HER2.
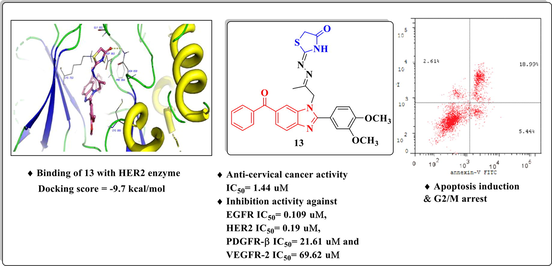
Abstract
New derivatives bearing 6-benzoyl benzimidazole core conjugated with different alicylic, aromatic and / or heterocyclic rings were synthesized. Utilizing cervical cancer as a putative target, the new compounds were evaluated in vitro as anti-cervical cancer agents. The promising compounds were examined against normal cell line WI-38. The promising compounds were examined as EGFR, HER2, PDGFR-β and VEGFR-2 inhibitors. As a representative example, compounds 9 and 13 were subjected to apoptosis induction and cell cycle analyses. Molecular modeling study was also performed to elucidate the binding modes of the active candidates with amino acid residues of HER2 and rationalize their pharmacological effects.
Abstract
Multi-target EGFR, HER2, VEGFR-2 and PDGFR is an improved strategy for the treatment of solid tumors. This work deals with synthesis of an array of new 6-benzoyl benzimidazole derivatives utlizing1-(6-benzoyl-2-(3,4-dimethoxyphenyl)-1H benzo[d] imidazol-1-yl)propan-2-one (1) as a starting compound. The new compounds were screened as cytotoxic agents against cervical cancer cells (Hela) and Doxorubicin served as a reference drug. Most of the tested compounds showed promising anticancer activity in addition to their safety towards the normal cell line. The most potent candidates were evaluated as EGFR, HER2, PDGFR-β and VEGFR2 inhibitors in comparison to Erlotinib. Compounds 9 and 13 exhibited promising suppression effects. Also, the latter compounds exhibited their ability to induce cellular apoptosis alongside cell cycle arrest at the G2/M phase and accumulation of cells in pre-G1 phase. Molecular docking analysis suggested that compounds 2c, 3f, 9, 12 and 13 tightly interacts with the amino acid residues in the active binding site of HER2 kinase.
Keywords
6-Benzoylbenzimidazole
Cervical cancer
EGFR
HER2
Apoptosis
Molecular docking

1 Introduction
Cervical cancer disease is assumed to be the fourth most common cancer affecting women worldwide. It has been estimated about 570 000 cases and 311 000 deaths in 2018, accounting for 3.3% of all cancer-related deaths (Vora and Gupta, 2018; Abbas, and Abd El-Karim, 2019). Metastasis develops in 15% to 61% of women with cervical cancer within the first 2 years of completing treatment (Šarenac and Mikov, 2019; Ueda et al., 2017). Cervical cancer has moderate to high levels of epidermal growth factor receptor (EGFR) protein expression. The percentage of cervical adenocarcinoma cases in which EGFR expression was detected varied from 19% to 67% in previous studies, and its overexpression was shown to be associated with poor prognosis (Pérez-Regadera et al., 2011). As single agents in the treatment of recurrent cervical cancer, Gefitinib and Erlotinib have shown minimal activity (Schilder et al., 2009; Goncalves et al., 2008).
The human epidermal growth factor receptor (HER) family is categorized into four candidates, which are HER1 (EGFR/ErbB1), HER2 (ErbB2/neu), HER3 (ErB3) and HER4 (Erb4) that are only expressed at low levels in normal human tissues (Soliman et al., 2019; Hou et al., 2019).
It has been reported that, several malignant tumors including breast, cervix, ovary, lung, colon, head, and neck are associated with deregulation of HER family signaling enhancing proliferation, invasion, metastasis, angiogenesis, and tumor cell survival (Zou et al., 2018). Additionally, the up regulation of EGFR and HER2 receptors is correlated with therapeutic resistance, which represents a significant a rationale for continuous urgent need for drug intervention (El-Sherief et al., 2018).
Molecular targeted medications represent one of the main strategies in the research of novel cancer targets and associated pathways (Oh et al., 2015). Many EGFR and HER2 targeted drugs were approved by the U.S. Food and Drug Administration (FDA) in recent years. They are currently divided into (i) first generation drugs that interact reversibly to EGFR (such as; Gefitinib, Erlotinib, Icotinib and Lapatinib), (ii) second generation drugs of irreversible dual EGFR/HER2 binding thereby, enhancing their potencies by suppressing EGFR signaling more effectively (such as Afatinib, Dacomitinib and Neratinib) and (iii) third-generation agents that overcome resistance to the previous two generations (such as; Nazartinib, Avitinib and Osimertinib) (Das et al., 2019; Kujtan and Subramanian, 2019) (Fig. 1).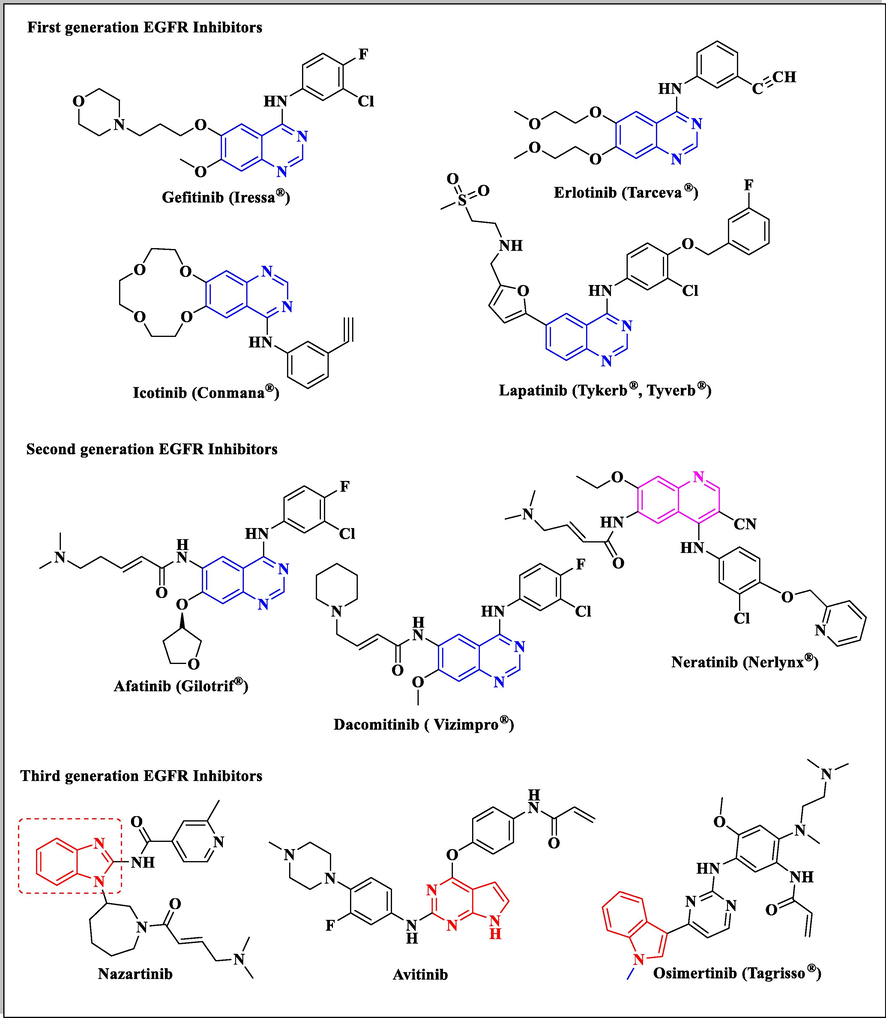
Various approved drugs of EGFR tyrosine kinase inhibiting activity.
Benzimidazole is an isostere of purine nucleosides so; it is widely used as a basic nucleus in the development of different anticancer agents (Jawaid Akhtar et al., 2018; Shrivastava et al., 2017). Various researches investigated that benzimidazole derivatives showing anticancer properties introduce their activities through different mechanisms via DNA intercalation, topoisomerases-I & II inhibition, androgen receptor antagonistic effect, poly (ADP-ribose) polymerase (PARP), dihydrofolate reductase (DHFR) enzymes and microtubule inhibiting activity. Moreover, different anticancer benzimidazole candidates were confirmed as potent EGFR inhibitors (Shrivastava et al., 2017). In addition, the third generation EGFR inhibitor Nazartinib has a benzimidazole structure and the other two candidates Avitinib and Osimertinib (Zhang, 2016) are indolopyrimidine and indole derivatives, which are bio-isosteres to benzimidazole ring.
Recently, a paradigm change have been confirmed in designing the significant selective agents targeting a single protein or gene to controlling a multiplicity of targets within related signaling pathways (Elzahhar et al., 2019). Hence, this effort was focused on seeking for new multi-targeting anti-cervical cancer leads of high activity and selectivity with low toxicity that can be reached to the clinical stages.
In continuation of our previous efforts to gain new compounds bearing various heterocyclic scaffolds of efficient anticancer activity targeting different enzymes (Abd El-Meguid et al., 2019; Abd El-Meguid, and Ali, 2016), in this work, Nazartinib structure helped for rational design and synthesis of new derivatives bearing 6-benzoyl benzimidazole scaffold hybridized at C-2 with 3,4-dimethoxyphenyl moiety and at position N-1 with substituted phenylbut-3-en-2-one I, substituted phenyl iminopropyl II, propan-2-ylidene-hydrazine-1-(thio)carboxamide side chains III and propan-2-ylidenehydrazine-thiazoline/thiazolidin-4-one side chain IV (Fig. 2). The newly prepared compounds were evaluated as anti-cervical cancer agents. Furthermore, the most active derivatives were evaluated as EGFR/HER2 inhibitors. Additionally, the most promising derivatives were further subjected to kinase inhibition assay against the other two receptor tyrosine kinases (RTKs); vascular endothelial growth factor receptor-2 (VEGFR-2) and platelet-derived growth factor receptor (PDGFR-β) as well as cell cycle analysis and apoptosis induction study. Molecular docking study was performed for the most promising structures inside the active site of HER2 to confirm their possible mechanism of action.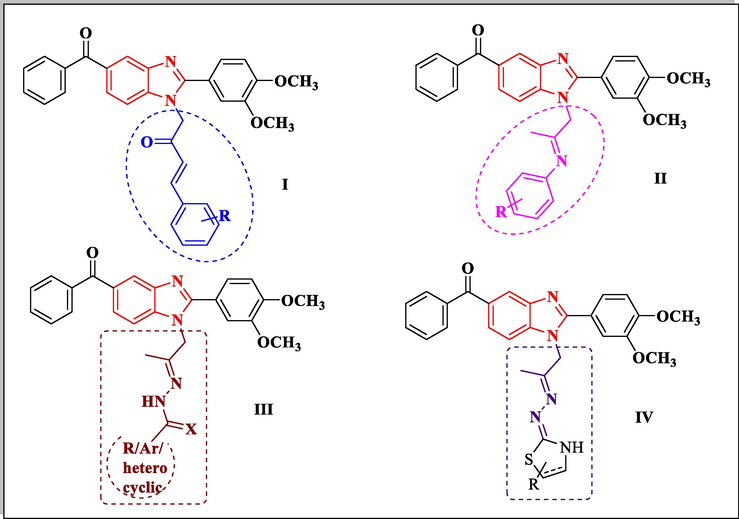
Newly designed benzimidazole derivatives as EGFR tyrosine kinase inhibitors.
