

Translate this page into:
Total synthesis and anticancer activity of a cyclic heptapeptide from marine sponge using water soluble peptide coupling agent EDC
⁎Corresponding author. Tel./fax: +91 1462 224500. vselva@aucev.edu.in (Vaithialingam Selvaraj) rajselva_77@yahoo.co.in (Vaithialingam Selvaraj)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Present investigation comprises of synthesis of a novel proline containing cyclic heptapeptide – Euryjanicin A, which was previously isolated from the marine sponge – Prosuberites laughlini. Naturally isolated cyclic heptapeptide was synthesized using solution phase peptide technique. All the coupling reactions were performed at room temperature via 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) – coupling reagent and triethylamine (Et3N) – base. The chemical structure of the finally synthesized cyclic heptapeptide was elucidated using FTIR, 1H & 13C NMR and mass spectral data, as well as elemental analyses. The newly synthesized peptide was subjected to anticancer activity against HeLa (Human Colon) cancer cell line and showed potent anticancer activity.
Keywords
Marine natural product
Euryjanicin A – cyclic heptapeptide
Solution phase synthesis
Anticancer activity

1 Introduction
Naturally isolated cyclic peptides have become increasingly important because of their wide range of pharmacological activities and interesting chemical structures (He et al., 1993; Wipf, 1995). A variety of active anticancer agents are derived from marine sponges, plants and terrestrial microorganisms (Blunt et al., 2009; Zheng et al., 2011).
Extensive literature survey has been done on anticancer cyclic peptides isolated from marine sponges. Examples of such biologically active peptides are Axinellins (Keyzers and Davies-Coleman, 2005), Kapakahines (Fusetani and Matsunaga, 1993), Microsclerodermins (Randazzo et al., 1998), Polytheonamides (Nakao et al., 1995), Papuamides (Yeung et al., 1996), Stylisins (Qureshi et al., 2000), Hymenamides (Hamada et al., 2005), Wainunuamide (Ford et al., 1999), dominican (Mohammed et al., 2006), etc.
A new proline containing cyclic heptapeptide, Euryjanicin A (8) has been isolated from the marine sponge Prosuberites laughlini indigenous to Puerto Rico, which showed potent cytotoxic activity against different cancer cell lines (Kobayashi et al., 1993; Tsuda et al., 1993, 1994). Indeed there is no systematic evidence on the naturally isolated cyclic peptide – Euryjanicin A. In this context, an attempt has been made to synthesize Euryjanicin A-cyclic heptapeptide. The molecule 1) Cyclo-L-[Phe-Ser-Ile-Pro-Try-Pro-Leu] was synthesized via solution phase technique of peptide synthesis using 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) as the coupling agent and triethylamine (Et3N) as the base. The synthesized cyclic heptapeptide was subjected to their anticancer activity against HeLa cancer cell lines using MTT assay.
2 Experimental
2.1 Materials
All the reactions requiring anhydrous conditions were conducted in flame dried apparatus. Melting point was determined using open capillary method and was uncorrected. Solvents and reagents were purified by standard methods. Organic extracts were dried over anhydrous sodium sulphate.
L Amino acids, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), trifluoroacetic acid (TFA), p-nitrophenol (PNP), triethylamine (TEA), di-tert-butylpyrocarbonate (Boc2O) and pyridine (C5H5N) were obtained from Spectrochem Limited (Mumbai, India). IR spectra were recorded on Thermo Nicolet 330 FTIR spectrophotometer (Thermo Nicolet, USA) using a thin film supported on KBr pellets for the synthesized cyclic heptapeptide – Euryjanicin A and Chloroform is used as a solvent for intermediate peptides.
1H and 13C NMR spectra were recorded on Bruker AC NMR spectrometer (500 and 125 MHz), (Bruker, USA) using CDCl3 as solvent and tetramethylsilane (TMS) as internal standard. Mass spectra were recorded on Joel Sx 102/DA-6000 mass spectrometer (Jeol, Tokyo, Japan) operating at 70 eV using fast atom bombardment technique. Elemental analyses for all the synthesized peptides were performed on Vario EL III elemental analyzer (Elementar, Germany). Purity of the synthesized cyclic peptide as well as their intermediates was checked by TLC on pre-coated silica gel G plates using CHCl3/MeOH as developing solvent system in different ratios (9:1/8:2 v/v) and brown spots were detected on exposure to iodine vapors in a tightly closed chamber (or) bluish spots were detected in UV chamber.
