

Translate this page into:
The study of the reaction paths of 4-aryl-4-oxobutanoic acids with terminal aliphatic N,N-diamines
⁎Corresponding author at: Institute of Chemistry, Chernyshevsky Saratov State University, Ulitsa Astrakhanskaya, 83, 410012, Russia. grinev@ibppm.ru (Vyacheslav S. Grinev)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Bicyclic pyrroloimidazolones and pyrrolopyrimidinones were synthesized by reacting aryl-substituted 4-oxobutanoic acids with aliphatic binucleophiles with different alkyl chain lengths using a sealed vessels reactor (SVR). By varying the synthesis conditions, the reaction path was studied and formed intermediates were isolated. The structures of the obtained compounds were proved using a set of FTIR, 1H, 13C NMR spectroscopy methods and X-ray diffraction data. Temperature and pressure changes data collected during the syntheses were analyzed and assigned to the processes which occurred inside the vials. It was shown that the reaction of 4-aryl-4-oxobutanoic acids with terminal aliphatic N,N-diamines starts from the formation of corresponded (AlkN2)2+An2- salts followed by two subsequent dehydration processes giving rise to the corresponding amides and, finally, heterocycles of pyrroloimidazolone and pyrrolopyrimidinone series.
Keywords
Synthesis
Pyrroloimidazolones
Pyrrolopyrimidinones
Intermediates
Process scheme
Reaction path

1 Introduction
The construction of heterocyclic systems containing a pyrrolo[1,2-a]imidazolone fragment or its analogs is an important step towards the creation of biologically active compounds with anti-inflammatory, antinociceptive, immunomodulatory and antioxidant (Ibrar et al., 2016), anticonvulsant effects (Lepri et al., 2010), antagonistic activity against melanocortin-4 receptor (MC4R) (Lee and Carpino, 2015). The pyrroloimidazolones and pyrrolopyrimidinones demonstrate the plant growth-regulating activity as well (Grinev et al., 2011). Bicyclic imidazolones are important intermediates in both synthetic and pharmaceutical chemistry. These compounds are promising building blocks for the creation of nitrogen-containing heterocycles, which constitute a huge class of biologically active compounds (Katritzky et al., 2000; Chimirri et al., 2001).
Modern and efficient methods of synthesis have been proposed, including flash vacuum pyrolysis (FVP), syntheses in the presence of basic catalysts, microwave (MW) activation. Previously, the methods used to construct pyrrolo[1,2-a]imidazolones were mainly variants of the classical methods of condensation of series of ketones or acids with aliphatic diamines under thermal heating conditions in various solvents. The reaction of maleic anhydride with cyclic N,N-acetals underlies the synthesis of 2- (7-aroyl-5-oxo-2,3,5,6-tetrahydro-1H-pyrrolo[1,2-a]imidazol-6-yl)acetic acid (Orlov et al., 2012). The interaction of heterocyclic ketene aminals with ethyl bromoacetate leads to the addition products - ethyl 3-aroyl-3-(2-imidazolidinylidene)propionates, which undergo further cyclocondensation with the formation of 7-aroyl-1,2,3,6-tetrahydro-5H-pyrrolo[1, 2-a]imidazol-1-ones (Liu et al., 2014). The use of the FVP technique (temperatures of about 600–800 °C, pressure in 0.01–0.05 Torr range) allows intramolecular condensations, as a result of which imidazole derivatives generate methylene ketenes followed by their rearrangements into more stable unsaturated ketenes that form the final products (McNab, 2004). In the case of methylimidazolyl acrylates, it is formally considered that the process goes through an antarasurface transition state with a subsequent 1,7-prototropic shift (McNab and Thornley, 1997; McNab and Tyas, 2007). Levulinic acid ethyl ester and 1,2-ethylenediamine on an Al2O3 as a substrate were introduced into the reaction under MW radiation conditions. The exposure time was varied within 6–28 min. range, and the radiation power was in 125–440 W range (Csende and Stájer, 2000).