2 Result and discussion
2.1 Synthesis
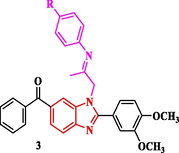
The synthetic approaches of the targeted benzimidazole compounds were described in Schemes 1-3. The key starting compound 1-(6-benzoyl-2-(3,4-dimethoxyphenyl)-1H-benzo[d]imidazol-1-yl)propan-2-one (1) was prepared by the reaction of o-phenylenediamine A with 3,4-dimethoxybenzaldeyde to give the corresponding (2-(3,4-dimethoxyphenyl)-1H-benzo[d]imidazol-6-yl)(phenyl)methanone derivative B which was alkylated with chloroacetone in dry acetone in the presence of potassium carbonate as a basic medium to give the target starting precursor 1. Compound 1 was allowed to react with different appropriate aromatic aldehydes in ethyl alcohol in the presence of a catalytic amount of piperidine as a basic medium to give the corresponding chalcone derivatives 2a-2e. In order to construct the Schiff bases 3a-3f, the starting 1 was heated with various aromatic amines in ethanol containing a few amounts of acetic acid at the refluxing temperature (Scheme 1).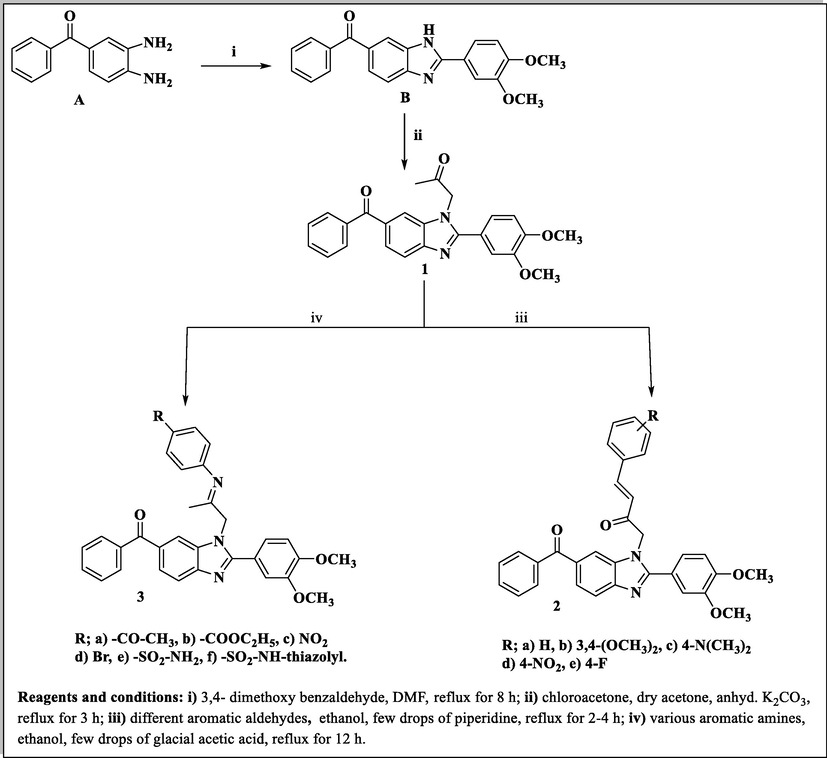
Synthesis of phenylbuten-2-one-benzimidazole and substituted phenyl iminopropyl – benzimidazole derivatives (2a-e and 3a-f).
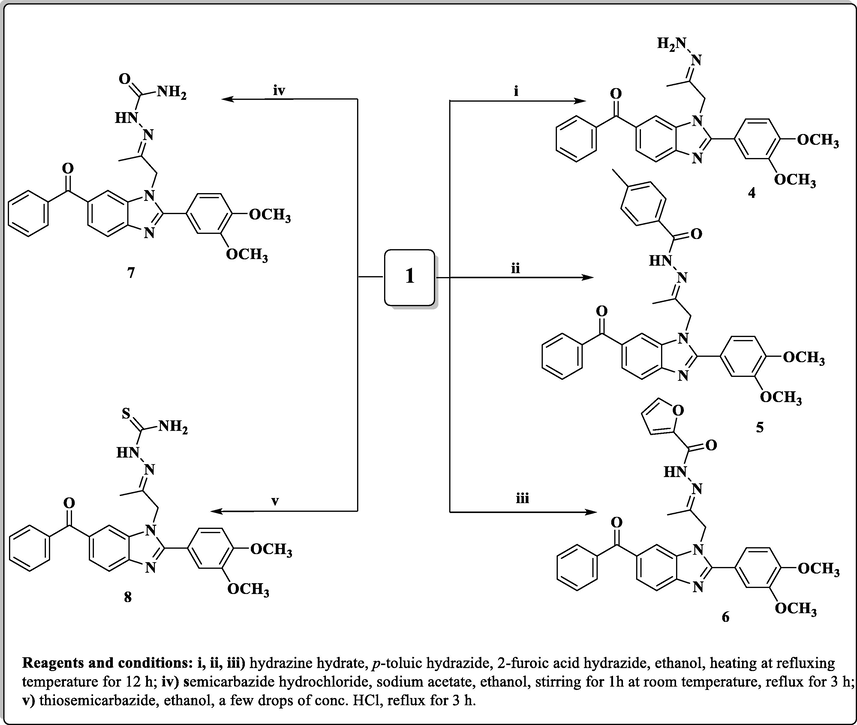
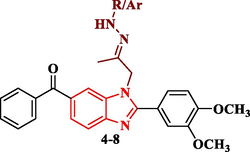
Synthesis of hydrazine-(thio)carboxamide-benzimidazole derivatives (4–8).
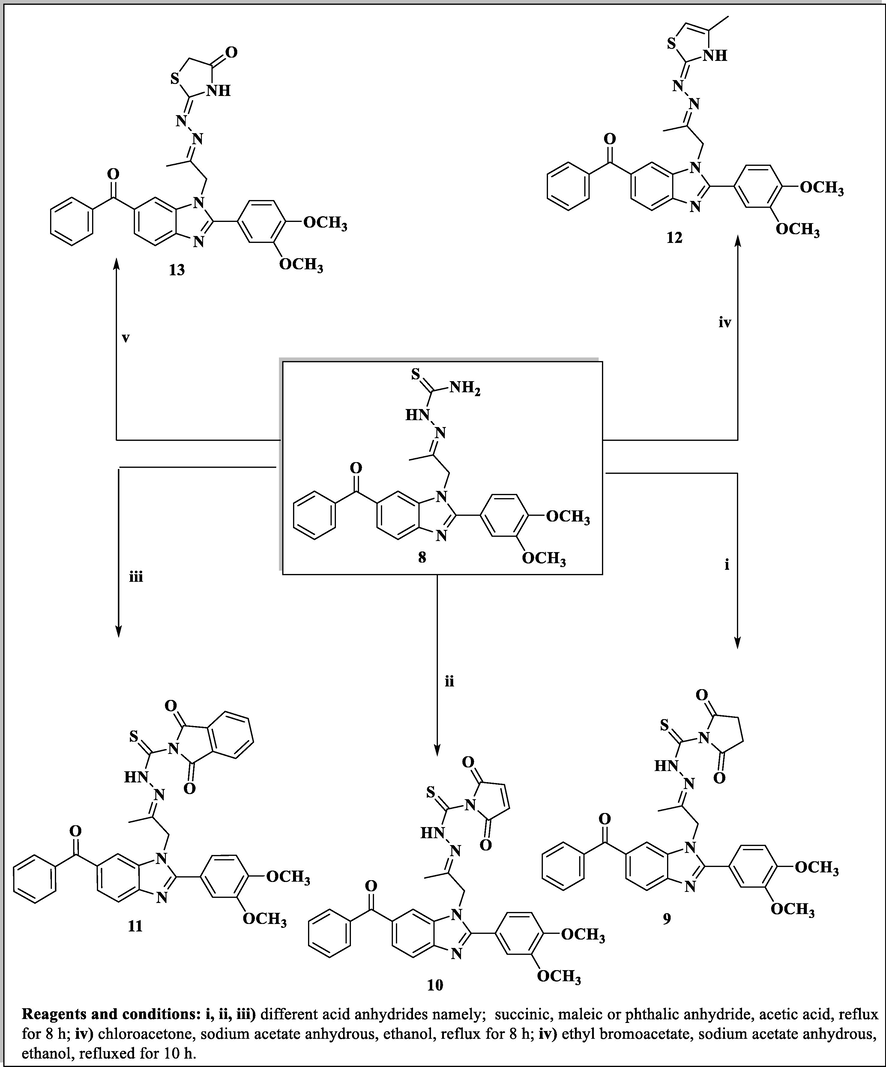
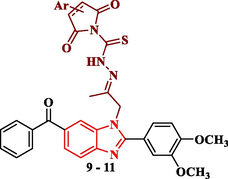
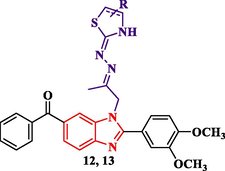
Synthesis of benzimidazole derivatives conjugated with 2,5-dioxopyrrolidine, 2,5-dioxopyrroline, 1,3-dioxoisoindoline, thiazoline and thiazolidine rings (9–13).
Also, compound 1 was allowed to react with hydrazine hydrate in absolute ethanol to give the corresponding hydrazone derivative 4, while its reaction with p-toluic hydrazide and 2-furoic acid hydrazide led to the formation of the corresponding hydrazone derivatives 5 and 6 (Scheme 2). When the acetyl compound 1 was condensed with semicarbazide hydrochloride in absolute ethanol in the presence of sodium acetate afforded the corresponding semicarbazone derivative 7, while condensation of 1 with thiosemicarbazide in ethanol containing a few drops of concentrated hydrochloric acid furnished the corresponding thiosemicarbazone derivative 8.
The thiosemicarbazone analogue 8 was utilized as a useful intermediate for further preparation of different benzimidazole-heterocyclic compounds. Thus, condensation of 8 with different acid anhydrides such as succinic acid anhydride, maleic acid anhydride and phthalic acid anhydride in acetic acid led to the formation of 2,5-dioxopyrrolidine, 2,5-dioxopyrroline and 1,3-dioxoisoindoline derivatives 9, 10 and 11, respectively. Also, cyclization of the 8 with haloketones such as chloroacetone and ethyl bromoacetate accomplished the thiazoline and thiazolidinone derivatives 12 and 13, respectively (Scheme 3).
2.2 In vitro anticancer activity
The present investigation deals with construction of new derivatives bearing 2-(3,4-dimethoxyphenyl)-1-substituted-benzo[d]imidazol-6-yl)(phenyl)methanone scaffold 2–13 for anti-cervical cancer evaluation. Accordingly, the new derivatives were subjected to MTT assay against Hela cell line using Doxorubicin as a reference drug (Thavamani et al., 2013; Prasetyaningrum et al., 2018) as represented in Fig. 3. The results were tabulated in Table 1. It has been detected that the 4-(dimethylamino)phenyl)but-3-en-2-one derivative 2c, the thiazolylsulfonyl derivative 3f, the 2,5-dioxopyrrolidine derivative 9, the 4-methylthiazoline derivative 12 and thiazolidin-4-one 13 exhibited the most potent cytotoxic activity that was higher than that gained by the reference drug (IC50 2c; 1.84, IC50 3f; 1.91; IC50 9; 1.62; IC50 12; 1.71; IC50 13; 1.44; IC50 Dox; 2.05 µM). A slight decrease in the potency was detected by the 4-nitrophenyl-iminopropyl derivative 3c of IC50 3.04 µM. It could be noted that the increase in the number of nitrogen atoms in addition to the incorporation of thiazole rings or pyrrolidine moiety contributed to more cytotoxic potency. This result comes in agreement with many researches which investigated the unique anticancer activity of different clinically applied anticancer drugs and compounds bearing thiazole and pyrrolidine scaffolds (Sharma et al., 2020; Smolobochkin, et al., 2019). A detectable drop-in the cytotoxic activity of the rest of the compounds was observed inducing IC50 values ranging from 6.20 − 28.12 µM. One of the most important drawbacks of the anticancer drugs is the deterioration of the normal cells. Accordingly, the cytotoxic activity of the potent tested compounds (2c, 3f, 9, 12 and 13) against cervical cancer cell (Hela) was also examined against the human normal fibroblast cells (WI-38) using Doxorubicin as a reference drug, and their IC50 values were determined by the MTT assay. The IC50 doses of all tested compounds against the normal cells were higher than their IC50 values against the Hela cells (Table 1). The most potent cytotoxic candidates 9 and 13 exhibited significant cytotoxic activity against Hela cell line and noteworthy safety profile against the normal cells.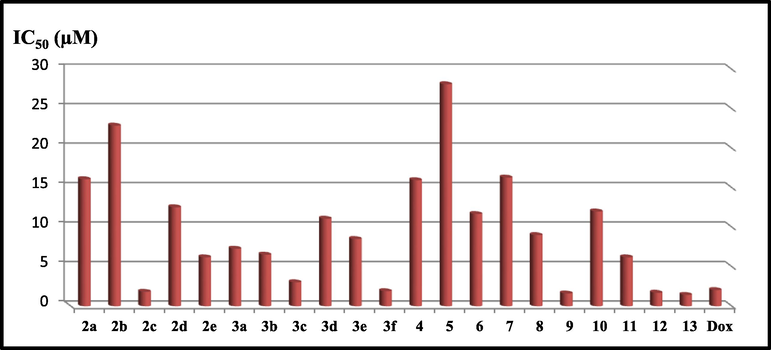
Anti-cervical cancer evaluation of compounds 2–13 against Hela cell line compared with Doxorubicin.