2.2 General procedure for the preparation of linear, tetra and tripeptide segments (Bodanszky et al., 1984)
Amino acid methyl ester hydrochloride (or) dipeptide methyl ester (10 mmol) was dissolved in chloroform (20 mL). To this, triethylamine (4 mL, 28.7 mmol) was added at 0 °C and the reaction mixture was stirred for about 15 min. Boc-amino acid (or) Boc-dipeptide (10 mmol) in CHCl3 (20 mL) and EDC (10 mmol) was added, while stirring the reaction mixture. After 8 h, the reaction mixture was filtered and the residue was washed with CHCl3 (30 mL) and added to the filtrate. The reaction filtrate was washed with 5% NaHCO3 (20 mL) and saturated NaCl (20 mL) solutions. The organic layer was filtered and evaporated in vacuum. The crude product was recrystallized using the solvent system – chloroform and petroleum ether (1:2 ratio) followed by cooling at 0 °C.
2.2.1 Tert-Butyloxycarbonyl-L-Tryptophanyl-L-Prolyl-L-Leucine-OMe (5)
Yield 87%, C28H40N4O6, 528.29. FTIR (KBr v cm−1): 3212 (N—H stretch), 2997–2989 (C—H stretch), 2965, 1752 (C⚌O stretch, ester), 1536 (N—H bend, 2° amide), 1388, 1375 (C—H bend, tert-butyl), 1269 (C—O stretch, ester) cm−1. 1H NMR (500 MHz, CDCl3): δ10.1 (1H, br.s, NH of Trp. Ring), 8.63 (1H, br.s, NH of Trp), 8.22 (1H, br.s, NH of Leu), 7.18–7.02 (4H, m, Phenyl ring of Trp), 4.40 (t, 1H, N—CH of Pro), 3.5–3.4 (m, 2H, —CH2 of Pro), 3.2–3.0 (t, 2H, N—CH2 of Pro), 2.9 (d, 2H, —CH2 of Trp) 2.8–2.5 (m, 2H, —CH2 of Pro), 4.42 (t, 1H, —CH of Leu), 3.63 (s, 3H, —OCH3 of Leu), 1.8 (d, 2H, —CH2 of Leu), 1.7 (m, 1H, —CH of Leu), 1.0 (s, 6H, —CH3 of Leu), 0.9 (s, 9H, —CH3 of Boc-group). Calcd. C28H40N4O6: C, 63.62; H, 7.63; N, 10.60; found: C, 63.65; H, 7.65; N, 10.63%.
2.2.2 Tert-Butyloxycarbonyl-L-Phenylanalyl-L-Seryl-L-Isoleucyl-L-Proline-OMe (6)
Yield 85%, C29H44N4O8, 576.36. FTIR (KBr v cm−1): v 3319 (N—H stretch), 3072, 3058 (Aromatic C—H stretch), 2998, 2987 (Aliphatic C—H stretch), 1751 (C⚌O stretch, ester), 1541, (N—H bend) cm−1. 1H NMR (CDCl3): δ8.63 (1H, s, —NH of Phe), 8.52 (1H, s, —NH of Ser), 8.43 (1H, s, —NH of Ile), 8.20 (1H, s, —NH of Pro), 7.20–7.02 (5H, m, Ar—H of Phe), 6.70 (1H, s, —OH of Ser), 4.92 (1H, t, —CH of Phe), 4.61 (1H, t, —CH of Ser), 4.53 (1H, d, —CH of Ile), 4.40 (1H, t, N—CH of Pro), 4.15–3.92 (2H, d, —CH2 of Ser), 3.64 (s, 3H, —OCH3 of Pro), 3.51–3.40 (2H, m, N—CH2 of Pro), 2.92 (2H, d, —CH2 of Phe), 2.50 (1H, m, —CH of Ile), 2.34–2.09 (2H, m, —CH2 of Pro), 2.02–1.92 (2H, m, —CH2 of Pro), 1.29 (2H, m, —CH2 of Ile), 1.06 (3H, d, —CH3 of Ile), 0.96 (3H, t, —CH3 of Ile), 0.89 (s, 9H, —CH3 of Boc group). Calcd. C29H44N4O8: C, 60.40; H, 7.69; N, 9.72; found C, 60.41; H, 7.70; N, 9.75%.