However, classical methods of synthesis under heating conditions in various solvents at the atmospheric pressure do not lose their relevance. The optimal conditions for the synthesis of a wide range of substituted bicyclic azoles based on 4-R-4-oxobutanoic acids or 5-R-furan-2(3H)-ones, which are their cyclic esters, and aliphatic binucleophiles is boiling in benzene or toluene with continuous removing water from the reaction mixture in the form of an azeotrope (Amal'chieva and Egorova, 2006; Grinev et al., 2010) (Scheme 1). Substituted 1,2-diamines with maleic anhydrides form benzopyrroloimidazolones through the intermediate stage of N-maleimides (Dawood et al., 2011).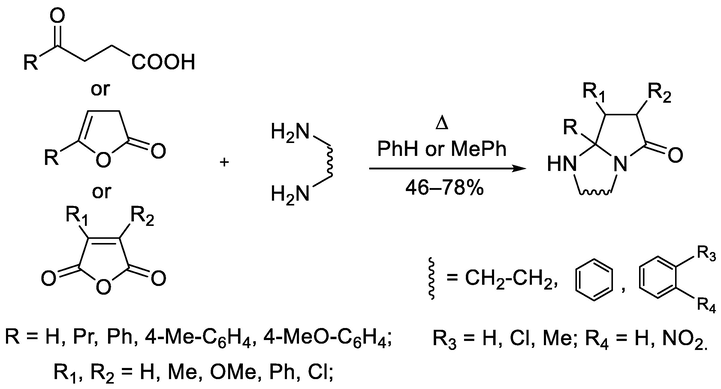
General scheme of syntheses of (benzo)pyrrolodiazabicycloalkanones under thermal heating conditions.
The most convenient and unambiguous method for preparing pyrrolodiazacycloalkanones is based on the use of 5-arylfuran-2(3H)-ones as synthons, the latter being internal esters of 4-oxobutanoic acids. The reaction scheme in this case is unambiguous, the reaction is carried out with the opening of the furanone ring, the formation of intermediate acid amides, and the subsequent heterocyclization leads to the formation of pyrroloimidazolones. However, the authors did not provide any evidence confirming proposed scheme.
It was shown that the use of 4-aryl-4-oxoalkanoic acids or 5-arylfuran-2(3H)-ones in the reaction with binucleophiles leads to the isolation of the same products. The use of 4-aryl-4-oxoalkanoic acids in the reaction with binucleophiles is promising, since it makes it possible to exclude one stage of the process as well as the isolation of intermediate furan-2(3H)-ones.
However, the presence of two unequal electrophilic centers in the structure of acids requires an explanation of the reaction mechanism. Previously, the most probable pathway of the reaction was considered through the formation of an γ-iminobutanoic acid, as a result of the attack of the nucleophile on the carbon atom of the oxo group of the acid, which has the greatest electron density deficit, followed by the formation of a heterocycle, the intramolecular acylation of which leads to the formation of reaction products (Grinev et al., 2010). The formation of final products is also possible with the initial formation of acid amides, as a consequence of the attack of the nucleophile on the carboxyl group, followed by additional heterocyclization.
Thus, the aim of this work is to establish the pathway for the reaction of 4-aryl-4-oxobutanoic acids with terminal aliphatic N,N-diamines by isolating individually the putative intermediates of this type of interaction and analyzing their structures using a set of spectral and XRD methods.
2 Materials and methods
2.1 General analytic techniques
All chemicals purchased from Sigma Aldrich (USA) were of reagent grade and were used without further purification. Analytical TLC was performed using Alugram Sil G UV254 plates (Macherey–Nagel GmbH & Co. KG, Germany) plates (hexane–ethyl acetate–chloroform, 2 : 2 : 1; development with iodine vapor). Syntheses were performed in the sealed vessels reactor (SVR) Monowave 50 (Anton Paar, Austria) in a borosilicate glass vial with magnetic PTFE coated stirrer sealed with a PTFE septum and a silicone cap. The vials were placed in the SVR and heated. Melting points were determined on a Stuart™ SMP10 melting point apparatus (Cole-Parmer, UK) in open capillaries. The elemental analyses were obtained on a Vario Micro cube Elementar CHNS analyzer (Elementar Analysensysteme GmbH, Germany). FTIR spectra were recorded as KBr pellets on a Nicolet 6700 FTIR spectrophotometer (Thermo Scientific, USA) in the 4000–400 cm−1 range with a spectral resolution of 4 cm−1. The 1H and 13C NMR spectra were recorded at rt on a Varian-400 (Agilent Technologies, USA) spectrometer (400 and 100 MHz, respectively) using CDCl3 or D2O as solvents and tetramethylsilane (TMS) or sodium trimethylsilylpropanesulfonate (DSS) as internal standards. X-ray diffraction of hexane-1,6-diaminium di{4-oxo-4-(p-tolyl)butanoate} 5b was performed on a four-circle diffractometer Xcalibur Ruby (Agilent Technologies, USA) with graphite monochromator [λ(MoKα) = 0.71073 Å, ω scan] at 295 K. The empirical absorption correction was introduced by multi-scan method using SCALE3 ABSPACK algorithm (CrysAlisPro, 2014), Agilent Technologies, USA. Using the Olex2 (ver. 1.2.8) (Dolomanov et al., 2009), the structure was solved with the SHELXS program (Sheldrick, 2008) and refined by the full-matrix least-squares method in the anisotropic approximation for all non-hydrogen atoms with the SHELXL program (https://www.shelxle.org/shelx/eingabe.php). Hydrogen atoms were located from the Fourier synthesis of the electron density and refined using a riding model.