Cpd No.
R/Ar
Hela IC50 (µM)
WI-38 IC50 (µM)
2a
H
16.08 ± 0.74
–
2b
3,4-(OCH3)2
22.87 ± 0.61
–
2c
4-N(CH3)2
1.84 ± 0.08
12.45 ± 1.41
2d
4-NO2
12.53 ± 0.27
–
2e
4-F
6.20 ± 0.18
–
3a
-CO-CH3
7.29 ± 0.25
–
3b
-COOC2H5
6.53 ± 0.47
–
3c
-NO2
3.04 ± 0.07
–
3d
-Br
11.07 ± 0.53
–
3e
-SO2-NH2
8.51 ± 0.33
–
3f
-SO2-NH-thiazolyl
1.91 ± 0.13
13.62 ± 1.08
4
-NH2
15.96 ± 0.62
–
5
-CO-p-tolyl
28.12 ± 0.11
–
6
-CO-2-furan
11.67 ± 0.46
–
7
-CO-NH2
16.29 ± 0.67
–
8
-CS-NH2
8.99 ± 0.29
–
9
2,5-dioxopyrrolidine
1.62 ± 0.16
57.38 ± 2.66
10
2,5-dioxopyrroline
6.99 ± 0.42
–
11
1,3-dioxoisoindoline
6.23 ± 0.18
–
12
4-methylthiazoline
1.71 ± 0.09
25.11 ± 1.94
13
thiazolidin-4-one
1.44 ± 0.06
77.34 ± 3.61
Doxorubicin
2.05 ± 0.03
79.7 ± 0.07
2.3 Evaluation of EGFR/HER2 inhibitory activity
The compounds showing promising cytotoxic potency 2c, 3f, 9, 12 and 13 were further evaluated for their dual EGFR/HER2 inhibitory activity referring to Erlotinib (Table 2, Fig. 4). The 2,5-dioxopyrrolidine derivative 9 and the thiazolidin-4-one 13 represented the most promising potential EGFR inhibiting activity but slightly less than that of the reference Erlotinib (IC50 9; 0.157, IC50 13; 0.109 µM, IC50 Erlotinib; 0.079 µM) and are more potent than that of the reference Nazartinib (IC50 Nazartinib; 0.16 µM). Further decrease in the suppression effect was investigated by the compounds 2c, 3f and 12 of IC50 values; 0.214, 0.342, 0.305 µM, respectively, which might be contributed to improper fitting in the EGFR active site. On the other hand, all of the examined compounds exhibited significant inhibition against HER2 enzyme. Compound 13 produced 6.5 folds more potency than Erlotinib producing IC50; 0.19 µM, IC50 Erlotinib; 1.23 µM. Furthermore, compound 9 represented 5 folds more inhibition potency of HER2 than the reference drug of IC50; 0.25 µM. Compounds 12, 3f and 2c were more potent suppressors against HER2 than Erlotinib by 5–3 folds, respectively. Scrutinizing the resultant data, the examined benzimidazole derivatives are more selective towards HER2 than EGFR enzyme and the most potent cytotoxic candidates 9 and 13 induced dual EGFR/HER2 inhibitory activity. Abbreviations: EGFR, epidermal growth factor receptor; HER, human epidermal growth factor receptor; PDGFR, platelet‐derived growth factor receptor; VEGFR, vascular endothelial growth factor receptor.
Cpd No.
EGFR IC50 (μM)
HER2 IC50 (μM)
PDGFR-β IC50 (µM)
VEGFR-2 IC50 (µM)
2c
0.214 ± 0.008
0.31 ± 0.007
95.31 ± 0.19
143.44 ± 1.1
3f
0.342 ± 0.005
0.29 ± 0.005
89.11 ± 0.17
138.15 ± 2.2
9
0.157 ± 0.007
0.25 ± 0.003
76.09 ± 2.04
123.27 ± 1.7
12
0.305 ± 0.009
0.26 ± 0.006
85.11 ± 0.17
133.54 ± 1.5
13
0.109 ± 0.014
0.19 ± 0.004
21.61 ± 0.47
69.62 ± 1.4
Erlotinib
0.079 ± 0.001
1.23 ± 0.005
83.11 ± 0.17
124.7 ± 1.1
Nazartinib
0.160 ± 0.014
–
–
–
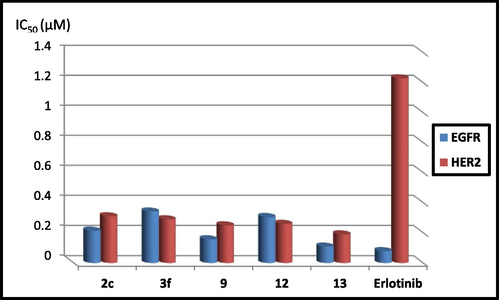
EGFR/HER2 inhibitory effects of compounds 2c, 3f, 9, 12 and 13 compared with Erlotinib.
2.4 In vitro kinase inhibition assay
Since it has been reported that the receptor tyrosine kinases (RTKs); EGFR, vascular endothelial growth factor receptor-2 (VEGFR-2) and platelet-derived growth factor receptor (PDGFR) participate significantly in regulating tumor cell proliferation, differentiation, survival, angiogenesis and apoptosis, they were successfully considered as attractive targets for anticancer therapies, particularly for producing multi-target anticancer drugs of improved therapeutic effectiveness (Li et al., 2011; Knight et al., 2010). Additionally, they also work in a synergistic pattern in regulating tumor response to anticancer RTK inhibitor drugs. Hence, multi-target RTK drugs that simultaneously inhibit EGFR, VEGFR-2 and PDGFR-β give substantially optimized anticancer therapeutic effect than drugs that inhibit individual RTKs (Wang et al., 2019). Accordingly, the promising cytotoxic compounds 2c, 3f, 9, 12 and 13 were subjected to kinase assay against PDGFR-β and VEGFR-2. As shown in Table 2, both compounds 9 and 13 exhibited promising inhibitory activity against both kinases (Fig. 5). Interestingly, the thiazoline compound 13 appeared about 3.8 times more potent than the reference drug Erlotinib against PDGFR-β (IC50; 21.61 µM, IC50 Erlotinib; 83.11 µM) and about 1.8 times more potent than Erlotinib against VEGFR-2 (IC50; 69.62 µM, IC50 Erlotinib; 124.7 µM). Furthermore, the 2,5-dioxopyrrolidine derivative 9 showed good inhibitory activities against PDGFR-β (IC50; 76.09 µM, IC50 Erlotinib; 83.11 µM) and against VEGFR-2 (IC50; 123.27 µM, IC50 Erlotinib; 124.7 µM). Further decrease in the suppression effects were investigated by the compounds 2c, 3f and 12 of IC50 values against PDGFR-β; 95.31, 89.11, 85.11 µM, respectively, and IC50 values against VEGFR-2; 143.44, 138.15, 133.54 µM, respectively.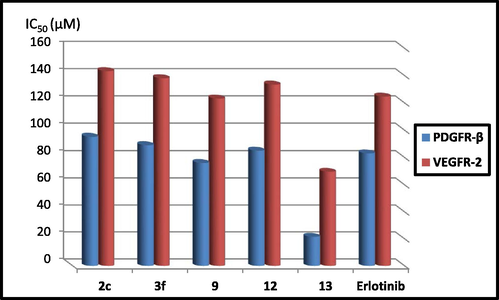
PDGFR-β and VEGFR-2 inhibitory effects of compounds 2c, 3f, 9, 12 and 13 compared with Erlotinib.
It could be noted that the potent anti-cancer activity of compound 13 against Hela cells comes as a companion to its significant multi-kinase suppression activity against the receptor tyrosine kinases (RTKs); EGFR, HER-2, PDGFR-β and VEGFR-2.
2.5 Cell cycle analysis and detection of apoptosis
The most promising compounds 9 and 13 were selected for further investigation to demonstrate their effects on the progression of cell cycle and apoptosis induction in Hela cell line. Exposure of Hela cells to both compounds 9 and 13 at their IC50 concentrations 1.62 and 1.44 µM for 24 h alongside induction of apoptosis were examined. Cells Exposure to both compounds led an interference with the normal cell cycle distribution. Both compounds 9 and 13 produced a pronounced increment in the cell percentages at pre-G1 phase by 13 and 27 folds, respectively and at G2/M phase by 7 and 10 folds, respectively compared to the untreated Hela cells (Table 3 and Fig. 6). Cumulation of cells in pre-G1 phase might be contributed to degradation of the genetic materials indicating a probable role the tested derivatives in inducing apoptosis in the cancer cells. Furthermore, cumulation of the cells at G2/M phase indicted cell cycle disruption at this stage preventing its mitotic stage.
Cpd No.
%G0-G1
%S
%G2/M
%Pre-G1
Comment
9
41.03
36.26
22.71
13.17
PreG1apoptosis&Cell growth arrest@G2/M
13
36.59
33.15
30.26
27.04
PreG1apoptosis&Cell growth arrest@G2/M
Cont. Hela cells
55.29
41.41
3.3
1.88
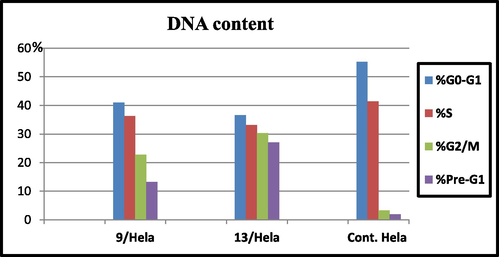
Cell Cycle analysis of compounds 9 and 13.
2.6 Apoptosis detection by annexin V-FITC assay
Apoptosis occurs due to the autonomous and organized destruction of genes-controlled cells (He et al., 2009). In addition, multiple external factors influence its startup. Thus, it was of interest to investigate the relationship between apoptosis of Hela cells and the compounds 9 and 13. The induction of apoptosis of the latter compounds was examined utilizing annexin V-FITC and propidium iodide (PI) double staining flow cytometry analysis (Cao et al., 2020; Yang et al., 2020). Based on the obtained results presented in Table 4 and Fig. 7, both derivatives effectively induced cell apoptosis at their IC50 concentrations of 1.62, 1.44 µM. Treatment of Hela cells with compounds 9 and 13 for 24 h led to 8.47% and 18.99% of apoptotic cells (late apoptosis), respectively, compared to 0.18% of apoptotic cells in the untreated control. Also, there was an increase in the early apoptosis from 0.47% (control) to 2.81% and 5.44% cause by 9 and 13, respectively with necrosis percent of 1.89 and 2.61% vs. 1.23% produced by DMSO control. It has been noted that the late apoptotic percentages caused by both examined derivatives were higher than that of the early phase which makes it more challenging to recover the apoptotic cells to safe ones. These results revealed that compounds 9 and 13 inhibited cell growth through cell apoptosis induction.
Apoptosis
Cpd No.
Total
Early
Late
Necrosis
9/Hela
13.17%
2.81%
8.47%
1.89%
13/Hela
27.04%
5.44%
18.99%
2.61%
Cont. Hela
1.88%
0.47%
0.18%
1.23%
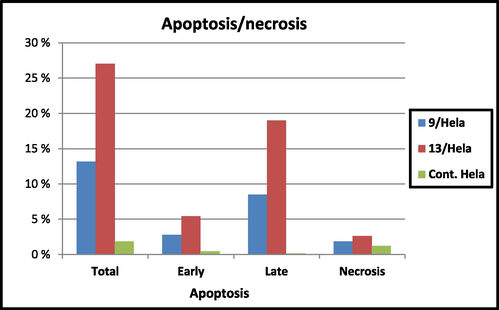
Apoptotic activity of benzimidazole compounds 9 and 13.
2.7 Molecular docking study
In order to better rationalize how the compounds 2c, 3f, 9, 12 and 13 contributed to the HER2 kinase inhibitory activities, molecular docking simulation studies were carried. The HER2 kinase domain complexed with a ligand was downloaded from the protein databank (PDB ID: 3RCD, http://www.rcsb.org). Protein preparation wizard of Schrödinger Suite (Sastry et al., 2013, Schrödinger, 2018) was used to correct the protein structural file by correcting bond orders, adding any missing hydrogen atoms, and completing unresolved side chains or loops.
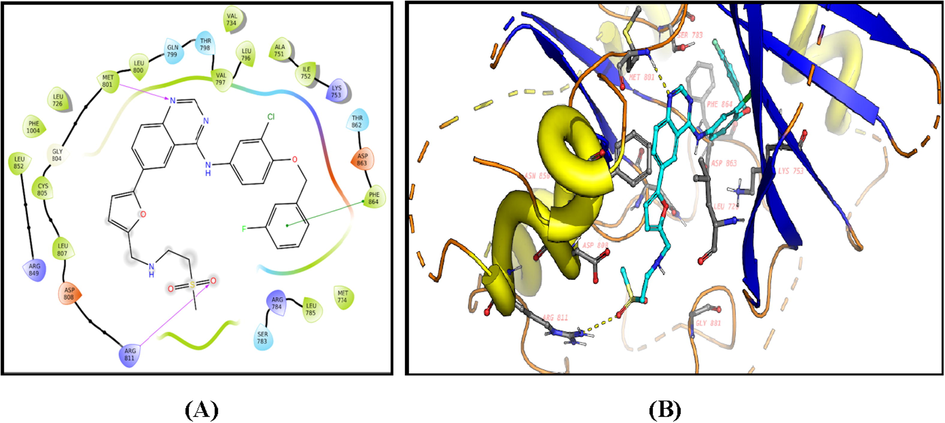
The ligands showed good fitting in the active site of HER2 with Glide docking scores in proportion to their biological activities. In general, the docking poses of the ligands demonstrated several good interactions with the DFG region, catalytic loop, activation loop, G-rich loop, phosphate binding loop and α-C helix of HER2 as shown in Table 5. Compound 2c was docked well in the ligand binding pocket with a Glide docking score of −8.03 kcal/mol. It shows strong hydrogen bonding and cation-π contacts with Lys 753. It has hydrophobic and electrostatic interactions with the surrounding amino acids in the binding site. Compound 3f showed a docking score of −9.1 kcal/mol (Fig. 8), and a strong hydrogen bonding with Gly 881 in the activation loop. The sulphonyl substituent shifted the structure to some extent inside the binding site. Other hydrophobic and polar contacted are observed with the kinase structural motifs. Compounds 9 (Fig. 9) and 12 (Fig. 10) have docking score of −9.4 and −9.3 kcal/mol, respectively. Both compounds demonstrated strong π-π contacts with Phe 864 in the DFG regions, and multiple polar/nonpolar contacts with the surrounding residues. Compound 13 exhibited strong fitting in the binding site with a Glide score of −9.7 kcal/mol. The compound is strongly interacting with Asn 850 of the catalytic loop, and Asp 863 of the DFG region via hydrogen bonding. It shows π-π contacts with Phe 864 of the DFG region (Fig. 11). It could be noted that the most potent HER2 inhibitors 9 and 13 showed the highest negative energy score of −9.4, −9.7 kcal/mol, respectively, with higher predicted binding affinity than the co-crystalized ligand Erlotinib which showed a docking score of −5.8 kcal/mol but lower binding affinity than Lapatinib (docking score = -12 kcal/mol) (Fig. 12).
Cpd No.
Docking Score (kcal/mol)
Hydrogen-Bond
Aromatic-Bond
pi-pi/ cation-pi
Hydrophobic
2c
−8.03
Lys753
Asp863
Lys753
Phe731, Val734, Ala751, Ile752, Leu796, Val797, Leu800, Met801, Phe864, Phe1004
3f
−9.1
Gly881
Asp863, Ser783
–
Ala730, Ala751, Leu800, Met801, Leu852
9
−9.4
–
Ser783, Asp863
Phe864
Phe731, Val734, Ala751, Ile752, Leu796, Leu800, Met801, Phe1004
12
−9.3
Asp863, Lys753
Ser783, Asp863
Phe864
Leu726, Ala730, Phe731, Ala751, Ile752, Met774, Leu785, Leu796, Leu800, Met801, Leu852, Phe1004
13
−9.7
Asn850, Asp863
Asp863, Ser783
Phe864
Ala730, Phe731, Ala751, Ile752, Leu796, Leu800, Met801, Phe1004
Erlotinib
−5.8
Cys805
Asp863
–
Leu726, Val734, Ala751, ILe752, Met774, Phe1004
Lapitanib
−12
Met801, Arg811
Ser783, Gln799, Asp808, Asp863
Phe864
Leu726, Val734, Ala751, ILe752, Met774, Phe1004
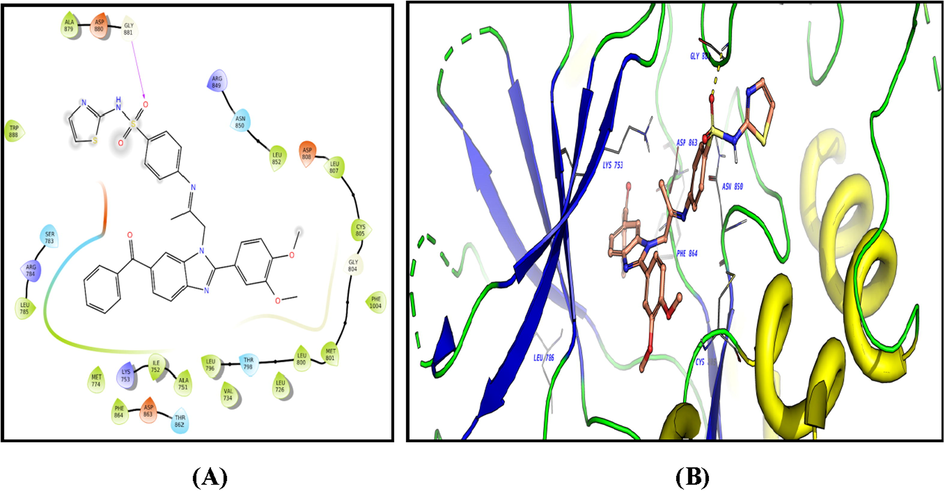
2D diagram (A) and 3D representation (B) of compound 3f showing its interaction with the HER2 kinase active site.
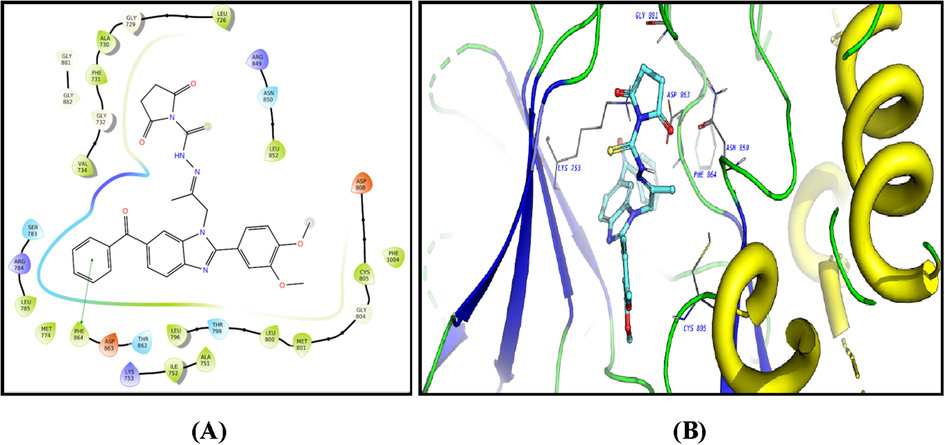
2D diagram (A) and 3D representation (B) of compound 9 showing its interaction with the HER2 kinase active site.
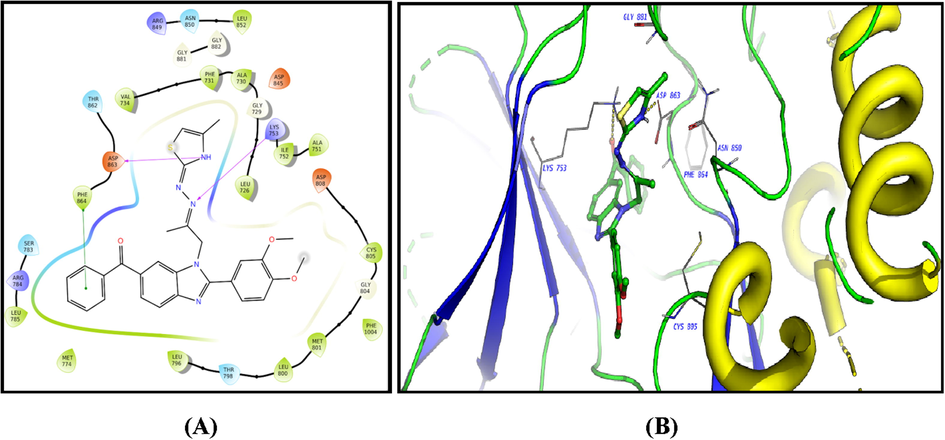
2D diagram (A) and 3D representation (B) of compound 12 showing its interaction with the HER2 kinase active site.
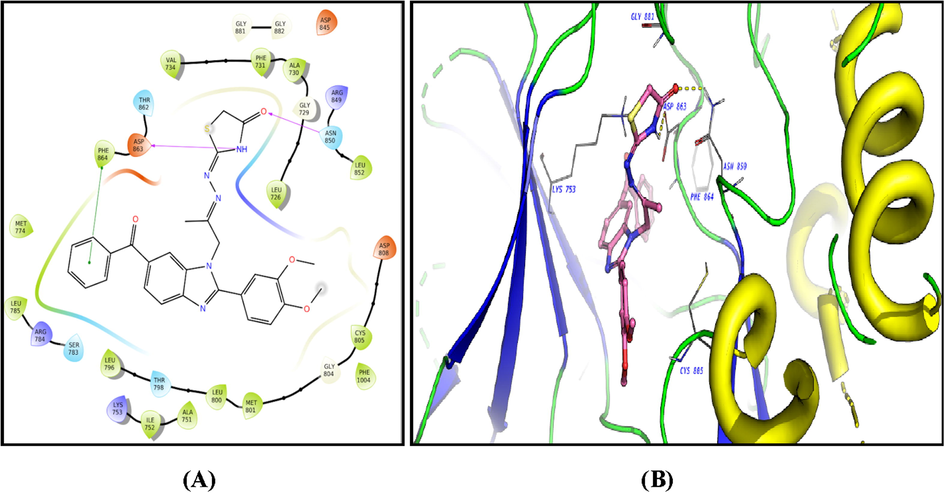
2D diagram (A) and 3D representation (B) of compound 13 showing its interaction with the HER2 kinase active site.