2.3 Deprotection of tripeptide unit at the carboxyl terminal
To the solution of tripeptide 5 (4.4 g, 0.01 mol) in THF-H2O (1:1, 36 mL), 0.36 g (0.015 mol) of LiOH was added at 0 °C. The reaction mixture was stirred at room temperature for about one hour and then acidified to pH 3.5 using 1 N H2SO4. The aqueous layer was extracted with diethyl ether (3 × 25 mL). The organic extracts were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude product was recrystallized using methanol and petroleum ether (1:2) to afford pure deprotected tripeptide unit.
2.4 Deprotection of tetrapeptide unit at amino terminal
The tetrapeptide 6 (6.36 g, 0.01 mol) was dissolved in CHCl3 (15 mL) and treated with CF3COOH (2.28 g, 0.02 mol). The resulting solution was stirred at room temperature for about one hour and washed with saturated NaHCO3 solution (25 mL). The organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude product was purified through crystallization using CHCl3 and petroleum ether (1:2) to afford pure deprotected tetrapeptide unit.
2.5 Procedure for the synthesis of the linear heptapeptide unit
The deprotected tetrapeptide (5.36 g, 0.01 mol) was dissolved in tetrahydrofuran (35 mL). To this reaction mixture, triethyl amine (2.8 mL, 0.021 mol) was added at 0 °C and the resulting solution was stirred for about 15 min. Furthermore, the deprotected tripeptide (4.25 g, 0.01 mol) was dissolved in tetrahydrofuran (35 mL) and then finally the coupling agent; EDC (1.26 g, 0.01 mol) was added at 0 °C. After 8 h, the reaction mixture was filtered and the filtrate was washed with 5% NaHCO3 and saturated NaCl solutions (30 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and evaporated in vacuum. The crude product was recrystallized using CHCl3 and petroleum ether (1:2) by cooling at 0 °C to afford the pure linear heptapeptide.
2.5.1 Tert-Butyloxycarbonyl-L-Phenylanalyl-L-Seryl-L-Isoleucyl-L-Prolyl-L-Tryptophanyl-L-Prolyl-L-Leucine-OMe (7)
Yield 81%, C51H72N8O11, 972.53. FTIR (KBr v cm−1): v 3375 (OH stretch, Tyr), 3132 (N—H stretch), 3069 (C—H stretch, ring), 2992–2984 (C—H stretch), 1758 (C⚌O stretch, ester), 1541 (N—H bend) cm−1. 1H NMR (CDCl3): δ10.4 (1H, br.s, NH of Trp. Ring), 8.65 (1H, br.s, NH of Trp), 8.62 (1H, s, —NH of Phe), 8.52 (1H, s, —NH of Ser), 8.43 (1H, s, —NH of Ile), 8.24 (1H, br.s, NH of Leu),8.22 (1H, s, —NH of Pro), 7.20–7.04 (5H, m, Ar—H of Phe), 7.18–7.01 (4H, m, Phenyl ring of Trp.), 6.72 (1H, s, —OH of Ser), 4.93 (1H, t, —CH of Phe), 4.61 (1H, t, —CH of Ser), 4.53 (1H, d, —CH of Ile), 4.42 (2H, t, N—CH of Pro), 4.17–3.91 (2H, d, —CH2 of Ser), 4.41 (t, 1H, —CH of Leu), 3.59 (s, 3H, —OCH3 of Leu), 3.50–3.41 (m, 4H, N—CH2 of Pro), 3.38–2.96 (t, 4H, N—CH2 of Pro), 2.94 (2H, d, —CH2 of Phe), 2.91–2.52 (m, 4H, —CH2 of Pro), 2.53 (1H, m, —CH of Ile), 2.34–2.09 (2H, m, —CH2 of Pro), 2.02–1.93 (2H, m, —CH2 of Pro), 1.24 (2H, m, —CH2 of Ile), 1.07 (3H, d, —CH3 of Ile), 0.97 (3H, t, —CH3 of Ile), 1.82 (d, 2H, —CH2 of Leu), 1.71 (m, 1H, —CH of Leu), 1.01 (s, 6H, —CH3 of Leu), 0.92 (s, 9H, —CH3 of Boc-group). Calcd. C51H72N8O11: C, 62.94; H, 7.46; N, 11.51; found C, 62.95; H, 7.48; N, 11.53%.