2.2 Synthesis of 3–5
Appropriate amounts of 4-aryl-4-oxobutanoic acid (1a,b) and aliphatic binucleophile (1,2-diaminoethane 2a or 1,3-diaminopropane 2b or 1,6-diaminohexane 2c) in a molar ratio of 1 : 1.2 (with the excess of corresponding diamine), 2 mL of absolute benzene were placed in the SVR and heated at 50 °C for 5 min. The formed precipitate was separated, washed with benzene, and dried in vacuo.
2.2.1 Ethane-1,2-diaminium di{4-oxo-4-phenylbutanoate} (3a)
Yield: 85%, white powder, m.p.: 164–165 °C; FTIR (KBr), ν, cm−1: 3430 (NH), 2854, 2793, 2693, 2626, 2572 (NH3+), 1725 (C⚌O), 1604, 1408 (COO−); 1H NMR (400 MHz, D2O): δ 2.81 (t, 4H, 2 × CH2, J = 6.3 Hz), 3.10 (s, 4H, CH2-CH2, diam.), 3.27 (t, 4H, 2 × CH2, J = 6.3 Hz), 7.55–7.99 (m, 10H, Ar). 13C NMR (100 MHz, D2O): δ 34.25, 37.76, 39.84, 40.13, 130.79, 131.47, 136.36, 139.15, 185.15 (C⚌O), 194.45 (C⚌O). Anal. calcd. for C22H28O6N2, %: C 63.45; H 6.78; N 6.73; found, %: C 63.74; H 6.55; N 6.48.
2.2.2 Ethane-1,2-diaminium di{4-oxo-4-(p-tolyl)butanoate} (3b)
Yield: 75%, white powder, m.p.: 167–168 °C. FTIR (KBr), ν, cm−1: 3433 (NH), 2856, 2792, 2694, 2625, 2572 (NH3+), 1723 (C⚌O), 1608, 1410 (COO−). 1H NMR (400 MHz, D2O): δ 2.41 (s, 6H, Ar), 2.82 (t, 4H, 2 × CH2, J = 6.3 Hz), 3.08 (s, 4H, CH2-CH2, diam.), 3.33 (t, 4H, 2 × CH2, J = 6.3 Hz), 7.38–7.91 (m, 8H, Ar). 13C NMR (100 MHz, D2O): δ 23.36, 34.35, 37.63, 41.41, 42.03, 131.01, 132.92, 136.09, 148.05, 185.15 (C⚌O), 194.45 (C⚌O). Anal. calcd. for C24H32O6N2, %: C 64.86; H 7.21; N 6.31; found, %: C 64.69; H 7.18; N 6.02.
2.2.3 Propane-1,3-diaminium di{4-oxo-4-phenylbutanoate} (4a)
Yield: 85%, white powder, m.p.: 170–171 °C. FTIR (KBr), ν, cm−1: 3433 (NH), 2854, 2792, 2694, 2624, 2571 (NH3+), 1724 (C⚌O), 1598, 1403 (COO−). 1H NMR (400 MHz, D2O): δ 2.06 (p, 4H, 2 × CH2, diam., J = 7.9 Hz), 2.81 (t, 4H, 2 × CH2, J = 6.7 Hz), 3.01 (t, 2H, CH2, diam., J = 8.3 Hz), 3.30 (t, 4H, 2 × CH2, J = 6.7 Hz), 7.55–7.99 (m, 10H, Ar). 13C NMR (100 MHz, D2O): δ 29.77, 30.54, 39.64, 39.76, 40.23, 130.79, 131.47, 136.50, 139.06, 184.29 (C⚌O), 207.07 (C⚌O). Anal. calcd. for C23H30O6N2, %: C 64.19; H 6.98; N 6.51; found, %: C 63.95; H 6.79; N 6.33.