2D diagram (A) and 3D representation (B) of Lapatinib showing its interaction with the HER2 kinase active site.
3 Conclusion
In conclusion, a new series of 6-benzoyl-2-(3,4-dimethoxyphenyl)-1H-substituted benzimidazole compounds have been synthesized and evaluated as anticancer agents against cervical Hela cancer cells. The potential inhibitory activity against EGFR/HER2 kinases was evaluated for the most promising cytotoxic derivatives 2c, 3f, 9, 12 and 13, which exhibited better suppression impact against HER2 than EGFR of IC50 values ranging from 0.19 to 0.31 µM comparing to Erlotinib of IC50; 1.23 µM. The 2,5-dioxopyrrolidine derivative 9 and the thiazolidin-4-one 13 represented promising dual inhibition activity against both EGFR and HER2 kinases. Multi-targeting RTK inhibition was also evaluated against PDGFR-β and VEGFR-2 by compounds 2c, 3f, 9, 12 and 13 as representative examples. Compound 13 demonstrated 3.8 and 1.8 folds more suppression potency than that obtained by Erlotinib as a reference drug. Molecular docking studies exhibited good fitting of compounds 2c, 3f, 9, 12 and 13 in the active site of HER2 with Glide docking scores in proportion to their biological activities. They exhibited strong hydrogen bonding, π-π contacts, hydrophobic and electrostatic interactions with the surrounding amino acids in the binding site of HER2. In addition, cell cycle data exhibited that compounds 9 and 13 caused a pronounced increase in the percentage of Hela cells at pre-G1 by 13 and 27 folds and at G2/M phase by 7 and 10 folds, respectively compared to the untreated Hela cells which confirm a possible role of apoptosis in compounds 9 and 13 induced cancer cell death and cytotoxicity. Compounds 9 and 13 exhibited significant apoptotic activity. Based on the results obtained, it is of interest to continue derivatization and optimization of various benzimidazole series for further kinases inhibition and anticancer studies.
4 Experimental
4.1 Synthesis
The instruments utilized in measuring the melting points, spectral data (IR, Mass, 1H NMR and 13C NMR) and elemental analysis are cited in details in the Supplementary material.
4.1.1 Synthesis of starting key 1-(6-benzoyl-2-(3,4-dimethoxyphenyl)-1H-benzo[d]imidazol-1-yl)propan-2-one (1)
4.1.1.1 Synthesis of (2-(3,4-dimethoxyphenyl)-1H-benzo[d]imidazol-6-yl)(phenyl) methanone (B)
A stirred mixture of o-phenylenediamine A (2.12 g, 10 mmol) and 3,4-dimethoxybenzaldeyde (1.67 g, 10 mmol) in dimethylformamide (20 mL) was refluxed for 8 hr. The mixture was then cooled and poured over ice, the precipitated product was collected by filtration, followed by crystallization from methanol to give the pure yellow product B (82%); m.p. 116–119 °C. IR (KBr, ν cm−1): 3438 (NH), 3051 (CH-aromatic), 1652 (C = O). 1H NMR (DMSO‑d6, 400 MHz) δ: 3.91 (s, 3H, OCH3), 3.93 (s, 3H, OCH3), 6.68–8.12 (m, 11H, ArH), 9.68 (s, 1H, NH). 13C NMR (DMSO‑d6, 100 MHz) δ: 56.5 (2OCH3), 111.2, 112.4, 114.9, 119.0, 122.1, 123.3, 128.9, 130.5, 130.7, 131.2, 132.7, 138.4, 145.5, 149.2, 150.9, 152.0, 194.2 (C = O). MS m/z (%): M+: 358 (94.5%), (M + H)+: 359 (1.1%); Anal. Calcd for C22H18N2O3 (358.4): C, 73.73; H, 5.06; N, 7.82. Found C, 73.73; H, 5.06; N, 7.82.
4.1.1.2 Synthesis of 1-(6-benzoyl-2-(3,4-dimethoxyphenyl)-1H-benzo[d]imidazol-1-yl)propan-2-one (1)
A stirred mixture of (2-(3,4-dimethoxyphenyl)-1H-benzo[d]imidazol-6-yl)(phenyl) methanone B (3.58 g, 10 mmol) and chloroacetone (0.93 mL, 10 mmol) in dry acetone (30 mL) in the presence of potassium carbonate anhydrous (1.38 g, 10 mmol) was refluxed for 12 hr. The mixture was then cooled and poured over ice, the precipitated product was collected by filtration after neutralization with drops of dilute hydrochloric acid, followed by crystallization from methanol to give the pure yellow product 1 (75%); m.p. 122–125 °C. IR (KBr, ν cm−1): 3051 (CH-aromatic), 1672, 1652 (2C = O). 1H NMR (DMSO‑d6, 400 MHz) δ: 2.89 (s, 3H, CH3), 3.91 (s, 3H, OCH3), 3.93 (s, 3H, OCH3), 4.71 (s, 2H, CH2), 7.1–8.12 (m, 11H, ArH). 13C NMR (DMSO‑d6, 100 MHz) δ: 28.3 (CH3), 56.5 (2OCH3), 65.4 (CH2), 111.2, 112.4, 114.9, 119.0, 122.1, 123.3, 128.9, 130.5, 130.7, 131.2, 132.7, 138.4, 145.5, 149.2, 150.9, 152.0, 194.2 (C = O), 196.2 (C = O). MS m/z (%): M+: 414 (91.5%), (M + H)+: 415 (2.1%); Anal. Calcd for C25H22N2O4 (414.4): C, 72.45; H, 5.35; N, 6.76. Found C, 72.45; H, 5.35; N, 6.76.
4.1.2 General method for the synthesis of (E)-1-(6-benzoyl-2-(3,4-dimethoxyphenyl)-1H-benzo[d]imidazol-1-yl)-4-(substituted)phenylbut-3-en-2-one compounds (2a-2e)
A mixture of compound 1 (4 g, 10 mmol) and the appropriate aromatic aldehydes, namely; benzaldehyde, 3,4-dimethoxybenzaldehyde, 4-N-dimethylbenzaldehyde, 4-nitro benzaldehyde and 4-fluorobenzaldehyde, (10 mmol) in absolute ethanol (30 mL) containing few drops of piperidine was refluxed for 2–4 h. After completion of the reaction, the solution mixture was cooled and triturated with ethanol, then filtered, and crystallized from methanol to give the corresponding chalcones 2a-2e.
4.1.2.1 (E)-1-(6-benzoyl-2-(3,4-dimethoxyphenyl)-1H-benzo[d]imidazol-1-yl)-4-phenylbut-3-en-2-one (2a)
Yellowish white solid, yield: 65%, m.p. 212–215 °C. IR (KBr, ν cm−1): 3051 (CH-aromatic), 1670, 1652 (2C = O). 1H NMR (DMSO‑d6, 400 MHz) δ: 3.93, 3.95 (2 s, 6H, 2OCH3), 5.18 (s, 2H, N-CH2), 6.92 (d, 1H, =CH a, J = 9.6 Hz), 7.56–7.77 (m, 17H, =CH β & ArH). 13C NMR (DMSO‑d6, 100 MHz) δ: 56.5 (2OCH3), 64.1 (CH2), 111.2, 112.4, 112.5, 113.1, 113.5, 117.9, 122.0, 122.3, 122.6, 124.9, 126.2, 128.9, 129.5, 130.5, 131.0, 133.7, 136.7, 138.4, 146.5, 149.2, 150.9, 155.0, 156.0 (Ar-C), 196.2 (C = O). MS m/z (%): M+: 502 (84.5%), (M + H)+: 503 (1.1%); Anal. Calcd for C32H26N2O4 (502.57): C, 76.48; H, 5.21; N, 5.57. Found C, 76.48; H, 5.20; N, 5.57.
4.1.2.2 (E)-1-(6-benzoyl-2-(3,4-dimethoxyphenyl)-1H-benzo[d]imidazol-1-yl)-4-(3,4-dimethoxy phenyl)but-3-en-2-one (2b)
Yellowish white solid, yield: 65%, m.p. 140–142 °C. IR (KBr, ν cm−1): 3049 (CH-aromatic), 1672, 1640 (2C = O). 1H NMR (DMSO‑d6, 400 MHz): δ 3.81, 3.93, 3.95 (3 s, 12H, 4OCH3), 5.28 (s, 2H, N-CH2), 7.00 (d, 1H, =CH a, J = 9.6 Hz), 7.22–8.21 (m, 15H, =CH β & ArH). 13C NMR (DMSO‑d6, 100 MHz) δ: 56.1 (4OCH3), 64.1 (CH2), 111.2, 112.4, 112.5, 113.1, 113.5, 117.9, 122.0, 122.3, 122.6, 124.9, 126.2, 128.9, 129.5, 130.5, 131.0, 133.7, 136.7, 138.4, 146.5, 149.2, 150.9, 155.0, 156.0 (Ar-C), 196.2 (C = O). MS m/z (%): M+: 562 (64.5%), (M + H)+: 563 (1.1%); Anal. Calcd for C34H30N2O6 (562.62): C, 72.58; H, 5.37; N, 4.98. Found C, 72.58; H, 5.37; N, 4.98.
4.1.2.3 (E)-1-(6-benzoyl-2-(3,4-dimethoxyphenyl)-1H-benzo[d]imidazol-1-yl)-4-(4-(dimethylamino)phenyl)but-3-en-2-one (2c)
Brownish white solid, yield: 65%, m.p. 197–199 °C. IR (KBr, ν cm−1): 3050 (CH-aromatic), 1670, 1650 (2C = O). 1H NMR (DMSO‑d6, 400 MHz): δ 2.41 (s, 6H, -N(CH3)2), 3.93, 3.95 (2 s, 6H, 2OCH3), 5.18 (s, 2H, N-CH2), 6.92 (d, 1H, =CH-a, J = 7.6 Hz), 7.22–8.59 (m, 16H, =CH-β & ArH). 13C NMR (DMSO‑d6, 100 MHz) δ 45.1 (N-(CH3)2), 56.1 (2OCH3), 64.1 (CH2), 111.2, 112.2, 112.5, 113.0, 113.4, 118.9, 122.0, 122.1, 122.2, 124.9, 126.2, 128.9, 129.9, 130.5, 131.7, 133.7, 136.7, 138.4, 146.1, 149.2, 150.9, 154.0, 156.0 (Ar-C), 196.2 (C = O). MS m/z (%): M+: 545 (44.5%); Anal. Calcd for C34H31N3O4 (545.64): C, 74.84; H, 5.73; N, 7.70. Found C, 74.84; H, 5.73; N, 7.70.
4.1.2.4 (E)-1-(6-benzoyl-2-(3,4-dimethoxyphenyl)-1H-benzo[d]imidazol-1-yl)-4-(4-nitrophenyl) but-3-en-2-one (2d)
Yellow solid, yield: 63%, m.p. 195–197 °C. IR (KBr, ν cm−1): 3049 (CH-aromatic), 1665, 1643 (2C = O), 1553, 1320 (NO2). 1H NMR (DMSO‑d6, 400 MHz): δ 3.92, 3.95 (2 s, 6H, 2OCH3), 5.12 (s, 2H, N-CH2), 7.23 (d, 1H, =CHα, J = 7.6 Hz), 7.62–8.45 (m, 16H, =CH β & ArH). 13C NMR (DMSO‑d6, 100 MHz) δ 56.1 (2OCH3), 64.1 (CH2), 111.2, 112.2, 112.5, 113.0, 113.4, 118.9, 122.0, 122.1, 122.2, 124.9, 126.2, 128.9, 129.9, 130.5, 131.7, 133.7, 136.7, 138.4, 146.1, 149.2, 150.9, 154.0, 156.0 (Ar-C), 196.2 (C = O). MS m/z (%): M+: 547 (44.5%); Anal. Calcd for C32H25N3O6 (547.57): C, 70.19; H, 4.60; N, 7.67. Found C, 70.19; H, 4.60; N, 7.67.
4.1.2.5 (E)-1-(6-benzoyl-2-(3,4-dimethoxyphenyl)-1H-benzo[d]imidazol-1-yl)-4-(4-fluorophenyl)but-3-en-2-one (2e)
Gray white solid, yield: 65%, m.p. 132–134 °C. IR (KBr, ν cm−1): 3052 (CH-aromatic), 1670, 1645 (2C = O). 1H NMR (DMSO‑d6, 400 MHz): δ 3.75, 3.87 (2 s, 6H, 2OCH3), 5.17 (s, 2H, N-CH2), 7.20 (d, 1H, =CHα, J = 7.6 Hz), 7.41–7.90 (m, 16H, =CH β & ArH). 13C NMR (DMSO‑d6, 100 MHz) δ 56.1 (2OCH3), 64.1 (CH2), 111.2, 112.2, 112.5, 113.0, 113.4, 118.9, 122.0, 122.1, 122.2, 124.9, 126.2, 128.9, 129.9, 130.5, 131.7, 133.7, 136.7, 138.4, 146.1, 149.2, 150.9, 154.0, 156.0 (Ar-C), 196.2 (C = O). MS m/z (%): M+: 520 (73.5%); Anal. Calcd for C32H25FN2O4 (520.56): C, 73.83; H, 4.84; N, 5.38. Found C, 73.83; H, 4.84; N, 5.38.
4.1.3 General method for the synthesis of Schiff bases (3a-3f)
A mixture of compound 1 (4 g, 10 mmol) and various aromatic amines such as 4-aminoacetophenone, ethyl 4-aminobenzoate, 4-nitoaniline, 4-bromoanline, 4-aminobenzenesulfonamide and sulfathiazole (10 mmol) in ethanol (30 mL) containing few drops of glacial acetic acid was heated at refluxing temperature for 12 h. The mixture solution was left to cool and the precipitated solid was filtered and crystallized from DMF/EtOH to give the corresponding Schiff compounds 3a-3f.
4.1.3.1 (E)-1-(4-((1-(6-benzoyl-2-(3,4-dimethoxyphenyl)-1H-benzo[d]imidazol-1-yl) propan-2-ylidene)amino)phenyl)ethan-1-one (3a)
Brownish white solid, yield: 70%, m.p. 125–127 °C. IR (KBr, ν cm−1): 3050 (CH-aromatic), 1704, 1665 (2C = O). 1H NMR (DMSO‑d6, 400 MHz): δ 2.25 (s, 3H, CH3), 2.41 (s, 3H, –COCH3), 3.92, 3.95 (2 s, 6H, 2OCH3), 5.18 (s, 2H, N-CH2), 6.92–8.59 (m, 15H, ArH). 13C NMR (DMSO‑d6, 100 MHz) δ 14.1 (CH3), 27.2 (CO-CH3), 56.0 (2OCH3), 57.6 (CH2), 112.2, 112.6, 112.9, 113.4, 118.9, 122.0, 122.1, 122.2, 124.9, 128.9, 129.9, 130.1, 131.7, 132.7, 136.7, 138.4, 146.1, 149.2, 150.9, 154.0 (Ar-C), 156.0 (C = N), 196.1, 203.1 (2C = O). MS m/z (%): M+: 531 (44.5%); Anal. Calcd for C33H29N3O4 (531.61): C, 74.56; H, 5.50; N, 7.90. Found C, 74.56; H, 5.50; N, 7.90.
4.1.3.2 Ethyl (E)-4-((1-(6-benzoyl-2-(3,4-dimethoxyphenyl)-1H-benzo[d]imidazol-1-yl) propan-2-ylidene)amino)benzoate (3b)
Gray white solid, yield: 65%, m.p. 143–145 °C. IR (KBr, ν cm−1): 3072 (CH-aromatic), 1680, 1665 (2C = O). 1H NMR (DMSO‑d6, 400 MHz): δ 1.52 (t, 3H, –CH2CH3, J = 7.6 Hz), 2.04 (s, 3H, CH3), 3.92, 3.95 (2 s, 6H, 2OCH3), 4.25 (q, 4H, –CH2CH3, J = 7.6 Hz, N-CH2), 5.18 (s, 2H, N-CH2), 6.92–8.59 (m, 15H, ArH). 13C NMR (DMSO‑d6, 100 MHz) δ 14.1 (CH3), 27.2 (CH2-CH3), 56.0 (2OCH3), 57.6 (CH2), 60.9 (CH2), 112.2, 112.6, 112.9, 113.4, 118.9, 122.0, 122.1, 122.2, 124.9, 128.9, 129.9, 130.1, 131.7, 132.7, 136.7, 138.4, 146.1, 149.2, 150.9, 154.0 (Ar-C), 156.0 (C = N), 196.1, 203.1 (2C = O). MS m/z (%): M+: 561 (66.5%); Anal. Calcd for C34H31N3O5 (561.64): C, 72.71; H, 5.56; N, 7.48. Found C, 72.71; H, 5.56; N, 7.48.
4.1.3.3 (E)-(2-(3,4-dimethoxyphenyl)-1-(2-((4-nitrophenyl)imino)propyl)-1H-benzo[d] imidazol-6-yl)(phenyl)methanone (3c)
Yellow solid, yield: 65%, m.p. 117–119 °C. IR (KBr, ν cm−1): 3055 (CH-aromatic), 1669 (C = O), 1550, 1325 (NO2). 1H NMR (DMSO‑d6, 400 MHz): δ 2.01 (s, 3H, CH3), 3.92, 3.95 (2 s, 6H, 2OCH3), 5.18 (s, 2H, N-CH2), 6.92–8.59 (m, 15H, ArH). 13C NMR (DMSO‑d6, 100 MHz) δ 26.0 (CH3), 56.3 (2OCH3), 57.5 (CH2), 112.2, 112.2, 113.0, 113.4, 119.9, 122.0, 122.3, 123.2, 124.9, 128.8, 129.9, 130.2, 131.7, 132.7, 136.7, 139.4, 145.1, 149.3, 152.9, 154.0, 156.0 (Ar-C), 196.1 (C = O). MS m/z (%): M+: 534 (44.5%); Anal. Calcd for C31H26N4O5 (534.57): C, 69.65; H, 4.90; N, 10.48. Found C, 69.65; H, 4.90; N, 10.48.
4.1.3.4 (E)-(1-(2-((4-bromophenyl)imino)propyl)-2-(3,4-dimethoxyphenyl)-1H-benzo[d] imidazol-6-yl)(phenyl)methanone (3d)
Orange solid, yield: 65%, m.p. 162–164 °C. IR (KBr, ν cm−1): 3155 (CH-aromatic), 1665 (C = O). 1H NMR (DMSO‑d6, 400 MHz): δ 2.01 (s, 3H, CH3), 3.92, 3.95 (2 s, 6H, 2OCH3), 5.15 (s, 2H, N-CH2), 6.92–8.59 (m, 15H, ArH). 13C NMR (DMSO‑d6, 100 MHz) δ 26.0 (CH3), 56.3 (2OCH3), 57.5 (CH2), 112.2, 112.2, 113.0, 113.4, 119.9, 122.0, 122.3, 123.2, 124.9, 128.8, 129.9, 130.2, 131.7, 132.7, 136.7, 139.4, 145.1, 149.3, 152.9, 154.0, 156.0 (Ar-C), 196.1 (C = O). MS m/z (%): M+: 568 (50%), (M + 2)+: 370 (49%); Anal. Calcd for C31H26BrN3O3 (568.47): C, 65.50; H, 4.61; N, 7.39. Found C, 65.50; H, 4.61; N, 7.39.
4.1.3.5 (E)-4-((1-(6-benzoyl-2-(3,4-dimethoxyphenyl)-1H-benzo[d]imidazol-1-yl)propan-2-ylidene)amino)benzenesulfonamide (3e)
Gray white solid, yield: 66%, m.p. 103–105 °C. IR (KBr, ν cm−1): 3051 (CH-aromatic), 1663 (C = O), 1357, 1134 (SO2). 1H NMR (DMSO‑d6, 400 MHz): δ 2.04 (s, 3H, CH3), 3.92, 3.95 (2 s, 6H, 2OCH3), 5.15 (s, 2H, N-CH2), 6.92–8.59 (m, 15H, ArH), 9.25 (s, 2H, NH2, D2O exchangeable). 13C NMR (DMSO‑d6, 100 MHz) δ 26.0 (CH3), 56.3 (2OCH3), 57.5 (CH2), 112.2, 112.2, 113.0, 113.4, 119.9, 122.0, 122.3, 123.2, 124.9, 128.8, 129.9, 130.2, 131.7, 132.7, 136.7, 139.4, 145.1, 149.3, 152.9, 154.0, 156.0 (Ar-C), 196.1 (C = O). MS m/z (%): M+: 568 (94%); Anal. Calcd for C31H28N4O5S (568.65): C, 65.48; H, 4.96; N, 9.85. Found C, 65.48; H, 4.96; N, 9.85.
4.1.3.6 (E)-(2-(3,4-dimethoxyphenyl)-1-(2-((4-(thiazol-2-ylsulfonyl)phenyl)imino)propyl) −1H-benzo[d]imidazol-6-yl)(phenyl)methanone (3f)
White solid, yield: 62%, m.p. 113–115 °C. IR (KBr, ν cm−1): 3435 (NH), 3065 (CH-aromatic), 1663 (C = O), 1355, 1130 (SO2). 1H NMR (DMSO‑d6, 400 MHz): δ 1.94 (s, 3H, CH3), 3.91, 3.95 (2 s, 6H, 2OCH3), 5.16 (s, 2H, N-CH2), 6.92–8.63 (m, 17H, ArH), 11.31 (s, 1H, NH, D2O exchangeable). MS m/z (%): M+: 651 (34%); Anal. Calcd for C34H29N5O5S2 (651.74): C, 62.66; H, 4.49; N, 10.75. Found C, 62.66; H, 4.49; N, 10.75.
4.1.4 General method for synthesis of the hydrazone derivatives (4–6)
A mixture of compound 1 (4 g, 10 mmol) and nucleophilic compounds such as hydrazine hydrate (98%), p-toluic hydrazide and 2-furoic acid hydrazide (10 mmol) in ethanol (30 mL) was heated at refluxing temperature for 12 h. The reaction mixture was left to cool. The obtained solid was filtered and crystallized from methanol to give the corresponding compounds 4–6, respectively.
4.1.4.1 (E)-(2-(3,4-dimethoxyphenyl)-1-(2-hydrazineylidenepropyl)-1H-benzo[d] imidazol-6-yl)(phenyl)methanone (4)
White solid, yield: 67%, m.p. 103–105 °C. IR (KBr, ν cm−1): 3423, 3252 (NH2), 3037 (CH-aromatic), 1650 (C = O). 1H NMR (DMSO‑d6, 400 MHz): δ 1.91 (s, 3H, CH3), 3.94, 3.85 (2 s, 6H, 2OCH3), 5.28 (s, 2H, N-CH2), 4.78 (s, 2H, NH2, D2O exchangeable), 7.03–8.31 (m, 11H, ArH). 13C NMR (DMSO‑d6, 100 MHz) δ: 18.3 (CH3), 56.5 (2OCH3), 65.4 (CH2), 111.2, 112.4, 114.9, 119.0, 122.1, 123.3, 128.9, 130.5, 130.7, 131.2, 132.7, 138.4, 146.5, 149.2, 152.9, 155.0, 194.2 (C = O). MS m/z (%): M+: 428 (29.5%); Anal. Calcd for C25H24N4O3 (428.495): C, 70.08; H, 5.65; N, 13.08. Found C, 70.08; H, 5.65; N, 13.07.
4.1.4.2 (E)-N'-(1-(6-benzoyl-2-(3,4-dimethoxyphenyl)-1H-benzo[d]imidazol-1-yl) propan-2-ylidene)-4-methylbenzohydrazide (5)
Gray white solid, yield: 65%, m.p. 195–197 °C. IR (KBr, ν cm−1): 3423 (NH), 3037 (CH-aromatic), 1650, 1643 (2C = O). 1H NMR (DMSO‑d6, 400 MHz): δ 2.25 (s, 3H, CH3), 2.41 (s, 3H, CH3), 3.93, 3.95 (2 s, 6H, 2OCH3), 5.18 (s, 2H, N-CH2), 6.92–8.59 (m, 15H, ArH), 11.50 (s, IH, NH, D2O exchangeable). 13C NMR (DMSO‑d6, 100 MHz) δ 21.4, 29.9 (2CH3), 56.0 (2OCH3), 58.9 (CH2), 85.8, 111.9, 112.1, 112.2, 112.8, 122.2, 122.2, 122.3, 128.1, 128.9, 129.2, 129.4, 130.9, 131.3, 142.2, 149.0, 150.7 (Ar-C), 178.3, 198.5 (2C = O). MS m/z (%): M+: 546 (44.5%); Anal. Calcd for C33H30N4O4 (546.63): C, 72.51; H, 5.53; N, 10.25. Found: C, 72.51; H, 5.53; N, 10.25.
4.1.4.3 (E)-N'-(1-(6-benzoyl-2-(3,4-dimethoxyphenyl)-1H-benzo[d]imidazol-1-yl) propan-2-ylidene) furan-2-carbohydrazide (6)
Gray white solid, yield: 60%, m.p. 235–237 °C. IR (KBr, ν cm−1): 3440 (NH), 3050 (CH-aromatic), 1655, 1645 (2C = O). 1H NMR (DMSO‑d6, 400 MHz): δ 2.41 (s, 3H, CH3), 3.93, 3.95 (2 s, 6H, 2OCH3), 5.18 (s, 2H, N-CH2), 7.21–7.95 (m, 14H, ArH), 10.42 (s, IH, NH, D2O exchangeable). MS m/z (%): M+: 522 (49%); Anal. Calcd for C30H26N4O5 (522.56): C, 68.95; H, 5.02; N, 10.72. Found: C, 68.95; H, 5.02; N, 10.73.
4.1.5 (E)-2-(1-(6-benzoyl-2-(3,4-dimethoxyphenyl)-1H-benzo[d]imidazol-1-yl)propan-2-ylidene)hydrazine-1-carboxamide (7)
A mixture of compound 1 (4.10 g, 10 mmol), semicarbazide hydrochloride (1.11 g, 10 mmol) and sodium acetate (1.64 g, 20 mmol) in ethanol (20 mL) was stirred for 1 h at room temperature, then refluxed for 3 h. The formed precipitate was filtered, washed several times with water, dried, and recrystallized from ethanol to give the compound 7 as white solid.
Yield: 70%, m.p. 282–284 °C. IR (KBr, ν cm−1): 3440, 3350, 3235 (NH2), 3050 (CH-aromatic), 1650, 1645 (2C = O). 1H NMR (DMSO‑d6, 400 MHz): δ 1.91 (s, 3H, CH3), 3.73, 3.84 (2 s, 6H, 2OCH3), 5.45 (s, 2H, NH2, D2O exchangeable), 5.65 (s, 2H, N-CH2), 7.11–7.85 (m, 11H, ArH), 9.52 (s, IH, NH, D2O exchangeable). MS m/z (%): M+: 471 (60%); Anal. Calcd for C26H25N5O4 (471.52): C, 66.23; H, 5.34; N, 14.85. Found: C, 66.24; H, 5.34; N, 14.85.
4.1.6 (E)-2-(1-(6-benzoyl-2-(3,4-dimethoxyphenyl)-1H-benzo[d]imidazol-1-yl)propan-2-ylidene) hydrazine-1-carbothioamide (8)
A mixture of 1 (4.10 g, 10 mmol), thiosemicarbazide (1 g, 10 mmol) in absolute ethanol (15 mL) containing a few drops of concentrated hydrochloric acid was refluxed for 3 h. The formed precipitate was filtered, dried, and crystallized from ethanol to give the compound 8 as yellowish white solid.
Yield: 73%, m.p. 233–235 °C. IR (KBr, ν cm−1): 3440, 3360, 3240 (NH2, NH), 3050 (CH-aromatic), 1665 (C = O), 1110 (C = S). 1H NMR (DMSO‑d6, 400 MHz): δ 1.91 (s, 3H, CH3), 3.91, 3.94 (2 s, 6H, 2OCH3), 5.45 (s, 2H, N-CH2), 5.75 (s, 2H, NH2, D2O exchangeable), 7.01–7.95 (m, 11H, ArH), 10.42 (s, IH, NH, D2O exchangeable). 13C NMR (DMSO‑d6, 100 MHz) δ 22.1 (CH3), 56.0 (2OCH3), 57.6 (CH2), 112.1, 112.6, 128.7, 128.8, 128.9, 129.0, 130.1, 130.3, 137.7, 143.1, 143.9, 149.2, 149.5, 150.9, 154.0, 156.0 (Ar-C), 185.1 (C = O), 187.2 (C = S). MS m/z (%): M+: 487 (90%); Anal. Calcd for C26H25N5O3S (487.58): C, 64.05; H, 5.17; N, 14.36. Found: 64.05; H, 5.18; N, 14.36.
4.1.7 General method for synthesis of 2,5-dioxopyrrolidine, 2,5-dioxopyrroline and 1,3-dioxoisoindoline derivatives (9–11)
To a solution of compound 8 (4.8 g, 10 mmol) in glacial acetic acid, succinic anhydride, maleic anhydride or phthalic anhydride (10 mmol) was added. The mixture was refluxed for 8 h, then poured onto ice/H2O. The formed precipitate was filtered, washed with water and recrystallized from dioxane to give the corresponding derivatives 9–11.
4.1.7.1 (E)-N'-(1-(6-benzoyl-2-(3,4-dimethoxyphenyl)-1H-benzo[d]imidazol-1-yl) propan-2-ylidene)-2,5-dioxopyrrolidine-1-carbothiohydrazide (9)
White solid, yield: 73%, m.p. 113–115 °C. IR (KBr, ν cm−1): 3335 (NH), 3150 (CH-aromatic), 1688, 1655 (3C = O), 1125 (C = S). 1H NMR (DMSO‑d6, 400 MHz): δ 1.92 (s, 3H, CH3), 2.75 (t, 4H, pyrrolidine-(CH2)2), 3.85, 3.90 (2 s, 6H, 2OCH3), 5.45 (s, 2H, N-CH2), 7.11–7.75 (m, 11H, ArH), 12.12 (s, IH, NH, D2O exchangeable). 13C NMR (DMSO‑d6, 100 MHz) δ 26.7 (CH3), 29.7 (pyrrolidine-(CH2)2), 56.3 (2OCH3), 57.6 (CH2), 112.0, 112.5, 112.9, 117.9, 120.5, 122.1, 127.8, 128.7, 129.9, 130.1, 131.9, 132.7, 146.3, 149.2, 153.9, 156.0, 159.0 (Ar-C), 186.1 (C = S), 196.5, 203.3 (2C = O). MS m/z (%): M+: 569 (40%); Anal. Calcd for: C30H27N5O5S (569.64): C, 63.26; H, 4.78; N, 12.29. Found: C, 63.25; H, 4.78; N, 12.29.
4.1.7.2 (E)-N'-(1-(6-benzoyl-2-(3,4-dimethoxyphenyl)-1H-benzo[d]imidazol-1-yl) propan-2-ylidene)-2,5-dioxo-2,5-dihydro-1H-pyrrole-1-carbothiohydrazide (10)
White solid, yield: 73%, m.p. 190–192 °C. IR (KBr, ν cm−1): 3440 (NH), 3150 (CH-aromatic), 1670, 1655 (3C = O), 1123 (C = S). 1H NMR (DMSO‑d6, 400 MHz): δ 1.81 (s, 3H, CH3), 3.92, 3.95 (2 s, 6H, 2OCH3), 5.41 (s, 2H, N-CH2), 7.01–7.70 (m, 13H, ArH), 12.21 (s, IH, NH, D2O exchangeable). MS m/z (%): M+: 567 (30%); Anal. Calcd for: C30H25N5O5S (567.62): C, 63.48; H, 4.44; N, 12.34. Found: C, 63.48; H, 4.45; N, 12.34.
4.1.7.3 (E)-N'-(1-(6-benzoyl-2-(3,4-dimethoxyphenyl)-1H-benzo[d]imidazol-1-yl) propan-2-ylidene)-1,3-dioxoisoindoline-2-carbothiohydrazide (11)
White solid, yield: 71%, m.p. 190–192 °C. IR (KBr, ν cm−1): 3440 (NH), 3155 (CH-aromatic), 1672, 1650 (3C = O), 1125 (C = S). 1H NMR (DMSO‑d6, 400 MHz): δ 1.91 (s, 3H, CH3), 3.80, 3.92 (2 s, 6H, 2OCH3), 5.65 (s, 2H, N-CH2), 7.01–7.70 (m, 15H, ArH), 12.21 (s, IH, NH, D2O exchangeable). MS m/z (%): M+: 617 (40%); Anal. Calcd for: C34H27N5O5S (617.68): C, 66.11; H, 4.41; N, 11.34. Found: C, 66.11; H, 4.41; N, 11.35.
4.1.8 General procedure for synthesis of 4-methylthiazoline and thiazolidin-4-one compounds (12, 13)
A mixture of compound 8 (4.8 g, 10 mmol) and chloroacetone or ethyl bromoacetate (10 mmol) in absolute ethanol (20 mL) containing sodium acetate anhydrous (1.64 g, 20 mmol) was heated at the refluxing temperature for 10 h. The formed precipitate was filtered, washed with water, dried, and recrystallized from the proper solvent to accomplish the target derivatives 12, 13, respectively.
4.1.8.1 (2-(3,4-Dimethoxyphenyl)-1-((E)-2-(((E)-4-methylthiazol-2(3H)-ylidene) hydrazineylidene)propyl)-1H-benzo[d]imidazol-6-yl)(phenyl)methanone (12)
Recrystallized from chloroform. Yellowish white solid, yield: 70%, m.p. 195–197 °C. IR (KBr, ν cm−1): 3445 (NH), 3055 (CH-aromatic), 1650 (C = O). 1H NMR (DMSO‑d6, 400 MHz): δ 1.91 (s, 3H, CH3), 2.24 (s, 3H, thiazoline-CH3), 3.93, 3.95 (2 s, 6H, 2OCH3), 5.18 (s, 2H, N-CH2), 6.93 (s, 1H, thiazoline-H5), 7.01–8.59 (m, 11H, ArH), 11.5 (s, IH, NH, D2O exchangeable). 13C NMR (DMSO‑d6, 100 MHz) δ 27.7, 29.7 (2CH3), 56.0 (2OCH3), 57.6 (CH2), 112.2, 112.6, 112.9, 118.9, 122.0, 122.1, 127.8, 128.9, 129.9, 130.1, 131.7, 132.7, 146.3, 149.2, 150.9, 156.0, 196.1 (Ar-C), 203.1 (C = O). MS m/z (%): M+: 525 (90%); Anal. Calcd for: C29H27N5O3S (525.63): C, 66.27; H, 5.18; N, 13.32. Found: C, 66.27; H, 5.19; N, 13.32.
4.1.8.2 (E)-2-(((E)-1-(6-benzoyl-2-(3,4-dimethoxyphenyl)-1H-benzo[d]imidazol-1-yl) propan-2-ylidene)hydrazineylidene)thiazolidin-4-one (13)
Recrystallized from methanol. Gray white solid, yield: 73%, m.p. 190–192 °C. IR (KBr, ν cm−1): 3437 (NH), 3055 (CH-aromatic), 1655, 1675 (2C = O). 1H NMR (DMSO‑d6, 400 MHz): δ 3.81 (s, 3H, CH3), 3.93, 3.95 (2 s, 6H, 2OCH3), 4.76 (s, 2H, thiazolidine-CH2), 5.28 (s, 2H, N-CH2), 7.00–7.76 (m, 11H, ArH), 11.53 (s, IH, NH, D2O exchangeable). 13C NMR (DMSO‑d6, 100 MHz) δ 26.7 (CH3), 29.7 (thizolidine-CH2), 56.1 (2OCH3), 57.6 (CH2), 112.2, 112.6, 112.9, 118.9, 122.5, 122.1, 127.8, 128.9, 129.9, 130.1, 131.7, 132.7, 146.3, 149.2, 150.9, 156.0, 186.1 (Ar-C), 196.5, 203.1 (2C = O). MS m/z (%): M+: 527 (70%); Anal. Calcd for: C28H25N5O4S (527.60): C, 63.74; H, 4.78; N, 13.27. Found: C, 63.74; H, 4.78; N, 13.27.
4.2 Biological evaluation
4.2.1 In vitro anticancer activity
MTT assay was used to evaluate the in vitro cytotoxicity of the new compounds against Hela cell line (Thavamani et al., 2013; Prasetyaningrum et al., 2018) and WI-38 normal fibroblast cell line. MTT assay depends on the reduction of the soluble 3-(4,5-methyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) into a blue purple formazan product, mainly by mitochondrial reductase activity inside the living cells. The cells used in cytotoxicity assay were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum.
4.2.2 Kinase assays
The activities of the examined compounds against EGFR, HER2, PDGFR-β and VEGFR2 were in vitro tested using abcam’s Human In cell ELISA Kit (ab 126419) for EGFR, ADP-Glo™ Kinase Assay for Her2, PDGFR- β, Active, Recombinant protein expressed in Sf9 cells for VEGFR2 (KDR) Kinase Assay Kit Catalog # 40325, respectively. The procedure of the used kits was done according to the manufacturer, s instructions.
4.2.3 Cell cycle analysis and apoptosis detection
Cell cycle analysis and apoptosis investigation were carried out by flow cytometry (He et al., 2009; Cao et al., 2020; Yang et al., 2020). Hela cells were seeded at 8 × 104 and incubated at 37 °C, 5% CO2 overnight. After treatment with the tested compound, for 24 h, cell pellets were collected and centrifuged (300g, 5 min).
4.2.4 Molecular docking study
The HER2 kinase domain complexed with a ligand was downloaded from the protein databank (PDB ID: 3RCD, http://www.rcsb.org). Protein preparation wizard of Schrödinger Suite (Sastry et al., 2013, Schrödinger, 2018) was used to correct the protein structural file by correcting bond orders, adding any missing hydrogen atoms, and completing unresolved side chains or loops. Water molecules which are beyond 5 Å of bound ligand and are not involved in at least two hydrogen bonds with non-water residues, were removed from the protein complex. The structure was then energetically minimized to remove atomic clashes. The ligand coordinates were designated as the center for docking. Glide receptor grid preparation (Friesner et al., 2006; Halgren et al., 2004; Friesner et al., 2004; Schrödinger, 2018) was used to define and construct the receptor for the docking step. To decrease penalties for close contacts between ligands and protein, 0.85 was used to scale van der Waals radii of nonpolar atoms. The hydroxyl groups of Ser, Thr, or Tyr residues in the active site were identified as rotatable groups to account for all possible hydrogen bonds with incoming ligands. Ligand structures were prepared by LigPrep (Schrödinger, 2018). Standard-precision (SP) docking option was chosen for the docking experiment, and Glide was directed to generate ligand conformations through the docking process.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Declaration of Competing Interest
The authors declared that there is no conflict of interest.
References
- Design, synthesis and anticervical cancer activity of new benzofuran–pyrazol-hydrazono- thiazolidin-4-one hybrids as potential EGFR inhibitors and apoptosis inducing agents. Bioorg. Chem.. 2019;89:103035.
- [CrossRef] [Google Scholar]
- Synthesis of New 1,3,4-Oxadiazole-benzimidazole Derivatives as Potential Antioxidants and Breast Cancer Inhibitors with Apoptosis Inducing Activity. Russ. J. Gen. Chem.. 2019;89(2):348-356.
- [Google Scholar]
- Synthesis of some novel 4-benzothiazol-2-yl-benzoyl-1H-pyrazoles, and evaluation as antiangiogenic agents. Res. Chem. Intermed.. 2016;42(2):1521-1536.
- [Google Scholar]
- Long Noncoding RNA UCA1 Regulates PRL-3 Expression by Sponging MicroRNA-495 to Promote the Progression of Gastric Cancer. Mol. Ther. Nucleic Acids. 2020;19:853-864.
- [Google Scholar]
- Discovery of new quinazoline derivatives as irreversible dual EGFR/HER2 inhibitors and their anticancer activities – Part 1. Bioorg. Med. Chem. Lett.. 2019;29(4):591-596.
- [Google Scholar]
- Synthesis, anticancer activity and molecular modeling studies of 1,2,4-triazole derivatives as EGFR inhibitors. Eur. J. Med. Chem.. 2018;156:774-789.
- [Google Scholar]
- Shooting three inflammatory targets with a single bullet: Novel multi-targeting anti-inflammatory glitazones. Eur. J. Med. Chem.. 2019;167:562-582.
- [Google Scholar]
- Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem.. 2004;47:1739-1749.
- [CrossRef] [Google Scholar]
- Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem.. 2006;49(21):6177-6196.
- [CrossRef] [Google Scholar]
- A phase II trial to evaluate gefitinib as second- or third-line treatment in patients with recurring locoregionally advanced or metastatic cervical cancer. Gynecol. Oncol.. 2008;108(1):42-46.
- [Google Scholar]
- Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem.. 2004;47(7):1750–1759
- [CrossRef] [Google Scholar]
- Cellular and nuclear degradation during apoptosis. Curr. Opin. Cell Biol.. 2009;21(6):900-912.
- [Google Scholar]
- Identification and Optimization of Novel Cathepsin C Inhibitors Derived from EGFR Inhibitors. J. Med. Chem.. 2019;62(12):5901-5919.
- [Google Scholar]
- Synthesis of stable benzimidazole derivatives bearing pyrazole as anticancer and EGFR receptor inhibitors. Bioorg. Chem.. 2018;78:158-169.
- [Google Scholar]
- Targeting the cancer kinome through polypharmacology. Nat. Rev. Cancer. 2010;10(2):130-137.
- [Google Scholar]
- Epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of non-small cell lung cancer. Expert Rev. Anticancer Ther.. 2019;19(7):547-559.
- [Google Scholar]
- Discovery of benzimidazole derivatives as novel multi-target EGFR, VEGFR-2 and PDGFR kinase inhibitors. Bioorg. Med. Chem.. 2011;19(15):4529-4535.
- [Google Scholar]
- HER2 as a novel therapeutic target for cervical cancer. Oncotarget. 2015;6(34):36219-36230.
- [Google Scholar]
- Impact of Epidermal Growth Factor Receptor Expression on Disease-Free Survival and Rate of Pelvic Relapse in Patients With Advanced Cancer of the Cervix Treated With Chemoradiotherapy:. Am. J. Clin. Oncol.. 2011;34(4):395-400.
- [Google Scholar]
- Prasetyaningrum, P.W., ID, A.B., Hayun, H., 2018. Synthesis and Cytotoxicity Evaluation of Novel Asymmetrical Mono-Carbonyl Analogs of Curcumin (AMACs) against Vero, HeLa, and MCF7 Cell Lines. Sci. Pharm., 86, 25; doi:10.3390/scipharm86020025.
- Šarenac, T. and Mikov, M., 2019. Cervical Cancer, Different Treatments and Importance of Bile Acids as Therapeutic Agents in This Disease, Frontiers in Pharmacology 10 Article 484. https://doi.org/10.3389/fphar.2019.00484.
- Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27(3):221-234.
- [Google Scholar]
- A Phase II Trial of Erlotinib in Recurrent Squamous Cell Carcinoma of the Cervix: A Gynecologic Oncology Group Study. Int. J. Gynecological Cancer. 2009;19(5):929-933.
- [Google Scholar]
- Schrödinger Release 2018-4: Protein Preparation Wizard; Epik, Schrödinger, LLC, New York, NY, 2016; Impact, Schrödinger, LLC, New York, NY, 2016; Prime, Schrödinger, LLC, New York, NY, 2018.
- Schrödinger Release 2018-4: Glide, Schrödinger, LLC, New York, NY, 2018.
- Schrödinger Release 2018-4: LigPrep, Schrödinger, LLC, New York, NY, 2018.
- Thiazole-containing compounds as therapeutic targets for cancer therapy. Eur. J. Med. Chem.. 2020;188:112016.
- [CrossRef] [Google Scholar]
- Benzimidazole Scaffold as Anticancer Agent: Synthetic Approaches and Structure-Activity Relationship: Benzimidazole Scaffold as Anticancer Agent. Arch. Pharm. Chem. Life Sci.. 2017;350(6):e201700040.
- [CrossRef] [Google Scholar]
- Smolobochkin, A., Gazizov, Almir, Sazykina, M. et al., 2019. Synthesis of Novel 2-(Het)arylpyrrolidine Derivatives and Evaluation of Their Anticancer and Anti-Biofilm Activity. Molecules. 24(17), 3086. doi: 10.3390/molecules24173086.
- Novel sulfonamide benzoquinazolinones as dual EGFR/HER2 inhibitors, apoptosis inducers and radiosensitizers. J. Enzyme Inhib. Med. Chem.. 2019;34(1):1030-1040.
- [Google Scholar]
- In vitro cytotoxic activity of menispermaceae plants against HeLa cell line. Ancient Sci. life. 2013;33(2):81-84.
- [CrossRef] [Google Scholar]
- Ueda, A., Takasawa, A., Akimoto, T. et al., 2017. Prognostic significance of the co-expression of EGFR and HER2 in adenocarcinoma of the uterine cervix, PLoS ONE 12(8) e0184123. https://doi.org/10.1371/journal.pone.0184123.
- Targeted therapy in cervical cancer. ESMO Open. 2018;3(Suppl 1):e000462.
- [CrossRef] [Google Scholar]
- Design, synthesis and biological evaluation of novel 4-anlinoquinazoline derivatives as EGFR inhibitors with the potential to inhibit the gefitinib-resistant nonsmall cell lung cancers. J. Enzyme Inhib. Med. Chem.. 2019;34(1):203-217.
- [Google Scholar]
- Silencing of microRNA-517a induces oxidative stress injury in melanoma cells via inactivation of the JNK signaling pathway by upregulating CDKN1C. Cancer Cell Int. 2020;20(1)
- [CrossRef] [Google Scholar]
- Osimertinib making a breakthrough in lung cancer targeted therapy. OncoTargets and Therapy. 2016;9:5489-5493.
- [CrossRef] [Google Scholar]
- Synthesis and Biological Evaluation of Some Novel Thiophene-bearing Quinazoline Derivatives as EGFR Inhibitors. LDDD. 2018;16(2):102-110.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2020.10.041.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary Data 1
Supplementary Data 1