2.6 Synthesis of the cyclic heptapeptide 8
The cyclization of linear heptapeptide unit was carried out via p-nitro phenyl ester method of Bodanszky (Dahiya, 2007a,b) with certain modifications. The ester group of the linear segment was removed using Lithium hydroxide and then the p-nitro phenyl ester group was introduced using the subsequent standard method (Dahiya, 2007a,b).
To synthesize Euryjanicin A (8), linear heptapeptide unit 7 (4.72 g, 0.005 mol) was deprotected at carboxyl end using LiOH (0.18 g, 0.0075 mol) to get Boc-L-Phenylalanyl-L-Seryl-L-Isoleucyl-L-Prolyl-L-Tryptophanyl-L-Prolyl-L-Leucine-OMe. The deprotected heptapeptide unit (4.65 g, 0.005 mol) was dissolved in CHCl3 (50 mL) at 0 °C. To this solution, p-nitrophenol (0.94 g or 1.23 g, 0.0067 mol) and EDC (0.63 g, 0.005 mol) were added and then the reaction mixture was stirred at room temperature for about 12 h. The reaction mixture was filtered and the filtrate was washed with 10% NaHCO3 solution (3 × 25 mL) and finally washed with 5% HCl (2 × 30 mL) to get the corresponding p-nitro-phenyl ester Boc-L-Phenylalanyl-L-Seryl-L-Isoleucyl-L-Prolyl-L-Tryptophanyl-L-Prolyl-L-Leucine-Opnp.
The p-nitro phenyl ester peptides (4.2 g 0.004 mol) were dissolved in CHCl3 (35 mL), and CF3COOH (0.91 g, 0.008 mol) was added and stirred at room temperature for about one hour. The resulting solution was washed with 10% NaHCO3 solution (2 × 25 mL). The organic layer was dried over anhydrous Na2SO4 to get L-Phenylalanyl-L-Seryl-L-Isoleucyl-L-Prolyl-L-Tryptophanyl-L-Prolyl-L-Leucine Opnp, which was dissolved in CHCl3 (25 mL) and pyridine (2.8 mL, 0.021 mol) was added. The final alkaline solution was kept at 0 °C for about 3 days. Once again, the reaction mixture was washed with 10% NaHCO3 (3 × 25 mL) and 5% HCl (2 × 30 mL) solutions. The organic layer was dried over anhydrous Na2SO4 and crude cyclized peptide product was crystallized using CHCl3 and n-hexane (1:2 ratios) to acquire pure cyclic peptide – Euryjanicin A.
2.6.1 Cyclo (L-Phenylalanyl-L-Seryl-L-Isoleucyl-L-Prolyl-L-Tryptophanyl-L-Prolyl-L-Leucine) (8)
Yield 87%, C45H60N8O8, 841.05. FTIR (KBr v cm−1): v 3134 (N—H stretch), 3070 (C—H stretch), 2927–2922 (C—H stretch, asym), 2876–2872, 1759 (C⚌O stretch, ester), 1539 (N—H bend) cm−1. 1H NMR (CDCl3): δ10.3 (1H, br.s, NH of Trp. Ring), 8.63 (1H, br.s, NH of Trp), 8.61 (1H, s, —NH of Phe), 8.53 (1H, s, —NH of Ser), 8.41 (1H, s, —NH of Ile), 8.23 (1H, br.s, NH of Leu), 8.21 (1H, s, —NH of Pro), 7.22–7.05 (5H, m, Ar—H of Phe), 7.19–7.02 (4H, m, Phenyl ring of Trp.), 6.73 (1H, s, —OH of Ser), 4.92 (1H, t, —CH of Phe), 4.62 (1H, t, —CH of Ser), 4.52 (1H, d, —CH of Ile), 4.43 (2H, t, N—CH of Pro), 4.16–3.92 (2H, d, —CH2 of Ser), 4.42 (t, 1H, —CH of Leu), 3.52–3.40 (m, 4H, N—CH2 of Pro), 3.39–2.95 (t, 4H, N—CH2 of Pro), 2.92 (2H, d, —CH2 of Phe), 2.90–2.53 (m, 4H, —CH2 of Pro), 2.54 (1H, m, —CH of Ile), 2.31–2.01 (2H, m, —CH2 of Pro), 2.02–1.93 (2H, m, —CH2 of Pro), 1.24 (2H, m, —CH2 of Ile), 1.07 (3H, d, —CH3 of Ile), 0.97 (3H, t, —CH3 of Ile), 1.83 (d, 2H, —CH2 of Leu), 1.72 (m, 1H, —CH of Leu), 1.03 (s, 6H, —CH3 of Leu). 13C NMR (CDCl3): δ173.5 (C⚌O, Ile), 172.6 (C⚌O, Ser),172.7, 171.5 (2C, C⚌O, Pro), 171.9 (C⚌O, Leu), 170.9 (C⚌O, Trp), 169.9 (C⚌O, Phe), 139.5–127.3 (6C, Aromatic carbon of Phe), 126.5–111.1 (7C, Carbons of Trp), 61.8 (1C, —CH2 OH of Ser), 60.4, 60.7 (2C, α-CH2 of Pro-1 & Pro-2), 56.9 (1C, —CH of Ile), 56.5 (1C, —CH of Leu), 55.4 (—CH of Trp), 54.9 (—CH of Ser), 52.7 (—CH of Leu),46.6, 45.8 (2C, δ-CH2, Pro-1 & Pro-2), 41.2 (—CH2 of Leu), 31.2, 30.5 (2C, β-CH2, Pro-1 & Pro-2), 22.6, 21.7 (2C, δ-CH2, Pro-1 & Pro-2), 37.6 (—CH2 of Leu), 37.1 (—CH of Ile), 24.7 (—CH2 of Ile), 22.3, 22.7 (2C, —CH3 of Leu), 14.6 (1C, —CH3 of Ile), 10.9 (1C, —CH3 of Ile); EI/MS m/z [EI]: 827 [(M)+, 100], Calcd. For C45H60N8O8: C, 65.09; H, 7.08; N, 12.08; found: C, 65.11; H, 7.05; N, 12.10%.
2.7 Anticancer activity
2.7.1 MTT [3-(4, 5-dimethylthiazole-2-yl)-2,5-diphenyltetrazoliumbromide] Assay – (Carnichael et al., 1987; Mosmann et al., 1983)
The Human colon cancer cell lines were obtained from ATCC and maintained in DMEM (Hi-Media Laboratories Pvt. Ltd., Mumbai, India) supplemented with 10% heat inactivated FBS (v/v), streptomycin (100 μg/ml) and penicillin (100 μg/ml). The cell line was maintained at 37 °C with 5% carbon dioxide in CO2 incubator. The MTT cell proliferation assay was used to evaluate the anticancer activity of cyclic heptapeptide using the Cell Quantification MTT cell viability assay kit (Bio assay Systems). The optical density was measured at 570 nm for each well on the absorbance plate reader. Trypan blue dye exclusion assay was also used to count the number of viable and non-viable HeLa cancer cells in the culture medium after drug treatment. Treatment with Cisplatin at the same concentration served as positive control.
3 Results and discussion
In order to carry out the first total synthesis of Euryjanicin A, disconnection strategy was employed. The cyclic heptapeptide molecule was detached into three dipeptide units Boc-L-Phe-L-Ser-OMe (1), Boc-L-Ile-L-Pro-OMe (2), Boc-L-Trp-L-Pro-OMe (3), and single amino acid unit L-Leu-OMe·HCl (4). The necessary dipeptides (1–3) were prepared by coupling of Boc-amino acids, viz. Boc-L-Phe, Boc-L-Ile and Boc-L-Trp with the corresponding amino acid methyl ester hydrochlorides viz. L-Ser-OMe·HCl, L-Pro-OMe·HCl and L-Leu-OMe·HCl employing EDC as coupling agent (Williams and Ibrahim, 1981). Ester group of the dipeptide (3) was removed by alkaline hydrolysis with Lithium hydroxide and the deprotected peptide was coupled with amino acid methyl ester hydrochloride (4) using EDC and triethylamine (TEA), to get the tripeptide unit Boc-L-Trp-L-Pro-Leu-OMe (5). Correspondingly, dipeptide (1) after deprotection at the carboxyl end was coupled with dipeptide (2) deprotected at amino terminal, to get the tetrapeptide unit Boc-L-Pro-L-Ser-L-Ile-L-Pro-OMe (6). Subsequent removal of the ester group of tetrapeptide (6) and the Boc group of tripeptide (5), these deprotected units were coupled to get the linear heptapeptide unit Boc-L-Phe-L-Ser-L-Ile-L-Pro-L-Trp-L-Pro-Leu-OMe (7). The methyl ester group of the linear peptide fragment was replaced by p-nitro phenyl (PnP) ester group. The Boc-group of the resulting compound was removed using trifluoroacetic acid and then the deprotected linear fragment was cyclized by the addition of chloroform and pyridine at 0 °C and the reaction mixture was kept for about 3 days in refrigerator to get cyclic heptapeptide (8) as described in the Scheme 1.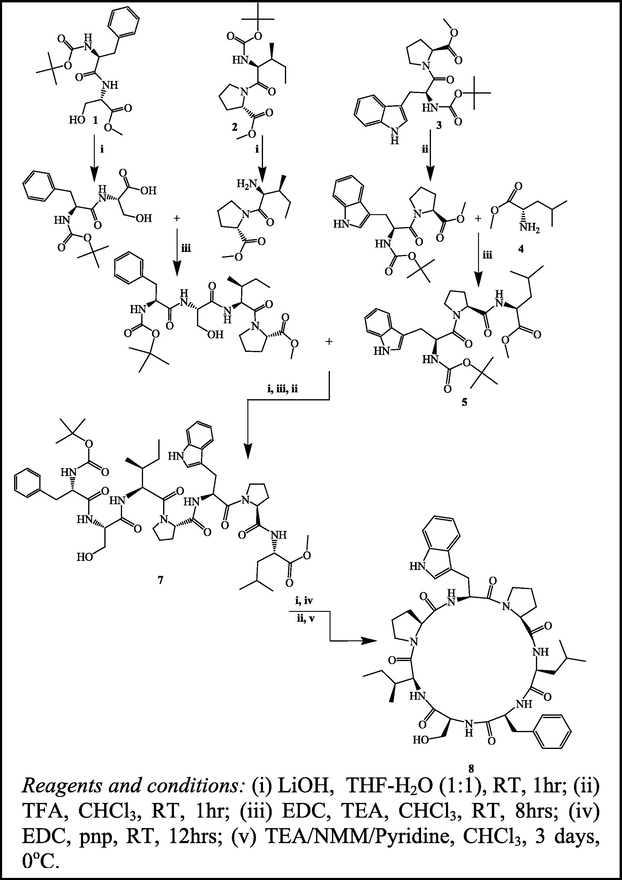
The synthetic route in the preparation of the cyclic heptapeptide Euryjanicin A (8).
Infrared spectrum of Euryjanicin A showed characteristic medium to strong bands corresponding to the carbonyl stretching at 1652.41 cm−1 and NH bending at 1528.35 cm−1, confirmed the coupling reaction of the cyclic heptapeptide. In the Proton NMR spectra of 8, formation of cyclic heptapeptide was confirmed by the disappearance of singlet at δ3.58 and δ1.52 ppm corresponding to the three protons of methyl ester group and nine protons of the Boc tert-butyl group. Furthermore, 1H & 13C NMR spectra of the synthesized cyclic heptapeptide showed characteristic peaks confirming the presence of all sixty methyl, methylene and methine protons and forty five carbon atoms. The mass spectral data clearly confirmed the presence of molecular ion peak (M)+ at m/z 827 for the synthesized compound. All these spectral data of Euryjanicin A clearly confirmed the coupling reaction of amino acids and peptides.
The effect of Euryjanicin A on the growth of HeLa cancer cells was tested under invitro conditions determined using five different concentrations (10, 20, 30, 40 and 50 μg/mL) and the cell survival after 48 h was illustrated in Fig. 1. Cyclic heptapeptide significantly reduced the growth of HeLa cancer cells and the results are shown in Table 1.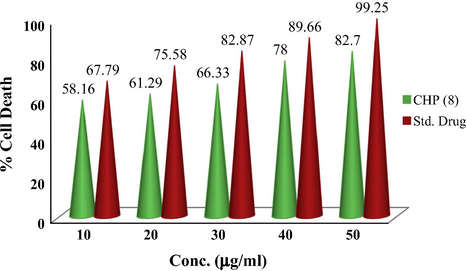
Anticancer activity of Euryjanicin A (8) cyclic heptapeptide.
Compd.
Conc. (μg/mL)
Hela cancer cell lines
Absorbance (OD)
% Cell death
Cyclic heptapeptide (CHP) 8
50
1.985
82.70
40
1.872
78.00
30
1.592
66.33
20
1.471
61.29
10
1.396
58.16
Standard (Cisplatin)
50
2.382
99.25
40
2.152
89.66
30
1.989
82.87
20
1.814
75.58
10
1.627
67.79
Screening concentrations of 50 and 40 μg/mL result to a reduction in viable cell of 17.3% and 22% respectively. At 30 μg/mL screening concentration of Euryjanicin A reduced viable cells of 33.67%. Cyclic heptapeptide at 10 and 20 μg/mL produced potent inhibitory effects [38.71% and 41.29% viable cells] against HeLa cancer cell line. The result of the study clearly indicates the positive control cisplatin to have the significant inhibition of tumor cell growth for the cell line in comparison with the cyclic heptapeptide. Cisplatin at 50 μM produced the considerable inhibitory result on the HeLa cell line resulting in a 99.25% cell death (see Fig. 2).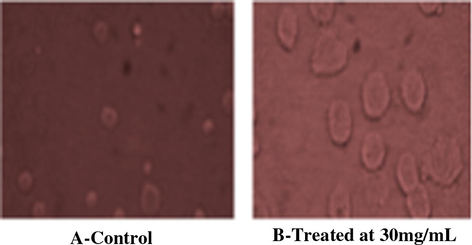
Anticancer activity of Euryjanicin A (8). (i) A-control and (ii) B-treated at 30 mg/mL.
4 Conclusion
In the present work, we have used an effective coupling reagent, EDC, for the coupling of peptide and amino acids in good to high yields. Euryjanicin A was naturally isolated from Caribbean marine peptide and the marine peptide was synthetically prepared in high yield using a new water soluble coupling agent EDC. The synthesized cyclic heptapeptide possessed high cytotoxic activity against HeLa cell lines with respect to standard drug cisplatin. The synthesized cyclic heptapeptide compound was used as effective anticancer drug in future after clinical trials.
Acknowledgments
The authors wish to thank Anna University, University College of Engineering Villupuram and Kingston Engineering Management for providing the research facilities. The authors are thankful to SAIF, IIT Chennai for spectral analysis.
References
- Nat. Prod. Rep.. 2009;26:170-244.
- Practice of Peptide Synthesis Vol vol. 91. (first ed.). New York: Springer-Verlag; 1984. p. :131.
- Cancer Res.. 1987;47:936-942.
- Acta Pol. Pharm.. 2007;64:509-516.
- Pak. J. Pharm. Sci.. 2007;20:317-323.
- J. Am. Chem. Soc.. 1999;121:5899-5909.
- Chem. Rev.. 1993;93:1793-1806.
- J. Am. Chem. Soc.. 2005;127:110-118.
- J. Am. Chem. Soc.. 1993;115:8066-8072.
- Chem. Soc. Rev.. 2005;34:355-365.
- Tetrahedron. 1993;49:2391-2402.
- Nat. Prod.. 2006;69:1739-1744.
- J. Immunol. Meth.. 1983;65:55-63.
- J. Am. Chem. Soc.. 1995;117:8271-8272.
- Tetrahedron. 2000;56:3679-3685.
- Eur. J. Org. Chem. 1998:2659-2665.
- Tetrahedron. 1993;49:6785-6796.
- Tetrahedron. 1994;50:4667-4680.
- J. Am. Chem. Soc.. 1981;103:7090-7095.
- Chem. Rev.. 1995;95:2115-2134.
- J. Org. Chem.. 1996;61:7168-7173.
- Mar. Drugs. 2011;9:1840-1859.
Appendix A
Supplementary material
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.arabjc.2014.05.037.
Appendix A
Supplementary material
Supplementary data 1
Supplementary data 1