2.2.4 Propane-1,3-diaminium di{4-oxo-4-(p-tolyl)butanoate} (4b)
Yield: 70%, white powder, m.p.: 172–173 °C. FTIR (KBr), ν, cm−1: 3432 (NH), 2853, 2793, 2695, 2626, 2573 (NH3+), 1722 (C⚌O), 1603, 1410 (COO−). 1H NMR (400 MHz, D2O): δ 2.28 (s, 6H, Ar), 2.08 (p, 4H, 2 × CH2, diam., J = 7.9 Hz), 2.82 (t, 4H, 2 × CH2, J = 6.7 Hz), 3.09 (t, 4H, CH2, diam., J = 8.3 Hz), 3.32 (t, 4H, 2 × CH2, J = 6.7 Hz), 7.41–7.89 (m, 8H, Ar). 13C NMR (100 MHz, D2O): δ 21.85, 34.25, 35.85, 39.89, 62.01, 130.35, 130.95, 136.74, 139.03, 177.93 (C⚌O), 197.89 (C⚌O). Anal. calcd. for C25H34O6N2, %: C 65.50; H 7.40; N 6.11; found, %: C 65.21; H 7.11; N 6.31.
2.2.5 Hexane-1,6-diaminium di{4-oxo-4-(p-tolyl)butanoate} (5b)
Yield: 76%, white powder, m.p.: 187–188 °C. FTIR (KBr), ν, cm−1: 3435 (NH), 2853, 2795, 2694, 2625, 2571 (NH3+), 1728 (C⚌O), 1609 (COO−). 1H NMR (400 MHz, D2O): δ 1.19–1.26 (m, 4H, 2 × CH2), 1.46–1.56 (m, 4H, 2 × CH2), 2.42 (s, 6H, Ar), 2.82 (t, 4H, 2 × CH2, J = 6.7 Hz), 2.89–2.92 (m, 4H, 2 × CH2), 3.32 (t, 4H, 2 × CH2, J = 6.7 Hz), 7.34–7.88 (m, 8H, Ar). 13C NMR (100 MHz, D2O): δ 21.13, 27.85, 31.63, 32.59, 38.45, 40.65, 41.33, 127.89, 129.25, 136.43, 144.09, 177.36, 177.58 (C⚌O), 198.04 (C⚌O). Anal. calcd. for C28H40O6N2, %: C 67.20; H 8.00; N 5.60; found, %: C 67.53; H 7.98; N 5.43.
2.3 Synthesis of 6, 7
The method described above for compounds 3–5 was used. The reactions were carried out for 5 min. and with temperatures in 80–100 °C range.
2.3.1 N-(2-Aminoethyl)-4-oxo-4-phenylbutanamide (6a)
Yield: 80%, yellow powder, m.p.: 170–172 °C. FTIR (KBr), ν, cm−1: 3250 (NH), 1689 (C⚌O), 1640 (N—C⚌O, amide I). 1H NMR (400 MHz, CDCl3): δ 2.84 (t, J = 6.4 Hz, 2H), 3.38 (t, 2H, CH2, J = 5.9 Hz), 3.48 (t, J = 6.4 Hz, 2H), 3.60 (t, 2H, CH2, J = 5.9 Hz), 7.34–7.79 (m, 5H, Ar). 13C NMR (100 MHz, CDCl3): δ 34.99, 36.53, 40.96, 41.84, 127.57, 127.94, 132.93, 136.93, 173.83 (C⚌O), 198.53 (C⚌O). Anal. calcd. for C12H16O2N2, %: C 65.43; H 7.32; N 12.72; found, %: C 65.21; H 7.11; N 12.31.
2.3.2 N-(2-Aminoethyl)-4-oxo-4-(p-tolyl)butanamide (6b)
Yield: 85%, yellow powder, m.p.: 174–175 °C. FTIR (KBr), ν, cm−1: 3240 (NH), 1700 (C⚌O), 1635 (N—C⚌O, amide I). 1H NMR (400 MHz, CDCl3): δ 2.35 (s, 3H, Ar), 2.82 (t, J = 6.5 Hz, 2H), 3.36 (t, 2H, CH2, J = 5.9 Hz), 3.49 (t, J = 6.5 Hz, 2H), 3.60 (t, 2H, CH2, J = 5.9 Hz), 7.41–7.80 (m, 4H, Ar). 13C NMR (100 MHz, CDCl3): δ 20.96, 34.87, 37.01, 40.97, 41.85, 128.01, 129.45, 137.01, 143.98, 175.72 (C⚌O), 200.01 (C⚌O). Anal. calcd. for C13H18O2N2, %: C 66.64; H 7.74; N 11.96; found, %: C 66.21; H 7.43; N 11.73.
2.3.3 N-(3-Aminopropyl)-4-oxo-4-phenylbutanamide (7a)
Yield: 76%, yellow powder, m.p.: 178–179 °C. FTIR (KBr), ν, cm−1: 3302 (NH), 1697 (C⚌O), 1632 (N—C⚌O, amide I). 1H NMR (400 MHz, CDCl3): δ 2.76 (t, J = 6.5 Hz, 2H), 3.40 (t, 2H, CH2, J = 5.9 Hz), 3.52 (t, J = 6.5 Hz, 2H), 3.58 (t, 2H, CH2, J = 5.9 Hz), 7.44–7.85(m, 5H, Ar). 13C NMR (100 MHz, CDCl3): δ 30.74, 34.99, 37.00, 37.86, 39.74, 127.76, 127.98, 133.12, 137.45, 174.72 (C⚌O), 199.05 (C⚌ O). Anal. calcd. for C13H18O2N2, %: C 66.64; H 7.74; N 11.96; found, %: C 66.31; H 7.95; N 11.58.
2.3.4 N-(3-Aminopropyl)-4-oxo-4-(p-tolyl)butanamide (7b)
Yield: 80%, yellow powder, m.p.: 182–183 °C. FTIR (KBr), ν, cm−1: 3312 (NH), 1698 (C⚌O), 1630 (N—C⚌O, amide I). 1H NMR (400 MHz, CDCl3): δ 2.35 (s, 3H, Ar), 2.84 (t, J = 6.2 Hz, 2H), 3.37 (t, 2H, CH2, J = 5.9 Hz), 3.47 (t, J = 6.2 Hz, 2H), 3.58 (t, 2H, CH2, J = 5.9 Hz), 7.50–7.90 (m, 4H, Ar). 13C NMR (100 MHz, CDCl3): δ 20.97, 31.23, 34.98, 36.95, 37.88, 38.96, 128.15, 129.33, 136.54, 143.87, 175.72 (C⚌O), 198.89 (C⚌O). Anal. calcd. for C14H20O2N2, %: C 67.72; H 8.12; N 11.28; found, %: C 67.34; H 8.40; N 11.44.
2.4 Synthesis of 8, 9
The method described above for compounds 3–7 was used. The reactions were carried out for 5 min. and with temperature of 160 °C. Physicochemical characteristics of synthesized compounds 8, 9 were in harmony with previously reported (Amal'chieva and Egorova, 2006; Grinev et al., 2020a,b).
Yields of all synthesized compounds as well as reaction parameters are given in Table S1.
3 Results and discussion
Firstly, in order to optimize the method a series of syntheses were carried out in the SVR on the basis of the interaction of 4-phenyl- and 4-(p-tolyl)-4-oxobutanoic acid (1a,b) with N,N-binucleophiles (1,2-diaminoethane 2a, 1,3-diaminopropane 2b, 1,6-diaminohexane 2c) in an aprotic solvent benzene (Scheme 2). Optimal conditions to obtain heterocycles smoothly in up to 85% yield were as follows: 5 min. at 130 °C. Physicochemical and spectral characteristics of the synthesized substances obtained in the SVR coincide with the corresponding characteristics of previously synthesized at atmospheric pressure (Grinev et al., 2020a,b).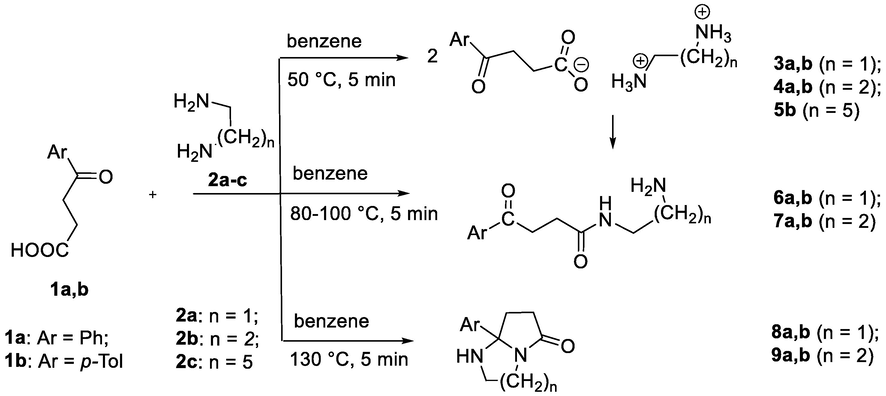
General scheme of syntheses based on 4-aryl-4-oxobutanoic acids and aliphatic N,N-binucleophiles.
In continuation of the study to establish the mechanism of the formation of bicyclic systems 8, 9, a series of reactions of 4-aryl-4-oxobutanoic acids (1a,b) with alkyl terminal N,N-binucleophiles (2a-c) in dry benzene. Varying reaction conditions, we were able to isolate intermediate interaction products – amides, and, especially, more labile salts, which weren’t previously describe in literature, probably due to their reactivity. Alkane diaminium di{4-oxo-4-arylbutanoate}s 3–5 were synthesized in yield up to 85% under mild conditions, at 50 °C for 5 min. Under such conditions, only the transfer of the protons from 4-oxobutanoic acids to amino groups of diamines occurs with the formation of the corresponding salts, which is not observed at room temperature. Spectral characteristics and crystal structures of salts were also unknown up to time. In FTIR spectra, there were a characteristic series of bands at 2853–2573 cm−1 assigned to the vibrations of NH3+ terminal groups of the doubly charged cation as well as bands assigned to the stretching vibrations of the carbonyl groups at 1728–1722 cm−1 along with antisymmeterical and symmetrical vibrations of the dissociated carboxyl groups at around 1600 and 1410 cm−1, respectively. In the 1H NMR spectra there were triplets of two methylene groups of the 4-oxobytanoic acids anions as well as multiplets of methylene units in the diaminium cations. In the 13C spectra there are two characteristic signals of carbons of carbonyl groups near 195 ppm as well as COO− at 185 ppm.
Hexane-1,6-diaminium di{4-oxo-4-(p-tolyl)butanoate} (5b) was investigated also in the crystal state by X-ray diffraction analysis (Fig. 1).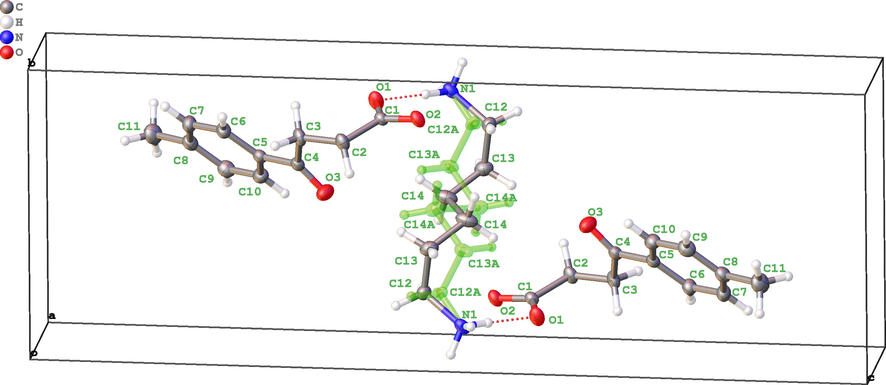
Crystal structure of hexane-1,6-diaminium di{4-oxo-4-(p-tolyl)butanoate} 5b. A fragment with a contribution of 27.5% is highlighted in green.
The XRD revealed the presence of two anions of 4-oxo-4-(p-tolyl)butanoic acid binding to double positively charged alkyl diamminium cation via hydrogen bonds. The alkyl chain of the diammonium fragment is disordered with contributions of 27.5% and 72.5% due to its high conformational mobility. The asymmetric unit includes one acid anion and a “half” of the alkyl diaminium cation, which corresponds to (AlkN2)2+An2- chemical composition.
The alkane diaminium di{4-oxo-4-arylbutanoate}s 3–5 are hygroscopic and unstable during the storage and spontaneously turn to corresponding amides. Probably, absorbed traces of water from air increase interaction between cations and anions. Obviously, the spatial proximity of the carboxylic acid anion and one of the alkane diaminium cations in crystal persists also in aqueous solution due to strong Coulomb interactions. This proximity largely predetermines the reactivity of the salts and the further course of the reaction of the formation of the corresponding acid amide by the elimination of the water molecule catalyzing further reaction.
To isolate amides in good yields, we have conducted syntheses in dry benzene starting from 4-oxobutanoic acids 1 and binucleophiles 2 at higher than for salts syntheses temperatures in 80–100 °C range. Dry benzene, the same liquid, acts on the one hand as a good solvent for synthons 1 and 2, on the other hand, it is a good water absorber. The walls of the vials act, thereby, as a condenser for benzene-water azeotrope.
The FTIR spectra clearly indicates the presence of two different carbonyl groups at 1690 and 1635 cm−1. Also, bands assigned to the antisymmetrical and symmetrical stretching vibrations of free secondary amino group at around 3310 and 3000 cm−1 and broadened band of stretching vibrations of amide NH group centered at 3425 cm−1 were found as well. In the 1H NMR spectra there were triplets of two methylene groups of the 4-oxobytanoic acids moieties along with multiplets of methylene units from the diamines. In the 13C spectra remain characteristic signals of carbons of carbonyl groups in 195–200 ppm range, but the signals of amide carbons are downfield shifted of about 10 ppm and are in 174–176 ppm range indicating the formation of amides 6, 7.
The use of the SVR allows us to monitor the course of each reaction and to analyze the conditions inside the vials (Fig. 2). Under mild conditions, during the syntheses of salts 3–5 the temperature curve rises smoothly up to about 60 °C that indicated slightly exothermic process with no pressure increment due to higher boiling point of the benzene and absence of any volatile reaction products. Formed salts 3–5 precipitate and the excess of diamine remains dissolved in benzene that does not create an excessive pressure. Syntheses of amides 6, 7 accompanies more significant enthalpy changes.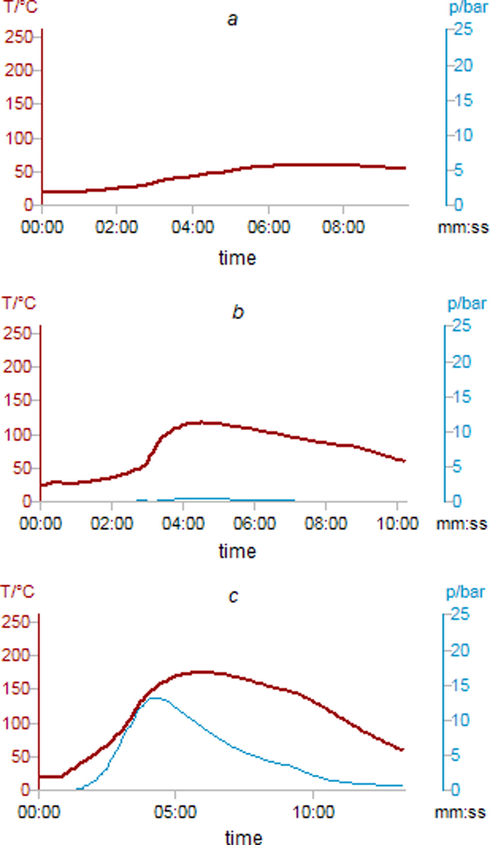
Temperature (red) and pressure (blue) inside the vial during the reactions, according to SVR built-in sensors. Syntheses of: (a) salts 3–5; (b) amides 6, 7; (c) heterocycles 8, 9.
The temperature curve rises smoothly up to 50 °C that correspond to dissolving process followed by a dramatical increase significantly above the initial setting clearly evidencing the exothermic reaction with the formation of an amide. The pressure curve reveals similar profile demonstrating firstly the boiling of benzene followed by the formation of gaseous water-benzene azeotrope during the reaction run. In contrast to salts 3–5, formed amides 6, 7 precipitate only after cooling the vial to the rt.
The use of a temperature more than boiling point of benzene (130 °C vs 80.1 °C) allows us to intense the interaction due to the formation of gaseous solvent that clearly indicated by significant rise of the pressure up to about 12 bars. Initially, the system consumes heat to turn the solvent into gaseous phase as well as for the activation of diamine 2a-c for nucleophilic attack. Moreover, heat is required for the overcoming conformational hindrances to make close a free terminal amino group to the electrophilic center with the closure of bicycles 8, 9 and with the elimination in total of two water molecules which successfully removed as an azeotrope with benzene. In contrast to classical method of synthesis under atmospheric pressure, the reaction completes in few minutes, according to the Arrhenius’ law.
4 Conclusion
Using the SVR, we were able to study the reactions between 4-aryl-4-oxobutanoic acids with aliphatic terminal N,N-binucleophiles under various conditions and to isolate intermediate (in syntheses of the bicycles) compounds, previously suggested speculatively. The reaction starts with the acid-base interaction with the formation of corresponding alkane diaminium di{4-oxo-4-arylbutanoate}s and proceeds further with the formation of the corresponding amides which undergoes a heterocyclization to bicycles of pyrroloimidazolone and pyrrolopyrimidinone series. One of the representatives of salts was studied in the crystal for the first time. Temperature and pressure changes data collected during the syntheses give us insight the processes occurred. It was found that under mild conditions at the temperatures lower or close to boiling point of the solvent benzene there is almost no pressure changes and the exothermic effects of reactions were moderate. Conducting syntheses under higher than bp of the solvent leads to a significant exothermic effect and an increase in pressure in the system, which increases the efficiency of synthesis.
Funding
Synthetic part of the study was supported by a grant from the Russian Foundation for Basic Research (Project 19-33-90157). Structural part of the study was supported by a grant from the Russian Foundation for Basic Research (Project 19-33-60038).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Reaction of furan-2(3H)-ones with 1,2-binucleophiles. Russ. J. Org. Chem.. 2006;42(9):1340-1343.
- [CrossRef] [Google Scholar]
- Synthesis and anticonvulsant properties of 2,3,3a,4-tetrahydro-1H-pyrrolo[1,2-a]benzimidazol-1-one derivatives. Il Farmaco.. 2001;56:821-826.
- [CrossRef] [Google Scholar]
- 4- and 5-Oxocarboxylic acids as versatile synthons for the preparation of heterocycles. Heterocycles. 2000;53:1379-1419.
- [CrossRef] [Google Scholar]
- Recent advances on the synthesis of azoles, azines and azepines fused to benzimidazole. ARKIVOC. 2011;i:111-195.
- [CrossRef] [Google Scholar]
- OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Cryst.. 2009;42:339-341.
- [CrossRef] [Google Scholar]
- Reaction of 4-oxocarboxylic acids and 5-substituted 3h-furan-2-ones with 1,2-binucleophiles of aromatic and alicyclic series. Russ. J. Org. Chem.. 2010;46(9):1378-1382.
- [CrossRef] [Google Scholar]
- Effect of benzo(2,3-b)-1,4-diaza- and benzo-1-aza-4-oxabicyclo[3.3.0]octane -8-ones on the regulation of wheat growth Triticum aestivum L. Agrochemistry.. 2011;3:46-50. (in Russian)
- [Google Scholar]
- Synthesis and conformational features of perhydropyrrolodiazacycloalkanones. . Izv. Saratov Univ. (N. S.), Ser. Chem. Biol. Ecol.. 2020;20(2):122-130.
- [CrossRef] [Google Scholar]
- Crystal structures, packing features, Hirschfeld surface analysis and DFT calculations of the hydrogen bond energy of two homologous 8a-aryl-2,3,4,7,8,8-hexahydropyrrolo[1,2-a]pyrimidin-6(1H)-ones. Acta Cryst. Sect. C.. 2020;C76:483-489.
- [CrossRef] [Google Scholar]
- Transition-metal-free synthesis of oxazoles: valuable structural fragments in drug discovery. RSC Adv.. 2016;6:93016-93047.
- [CrossRef] [Google Scholar]
- Novel syntheses of hexahydro-1H-pyrrolo[1,2-a]imidazoles and octahydroimidazo[1,2-a]pyridines. J. Org. Chem.. 2000;65(12):3683-3689.
- [CrossRef] [Google Scholar]
- Melanocortin-4 receptormodulators for the treatment of obesity: a patent analysis (2008–2014) Pharm. Pat. Anal.. 2015;4(2):95-107.
- [CrossRef] [Google Scholar]
- substituent effects on retention and chiral resolution of ketones and alcohols on microcrystalline cellulose triacetate plates. Chromatographia. 2010;71(7/8):685-694.
- [CrossRef] [Google Scholar]
- Catalyst-free cascade reaction of heterocyclic ketene aminals with N-substituted maleimide to synthesise bicyclic pyrrolidinone derivatives. RSC Adv.. 2014;4:27582-27590.
- [CrossRef] [Google Scholar]
- Chemistry without reagents: synthetic applications of flash vacuum pyrolysis. Aldrichimica Acta. 2004;37(1):19-26.
- [Google Scholar]
- New synthetic routes to pyrrolo-[1,2-a]- and -[1,2-c]-imidazol-5-ones by flash vacuum pyrolysis. J. Chem. Soc., Perkin Trans.. 1997;1:2203-2210.
- [CrossRef] [Google Scholar]
- A thermal cascade route to pyrroloisoindolone and pyrroloimidazolones. J. Org. Chem.. 2007;72:8760-8769.
- [CrossRef] [Google Scholar]
- Reactions of ketenaminals with N-arylmaleimides and dimethylacethylenedicarboxylate - a direct path to the derivatives of pyrrolo[1,2-a]imidazole and imidazo[1,2-a]pyridine. Chem. Heterocycl. Compd.. 2012;48(8):1204-1212.
- [Google Scholar]
- CrysAlisPro, Version 1.171.37.33 (release 27-03-2014 CrysAlis171 .NET) Agilent Technologies (USA) 2014.
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2021.103350.
Appendix A
Supplementary material
The following are the Supplementary data to this article:




