

Translate this page into:
Synthesis and antitumor activity of novel indole derivatives containing α-aminophosphonate moieties
⁎Corresponding author. cheng@gdupt.edu.cn (Huicheng Cheng)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
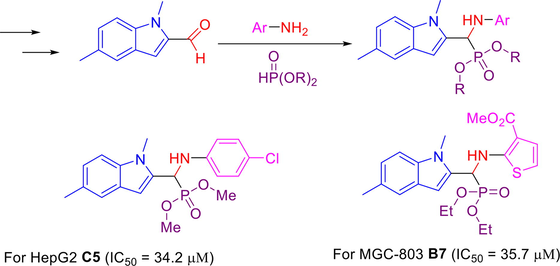
A series of novel indole derivatives containing α-aminophosphonate moieties were synthesized as antitumor agents. The in vitro cytotoxic activity of the compounds was evaluated against human hepatoma cells (HepG2) and human gastric cancer cells (MGC-803) by MTT assay, revealing that most of target compounds exhibited moderate to high antitumor activities. Among them, compound C5 (IC50 = 34.2 µM) demonstrated superior inhibitory activities against HepG2 compared with 5-fluorouracil (IC50 = 78.7 µM). It is noteworthy that compound B7 (IC50 = 35.7 µM) displayed higher inhibitory activities against MGC-803 than that of 5-fluorouracil (IC50 = 82.0 µM).
Keywords
Indole derivatives
α-Amino phosphonate moieties
Synthesis
Antitumor
In vitro

1 Introduction
Cancer has been considered as one of the most important public health problems for a long time (Fidler et al., 2018), accounting for nearly 10 million deaths worldwide in 2020 (Ferlay et al., 2020). Despite many efforts to fight against cancer, the successful treatment of certain tumor types continues to be a challenge owing to their aggressiveness, the complex mechanisms of malignant cell metastasis, chemoresistance and the lack of selectivity for some drugs (Colombano et al., 2010). As the continuous progress of therapies, such as hyperthermia, photodynamic therapy and immunotherapy, chemotherapy is still the most commonly used method to cure cancer and prolong the life of patients (Priestman, 2012). Therefore, the development of novel anticancer agents with low toxicity, low cost and high efficiency is the key way to solve this problem.
Heterocyclic compounds are key structural motifs prevalent in numerous agrochemicals, natural products and pharmaceuticals. As privileged N-heterocyclic compounds, the indole derivatives exhibit unique physic-chemical and biological properties as well as important biological and pharmaceutical activities. They have been widely existed in many natural products and bioactive molecules, such as the antibacterial (Osawa and Namiki, 1983; Ryu et al., 2007), the anti-inflammatory (Jiang et al., 2013) and the anti-virus (Chen et al., 2000; Lu et al., 2007; Yeung et al., 2013). It is worth noting that the indole derivatives have been used as a core scaffold in the anti-cancer agents design (de Sá Alves et al., 2009; Dadashpour and Emami, 2018; Friberg et al., 2013; Garg et al., 2019; Panathur et al., 2013; Rathi et al., 2017; Wan et al., 2019). As phosphorus analogs of a-amino acids and their esters, α-aminophosphonates present unique biological activities (Kukhar and Hudson, 2000; Naydenova et al., 2010; Orsini et al., 2010) and can act as enzyme inhibitors (Sieńczyk and Oleksyszyn, 2009), anti-inflammatory (Sujatha et al., 2017), antimicrobials (Abdel-Megeed et al., 2012; Abdel-Rahman and Ali, 2013, Sujatha et al., 2017), antivirals (Xu et al., 2006; Zhang et al., 2010), anti-HIV (Bhattacharya et al., 2012), antitubercular agents (Li et al., 2017), anti-cancer (Bhattacharya et al., 2013; Guo et al., 2015; Ma et al., 2013; Rezaei et al., 2009; Zhu et al., 2017) antioxidants (Devineni et al., 2013) and antitumor (Gu and Cheng, 2012). The introduction of aminophosphonate group to a pharmacophore can further enhance the anticancer activity against human tumors (Chinthaparthi et al., 2013; Huang et al., 2013; Reddy et al., 2012).
Since both indole derivatives and α-aminophosphonates showed unique biological and pharmaceutical activities, structural modifications of the indole derivatives by introducing α-aminophosphonates groups to improve the pharmacological potency have been recognized as a cutting-edge strategy. Recently, we found that α-aminophosphonate groups could enhance the antitumor activity of heterocyclic compounds (Ma et al., 2013; Guo et al., 2015; Zhu et al., 2017). As a continuation of our previous research, we presently designed and synthesized indole derivatives containing α-aminophosphonate moieties to establish a new strategy for human hepatoma and human gastric cancer therapy (Scheme 1).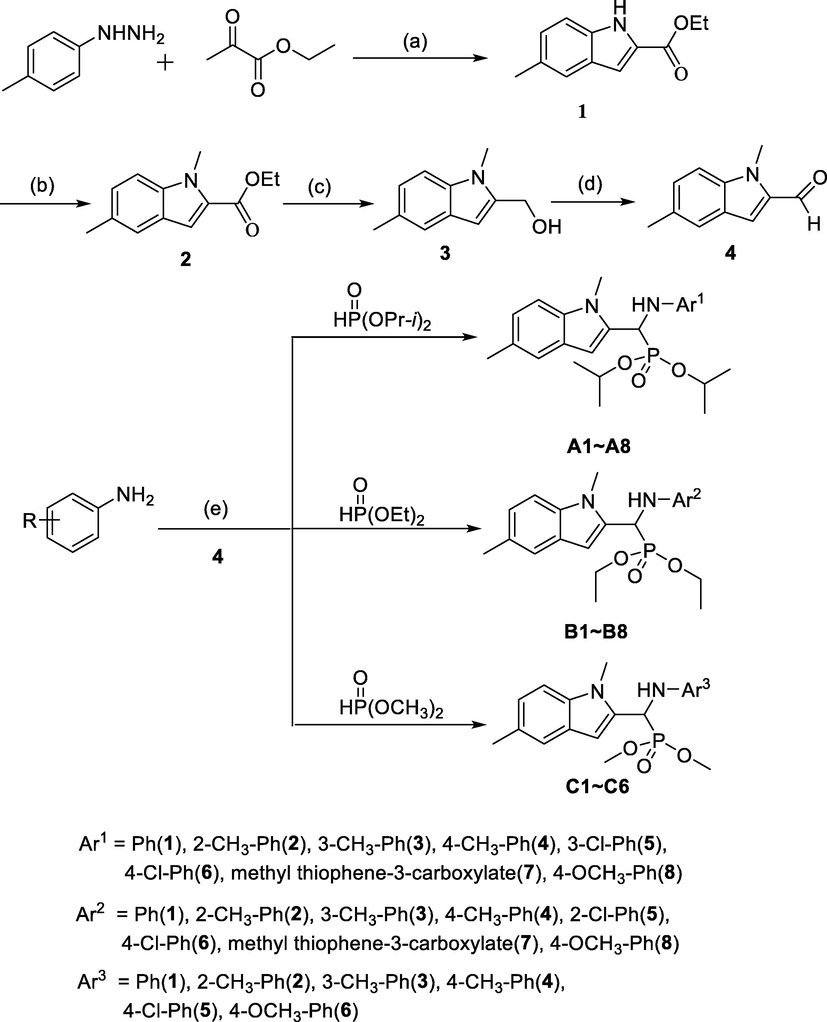
Synthetic route of target compounds. aReagents and conditions: (a) CH3SO3H, EtOH, 80 °C; CH3SO3H, CH3COOH, 115 °C, 85% for two steps; (b and c) CH3I, NaH, anhydrous THF, 0 °C to r.t.; NaBH4, CaCl2, anhydrous EtOH, r.t. to 50 °C, 79% for two steps; (d) MnO2,CH2Cl2, r.t., 61%; (e) anhydrous PhCH3, reflux, 45–88%.
Based on the concept of bioisosterism and assorted mechanisms, we proposed to graft α-aminophosphonate moieties onto indole nucleus. Herein, we synthesize 22 novel indole derivatives containing α-aminophosphonate moieties and the structures of these compounds (A1-A8, B1-B8 and C1-C6) were characterized by 1H NMR, 13C NMR, 31P NMR, IR, MS and HRMS. Moreover, the absolute configurations of A2, B1 and C3 were further confirmed by X-ray single crystal diffraction test. These target compounds were tested for their anti-proliferation activity against HepG2 and MGC-803 at different concentrations. Our work represents the first report about the synthesis and in vitro antitumor activity evaluation of indole derivatives containing α-aminophosphonate moieties.
2 Results and discussion
2.1 Chemistry
The synthetic routes of the target compounds are depicted in Scheme 1. In this experiment, compound 1 was synthesized by the Fischer indole synthesis with p-methylphenylhydrazine and ethyl pyruvate as raw materials. Compound 2 underwent nucleophilic methylation, reduction and oxidation, producing compound 4 (Li et al., 2015; Li et al., 2017). Finally, indole formaldehyde, aromatic amine and phosphite via the “one-pot cooking” reaction in toluene under reflux conditions gave the target compounds. All the new compounds were characterized using 1H NMR and 13C NMR, mass spectrometry and high resolution mass spectrometry.
In anhydrous THF, 1,5-dimethyl-1H-indole 1,5-dimethyl-1H-indole-2-carboxylic acid ethyl ester 2 originating from 5-methyl-1H-indole-2-carboxylic acid ethyl ester 1 was obtained in a high yield. Otherwise, the ester group was hydrolyzed to carboxylic acid. When reducing the ester group in compound 2 to alcohol 3, the combination of CaCl2 and NaBH4 in batches under stirring at room temperature was used. For a period of time, the reaction temperature is increased to 50 °C, the reaction proceeds smoothly. The one-pot three-component Mannich reaction was used for the direct synthesis of the target compounds, which not only greatly improved the conversion rate of raw materials, but also accelerated the reaction speed and simplified the reaction steps.
Fortunately, single crystals of compound A2, B1 and C3 were successfully obtained. High quality and colorless single crystal of the compound A2 (0.25 mm × 0.2 mm × 0.2 mm) was carefully selected for single crystal X-ray diffraction test. The data were recorded at room temperature on an Agilent xcalibur Eos-II-CCD-diffractometer equipped with a graphite-monochromatic Cu Kα radiation (λ = 1.54184 Å). The entire structure was solved by the direct methods using the SHELXS-97 program and refined by the full-matrix least-squares method on F2 with anisotropic thermal parameters for all non-hydrogen atoms using SHELXL-97. The hydrogen atoms on organic ligands were generated by the riding mode.
For compound A2, 8723 reflections were collected and 4279 were independent reflections (Rint = 0.0190, Rsigma = 0.0275), among which 4279 (−11 ≤ h ≤ 12, −12 ≤ k ≤ 9, −13 ≤ l ≤ 15) were observed. X-ray diffraction analysis revealed that the molecular structure of A2 crystallizes in triclinic space group P-1 and is illustrated in Figs. 1 and 2. It is found that the structure is stabilized by intermolecular hydrogen bonding interaction (N2-H2⋯O3 = 2.234(15) Å). CCDC2064712, 2064713 and 2064714 contain the supplementary crystallographic data of compound A2, B1 and C3, respectively. They can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.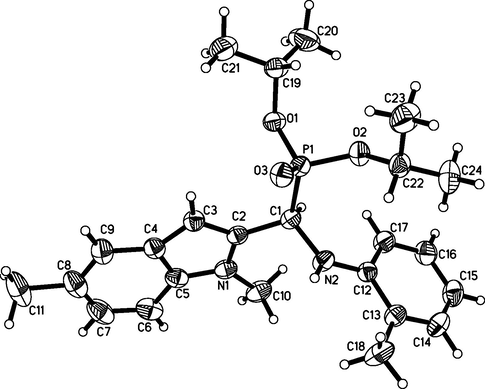
Molecular structure of Compound A2.
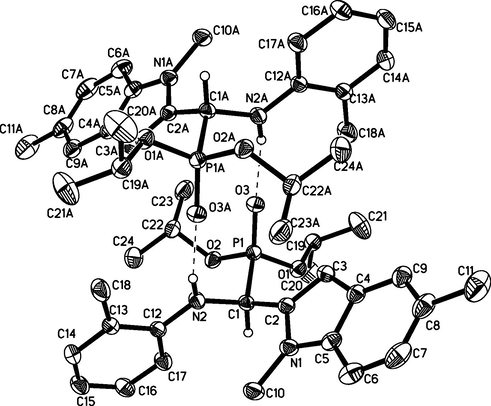
Single crystal diffraction diagram of compound A2 with two molecules interacting through hydrogen bonds.
2.2 Biological activity evaluation against HepG2 and MGC-803
In this paper, 5-fluorouracil was used as a positive control drug to study the growth inhibitory activity of target compounds on HepG2 and MGC-803 by MTT assay. The growth inhibitory activity of the target product on HepG2 and MGC-803 was determined by using 100 μg/mL as the primary screening concentration. The corresponding inhibition rate was calculated according to the related formula, as shown in Table 1.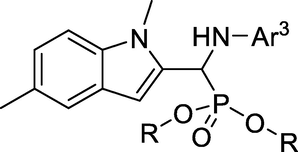
comp.
Ar
R
Inhibition rate (%)
HepG2
MGC-803
A1
Ph
i-Pr
11.7
21.5
A2
2-Me-Ph
i-Pr
78.7
−27.4
A3
3-Me-Ph
i-Pr
60.2
−8.8
A4
4-Me-Ph
i-Pr
48.8
−24.7
A5
3-Cl-Ph
i-Pr
60.7
−17.2
A6
4-Cl-Ph
i-Pr
73.7
79.2
A7
methyl thiophene-3-carboxylate
i-Pr
78.0
80.1
A8
4-MeO-Ph
i-Pr
42.9
−11.7
B1
Ph
Et
11.0
−31.7
B2
2-Me-Ph
Et
77.7
1.3
B3
3-Me-Ph
Et
19.1
−29.5
B4
4-Me-Ph
Et
9.9
9.8
B5
2-Cl-Ph
Et
35.0
−6.0
B6
4-Cl-Ph
Et
46.7
1.3
B7
methyl thiophene-3-carboxylate
Et
75.7
89.3
B8
4-MeO-Ph
Et
48.3
−32.2
C1
Ph
Me
26.8
−121.3
C2
2-Me-Ph
Me
76.6
8.1
C3
3-Me-Ph
Me
46.7
47.8
C4
4-Me-Ph
Me
84.4
8.2
C5
4-Cl-Ph
Me
84.7
67.4
C6
4-MeO-Ph
Me
84.0
82.3
With different substituents on the phenyl ring of aromatic amine, compounds A1-A8, B1-B8, and C1-C6 showed various degrees of inhibitory activity against HepG2 and MGC-803 cells. The target compounds had different degrees of inhibition on HepG2 at a concentration of 100 μg/mL, among which compounds A2, A7, B2, B7, C2 and C4-C6 inhibited the proliferation of HepG2 by more than 75%, and the anticancer activities range from moderate to excellent. The best activity was from compound C5 with inhibition rate of 84.7%. Compared to the antitumor activity of all the target compounds, it can be found that the introduction of substituents to the benzene ring was beneficial for increasing the antitumor activities. The different substituents R on the compounds A2, B2 and C2 had no obvious influence on the antitumor activity. When electron-withdrawing group chlorine was at the para position of the benzene ring, compounds A6 and C5 showed good activity. It was of interest that the introduction of a methyl group to the benzene ring of Ar in compounds A2-A4 and B2-B4 was important for improving antitumor activities with the order of ortho- > meta- > para-.
The results also revealed that at a concentration of 100 μg/mL, compounds A2-A5, A8, B1, B3, B5, B8 and C1 showed a negative inhibitory effect and promoted the proliferation of MGC-803 cells, which indicates that these compounds may activate the MGC-803 cells. Other target compounds exhibited different degrees of inhibition on MGC-803. Among them, compounds A6, A7, B7 and C6 inhibited the proliferation of MGC-803 by more than 75%, and the best inhibition rate 89.3% was from compound B7. The antitumor data indicated that the substitutes of phosphonate have apparently no influence on antitumor activity. For example, compounds A2, B2 and C2 against HepG2 cells exhibited similar degree of antitumor activity. This same trend was observed for compounds A7 and B7 against MGC-803 cells. It was of interest that compound A6 with a chlorine group to the para position of benzene ring as Ar exhibited good antitumor activities against MGC803, and similar results can also be observed in compound C5. To our delight, methyl thiophene-3-carboxylate as Ar was crucial for inhibitory activities against MGC-803, while other compounds bearing the methyl substituent to the benzene ring showed weak or negative inhibitory effect.
For compounds with better activity, the samples were formulated into 6 different concentrations (64 μg/mL, 32 μg/mL, 16 μg/mL, 8 μg/mL, 4 μg/mL and 2 μg/mL). According to the measured OD value, the half effective inhibitory concentration IC50 of each compound was calculated by SPSS 17.0 software. The results are summarized in Table 2. Note: Indicates that there is no second screening inhibitory activity.
comp.
Ar
R
IC50(µM)
HepG2
MGC-803
A2
2-Me-Ph
i-Pr
47.1
–
A7
methyl thiophene-3-carboxylate
i-Pr
40.6
45.1
B2
2-Me-Ph
Et
37.1
–
B7
methyl thiophene-3-carboxylate
Et
43.1
35.7
C2
2-Me-Ph
Me
67.4
–
C4
4-Me-Ph
Me
44.4
–
C5
2-Cl-Ph
Me
34.2
44.8
C6
4-MeO-Ph
Me
49.3
90.3
5-Fluorouracil
78.7
82.0
The anticancer activity data in Table 2 showed that all the tested compounds have better inhibitory activities against HepG2 than the positive control 5-fluorouracil. Among them, compound C5 (IC50 = 34.2 µM) and B2 (IC50 = 37.1 µM) exhibited excellent inhibitory activity on HepG2. From the above results, some structure-activity relationships could be concluded: (1) methyl thiophene-3-carboxylate as Ar in compounds A7 and B7 was beneficial for improving inhibitory activities against MGC-803. (2) compound C5 with chlorine atom exerted excellent inhibitory effect on HepG2, when the other compounds bearing the methyl and methoxy group to the benzene ring showed a positive inhibitory effect.
The anticancer activity data in Table 2 showed that compounds A7, B7 and C5 have better inhibitory activities against MGC-803 than 5-fluorouracil. The IC50 values of compound A7, B7 and C5 on MGC-803 were 45.1 µM, 35.7 µM and 44.8 µM, respectively. More significantly, compound B7 (IC50 = 35.7 µM) displayed superior inhibitory activity on MGC-803 compared with that of 5-fluorouracil (IC50 = 82.0 µM). Methyl thiophene-3-carboxylate as Ar was conducive for inhibitory activities against MGC-803. Compound C5 bearing an electron-withdrawing chlorine on the benzene ring showed high inhibitory effect, while compounds A2, B2, C2, C4 and C6 with electron-donating methyl group or methoxy group on the benzene ring showed negative or weak inhibitory effect.
3 Conclusion
In conclusion, we designed and synthesized a series of novel indole derivatives containing α-aminophosphonate moieties. The in vitro cytotoxic activity of the novel compounds was evaluated by the MTT method, revealing that most of target compounds exhibited moderate to high antitumor activities against HepG2 and MGC-803. Among them, compound C5 (IC50 = 34.2 µM) demonstrated more potent inhibitory activities against HepG2 compared with 5-fluorouracil (IC50 = 78.7 µM). It is noteworthy that compound B7 (IC50 = 35.7 μM) displayed higher inhibitory activities against MGC-803 than that of 5-fluorouracil (IC50 = 82.0 µM). The findings demonstrated that these compounds could be promising and leading bioactive compounds as novel antitumor drugs. Further studies will focus on influence of different substituents, minor structural modification and steric parameters on structure-activity relationships in our lab.
4 Experimental
All starting materials were commercially available and analytically pure. Commercial reagents and solvents were used as received without further purification unless otherwise specified. Toluene was freshly distilled over sodium with the use of diphenyl ketone as an indicator under nitrogen. All manipulations were performed in air atmosphere. All chemicals and reagents were purchased from JK Scientific Co., 9 dingchem. Scientific Co., and used without further purification. 1H NMR and 13C NMR were recorded on a Bruker AVANCE AV 400 (400 MHz for 1H, 100 MHz for 13C) instrument in CDCl3, and 31P chemical shifts were acquired in CDCl3 with H3PO4 as the internal standard. Data were reported as follows: chemical shift in ppm (δ), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad signal), coupling constant (Hz), integration. TLC were performed on silica gel Huanghai HSGF254 plates and visualized by quenching of UV fluorescence (λmax = 254 nm). Silica gel (200–300 mesh) was purchased from Qingdao Haiyang Chemical Co., China. Electron-impact-ionisation mass spectra (EI) were recorded with an Aligent 7890A/5975C GC–MS instrument. High resolution mass spectra (HRMS) were acquired on Varian 7.0 T FTMS. FTIR spectra were obtained with a Bruker Tensor 27 instrument. All IR samples were prepared as thin films and reported in wave numbers (cm−1). 1,5-dimethyl-1H-indole-2-carbaldehydewas synthesized according to the reference (Li et al., 2015; Li et al., 2017).
4.1 General procedure for the synthesis of compound A1
A 50 mL dry round bottom flask was equipped with a magnetic stir bar, and charged with 20 mL of anhydrous toluene, 1,5-dimethyl-1H-indole 2-carboxaldehyde (294 mg, 1.7 mmol), aniline (177 mg, 1.9 mmol), and diisopropyl phosphite (382 mg, 2.3 mmol). The mixture was stirred at 110 °C for 4 h. The resulting solution was then cooled to room temperature, and extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine, and dried over anhydrous Na2SO4. The volatiles were removed under vacuum, and the residue was purified by column chromatography to give the product. The other target compounds A2-A8, B1-B8 and C1-C6 were prepared by similar methods.
4.2 Characterization data for products
Diisopropyl ((1,5-dimethyl-1H-indol-2-yl)(phenylamino)methyl)phosphonate A1: Off-white solid, Yield: 88%, m.p. 158–159 °C; 1H NMR (CDCl3, 400 MHz) δ: 7.34 (s, 1H, ArH), 7.21 (d, J = 8.4 Hz, 1H, ArH), 7.16–7.12 (m, 2H, ArH), 7.04 (d, J = 8.3 Hz, 1H, ArH), 6.73 (t, J = 7.3 Hz, 1H, ArH), 6.66 (d, J = 7.9 Hz, 2H, ArH), 6.59 (d, J = 3.1 Hz, 1H, ArH), 4.97 (d, J = 23.6 Hz, 1H, PC.), 4.82–4.74 (m, 1H, CH(CH3)2), 4.62–4.54(m, 1H, CH(CH3)2), 3.84 (s, 3H, NCH3), 2.44 (s, 3H, ArCH3), 1.37 (d, J = 6.2 Hz, 3H, CH(CH3)2), 1.31–1.27 (m, 6H, 2 × CH(CH3)2), 0.93 (d, J = 6.2 Hz, 3H, CH(CH3)2); 13C NMR (CDCl3, 100 MHz) δ: 146.5, 136.4, 134.7, 129.2, 128.6, 127.7, 123.1, 120.3, 118.6, 113.8, 108.7, 101.6, 72.4, 72.3, 50.2 (d, CH, 1JC,P = 159.1 Hz), 30.3, 24.3, 23.8, 23.3, 21.3; 31P NMR (CDCl3, 162 MHz) δ: 19.44; IR (KBr) ν: 3276, 2975, 1602, 1537, 1498, 1383, 1327, 1236, 1104, 977, 791, 746, 694; ESI-MS m/z: 415.1 [M+H]+; HRMS (pos.): 415.2163 ([M+H]+, C23H31N2O3PH; calc. 415.2151).
Diisopropyl ((1,5-dimethyl-1H-indol-2-yl)(o-tolylamino)methyl)phosphonate A2: Brown yellow solid, Yield: 85%, m.p. 108–110 °C; 1H NMR (CDCl3, 400 MHz) δ: 7.33 (s, 1H, ArH), 7.22 (d, J = 8.4 Hz, 1H, ArH), 7.09–6.97 (m, 3H, ArH), 6.68 (t, J = 7.4 Hz, 1H, ArH), 6.55 (s, 1H, ArH), 6.50 (d, J = 8.0 Hz, 1H, ArH), 4.99 (d, J = 23.5 Hz, 1H, PCH), 4.80–4.72 (m, 1H, CH(CH3)2), 4.63–4.55 (m, 1H, CH(CH3)2), 3.85 (s, 3H, NCH3), 2.44 (s, 3H, ArCH3), 2.28 (s, 3H, ArCH3), 1.37 (d, J = 6.2 Hz, 3H, CH(CH3)2), 1.32–1.28 (m, 6H, 2 × CH(CH3)2), 0.96 (d, J = 6.2 Hz, 3H, CH(CH3)2); 13C NMR (CDCl3, 100 MHz) δ: 144.5, 136.5, 134.9, 130.2, 128.6, 127.8, 127.1, 123.1, 122.9, 120.3, 118.2, 111.1, 108.7, 101.4, 72.4, 72.3, 50.2 (d, CH, 1JC,P = 159.1 Hz), 30.4, 24.3, 24.3, 23.9, 23.4, 21.4, 17.6; 31P NMR (CDCl3, 162 MHz) δ: 19.37; IR (KBr) ν: 3423, 3325, 2975, 1604, 1527, 1454, 1383, 1311, 1246, 1104, 997, 872, 788, 743; ESI-MS m/z: 429.2 [M+H]+; HRMS (pos.): 429.2303 ([M+H]+, C24H33N2O3PH; calc. 429.2307).
Diisopropyl ((1,5-dimethyl-1H-indol-2-yl)(m-tolylamino)methyl)phosphonate A3: White solid, Yield: 88%, m.p. 163–165 °C; 1H NMR (CDCl3, 400 MHz) δ: 7.34 (s, 1H, ArH), 7.21 (d, J = 8.4 Hz, 1H, ArH), 7.05–6.99 (m, 2H, ArH), 6.59–6.54 (m, 2H, ArH), 6.51 (s, 1H, ArH), 6.44 (d, J = 8.0 Hz, 1H, ArH), 4.96 (d, J = 23.2 Hz, 1H, PCH), 4.81–4.73 (m, 1H, CH(CH3)2), 4.60–4.49(m, 1H, CH(CH3)2), 3.83 (s, 3H, NCH3), 2.44 (s, 3H, ArCH3), 2.25 (s, 3H, ArCH3), 1.37 (d, J = 6.2 Hz, 3H, CH(CH3)2), 1.29 (t, J = 13.4 Hz, 6H, 2 × CH(CH3)2), 0.92 (d, J = 6.2 Hz, 3H, CH(CH3)2); 13C NMR (CDCl3, 100 MHz) δ: 146.6, 138.9, 136.4, 134.9, 129.1, 128.6, 127.8, 123.1, 119.7, 114.8, 110.8, 108.7, 101.6, 72.4, 72.3, 50.2 (d, CH, 1JC,P = 159.2 Hz), 30.4, 24.4, 24.3, 23.9, 23.4, 21.6, 21.4; 31P NMR (CDCl3, 162 MHz) δ: 19.44; IR (KBr) ν: 3440, 3286, 2973, 1605, 1590, 1543, 1492, 1383, 1333, 1235, 1180, 1104, 976, 871, 791, 763, 695; ESI-MS m/z: 429.1 [M+H]+; HRMS (pos.): 429.2305 ([M+H]+, C24H33N2O3PH; calc. 429.2307).
Diisopropyl ((1,5-dimethyl-1H-indol-2-yl)(p-tolylamino)methyl)phosphonate A4: Light yellow solid, Yield: 86%, m.p. 132–134 °C; 1H NMR (CDCl3, 400 MHz) δ: 7.33 (s, 1H, ArH), 7.20 (d, J = 8.4 Hz, 1H, ArH), 7.03 (d, J = 8.3 Hz, 1H, ArH), 6.94 (d, J = 8.2 Hz, 2H, ArH), 6.57 (d, J = 8.2 Hz, 3H, ArH), 4.92 (d, J = 23.6 Hz, 1H, PCH), 4.82–4.74 (m, 1H, CH(CH3)2), 4.61–4.54(m, 1H, CH(CH3)2), 3.82 (s, 3H, NCH3), 2.43 (s, 3H, ArCH3), 2.20 (s, 3H, ArCH3), 1.37 (d, J = 6.2 Hz, 3H, CH(CH3)2), 1.29 (t, J = 10.1 Hz, 6H, 2 × CH(CH3)2), 0.93 (d, J = 6.2 Hz, 3H, CH(CH3)2); 13C NMR (CDCl3, 100 MHz) δ: 144.2, 136.3, 134.9, 129.7, 128.5, 127.9, 127.7, 123.0, 120.2, 114.0, 108.7, 101.5, 72.3, 72.2, 50.6 (d, CH, 1JC,P = 168.7 Hz), 30.3, 24.3, 24.2, 23.8, 23.3, 21.3, 20.3; 31P NMR (CDCl3, 162 MHz) δ: 19.48; IR (KBr) ν: 3272, 2978, 2922, 1616, 1530, 1384, 1237, 1007, 984, 796; ESI-MS m/z: 429.2 [M+H]+; HRMS (pos.): 429.2306 ([M+H]+, C24H33N2O3PH; calc. 429.2307).
Diisopropyl(((3-chlorophenyl)amino)(1,5-dimethyl-1H-indol-2-yl)methyl)phos-phornate A5: Light yellow solid, Yield: 62%, m.p. 175–176 °C; 1H NMR (CDCl3, 400 MHz) δ: 7.35 (s, 1H, ArH), 7.21 (d, J = 8.4 Hz, 1H, ArH), 7.06–7.01 (m, 2H, ArH), 6.70–6.67 (m, 2H, ArH), 6.58 (d, J = 2.8 Hz, 1H, ArH), 6.52–6.49(m, 1H, ArH), 4.92 (d, J = 23.4 Hz, 1H, PCH), 4.80–4.72 (m, 1H, CH(CH3)2), 4.60–4.52(m, 1H, CH(CH3)2), 3.83 (s, 3H, NCH3), 2.44 (s, 3H, ArCH3), 1.37 (d, J = 6.1 Hz, 3H, CH(CH3)2), 1.29 (t, J = 13.6 Hz, 6H, 2 × CH(CH3)2), 0.91 (d, J = 6.1 Hz, 3H, CH(CH3)2); 13C NMR (CDCl3, 100 MHz) δ: 147.7, 136.4, 134.9, 134.2, 130.2, 128.8, 127.7, 123.3, 120.4, 118.5, 113.7, 111.9, 108.8, 101.7, 72.6, 72.4, 49.9 (d, CH, 1JC,P = 159.3 Hz), 30.4, 24.3, 23.9, 23.3, 21.4; 31P NMR (CDCl3, 162 MHz) δ: 18.88; IR (KBr) ν: 3272, 2975, 2923, 1605, 1594, 1537, 1485, 1384, 1235, 1102, 999, 793, 774; ESI-MS m/z: 449.2 [M+H]+; HRMS (pos.): 449.1762 ([M+H]+, C23H30ClN2O3PH; calc. 449.1761).
Diisopropyl (((4-chlorophenyl)amino)(1,5-dimethyl-1H-indol-2-yl)methyl)phos-
phornate A6: White solid, Yield: 45%, m.p. 152–153 °C; 1H NMR (CDCl3, 400 MHz) δ: 7.34 (s, 1H, ArH), 7.22 (d, J = 8.4 Hz, 1H, ArH), 7.09–7.04 (m, 3H, ArH), 6.58 (t, J1 = 8.7 Hz, 3H, ArH), 4.91 (d, J = 23.4 Hz, 1H, PCH), 4.82–4.72 (m, 1H, CH(CH3)2), 4.63–4.54(m, 1H, CH(CH3)2), 3.82 (s, 3H, NCH3), 2.44 (s, 3H, ArCH3), 1.38 (d, J = 7.7 Hz, 3H, CH(CH3)2), 1.30 (t, J = 6.3 Hz, 6H, 2 × CH(CH3)2), 0.92 (d, J = 6.2 Hz, 3H, CH(CH3)2); 13C NMR (CDCl3, 100 MHz) δ: 145.1, 136.4, 134.2, 129.1, 128.8, 127.7, 123.3, 120.3, 114.9, 108.8, 101.7, 101.64, 72.6, 72.4, 50.3 (d, CH, 1JC,P = 159.3 Hz), 30.4, 24.3, 24.3, 23.9, 23.3, 21.4; 31P NMR (CDCl3, 162 MHz) δ: 19.12; IR (KBr) ν: 3286, 2980, 2935, 1597, 1528, 1489, 1384, 1318, 1235, 1178, 1104, 1012, 988, 875, 793; ESI-MS m/z: 449.2 [M+H]+; HRMS (pos.): 449.1765 ([M+H]+, C23H30ClN2O3PH; calc. 449.1761).
Methyl2-(((diisopropoxyphosphoryl)(1,5-dimethyl-1H-indol-2-yl)methyl)amino)-thiophene-3-carboxylate A7: Light yellow solid, Yield: 83%, m.p. 120–123 °C; 1H NMR (CDCl3, 400 MHz) δ: 7.58 (t, J = 7.0 Hz, 1H, NH), 7.35 (s, 1H, ArH), 7.24–7.20 (m, 2H, ArH), 7.05 (d, J = 8.3 Hz, 1H, ArH), 6.57–6.55 (m, 2H, ArH), 5.02 (d, J = 23.4 Hz, 1H, PCH), 4.74–4.65 (m, 2H, 2 × CH(CH3)2), 3.88 (s, 3H, NCH3), 3.85 (s, 3H, OCH3), 2.45 (s, 3H, ArCH3), 1.35–1.33 (m, 6H, 2 × CH(CH3)2), 1.27 (d, 3H, J = 6.2 Hz, CH(CH3)2), 1.15 (d, J = 6.2 Hz, 3H, CH(CH3)2); 13C NMR (CDCl3, 100 MHz) δ: 164.9, 137.2, 136.5, 134.1, 131.8, 128.7, 127.5, 124.1, 123.3, 116.8, 108.8, 102.0, 72.7, 72.5, 51.9 (d, CH, 1JC,P = 158.8 Hz), 51.3, 30.5, 24.2, 24.1, 23.7, 23.6, 21.3; 31P NMR (CDCl3, 162 MHz) δ: 17.17; IR (KBr) ν: 3364, 2977, 2945, 1675, 1567, 1446, 1254, 1206, 983, 777, 646; ESI-MS m/z: 479.1 [M+H]+; HRMS (pos.): 479.1768 ([M+H]+, C23H31N2O5PSH; calc. 479.1770).
Diisopropyl((1,5-dimethyl-1H-indol-2-yl)((4-methoxyphenyl)amino)methyl)phosphonate A8: Brown yellow solid, Yield: 49%, m.p. 136–138 °C; 1H NMR (CDCl3, 400 MHz) δ: 7.31 (s, 1H, ArH), 7.18 (d, J = 8.4 Hz, 1H, ArH), 7.03–6.99 (m, 2H, ArH), 6.56 (t, J = 3.1 Hz, 1H, ArH), 6.28–6.23 (m, 2H, ArH), 6.19–6.18 (m, 1H, ArH), 4.99 (d, J = 23.5 Hz, 1H, PCH), 4.79–4.71 (m, 1H, CH(CH3)2), 4.59–4.50 (m, 1H, CH(CH3)2), 3.80 (s, 3H, NCH3), 3.70 (s, 3H, OCH3), 2.41 (s, 3H, ArCH3), 1.34 (d, J = 6.2 Hz, 3H, CH(CH3)2), 1.27–1.25 (m, 6H, 2 × CH(CH3)2), 0.89 (d, J = 6.2 Hz, 3H, CH(CH3)2); 13C NMR (CDCl3, 100 MHz) δ: 160.6, 147.8, 136.3, 134.7, 129.9, 128.6, 127.7, 123.1, 120.2, 108.7, 106.7, 103.6, 101.5, 100.0, 72.4, 72.3, 55.0, 50.1(d, CH, 1JC,P = 159.5 Hz), 30.3, 24.3, 24.2, 23.8, 23.2, 21.3, 17.55; 31P NMR (CDCl3, 162 MHz) δ: 19.26; IR (KBr) ν: 3423, 3287, 2974, 2928, 1610, 1498, 1236, 1001, 980, 791; ESI-MS m/z: 445.2 [M+H]+; HRMS (pos.): 445.2257 ([M+H]+, C24H33N2O4PH; calc. 445.2256).
Diethyl ((1,5-dimethyl-1H-indol-2-yl)(phenylamino)methyl)phosphonate B1: Light brown solid, Yield: 84%, m.p. 128–131 °C; 1H NMR (CDCl3, 400 MHz) δ: 7.35 (s, 1H, ArH), 7.22 (d, J = 8.4 Hz, 1H, ArH), 7.17–7.13 (m, 2H, ArH), 7.06 (d, J = 8.4 Hz, 1H, ArH), 6.75 (t, J = 7.3 Hz, 1H, ArH), 6.67 (d, J = 8.3 Hz, 2H, ArH), 6.62 (d, J = 3.0 Hz, 1H, ArH), 5.04 (d, J = 23.1 Hz, 1H, PCH), 4.25–4.12 (m, 2H, CH2CH3), 4.07–4.01 (m, 1H, CH2CH3), 3.83 (s, 3H, NCH3), 3.81–3.74 (m, 1H, CH2CH3), 2.45 (s, 3H, ArCH3), 1.33 (t, J = 7.0 Hz, 3H, CH2CH3), 1.15 (t, J = 7.0 Hz, 3H, CH2CH3); 13C NMR (CDCl3, 100 MHz) δ: 146.2, 136.4, 134.2, 129.2, 128.8, 127.6, 123.3, 120.3, 118.8, 113.8, 108.8, 101.7, 63.6, 63.5, 49.7 (d, CH, 1JC,P = 155.0 Hz), 30.3, 21.3, 16.5, 16.3; 31P NMR (CDCl3, 162 MHz) δ: 21.11; IR (KBr) ν: 3277, 2978, 2907, 1602, 1533, 1498, 1488, 1323, 1240, 1048, 1018, 973, 792, 745; ESI-MS m/z: 387.2 [M+H]+; HRMS (pos.): 387.1839 ([M+H]+, C21H27N2O3PH; calc. 387.1838).
Diethyl ((1,5-dimethyl-1H-indol-2-yl)(o-tolylamino)methyl)phosphonate B2: White solid, Yield: 82%, m.p. 97–99 °C; 1H NMR (CDCl3, 400 MHz) δ: 7.35 (s, 1H, ArH), 7.24 (d, J = 8.4 Hz, 1H, ArH), 7.10–7.00 (m, 3H, ArH), 6.70 (t, J = 7.4 Hz, 1H, ArH), 6.60 (d, J = 3.2 Hz, 1H, ArH), 6.55 (d, J = 8.0 Hz, 1H, ArH), 5.08 (d, J = 23.2 Hz, 1H, PCH), 4.51–4.49(m, 1H, NH), 4.24–4.14 (m, 2H, CH2CH3), 4.08–4.00 (m, 1H, CH2CH3), 3.85 (s, 3H, NCH3), 3.81–3.74 (m, 1H, CH2CH3), 2.45 (s, 3H, ArCH3), 2.29 (s, 3H, ArCH3), 1.34 (t, J = 7.0 Hz, 3H, CH2CH3), 1.17 (t, J = 7.0 Hz, 3H, CH2CH3); 13C NMR (CDCl3, 100 MHz) δ: 144.3, 136.5, 134.5, 130.3, 128.8, 127.7, 127.1, 123.3, 123.1, 120.4, 118.5, 111.2, 108.8, 101.6, 63.7, 63.5, 49.8 (d, CH, 1JC,P = 156.7 Hz), 30.3, 21.4, 17.6, 16.5, 16.3; 31P NMR (CDCl3, 162 MHz) δ: 21.11; IR (KBr) ν: 3329, 2979, 2907, 1603, 1524, 1479, 1309, 1249, 1020, 967, 791; ESI-MS m/z: 401.2 [M+H]+; HRMS (pos.): 401.1982 ([M+H]+, C22H29N2O3PH; calc. 401.1994).
Diethyl ((1,5-dimethyl-1H-indol-2-yl)(m-tolylamino)methyl)phosphonate B3: White solid, Yield: 83%, m.p. 138–140 °C; 1H NMR (CDCl3, 400 MHz) δ: 7.35 (s, 1H, ArH), 7.22 (d, J = 8.4 Hz, 1H, ArH), 7.06–7.01 (m, 2H, ArH), 6.62 (d, J = 3.1 Hz, 1H, ArH), 6.57 (d, J = 7.4 Hz, 1H, ArH), 6.52 (s, 1H, ArH), 6.47 (dd, J1 = 1.8 Hz, J2 = 1.8 Hz, 1H, ArH), 5.53 (d, J = 23.5 Hz, 1H, PCH), 4.49 (s, 1H, NH), 4.24–4.12 (m, 2H, CH2CH3), 4.07–3.99 (m, 1H, CH2CH3), 3.83 (s, 3H, NCH3), 3.81–3.74 (m, 1H, CH2CH3), 2.45 (s, 3H, ArCH3), 2.26 (s, 3H, ArCH3), 1.33 (t, J = 7.1 Hz, 3H, CH2CH3), 1.15 (t, J = 7.1 Hz, 3H, CH2CH3); 13C NMR (CDCl3, 100 MHz) δ: 146.3, 139.1, 136.4, 134.5, 129.2, 128. 8, 127.7, 123.3, 120.4, 119.8, 114.8, 110.8, 108.8, 101.7, 63.6, 63.4, 51.0 (d, CH, 1JC,P = 155.1 Hz), 30.3, 21.63, 21.4, 16.5, 16.3; 31P NMR (CDCl3, 162 MHz) δ: 21.09; IR (KBr) ν: 3426, 3288, 2978, 2925, 1605, 1542, 1493, 1332, 1239, 1047, 1017, 972, 792, 761; ESI-MS m/z: 401.2 [M+H]+; HRMS (pos.): 401.1992 ([M+H]+, C22H29N2O3PH; calc. 401.1994).
Diethyl ((1,5-dimethyl-1H-indol-2-yl)(p-tolylamino)methyl)phosphonate B4: Brown yellow solid, Yield: 81%, m.p. 126–129 °C; 1H NMR (CDCl3, 400 MHz) δ: 7.34 (s, 1H, ArH), 7.21 (d, J = 8.4 Hz, 1H, ArH), 7.04 (d, J = 8.4 Hz, 1H, ArH), 6.95 (d, J = 8.2 Hz, 2H, ArH), 6.60–6.58 (m, 3H, ArH), 5.00 (d, J = 23.1 Hz, 1H, PCH), 4.24–4.13 (m, 2H, CH2CH3), 4.08–4.01 (m, 1H, CH2CH3), 3.82 (s, 3H, NCH3), 3.79–3.75 (m, 1H, CH2CH3), 2.44 (s, 3H, ArCH3), 2.21 (s, 3H, ArCH3), 1.33 (t, J = 7.0 Hz, 3H, CH2CH3), 1.15 (t, J = 7.1 Hz, 3H, CH2CH3); 13C NMR (CDCl3, 100 MHz) δ: 144.0, 136.4, 134.4, 129.7, 128.7, 128.1, 127.7, 123.2, 120.3, 114.0, 108.8, 101.7, 63.6, 63.4, 50.1 (d, CH, 1JC,P = 168.3 Hz), 30.3, 21.3, 20.4, 16.5, 16.3; 31P NMR (CDCl3, 162 MHz) δ: 21.17; IR (KBr) ν: 3284, 2990, 2918, 1614, 1524, 1487, 1235, 1049, 1020, 973, 792; ESI-MS m/z: 401.2 [M+H]+; HRMS (pos.): 401.1991 ([M+H]+, C22H29N2O3PH; calc. 401.1994).
Diethyl(((2-chlorophenyl)amino)(1,5-dimethyl-1H-indol-2-yl)methyl)phosphor-nate B5: Yellow solid, Yield: 60%, m.p. 115–117 °C; 1H NMR (CDCl3, 400 MHz) δ: 7.35 (s, 1H, ArH), 7.29 (d, J = 5.7 Hz, 1H, ArH), 7.23 (d, J = 8.4 Hz, 1H, ArH), 7.07–7.01 (m, 2H, ArH), 6.68 (t, J = 7.6 Hz, 1H, ArH), 6.61–6.58 (m, 2H, ArH), 5.30–5.26 (m, 1H, NH), 5.06 (d, J = 23.2 Hz, 1H, PCH), 4.24–4.04 (m, 3H, CH2CH3), 3.92–3.86 (m, 4H, CH2CH3, NCH3), 2.45 (s, 3H, ArCH3), 1.33 (t, J = 7.0 Hz, 3H, CH2CH3), 1.21 (t, J = 7.0 Hz, 3H, CH2CH3); 13C NMR (CDCl3, 100 MHz) δ: 142.3, 136.6, 133.7, 129.3, 128.9, 127.8, 127.6, 123.4, 120.4, 120.2, 118.8, 112.5, 108.9, 101.9, 63.9, 63.6, 49.8 (d, CH, 1JC,P = 159.2 Hz), 30.4, 21.4, 16.5, 16.3; 31P NMR (CDCl3, 162 MHz) δ: 20.01; IR (KBr) ν: 3405, 2983, 2927, 1597, 1503, 1486, 1320, 1242, 1028, 977, 802, 745; ESI-MS m/z: 421.1 [M+H]+; HRMS (pos.): 421.1447 ([M+H]+, C21H26ClN2O3PH; calc. 421.1448).
Diethyl(((4-chlorophenyl)amino)(1,5-dimethyl-1H-indol-2-yl)methyl)phosphor-nate B6: Light brown solid, Yield: 73%, m.p. 145–147 °C; 1H NMR (CDCl3, 400 MHz) δ: 7.35 (s, 1H, ArH), 7.22 (d, J = 8.4 Hz, 1H, ArH), 7.10–7.05 (m, 3H, ArH), 6.60–6.58 (m, 3H, ArH), 4.97 (d, J = 23.0 Hz, 1H, PCH), 4.23–4.17 (m, 2H, CH2CH3), 4.06–3.99 (m, 1H, CH2CH3), 3.81–3.73 (m, 4H, CH2CH3, NCH3), 2.44 (s, 3H, ArCH3), 1.33 (t, J = 7.0 Hz, 3H, CH2CH3), 1.15 (t, J = 7.1 Hz, 3H, CH2CH3); 13C NMR (CDCl3, 100 MHz) δ: 144.5, 136.5, 134.9, 130.2, 128.6, 127.8, 127.1, 123.1, 122.9, 120.3, 118.2, 111.1, 108.7, 101.4, 72.4, 51.0, 49.4 (d, CH, 1JC,P = 159.2 Hz), 30.4, 21.3, 16.5, 16.3; 31P NMR (CDCl3, 162 MHz) δ: 20.77; IR (KBr) ν: 3441, 3280, 2920, 1594, 1492, 1390, 1285, 1231, 1052, 1021, 974, 654; ESI-MS m/z: 443.1 [M+H]+; HRMS (pos.): 443.1262 ([M+H]+, C21H26ClN2O3PH; calc. 443.1267).
Methyl2-(((diethoxyphosphoryl)(1,5-dimethyl-1H-indol-2-yl)methyl)amino)thiophene-3-carboxylate B7: Light yellow solid, Yield: 78%, m.p. 106–108 °C; 1H NMR (CDCl3, 400 MHz) δ: 7.63–7.60 (m, 1H, NH), 7.36 (s, 1H, ArH), 7.25–7.21 (m, 2H, ArH), 7.06 (d, J = 8.4 Hz, 1H, ArH), 6.58–6.55 (m, 2H, ArH), 5.09 (d, J = 23.6 Hz, 1H, PCH), 4.19–4.06 (m, 3H, CH2CH3), 4.05–3.95 (m, 1H, CH2CH3), 3.88 (m, 4H, NCH3), 3.84 (s, 3H, OCH3), 2.44 (s, 3H, ArCH3), 1.34–1.24 (m, 6H, CH2CH3); 13C NMR (CDCl3, 100 MHz) δ: 165.0, 154.2, 136.5, 133.7, 131.9, 128.8, 127.5, 123.4, 120.3, 116.7, 108.9, 102.0, 64.0, 63.6, 51.5 (d, CH, 1JC,P = 164.5 Hz), 51.4, 30.5, 21.3, 16.5, 16.4; 31P NMR (CDCl3, 162 MHz) δ: 19.03; IR (KBr) ν: 3394, 3321, 2989, 1675, 1586, 1478, 1441, 1392, 1245, 1083, 1021, 974, 805, 777; ESI-MS m/z: 473.2 [M+H]+; HRMS (pos.): 473.1271 ([M+H]+, C21H27N2O5PH; calc. 473.1276).
Diethyl((1,5-dimethyl-1H-indol-2-yl)((4-methoxyphenyl)amino)methyl)phosphonate B8: Light yellow solid, Yield: 53%, m.p. 128–131 °C; 1H NMR (CDCl3, 400 MHz) δ: 7.35 (s, 1H, ArH), 7.22 (d, J = 8.4 Hz, 1H, ArH), 7.07–7.03 (m, 2H, ArH), 6.62 (d, J = 3.2 Hz, 1H, ArH), 6.32–6.27 (m, 2H, ArH), 6.24–6.23 (m, 1H, ArH), 5.02 (d, J = 23.6 Hz, 1H, PCH), 4.58 (t, J = 7.6 Hz, 1H, NH), 4.25–4.13 (m, 2H, CH2CH3), 4.08–3.98 (m, 1H, CH2CH3), 3.83 (s, 3H, NCH3), 3.81–3.73 (m, 4H, CH2CH3, OCH3), 2.44 (s, 3H, ArCH3), 1.33 (t, J = 7.1 Hz, 3H, CH2CH3), 1.14 (t, J = 7.1 Hz, 3H, CH2CH3); 13C NMR (CDCl3, 100 MHz) δ: 160.6, 147.6, 136.4, 134.2, 130.0, 128.8, 127.6, 123.3, 120.3, 108.8, 106.6, 103.8, 101.7, 100.0, 63.6, 63.5, 55.0, 49.7 (d, CH, 1JC,P = 159.6 Hz), 30.3, 21.3, 16.5, 16.3; 31P NMR (CDCl3, 162 MHz) δ: 20.94; IR (KBr) ν: 3291, 2977, 2927, 1610, 1599, 1499, 1445, 1236, 1165, 1045, 974, 845, 792, 756; ESI-MS m/z: 417.2 [M+H]+; HRMS (pos.): 417.1941 ([M+H]+, C22H29N2O4PH; calc. 417.1943).
Dimethyl ((1,5-dimethyl-1H-indol-2-yl)(phenylamino)methyl)phosphonate C1: Brown yellow solid, Yield: 76%, m.p. 140–143 °C; 1H NMR (CDCl3, 400 MHz) δ: 7.36 (s, 1H, ArH), 7.23 (d, J = 8.4 Hz, 1H, ArH), 7.18–7.14 (m, 2H, ArH), 7.06 (d, J = 8.4 Hz, 1H, ArH), 6.76 (t, J = 7.38 Hz, 1H, ArH), 6.69–6.65 (m, 3H, ArH), 5.07 (d, J = 23.0 Hz, 1H, PCH), 3.83–3.81 (m, 6H, NCH3, OCH3), 3.55 (d, J = 10.6 Hz, 3H, OCH3), 2.44 (s, 3H, ArCH3); 13C NMR (CDCl3, 100 MHz) δ: 146.0, 136.4, 133.9, 129.3, 128.9, 127.6, 123.5, 120.4, 119.0, 113.8, 108.9, 101.9, 54.2, 53.9, 49.4 (d, CH, 1JC,P = 159.2 Hz), 30.2, 21.3; 31P NMR (CDCl3, 162 MHz) δ: 23.41; IR (KBr) ν: 3293, 2949, 1603, 1498, 1314, 1242, 1056, 1027, 840, 754; ESI-MS m/z: 359.2 [M+H]+; HRMS (pos.): 359.1520 ([M+H]+, C19H23N2O3PH; calc. 359.1525).
Dimethyl ((1,5-dimethyl-1H-indol-2-yl)(o-tolylamino)methyl)phosphonate C2: Light yellow solid, Yield: 79%, m.p. 152–155 °C; 1H NMR (CDCl3, 400 MHz) δ: 7.35 (s, 1H, ArH), 7.23 (d, J = 8.4 Hz, 1H, ArH), 7.10–7.01 (m, 3H, ArH), 6.71 (t, J = 7.4 Hz, 1H, ArH), 6.63 (d, J = 3.2 Hz, 1H, ArH), 6.57 (d, J = 8.0 Hz, 1H, ArH), 5.10 (d, J = 22.9 Hz, 1H, PCH), 3.82 (m, 6H, NCH3, OCH3), 3.56 (d, J = 10.5 Hz, 3H, OCH3), 2.45 (s, 3H, ArCH3), 2.28 (s, 3H, ArCH3); 13C NMR (CDCl3, 100 MHz) δ: 144.1, 136.5, 134.2, 130.4, 129.0, 127.6, 127.2, 123.5, 123.2, 120.4, 118.7, 111.1, 108.9, 101.8, 54.2, 53.9, 49.5 (d, CH, 1JC,P = 158.1 Hz), 30.3, 22.0, 17.6; 31P NMR (CDCl3, 162 MHz) δ: 23.43; IR (KBr) ν: 3392, 2952, 2851, 1603, 1510, 1484, 1450, 1316, 1246, 1080, 1028, 870, 835, 749; ESI-MS m/z: 373.2 [M+H]+; HRMS (pos.): 373.1678 ([M+H]+, C20H25N2O3PH; calc. 373.1681).
Dimethyl ((1,5-dimethyl-1H-indol-2-yl)(m-tolylamino)methyl)phosphonate C3: Brown solid, Yield: 73%, m.p. 133–136 °C; 1H NMR (CDCl3, 400 MHz) δ: 7.36 (s, 1H, ArH), 7.23 (d, J = 8.4 Hz, 1H, ArH), 7.07–7.02 (m, 2H, ArH), 6.66 (d, J = 2.5 Hz, 1H, ArH), 6.59 (d, J = 7.4 Hz, 1H, ArH), 6.53 (s, 1H, ArH), 6.48 (d, J = 8.0 Hz, 1H, ArH), 5.06 (d, J = 23.0 Hz, 1H, PCH), 3.83–3.81 (m, 6H, NCH3, OCH3), 3.55 (d, J = 10.5 Hz, 3H, OCH3), 2.45 (s, 3H, ArCH3), 2.26 (s, 3H, ArCH3); 13C NMR (CDCl3, 100 MHz) δ: 146.1, 139.2, 136.5, 134.1, 129.2, 128.9, 127.6, 123.5, 120.4, 120.0, 114.8, 110.7, 108.9, 101.9, 54.2, 53.9, 49.4 (d, CH, 1JC,P = 158.6 Hz), 30.3, 21.6, 21.4; 31P NMR (CDCl3, 162 MHz) δ: 23.38; IR (KBr) ν: 3313, 2949, 2917, 1608, 1589, 1489, 1458, 1326, 1245, 1179, 1060, 1028, 834, 768; ESI-MS m/z: 373.2 [M+H]+; HRMS (pos.): 373.1679 ([M+H]+, C20H25N2O3PH; calc. 373.1681).
Dimethyl ((1,5-dimethyl-1H-indol-2-yl)(p-tolylamino)methyl)phosphonate C4: Light yellow solid, Yield: 80%, m.p. 131–133 °C; 1H NMR (CDCl3, 400 MHz) δ: 7.36 (s, 1H, ArH), 7.22 (d, J = 8.4 Hz, 1H, ArH), 7.06 (d, J = 8.4 Hz, 1H, ArH), 6.97 (d, J = 8.2 Hz, 2H, ArH), 6.65–6.60 (m, 3H, ArH), 5.04 (d, J = 23.0 Hz, 1H, PCH), 3.84–3.81 (m, 6H, NCH3, OCH3), 3.56 (d, J = 10.6 Hz, 3H, OCH3), 2.45 (s, 3H, ArCH3), 2.23 (s, 3H, ArCH3); 13C NMR (CDCl3, 100 MHz) δ: 143.7, 136.4, 134.1, 129.8, 128.9, 128.3, 127.6, 123.4, 120.4, 114.0, 108.8, 101.9, 54.2, 53.8, 49.8 (d, CH, 1JC,P = 161.17 Hz), 30.2, 21.3, 20.4; 31P NMR (CDCl3, 162 MHz) δ: 23.44; IR (KBr) ν: 3317, 2951, 2850, 1616, 1521, 1486, 1241, 1184, 1063, 1029, 834, 794, 755; ESI-MS m/z: 373.2 [M+H]+; HRMS (pos.): 373.1683 ([M+H]+, C20H25N2O3PH; calc. 373.1681).
Dimethyl(((4-chlorophenyl)amino)(1,5-dimethyl-1H-indol-2-yl)methyl)phosphonate C5: Light pink solid, Yield: 63%, m.p. 140–142 °C; 1H NMR (CDCl3, 400 MHz) δ: 7.36 (s, 1H, ArH), 7.23 (d, J = 8.4 Hz, 1H, ArH), 7.11–7.06 (m, 3H, ArH), 6.62–6.59 (m, 3H, ArH), 5.00 (d, J = 22.8 Hz, 1H, PCH), 4.60 (s, 1H, NH), 3.83–3.80 (m, 6H, NCH3, OCH3), 3.54 (d, J = 10.6 Hz, 3H, OCH3), 2.44 (s, 3H, ArCH3); 13C NMR (CDCl3, 100 MHz) δ: 144.6, 136.4, 133.3, 129.1, 127.5, 123.6, 120.4, 114.9, 108.9, 102.0, 54.1, 54.0, 49.5 (d, CH, 1JC,P = 159.2 Hz), 30.2, 21.3; 31P NMR (CDCl3, 162 MHz) δ: 22.99; IR (KBr) ν: 3280, 2952, 1598, 1517, 1491, 1320, 1241, 1055, 1027, 834, 793; ESI-MS m/z: 393.1 [M+H]+; HRMS (pos.): 393.1136 ([M+H]+, C19H22ClN2O3PH; calc. 393.1135).
Dimethyl((1,5-dimethyl-1H-indol-2-yl)((4-methoxyphenyl)amino)methyl)phosphornate C6: Yellow solid, Yield: 56%, m.p. 127–128 °C; 1H NMR (CDCl3, 400 MHz) δ: 7.36 (s, 1H, ArH), 7.22 (d, J = 8.4 Hz, 1H, ArH), 7.08–7.04 (m, 2H, ArH), 6.65 (d, J = 3.0 Hz, 1H, ArH), 6.33–6.27 (m, 2H, ArH), 6.25–6.24 (m, 1H, ArH), 5.04 (d, J = 23.0 Hz, 1H, PCH), 4.55 (s, 1H, NH), 3.84–3.81 (m, 6H, NCH3, POCH3), 3.74 (s, 3H, ArOCH3), 3.54 (d, J = 10.6 Hz, 3H, POCH3), 2.44 (s, 3H, ArCH3); 13C NMR (CDCl3, 100 MHz) δ: 147.4, 136.4, 133.8, 130.1, 128.9, 127.6, 123.5, 108.9, 106.6, 104.0, 101.9, 100.0, 55.0, 54.1, 53.9, 49.3 (d, CH, 1JC,P = 159.2 Hz), 30.2, 21.3; 31P NMR (CDCl3, 162 MHz) δ: 23.24; IR (KBr) ν: 3450, 3297, 2952, 1613, 1492, 1250, 1163, 1022, 837, 754; ESI-MS m/z: 389.2 [M+H]+; HRMS (pos.): 389.1623 ([M+H]+, C20H25N2O4PH; calc. 389.1630).
4.3 In vitro anti-proliferative assay
The in vitro cytotoxic activity of compounds A1-A8, B1-B8 and C1-C6 against HepG2 and MGC-803 was evaluated by the MTT method and the potency was expressed as inhibition rate. A 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was performed to investigate in vitro cytotoxic activity. HepG2 and MGC-803 seeded into the 96-well plate (100 μL each well) were incubated at 37 °C under 5% CO2 and 95% O2 until cell adherence was observed. Cells were exposed to solutions of compounds A1-A8, B1-B8 and C1-C6 at different concentrations. After 48 h, 20 mL of MTT with a concentration of 5 mg/mL was added into the 96-well plate and incubated for 4 h at 37 °C. Then, the supernatant was aspirated, 150 μL of DMSO was added to each well, and the absorbance was measured at 570 nm. The formula for calculating inhibition of cell proliferation was as follows: inhibition of cell proliferation rate (%) = (OD value of the control group - OD value of drug group)/ control OD value 100%.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21171149), Science and Technology Plan of Maoming (2019401, 2020581), Doctor Startup Project of Guangdong University of Petrochemical Technology (517152, 2019rc053), and Young Creative Talents Training Project of Guangdong University of Petrochemical Technology (517136).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Synthesis and antimicrobial activities of a novel series of heterocyclic α-aminophosphonates. Arch. Pharm.. 2012;345(10):784-789.
- [CrossRef] [Google Scholar]
- Synthesis and biological evaluation of some new polyfluorinated 4-thiazolidinone and α-aminophosphonic acid derivatives. Monatsh. Chem.. 2013;144:1243-1252.
- [CrossRef] [Google Scholar]
- An efficient synthesis of a hydroxyethylamine (HEA) isostere and its α-aminophosphonate and phosphoramidate derivatives as potential anti-HIV agents. ChemMedChem. 2012;7(9):1601-1611.
- [CrossRef] [Google Scholar]
- Diversity-oriented synthesis of α-aminophosphonates: a new class of potential anticancer agents. Eur. J. Med. Chem.. 2013;66:146-152.
- [CrossRef] [Google Scholar]
- A novel potent nicotinamide phosphoribosyltransferase inhibitor synthesized via click chemistry. J. Med. Chem.. 2010;53(2):616-623.
- [CrossRef] [Google Scholar]
- Synthesis and antiviral evaluation of trisubstituted indole N-nucleosides as analogues of 2,5,6-trichloro-1-(beta-D-ribofuranosyl)benzimidazole (TCRB) J. Med. Chem.. 2000;43(12):2449-2456.
- [CrossRef] [Google Scholar]
- Green synthesis of α-aminophosphonate derivatives on a solid supported tio2-sio2catalyst and their anticancer activity. Archive. Pharm Chem. Life Sci.. 2013;346(9):667-676.
- [Google Scholar]
- From nature to drug discovery: the indole scaffold as a 'privileged structure' Mini Rev. Med. Chem.. 2009;9(7):782-793.
- [CrossRef] [Google Scholar]
- CeCl3-7H2O-SiO2: catalyst promoted microwave assisted neat synthesis, antifungal and antioxidant activities of α-diaminophosphonates. Chin. Chem. Lett.. 2013;24:759-763.
- [CrossRef] [Google Scholar]
- Indole in the target-based design of anticancer agents: A versatile scaffold with diverse mechanisms. Eur. J. Med. Chem.. 2018;150:9-29.
- [CrossRef] [Google Scholar]
- Ferlay, J., Ervik, M., Lam, F., Colombet, M., Mery, L., Piñeros, M., et al., 2020. Global Cancer Observatory: Cancer Today. Lyon: International Agency for Research on Cancer; (https://gco.iarc.fr/today, accessed February 2021).
- The global cancer burden and human development: A review. Scand. J. Public Health.. 2018;46(1):27-36.
- [CrossRef] [Google Scholar]
- Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J. Med. Chem.. 2013;56(1):15-30.
- [CrossRef] [Google Scholar]
- Synthesis and antitumor activities of novel α-aminophosphonates dehydroabietic acid derivatives. Bioorg. Med. Chem. Lett.. 2013;23(19):5283-5289.
- [Google Scholar]
- Synthesis and antitumor activity of α-aminophosphonate derivatives containing thieno[2, 3-d]pyrimidines. Chin. Chem. Lett.. 2015;26:755-758.
- [CrossRef] [Google Scholar]
- An insight into the medicinal perspective of synthetic analogs of indole: A review. Eur. J. Med. Chem.. 2019;180:562-612.
- [CrossRef] [Google Scholar]
- Synthesis and antitumor activity of α-aminophosphonates containing thiazole[5,4-b]pyridine moiety. Org. Biomol. Chem.. 2012;10(35):7098-7102.
- [Google Scholar]
- Jiang, J., Kang, T. B., Shim, d., Oh, N. H., Kim, T. J., Lee, K. H., 2013. Indole-3-carbinol inhibits LPS-induced inflammatory response by blocking TRIF-dependent signaling pathway in macrophages. Food Chem. Toxicol. 57, 256–261. https://doi.org/10.1016/j.fct.2013.03.040.
- Aminophosphonic and aminophosphinic acids: Chemistry and biological activity. New York: Wiley; 2000.
- Design and synthesis of human immunodeficiency virus entry inhibitors: sulfonamide as an isostere for the alpha-ketoamide group. J. Med. Chem.. 2007;50(26):6535-6544.
- [CrossRef] [Google Scholar]
- Zinc-catalyzed alkyne oxidation/C-H functionalization: highly site-selective synthesis of versatile isoquinolones and β-carbolines. Angew. Chem. Int. Edit.. 2015;54:8245-8249.
- [CrossRef] [Google Scholar]
- Reversal of regioselectivity in catalytic arene-ynamide cyclization: direct synthesis of valuable azepino[4,5-b]indoles and β–carbolines and DFT calculations. ACS Catal.. 2017;7:4004-4010.
- [CrossRef] [Google Scholar]
- Synthesis and Biological Activities of α-Aminophosphonates Derivatives Containing Thieno[3,2-c]pyridine. Chin. J. Org. Chem.. 2013;33:1472-1477.
- [Google Scholar]
- Recent synthesis of aminophosphonic acids as potential biological importance. Amino Acids. 2010;38(1):23-30.
- [CrossRef] [Google Scholar]
- Structure elucidation of streptindole, a novel genotoxic metabolite isolated from intestinal bacteria. Tetrahedron Lett.. 1983;24:4719-4722.
- [CrossRef] [Google Scholar]
- Aminophosphonic acids and derivatives. Synthesis and biological applications. Curr. Med. Chem.. 2010;17(3):264-289.
- [CrossRef] [Google Scholar]
- Cancer Chemotherapy in Clinical Practice. London: Springer; 2012.
- Identification and characterization of novel indole based small molecules as anticancer agents through SIRT1 inhibition. Eur. J. Med. Chem.. 2013;69:125-138.
- [CrossRef] [Google Scholar]
- Kinase Inhibitor Indole Derivatives as Anticancer Agents: A Patent Review. Recent Pat. Anticancer Drug Discov.. 2017;12(1):55-72.
- [CrossRef] [Google Scholar]
- Design and one-pot synthesis of alpha-aminophosphonates and bis(alpha-aminophosphonates) by iron(III) chloride and cytotoxic activity. Eur. J. Med. Chem.. 2009;44(11):4266-4275.
- [CrossRef] [Google Scholar]
- PEG-SO3H catalyzed synthesis and cytotoxicity of α-aminophosphonates. Eur. J. Med. Chem.. 2012;47(1):553-559.
- [Google Scholar]
- Synthesis and antifungal activity of 1H-indole-4,7-diones. Bioorg. Med. Chem. Lett.. 2007;17(1):127-131.
- [CrossRef] [Google Scholar]
- Irreversible inhibition of serine proteases - design and in vivo activity of diaryl alpha-aminophosphonate derivatives. Curr. Med. Chem.. 2009;16(13):1673-1687.
- [Google Scholar]
- Microwave-assisted synthesis and anti-inflammatory activity evaluation of some novel α-aminophosphonates. Phosphorus Sulfur Silicon Relat. Elem.. 2017;192(10):1110-1113.
- [Google Scholar]
- Indole: A privileged scaffold for the design of anti-cancer agents. Eur. J. Med. Chem.. 2019;183(1):111691
- [CrossRef] [Google Scholar]
- Synthesis and antiviral bioactivities of alpha-aminophosphonates containing alkoxyethyl moieties. Molecules (Basel, Switzerland). 2006;11(9):666-676.
- [CrossRef] [Google Scholar]
- Inhibitors of HIV-1 attachment. Part 8: the effect of C7-heteroaryl substitution on the potency, and in vitro and in vivo profiles of indole-based inhibitors. Bioorg. Med. Chem. Lett.. 2013;23(1):203-208.
- [CrossRef] [Google Scholar]
- Solvent-free synthesis of pyrimidine nucleoside aminophosphonate hybrids and their biological activity evaluation. Nucleosides Nucleotides Nucleic Acids. 2010;29(8):616-627.
- [CrossRef] [Google Scholar]
- Zhu, X. F., Zhang, j., Sun, S., et al., 2017. Synthesis and structure-activity relationships study of α-aminophosphonate derivatives containing a quinoline moiety. Chin. Chem. Lett. 28, 1514-1518. http://dx.doi.org/10.1016/j.cclet.2017.02.012.
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2021.103256.
Appendix A
Supplementary material
The following are the Supplementary data to this article:




