

Translate this page into:
Synthesis and study on aroylhydrazones having cyanovinylpyrrole
⁎Corresponding author. Tel.: +91 9451308205. rnsvk.chemistry@gmail.com (R.N. Singh)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
A series of C-cyanovinylpyrrole containing aroylhydrazones (3a–c), derived from ethyl 2-cyano-3-(5-formyl-1H-pyrrol-2-yl)-acrylate and acid hydrazides: salicylhydrazide, isoniazid and 3,5-dinitrobenzohydrazide have been characterized by various spectroscopic techniques (1H NMR, 13C NMR, Mass, UV–Visible, Emission, FT-IR) and quantum chemical calculations. TD-DFT has been used to calculate the various electronic excitations and their nature. The emission spectra show that (3a–c) are good photoluminescent material due to intense emission at higher wavelength in yellow, green and blue region with high Stoke’s shift in the region 80–328 nm. The molar refractivity (MR) for (3a–c) is calculated as 109.28, 102.96, 119.14 esu, respectively. Natural bond orbital (NBO) analysis has been carried out to explore the various conjugative and hyperconjugative interactions within molecule and their second order stabilization energy (E(2)). Global electrophilicity index (ω = 5.41–8.11 eV) shows that (3a–c) molecules work as strong electrophiles. The local reactivity descriptors analyses such as Fukui functions (fk+, fk−), local softnesses (sk+, sk−) and electrophilicity indices (ωk+, ωk−) have been performed to determine the reactive sites within molecules. The first hyperpolarizabilities (β0) of (3a–c) have been computed and found to increase with electron withdrawing substituents.
Keywords
Emission
TD-DFT
NBO
First hyperpolarizabilities

1 Introduction
Aroylhydrazones (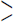 C⚌N—NH—CO—) are special group of compounds in the Schiff bases family that have wide applications in chemistry and biology and are characterized by the presence of two inter-linked nitrogen atoms as Nsp2–Nsp3. They are good synthons for the target syntheses of 1,3,4-oxadiazine, 1,2,4-triazine, pyrazole derivatives (Mohareb et al., 2010) and 1,3,4-oxadiazolines, 2-azetidinones, 4-thiazolidinones via heterocyclic transformations (Rollas and Küçükgüzel, 2007; Armbruster et al., 2006). Several aroylhydrazones have wide applications in the field of analytical chemistry as a selective metal extracting agent as well as in spectrophotometric determination of certain transition metals (Tang et al., 2004; Kavlentis, 1998; Chalapathi et al., 2011). However, the most valuable property of aroylhydrazones is their great physiological activity due to the presence of the active pharmacophore (C⚌N—NH—CO—) and provides a wide range of application in medicinal and pharmaceutical fields with various biological applications. Therefore, a number of hydrazone derivatives have been used for the treatment of diseases such as convulsant (Dimmock et al., 2000), malaria (Melnyk et al., 2006), HIV, cancer (Savini et al., 2004), microbial (Rallas et al., 2002; Gursoy et al., 1997; Ajani et al., 2010; Rawat and Singh, 2014a, 2014b, 2015a, 2015b) and tuberculosis (Zheng et al., 2009; Richardson and Bernhardt, 1999; Bedia et al., 2006; Darnell and Richardson, 1999; Murukan and Mohanan, 2007). Many of hydrazones are used as prospective new materials for the development of potential chemosensors (Yu et al., 2007), opto-electronic (Szczesna and Lipkowska, 2001) and nonlinear optical (NLO) (Vijayakumar et al., 2010, 2011; Shirota and Kageyama, 2007; Blau, 1987; Vogt et al., 2008; Genger et al., 2008) applications.
C⚌N—NH—CO—) are special group of compounds in the Schiff bases family that have wide applications in chemistry and biology and are characterized by the presence of two inter-linked nitrogen atoms as Nsp2–Nsp3. They are good synthons for the target syntheses of 1,3,4-oxadiazine, 1,2,4-triazine, pyrazole derivatives (Mohareb et al., 2010) and 1,3,4-oxadiazolines, 2-azetidinones, 4-thiazolidinones via heterocyclic transformations (Rollas and Küçükgüzel, 2007; Armbruster et al., 2006). Several aroylhydrazones have wide applications in the field of analytical chemistry as a selective metal extracting agent as well as in spectrophotometric determination of certain transition metals (Tang et al., 2004; Kavlentis, 1998; Chalapathi et al., 2011). However, the most valuable property of aroylhydrazones is their great physiological activity due to the presence of the active pharmacophore (C⚌N—NH—CO—) and provides a wide range of application in medicinal and pharmaceutical fields with various biological applications. Therefore, a number of hydrazone derivatives have been used for the treatment of diseases such as convulsant (Dimmock et al., 2000), malaria (Melnyk et al., 2006), HIV, cancer (Savini et al., 2004), microbial (Rallas et al., 2002; Gursoy et al., 1997; Ajani et al., 2010; Rawat and Singh, 2014a, 2014b, 2015a, 2015b) and tuberculosis (Zheng et al., 2009; Richardson and Bernhardt, 1999; Bedia et al., 2006; Darnell and Richardson, 1999; Murukan and Mohanan, 2007). Many of hydrazones are used as prospective new materials for the development of potential chemosensors (Yu et al., 2007), opto-electronic (Szczesna and Lipkowska, 2001) and nonlinear optical (NLO) (Vijayakumar et al., 2010, 2011; Shirota and Kageyama, 2007; Blau, 1987; Vogt et al., 2008; Genger et al., 2008) applications.
The functionalized C-vinylpyrroles are prospective new materials for molecular optical switches, nano-devices, photo- and electro-conducting applications and also used as ligands for new photo catalysts, biologically active complexes (Hayes et al., 2000; Harmjanz et al., 2000; Sour et al., 1999). Therefore, the title compounds (3a–c) have been synthesized and characterized using various experimental and theoretical spectroscopic methods. In present paper, we report the spectroscopic, structural and NLO properties of these molecules. Furthermore, quantum chemical calculations have also been performed to determine the intramolecular conjugative and hyperconjugative interactions, hydrogen bonding, chemical reactivity and NLO properties of investigated molecules.
2 Experimental details
All the chemicals were used of analytical grade. The pyridine-4-carboxylic acid hydrazide or isoniazid was purchased from sigma Aldrich company. Ethyl 2-cyano-3-(5-formyl-1H-pyrrol-2-yl)-acrylate (1) was prepared by an earlier reported method (Kjell and Per-Åke, 1979). The salicylhydrazide and 3,5–dinitrobenzohydrazide (2a, 2c) were prepared according to the literature procedure (Gup and Kırkan, 2005; Wild, 1947). All spectra were recorded at 25 °C. The Mass spectra of (3a–c) were recorded on JEOL-Acc TDF JMS-T100LC, Accu TOF mass spectrometer. The 1H NMR spectra of (3a–c) were recorded in DMSO-d6 on Bruker DRX-300 spectrometer using TMS as an internal reference. The 13C NMR spectra of (3a–c) were recorded in DMSO-d6 on Bruker DRX-300 spectrometer using TMS as an internal reference. The FT-IR spectra were recorded in KBr medium on a Bruker spectrometer. The UV–Visible absorption spectra of (3a–c), (1 × 10–5 M in DMSO) were recorded on V-670 JASCO spectrophotometer. The emission spectra of (3a–c), (1 × 10–5 M in DMSO) were recorded on PL Spectrometer LS-55, Perkin Elmer, UK.
2.1 Preparation of ethyl 3,5-dinitrobenzoate (Vogel, 1956)
3,5-dinitrobenzoic acid (5.320 g) was dissolved in 15 ml ethanol and 1.0 ml of conc. H2SO4 was added as catalyst and solution was refluxed for 20 h. The solution was cooled down to room temperature and refrigerated for 8 h to precipitation. The precipitate was recrystallized in ethanol to give the product ethyl 3,5-dinitrobenzoate. m.p. – 90 °C.
2.2 Preparation of 3,5-dinitrobenzohydrazide (Wild, 1947)
Ethyl 3,5-dinitrobenzoate (1.60 g, 6.6633 mmol) and hydrazine hydrate (0.48 ml, 0.01 mol) of 100% in 10 ml of methanol were refluxed for 10 h. The solution was cooled to room temperature to obtain yellow colored precipitate. The precipitate was filtered off and recrystallized from methanol to give pure product 3,5-dinitrobenzohydrazide.
Color: dark yellow, yield: (0.9422 g, 62.54%), m.p.: 145 °C.
2.3 Preparation of 2-hydroxybenzohydrazide (salicylhydrazide) (Gup and Kırkan, 2005)
A solution of hydrazine hydrate (0.1645 g, 0.16 ml, 3.286 mmol) in 10 ml methanol was added dropwise to the stirring solution of methyl 2-hydroxybenzoate (0.500 g, 0.42 ml, 3.286 mmol) in 25 ml methanol. Reaction mixture was refluxed for 6 h. After refluxing, cream color precipitate was obtained. The precipitate was filtered off, washed with methanol and dried in air.
Color: creamy white, yield: 0.3020 g, 72.88%, m.p.: 140 °C.
2.4 Ethyl 2-cyano-3-[5-{(2-hydroxy-benzoyl)-hydrazonomethyl}-1H-pyrrol-2-yl]-acrylate (3a)
To the equimolar solution of ethyl 2-cyano-3-(5-formyl-1H-pyrrol-2-yl)-acrylate (1) (0.200 g, 0.9171 mmol) and 2-hydroxy-benzoic acid hydrazide (2a) (0.1394 g, 0.9171 mmol) in 20 ml methanol, 0.01 ml of conc. HCl acid was added as catalyst. The reaction mixture was stirred at room temperature for overnight to obtain yellow color precipitate. The precipitate was filtered off, washed with MeOH and dried in air, afforded (3a), as dark yellow solid. Yield: 0.1582 g, 48.96%. M.p.: 278–280 °C. Anal. calcd. for C18H16N4O4: C 61.34%, H 4.57%, N 15.90%; obs.: C 61.38%, H 4.54%, N 15.92%. MS (m/z): calcd. 352.1171, obs. 353 [M++1]. 1H NMR (300 MHz, DMSO-d6) δexp/calcd 12.680/12.545 (s, 1H, —OH), δ 12.050/10.313 (s, 1H, pyrrole-NH), 12.039/9.582 (s, 1H, ArCONH), 8.367/8.085 (s, 1H, vinyl-CH⚌C), 7.850–7.876/7.872–7.169 (d, J = 7.8 Hz, 2H, Ar-CH), 6.953–6.980/7.872–7.169 (t, J = 9.75 Hz, 2H, Ar-CH), 7.407/7.97 (s, 1H, —CH⚌N), 7.444–7.457, 6.887–6.875/6.966–7.253 (m, 2H, J = 3.9, 3.6 Hz, pyrrole-CH), 4.231–4.301/4.347 (q, J = 7.00 Hz, 2H, ester-CH2), 1.263–1.310/1.473–1.236 (t, J = 7.05 Hz, 3H, ester-CH3). 13C NMR (300 MHz, DMSO-d6) δexp/calcd C2-123.22/128.08, C3-122.26/126.82, C4-113.98/115.7, C5-132.39/137.05, C6-141.95/141.44, C7-108.27/91.89, C8-114.34/116.08, C10-163.68/161.23, C13-50.28/65.16, C14-15.23/16.72, C15-145.38/132.42, C18-168.54/162.88, C20-111.29/112.53, C21-120.42/124.33, C22-120.42/116.25, C23-129.53/133.29, C24-129.53/115.99, C25-158.48/160.96.
2.5 Synthesis of ethyl 2-cyano-3-[5-{(4-nitro-benzoyl)-hydrazonomethyl}-1H-pyrrol-2-yl]-acrylate (3b)
To the equimolar solution of ethyl 2-cyano-3-(5-formyl-1H-pyrrol-2-yl)-acrylate (0.250 g, 1.1464 mmol) and isoniazid (0.1572 g, 1.1464 mmol) in 25 ml methanol, 0.01 ml of conc. HCl acid was added as catalyst. The reaction mixture was stirred at room temperature for 8 h and yellow color precipitate was obtained. The precipitate was filtered off, washed with methanol and dried in air, afforded (3b), as yellow solid. Yield: 0.3464 g, 89.63%. The compound decomposes above 280 °C. Anal. calcd. for C17H15N5O3: C 60.51%, H 4.48%, N 20.76%; obs.: C 60.54%, H 4.50%, N 20.72%. MS (m/z): calcd. 337.117; obs. 338.14 [M++1]. 1H NMR (300 MHz, DMSO-d6) δexp/calcd 12.749/10.3155 (s, 1H, pyrrole-NH), 12.707/9.5591 (s, 1H, ArCONH), 8.790/8.0801 (s, 1H, vinyl-CH⚌C), 8.369–8.343/9.0865–9.1345 (d, J = 7.8 Hz, 2H, pyridine-CH), 8.463/7.9717 (s, 1H, —CH⚌N—), 6.974–6.995/7.8626–8.1343 (d, J = 6.3 Hz, 2H, pyridine-CH), 6.779–6.769/7.2548(d, 2H, J = 3.6, 2.7 Hz, pyrrole-CH), 8.596–8.587/6.9616 (d, J = 2.7 Hz, 1H, pyrrole-CH), 4.247–4.314/4.3452–4.3542 (q, J = 6.70 Hz, 2H, ester-CH2), 1.283–1.243/1.2348–1.4826 (t, J = 6.0 Hz, 3H, ester-CH3). 13C NMR (300 MHz, DMSO-d6) δexp/calcd C2-125.24/128.1798, C3-123.60/126.8273, C4-116.96/115.9032, C5-133.84/136.9796, C6-138.68/141.5059, C7-114.98/92.0105, C8-113.94/116.0699, C10-157.56/161.2264, C13-52.20/65.1485, C14-15.28/16.6665, C15-141.82/132.6996, C18-159.92/158.5427, C20-129.78/139.4344, C21-117.68/118.2875, C22-142.67/148.5444, C24-142.67/148.8017, C25-117.68/122.2078.
2.6 Synthesis of ethyl 2-cyano-3-[5-{(3,5-dinitro-benzoyl)-hydrazonomethyl}-1H-pyrrol-2-yl]-acrylate (3c)
To the equimolar solution of ethyl 2-cyano-3-(5-formyl-1H-pyrrol-2-yl)-acrylate (1) (0.250 g, 1.1464 mmol) and 3,5-dinitro-benzohydrazide (2c) (0.2592 g, 1.1464 mmol) in 25 ml methanol, 0.01 ml of conc. HCl acid was added as catalyst. The reaction mixture was stirred at room temperature. After stirring for 4 h, dark yellow color precipitate was obtained. The precipitate was filtered off, washed with methanol and dried in air, afforded (3c), as yellow solid. Yield: 0.3844 g, 78.68%. Anal. calcd. for C18H14N6O7: C 50.69%, H 3.31%, N 19.71%; obs.: C 50.72%, H 3.28%, N 19.74%. MS (m/z): calcd. 426.0924; obs. 427.18 [M++1]. 1H NMR (300 MHz, DMSO-d6) δ exp/calcd 12.675/10.4148, (s, 1H, pyrrole-NH), 12.110/9.0539, (s, 1H, ArCONH), 8.366/9.4455, (s, 1H, Ar-CH), 7.867/9.3966, (s, 1H, Ar-CH), 7.566/9.0367, (s, 1H, Ar-CH), 7.496/7.9468, (s, 1H, vinyl-CH⚌C), 7.447/7.8184, (s, 1H, —CH⚌N), 6.877/7.0459, (s, 1H, pyrrole-CH), 6.127/6.8171, (s, 1H, pyrrole-CH), 4.230–4.299/4.3266–4.3243, (q, J = 6.90 Hz, 2H, ester-CH2), 1.263–1.310/1.2642–1.4851, (t, J = 7.05 Hz, 3H, ester-CH3). 13C NMR (300 MHz, DMSO-d6) δexp/calcd C2-126.04/128.6533, C3-122.58/126.153, C4-117.98/116.3081, C5-133.04/135.7931, C6-138.96/141.43, C7-116.64/93.2815, C8-113.89/115.4622, C10-158.74/160.6803, C13-53.03/65.6575, C14-15.94/16.7965, C15-144.05/133.6347, C18-157.66/155.1949, C31-128.88/136.2025, C32-127.15/130.9513, C33-139.64/148.7423, C34-119.57/122.1914, C35-139.64/147.9564, C36-127.15/125.8432.
3 Quantum chemical calculations
The quantum chemical calculations have been carried out using Gaussian 09 program package (Frisch et al., 2010) to predict the optimized geometry of molecular structure, 1H NMR chemical shifts, vibrational wave numbers, global and local reactivity descriptors, first hyperpolarizability at B3LYP functional and 6-31G(d, p) basis set. To estimate the thermodynamic parameters as enthalpy (H) and Gibbs free energy (G), the thermal corrections to these parameters are added to the calculated total energies. The 1H NMR chemical shifts are calculated using gauge including atomic orbital (GIAO) approach using IEFPCM model in appropriate solvent. Time dependent density functional theory (TD-DFT) is used to find the various electronic excitations and their nature within molecule. The optimized geometrical parameters are used in the vibrational wave numbers calculation to characterize all stationary points as minima and their harmonic vibrational wave numbers are positive. Gauss–View program is used for visualization of optimized geometry of molecule (Gauss-View).
4 Results and discussion
4.1 Molecular geometry and stability of conformers
The route for the formation of products (3a–c) involved in chemical reactions is shown in Scheme 1. The optimized geometry for the ground state lower energy conformer of (3a–c) is shown in Fig. 1. Selected optimized geometrical parameters of (3a–c), calculated at B3LYP/6-31G(d, p) are listed in Supplementary material (S Table 1). The ground state lower energy conformer of all the molecules (3a–c) possesses C1 symmetry. The asymmetry of the N1—C2 and N1—C5 bonds i.e. difference between their bond lengths can be explained due to the presence of the two different groups as ‘ethyl 2-cyano-acrylate’ and ‘aroyl hydrazonomethyl’ at C2, C5 of pyrrole ring, respectively. The E–configuration about the vinyl C6⚌C7 bond with respect to the higher priority group ethoxycarbonyl and pyrrole gives lower energy conformer. The molecule also exists in E-configuration with respect to the pyrrole and —NH—CO—Ar group located on the opposite side of the Schiff base C15⚌N16 bond. The E-configuration about Schiff base C15⚌N16 bond is not only observed in our theoretical study but also reported in crystal structure of various aroylhydrazone derivatives (Wei, 2012; Zong, 2012). In (3a–c), due to the presence of intramolecular hydrogen bonding (N1—H27/26/20⋯N9) the pyrrole N—H bond is elongated than free N—H bond (Rawat and Singh, 2014a, 2014b, 2014c, 2015a, 2015b; Singh et al., 2013a, 2013b, 2013c, 2013e, 2013e, 2014a, 2014b).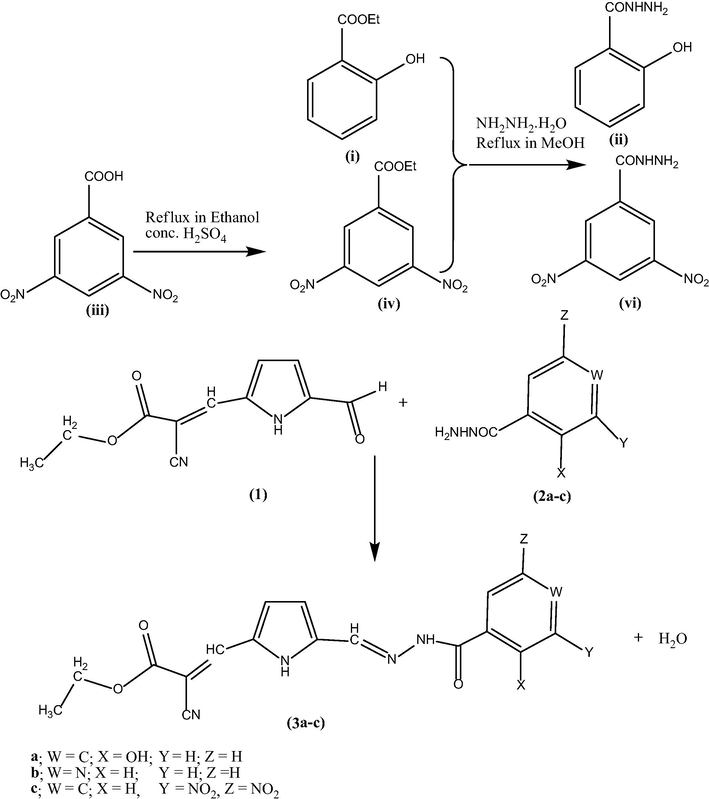
Optimized geometry of the reactants (1, 2a–c) and products (3a–c).
Optimized geometry for the ground state lower energy conformer of (3a–c).
4.2 1H and 13C NMR spectroscopy
NMR calculations are now attainable and accurate enough to be useful exploring the relationship between chemical shift and molecular structure. Density functional theory (DFT) has emerged in recent years as a promising alternative to conventional ab initio methods in quantum chemistry. It therefore seems reasonable to investigate in detail how well DFT predicts magnetic response properties, in particular shielding tensors (Kim et al., 2000). A number of methods have been developed for the calculation of molecular second-order magnetic response properties. It is generally accepted that accurate prediction of these properties within the finite basis approximation, requires gauge-invariant procedures (Wolinski et al., 1990). The geometry of (3a–c) compounds, together with that of tetramethylsilane (TMS) is fully optimized. 1H and 13C NMR chemical shifts are calculated with GIAO approach at B3LYP/6-31G(d, p) method. The experimental 1H NMR spectra of products (3a–c) in DMSO-d6 are given in Supplementary Fig. S1a–c. The 1H NMR chemical shifts (δ/ppm) of (3a–c) are assigned in Sections 2.4-2.6. In the 1H NMR spectra of (3a–c), a quartet and a triplet chemical shift designate the presence of ester —CH2, —CH3 group in all the molecules. In (3a–c), the observed chemical shifts as a singlet at 7.404, 8.463, 7.447 ppm indicate presence of the hydrazone linkage (CH⚌N—NH—) in these molecules. The big difference in the certain experimental and calculated chemical shift of the compound is due to the formation of intermolecular hydrogen bonding with solvent. The pyrrolic NH and hydrazide NH appear at downfield in experimental spectra due to the formation of intermolecular hydrogen bonding with DMSO solvent. The higher chemical shift for ArCONH is observed due to intermolecular hydrogen bonding with solvent than calculated in gaseous state. In order to compare the chemical shifts, correlation graph between the experimental and calculated 1H NMR chemical shifts is shown in Supplementary Fig. S2. The correlation coefficients (R2 = 0.937, 0.893, 0.927) for (3a–c), show that experimental 1H NMR data are consistent with the calculated data from optimized structure of probed molecules.
The experimental 13C NMR spectra of (3a–c) in DMSO-d6 are given in Supplementary Fig. S3a–c. Additional support for molecular structures of (3a–c) is provided by their 13C NMR spectra, in which chemical shifts of the C15 carbon atom at 145.38, 143.39, 144.05 ppm for —CH⚌N—NH and C18 carbon atom at 168.54, 160.46, 157.66 ppm for —NH—CO—Ar confirm the aroylhydrazone character of these molecules. In order to compare the chemical shifts, correlation graph between the experimental and calculated 13C NMR chemical shifts is shown in Supplementary Fig. S4. The correlation coefficients (R2 = 0.937, 0.952, 0.967) show that there is a good agreement between experimental and calculated 13C NMR chemical shifts.
The mass spectra of (3a–c) showed [M + 1] peaks with agreement of their molecular formula. The molecular ion M + 1 peaks observed at m/z: 353, 338 and 427 corresponding to their relative molecular mass peaks in 3a, 3b and 3c, mass spectra respectively are shown in Supplementary material Fig. S5a–c.
4.3 Electronic absorption (UV–Visible) and natural bond orbitals (NBO) analysis
The nature of excitations in the observed UV–Visible spectra of (3a–c) compounds has been studied by the time dependent density functional theory (TD-DFT). The calculated and experimental electronic excitations of high oscillatory strength are listed in Table 1. The experimental UV–Visible spectra of (3a–c) are shown in Fig. 2 and molecular orbital plots are shown in Supplementary material Fig. S6. The vicinal orbitals of HOMO and LUMO play the same role of electron donor and electron acceptor, respectively. The HOMO–LUMO energy gap is an important reactivity index. The HOMO–LUMO energy gap of 3.25, 3.26, 2.74 eV for (3a–c), reflects the chemical reactivity of these molecules.
Excitations
E (eV)
(f)
λ calcd.
λ obs.
Assignment
(3a)
H-2 → L
4.1973
0.1662
295.39
244
π → π∗
H → L
2.9078
0.9548
426.38
412
π → π∗
(3b)
H → L + 2
4.9148
0.4711
252.27
233
n → π∗
H → L
2.980
0.9518
416.01
389
n → π∗
(3c)
H-6 → L + 1
4.910
0.5936
252.47
242
π → π∗
H → L + 2
3.002
0.8984
413.00
402
n → π∗
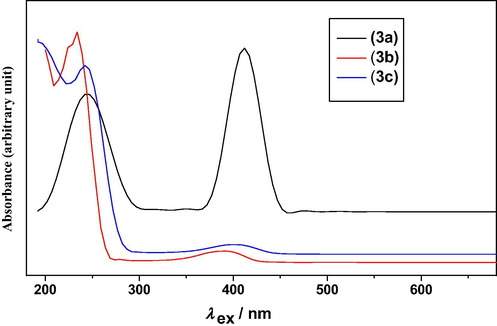
The experimental UV–Visible spectra of (3a-c).
A combined experimental and theoretical UV–Visible spectrum analysis of (3a) indicates that the first observed electronic transitions at λ = 244 nm correspond to the electronic excitations calculated at λ = 295.39 nm, f = 0.1662. The second observed λmax at 412 nm is in good agreement with the calculated λmax = 426.38 nm, f = 0.9548. For (3b), the observed λ at 233, 389 nm agrees with the calculated electronic excitations at λ = 252.27 nm, f = 0.4711, λ = 416.01 nm, f = 0.9518, respectively. The UV–Visible spectrum analysis of (3c) indicates that the observed λ at 242, 402 nm matches well with the electronic excitations calculated at λ = 252.47 nm, f = 0.5936; λ = 413.00 nm, f = 0.8984, respectively. Therefore, the observed λ is slightly blue shifted compared with the calculated λ. On the basis of molecular orbital coefficients and molecular orbital plots the nature of electronic excitations for (3a) is assigned as π → π∗, whereas for (3b), (3c) as n → π∗.
A useful aspect of the NBOs is that it provides an accurate method for studying intramolecular bonding and interaction among bonds and also gives an efficient basis for investigating charge transfer or conjugative interaction in various molecular systems. Second-order perturbation theory analysis of the Fock matrix in NBO basis for (3a–c) is presented in Supplementary material (S Table 2a–c). In (3a–c), the interactions π(C2—C3) → π∗(C4—C5), π(C4—C5) → π∗(C2—C3) are responsible for the conjugation of respective π-bonds in pyrrole ring due to the high electron density at conjugated π bonds (1.666–1.678) and low electron density at π∗ bonds (0.420–0.433) and stabilized the molecules with energy 83.01–95.60 kJ/mol. The interaction n1(N1) → π∗(C2—C3)/π∗(C4—C5) shows that loan pair of pyrrole N atom takes part in electron delocalization within ring. In the same manner, the π → π∗ interactions of benzene or pyridine ring designate the conjugation of respective π-bonds within ring and stabilized the molecules in a broad region 57.43–110.52 kJ/mol, due to presence of the different substituent at benzene ring as 2-OH, 3,5-NO2, for (3a), (3c) and pyridine ring in (3b), respectively. The interactions π(C2—C3) → π∗(C6—C7), π(C6—C7) → π∗(C2—C3) are responsible for the conjugation of bonds C2—C3, C6—C7 with C2—C6 and stabilized the molecule with energy 50.70–104.20 kJ/mol. In the same manner, the interactions π(C4—C5) → π∗(C15—N16) and π(C15—N16) → π∗(C4—C5) are responsible for the conjugation of these bonds with C5—C15. It is to be noticed that the charge transfer interactions are formed by the orbital overlap between bonding (π) and antibonding (π∗) orbitals, which results in intramolecular charge transfer (ICT) causing stabilization of the system. The movement of π-electron cloud from donor to acceptor i.e. intramolecular charge transfer (ICT) can make the molecules more polarized, which must be responsible for the NLO properties of molecules. Therefore, the titled compounds may be used for non–linear optical materials. The primary hyperconjugative interaction π1(C8—N9) → σ∗(N1—H27)/σ∗(N1—H26)/σ∗(N1—H20) designates the intramolecular hydrogen bonding as N1—H27⋯N9, N1—H26⋯N9, N1—H20⋯N9 in molecules (3a–c), respectively. In (3a), the interaction n2(O19) → σ∗(O26—H42) shows existence of intramolecular hydrogen bonding as O26—H42⋯O19. The presence of nitro group in (3c) stabilized the molecule to a greater extent of 679.30, 693.10 kJ/mol due to the interactions n3(O41) → π∗(N40—O42), n3(O38) → π∗(N37—O39), respectively. In (3a–c), the secondary hyperconjugative interactions σ → σ∗ associated with pyrrole and benzene ring stabilized the molecules with energy 9.57–19.48 kJ/mol. Selected Lewis orbitals (occupied bond or lone pair) of (3a–c) with their NBO hybrid orbitals are listed in Supplementary material (S Table 3a–c). The NBO hybrid orbitals analysis shows that all the N—H/C—N bond orbitals are polarized toward the nitrogen atom (ED = 57.39 − 74.75% at N), whereas C—O/N—O bond orbitals toward oxygen atom (ED = 51.34 − 79.02% at O). The electron density distribution (occupancy) around the imino group (N—H) also influences the polarity of the compound. Therefore, they consist with the maximum electron density on the oxygen and nitrogen atoms, which is responsible for the polarity of the molecule.
4.4 Emission (photoluminescence) spectroscopy
The optical properties of (3a–c) have been explored using electronic absorption and photoluminescence spectra. Quantum mechanically, Photoluminescence (PL) can be described as an excitation to a higher energy state and then a return to a lower energy state accompanied by the emission of a photon. The emitted radiation is generally of a longer wavelength than the wavelengths imposed on the photoluminescent material and difference between wavelength absorption maxima and wavelength emission maxima is known as the Stoke’s shift.
For any photoluminescent species, the quantum yield (ΦF) of its luminescence is a basic property, and its measurement is an important step in the characterization of the species. According to the definition of the ΦF (Brouwer, 2011), only two quantities need to be known, viz. the number of photons absorbed and the number of photons emitted per unit of time. The quantum yields (ΦF) of (3a–c) were determined in dilute solutions with an absorbance below 0.1 at the excitation wavelength. Quinine sulfate (in 0.1 M H2SO4 λex = 347 nm, Φ = 0.57) was used as a standard (Lakowicz, 2006). Quantum yields were calculated using the following equation: where Fs and Fr denote the area under the fluorescence band of sample and reference, fr and fs denote the absorbance at the excitation wavelength of sample and reference, and η denotes the refractive index. Refractive index was calculated by the Lorentz–Lorentz equation ((4π/3) ∗ (αλ/Vmol)). Integration of the emission bands was performed using Origin 7.1.
The experimental emission spectra of (3a–c) were recorded in DMSO, at excitation λex.max, and are shown in Fig. 3. The experimental PL spectral data of (3a–c) are listed in Table 2 with quantum yield. The most striking feature is that the (3a) gives an intense emission at λem.max = 572 nm in yellow region upon irradiation by ultraviolet light λex.max = 244 nm, whereas another intense emission at λem.max = 521 nm in green region upon irradiation by visible light λex.max = 412 nm. The compound (3b) gives two intense emissions at λem.max = 480, 522 nm in blue and green region, respectively, upon irradiation by ultraviolet light λex.max = 233 nm. Another ultraviolet irradiation at λex.max = 389 nm gives also an intense emission at same λem.max = 480 nm in blue region. The compound (3c) gives an intense emission at same λem.max = 520 nm in green region upon irradiation by λex.max = 242, 440 nm. Therefore, the emission spectra show that (3a–c) are good photoluminescent material due to intense emission at higher wavelength in yellow, green and blue region with high Stoke’s shift in the region 80–328 nm. s–strong, m–medium, w–weak, S = [λem.max − λex.max].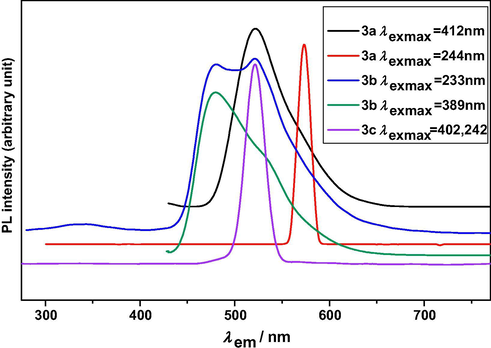
The experimental emission spectra of (3a–c).
λex.max (nm)
λem.max (nm)
S (nm)
(ΦF)
(3a)
244
572(s) [yellow]
328
0.563
412
521(s) [green]
109
0.269
(3b)
233
480(s) [blue]
247
0.411
522(s) [green]
289
389
480(s) [blue]
91
0.432
(3c)
242, 402
520(s) [green]
278, 80
0.613
The data with the model compounds have shown that the quantum yield of aromatic hydrazone of 3a is essentially affected by an ortho-hydroxyl substituent. Furthermore, the compound 3c shows only one excitation peak at 402 and 442 excitation wavelengths.
4.5 Vibrational band assignments
The aim of the vibrational analysis is to decide which of the vibrational modes in the molecule give rise to each of the observed bands at specific wave numbers in the FTIR spectra. The functional groups present in the molecule were identified and a satisfactory vibrational band assignment has been made for the fundamental modes of vibration by observing the position, shape and intensity of the bands. Vibrational frequencies of similar pyrrole hydrazones (Singh et al., 2013a, 2013b, 2013c, 2013d, 2013e, 2014a, 2014b; Rawat and Singh, 2015a, 2015b), compounds and their derivatives have been taken into consideration for the assignment of fundamental vibrations of studied compounds (3a–c). The theoretical (selected) and experimental vibrational modes of (3a–c), calculated at B3LYP/6-31G(d, p) method and their approximate assignments are given in Table 3. Comparison between experimental and theoretical IR spectra in the region 4000–400 cm−1 is shown in Supplementary Fig. 7a–c. The calculated vibrational wave numbers are higher than their experimental values for the majority of the normal modes. Two factors may be responsible for the discrepancies between the experimental and computed wave numbers. The first is caused by the environment (gas and solid phase) and the second is due to the fact that the experimental values are an anharmonic wave numbers while the calculated values are harmonic ones. Therefore, calculated wave numbers are scaled down using scaling factor 0.9608 (Rawat and Singh, 2014a, 2014b, and 2019), to discard the anharmonicity present in real system. Proposed assignment for different vibrational modes: Types of vibrations: νs – symmetric stretching, νas – asymmetric stretching, δsc – scissoring, ρ – rocking, ω – wagging, δ – deformation, δas – asymmetric deformation, R1 – benzene/pyridine ring.
(3a)
(3b)
(3c)
Approximate assignment
calcd.
exp.
calcd.
exp.
calcd.
exp.
3448
3463
3451
3422
3450
3460
νN—H-pyrrole
3397
3286
3375
3237
3376
3293
νN—H-ArCONH
3164
3286
–
–
–
–
νO—H-hydroxyl
3140
3131
3129
–
3141
3069
νC—H-pyrrole
3066
3071
3071
3070
3070
3069
νC—H-benzene
3017
3024
3010
2989
3011
2982
νas-CH ester Me
2983
2960
2987
2957
2987
2955
νas-CH ester CH2
2943
2851
2947
2838
2947
2869
νC—H-Schiff base
2945
–
2946
–
2946
–
νs-CH ester CH2
2936
–
2940
–
2939
–
νs-CH ester Me
2226
2216
2229
2217
2228
2225
νC8N9-cyanide
1722
1709
1724
1707
1729
1694
νC10⚌O11-ester carbonyl
1667
1645
1730
1670
1725
1655
νC18⚌O19-ArCONH
1608
1591
1610
1588
1623
1640
νC15⚌N16-Schiff base
–
–
–
–
1598
1585
νas-NO2
1598
–
1582
–
1598
–
νC⚌C-benzene
1575
–
1577
1554
1578
1553
νC6⚌C7-vinyl
1534
1555
1533
–
1534
1530
νC⚌C-pyrrole
1504
1493
1497
1480
1500
1470
ρNH-ArCONH
1475
–
1471
–
1490
–
δsc-ester CH2
1470
1424
1464
1424
1464
1423
ν[CC+CN]-pyrrole ring
1442
–
1441
–
1440
–
δas-ester Me
1406
1408
1406
1407
1405
1405
ν[CC+CN]-pyrrole ring
1382
–
1383
–
1383
–
δas-ester Me
1350
–
1350
–
1350
–
ω-ester CH2
–
–
–
–
1341
1343
νs-NO2
1326
1326
1319
1329
1303
1321
ν[CC+CN]-pyrrole ring
1289
1294
1289
1287
1287
1286
δCCH-pyrrole
1242
1236
1235
1235
1241
1238
νC10O12-ester + δOCO-ester
1229
1191
1213
1196
1207
1199
νC18—N17 —HN—
COAr
1167
1145
1167
1148
1184
1139
[δCCH + δCNH]-pyrrole
1131
1095
1134
1093
1137
1098
νN16—N17 —CH⚌N—
NH—
1083
1057
1084
1064
1083
1059
νO12—C13-ester O—
C—C
1035
1020
1036
1015
1037
1012
[δCCH + δCNH]-pyrrole
1008
–
1008
–
1008
–
νC13—C14 + νO12—C13
992
942
973
944
978
961
δtrigonal-R1
905
905
907
876
933
938
ωC15H38-Schiff base
884
884
887
842
897
875
[δO19C18N17 + δN16N17C18]
869
–
870
–
869
–
ωC—H-pyrrole
–
–
–
–
828
828
δNO2 + ωC—H-benzene
826
793
826
791
827
793
ωC—H-benzene + δNO2
767
745
768
756
769
759
δ-pyrrole ring + δC5C15N16 + δC15N16N17
725
696
724
720
740
738
δas–R1 + δoop-NO2
655
–
655
–
647
–
R1-puckering
626
668
628
648
628
663
ωN—H-pyrrole
514
461
506
493
529
497
ωN—H-ArCONH
4.5.1 N—H and O—H vibrations
The N—H stretching of C⚌N—H group occurs in the region 3400–3100 cm−1. Usually the frequency of this vibration is decreased in the presence of hydrogen bond (Bellamy, 1975. In the FT-IR spectra of (3a–c), the N—H stretches of pyrrole (νN1—H27/H26/H20) are observed at 3463, 3422, 3460 cm−1, whereas these are calculated at 3448, 3451, 3450 cm−1, respectively. The observed pyrrole νN—H deviates slightly from the free νN—H of pyrrole at 3475 cm−1, reported in the literature (Giuliano et al., 2010). Therefore, the maximum red shift of 53 cm−1 in observed νN1—H27/H26/H20 compared to the reported free νN–H indicates the involvement of the pyrrole νN—H group in hydrogen bonding. The theoretical IR spectra show that N—H wagging modes of pyrrole (ωN1—H27/26/20) are moderately active at 626–8 cm−1, whereas these are observed at 668, 648, 663 cm−1, respectively. The N—H stretch of —CH⚌N—NH— part (νN17—H37/36/30) is observed at 3237–3293 cm−1, whereas these are calculated at 3375–3397 cm−1 in theoretical IR spectra. The N—H rocking and wagging modes of —CH⚌N—NH— assigned at 1497–1504, 506–529 cm−1 agree well with the observed band at 1470–1493, 461–497 cm−1, experimentally.
The non-hydrogen bonded or free hydroxyl group of phenols absorbs strongly in the 3700–3584 cm−1 region. In o-hydroxy aryl acids and esters, the vibrational wave number of hydrogen-bonded O—H group shifts toward lower wave number up to 300 cm−1 due to the presence of intramolecular hydrogen bonding (Silverstein and Webster, 1963). In (3a), the observed O—H stretching vibration (νOH) at 3286 cm−1 agrees well with the hydrogen-bonded O—H group. Therefore, solid state spectrum of (3a) attributes to the vibration of hydrogen-bonded O—H group.
4.5.2 C—H vibrations
According to the Internal coordinate system recommended by Pulay et al. (1979), CH2 group associates with six types of vibrational frequencies. The scissoring and rocking deformations belong to polarized in-plane vibration, whereas wagging and twisting deformations belong to depolarized out-of-plane vibration. In (3a–c), a weak band for νasCH2 calculated at 2983–2987 cm−1 corresponds to the observed wave number at 2955–2960 cm−1. The observed band at 2955–2960 cm−1 also matches with the reported band in the literature in the region 3000 ± 50 (Gambino et al., 2007). The calculated νsCH2 at 2945–6 cm−1 matches well with the reported band in the literature in the region 2965 ± 30, for symmetric C—H stretching vibrations of CH2 (Gambino et al., 2007). The (δsc) and (ω) deformation modes for ester CH2 assigned at 1471–1490, 1350 cm−1 match well with the reported bands in the literature in the region 1455 ± 55, 1350 ± 85, respectively (Gambino et al., 2007). The calculated νasCH3 and νsCH3 at 3010–3017, 2936–2930 cm−1 correspond to the reported band in the literature in the region 2985 ± 25, 2970 ± 30 for asymmetric, symmetric C—H stretching vibrations of CH3, respectively. The calculated ester νasCH3 at 3010–3017 cm−1 also corresponds to the observed wave number at 2982–3024 cm−1, experimentally. In theoretical IR spectra, the wave numbers at 1440–2, 1382–3 cm−1 display the δas and δs deformation modes of ester methyl, respectively. The calculated weak bands for νC—H of benzene at 3066–3071 cm−1 match well with the observed wave numbers at 3069–3071 cm−1. The Schiff base νC—H stretches are assigned at 2943–7 cm−1, whereas these are observed at 2838–2869 cm−1 in FT-IR spectrum. The observed Schiff base νC—H stretches are responsible for the formation of hydrazone linkage in (3a–c).
4.5.3 C≡N vibrations
In theoretical IR spectra of (3a–c), the calculated wave numbers at 2226–2229 cm−1 designate the presence of C≡N stretching vibrations and match well with the observed wave numbers at 2216–2225 cm−1. The free C≡N stretching vibrations are reported in the literature in the region 2240–2260 cm−1 (Silverstein and Webster, 1963). Therefore, the red shift in the observed C≡N stretches compared with the free C≡N stretches indicates involvement of the C≡N group in intramolecular hydrogen bonding.
4.5.4 C⚌O and C—O vibrations
The ester carbonyl stretching vibration νC⚌O is expected in the region 1750–1715 cm−1 (Silverstein and Webster, 1963) and in the present study these modes are assigned at 1722–1729 cm−1 using DFT calculation. The experimental FT-IR spectra of (3a–c) give this mode at 1694–1709 cm−1. The “C—O stretching vibrations” of esters actually consist of two asymmetrical coupled vibrations as O— C(⚌O)—C, O— C—C and these bands occur in the region 1300–1000 cm–1 (Silverstein and Webster, 1963). The calculated wave number at 1083–4 cm−1 demonstrates the O— C—C stretching vibration (νO12—C13) and corresponds to the observed wave numbers at 1057–1064 cm−1, experimentally. In theoretical IR spectra, a combination band of the ‘O— C(⚌O)—C stretching vibration (νC10—O12)’ and ‘ester O—C—O deformation’ at 1235–1242 cm−1 is in good agreement with the observed bands at 1235–8 cm−1 in experimental FT-IR spectra. In (3a), (3b), (3c), ArCONH carbonyl stretches (νC18⚌O19) are observed at 1645, 1670, 1655 cm−1, respectively. Therefore, in (3a) the red shift in the observed νC18⚌O19 stretches compared with the (3b), (3c) indicates involvement of the C18⚌O19 group of (3a) in hydrogen bonding.
4.5.5 C⚌C, C—C vibrations
The calculated C⚌C stretches of benzene at 1582–1598 cm−1 match well with the reported band in the literature in the region 1600–1585 cm−1 (Gambino et al., 2007). The C⚌C stretches of pyrrole are assigned at 1533–4, 1464–1470 cm−1, whereas these DFT modes are observed at 1530–1555, 1423–4 cm−1, respectively. In DFT calculation, bands at 973–992 cm−1 display the trigonal deformation of benzene ring (δ-R1), whereas these are observed at 942–961 cm−1. The calculated modes at 1035–1037 cm−1 designate to the δC—C—H associated with pyrrole and correspond to the observed wave numbers at 1012–1020 cm−1. The observed bands at 1553–4 cm−1 designate the presence of the vinyl group (νC6⚌C7). The DFT mode at 1008 cm–1 assigns to the C—C stretching vibration of ester (νC13—C14). In theoretical IR spectrum of (3a–c), a weak band for ring puckering vibration (a torsional mode) of benzene is assigned at 647–655 cm−1. The DFT modes for asymmetric deformation of benzene (δas-R1) at 724–740 cm−1 agree well with the observed bands at 696–738 cm−1, experimentally.
4.5.6 C⚌N, C—N and N—N vibrations
For (3a–c), Schiff base υC⚌N modes assigned at 1608–1623 cm−1 correspond to the observed wave numbers at 1588–1640 cm−1, experimentally. The presence of the υC⚌N bands in (3a–c) confirms hydrazone linkage in all the investigated molecules. The calculated bands for υC⚌N also match with the reported band at 1602 cm−1 in the literature (Gambino et al., 2007). The observed C—N stretches as νC18—N17 at 1191–1199 cm−1 are in agreement with the calculated wave numbers at 1207–1229 cm−1. The C—N stretching vibration is also active in the region 1275 ± 55 cm−1. The DFT modes for N—N stretches as νN16—N17 at 1131–1137 cm−1 correspond to the observed wave numbers at 1093–1098 cm−1.
4.5.7 N⚌O vibrations
The molecules under investigation (3c) possess two nitro groups. The nitro groups show two types of stretching vibrations as asymmetric (νas) and symmetric (νs). The νas stretches are always observed at higher wave number than νs stretches. In (3c), the νas and νs stretches of nitro groups calculated at 1598, 1341 cm−1 are in agreement with the observed wave numbers at 1585, 1343 cm−1, respectively. The νas and νs stretching vibrations of nitro group are also reported in the literature at 1600, 1319 cm−1, respectively (Silverstein and Webster, 1963). The calculated deformation modes of nitro groups at 827 cm−1 are observed at the same wave number in experimental FT-IR spectrum.
4.6 Quantum theory of atoms in molecules (QTAIM) analysis
Topological as well as geometrical parameters are useful tool to characterize the strength and nature of hydrogen bond (Bader, 1990; Lee et al., 1994). Molecular graphs of (3a–c) using AIM program at B3LYP functional are shown in Supplementary Fig. S8. Topological as well as geometrical parameters calculated at B3LYP/6-31G(g, p) and ωB97X/6-31G(d, p) level for bonds of interacting atoms for 3a, 3b and 3c are given in Tables 4 and 5, respectively. For the intramolecular interactions N1—H27/H26/H20⋯N9 electron density (ρH…A) and its Laplacian (∇2ρBCP) follow the Koch and Popelier (1995) criteria and the distance between interacting atoms (dH27/H26/H20⋯N9 = 2.44–2.45 Å) is less than the sum of van der Waals radii of these atoms. Therefore, these interactions are in the category of hydrogen bonds. The nature of N1—H27/H26/H20⋯N9 hydrogen bonds is weak due to (∇2ρBCP) > 0 and HBCP > 0, whereas O26—H42⋯O19 is a medium hydrogen bond due to (∇2ρBCP) > 0 and HBCP < 0. In this article, QTAIM theory is used to estimate hydrogen bond energy (E), and the energy of intramolecular hydrogen bonds N1—H27/H26/H20⋯N9, O26—H42⋯O19 is calculated as 9.23, 9.23, 9.41, 55.72 kJ/mol, respectively. In (3a), the presence of bond critical point (BCP) at H37···H38 contact designates presence of the dihydrogen bonding due to short dH37⋯H38 of 1.97 Å. Energy of hydrogen bonding is calculated using AIM and DFT calculations. With respect to change of both the method used and the basis set the reliability and stability of values of QTAIM parameters have been studied and found that they were almost independent of basis set in case of used B3LYP functional in DFT (Jablonski and Palusiak, 2010). Energy of hydrogen bonding has been calculated using both B3LYP functional and ωB97X with 6-31G(d, p) basis set for describing hydrogen bond interactions in molecules. The calculated values tabulated in Tables 4 and 5 found to vary only slightly. ρBCP, ∇2ρBCP, GBCP, VBCP, HBCP in a.u. and EHB in (kJ/mol).
B3LYP/6-31G(d, p)
Interactions
ρBCP
∇2ρBCP
GBCP
VBCP
HBCP
EHB
(3a)
O26—H42⋯O19
0.0515
0.1487
0.0398
−0.0424
−0.0026
−13.30
N1—H27⋯N9
0.0118
0.0428
0.0088
−0.0070
0.00184
−2.196
H37⋯H38
0.0128
0.0543
0.0109
−0.0082
0.00267
−2.572
(3b)
N1—H26⋯N9
0.0118
0.0428
0.0088
−0.0070
0.0018
−2.196
(3c)
N1—H20⋯N9
0.0120
0.0435
0.0090
−0.0071
0.0018
−2.227
ωB97X/6-31G(d, p)
(3a)
O26—H42⋯O19
0.0475
0.1439
0.0375
−0.0390
−0.0015
−12.26
N1—H27⋯N9
0.0121
0.0445
0.0092
−0.0074
0.0018
−2.329
H37⋯H38
0.0126
0.0556
0.0110
−0.0081
0.00287
−2.564
(3b)
N1—H26⋯N9
0.01212
0.0443
0.0092
−0.0073
0.0018
−2.315
(3c)
N1—H20⋯N9
0.01213
0.0443
0.0092
−0.0074
0.0018
−2.323
B3LYP/6-31G(d, p)
Interactions (D—H⋯A)
dD—H (Å)
dH⋯A (Å)
dD⋯A (Å)
D—H⋯A (°)
(rH + rA) (Å)
(3a)
N1—H27⋯N9
1.01
2.45
3.31
141.90
2.75
O26—H42⋯O19
0.99
1.66
2.56
148.17
2.72
H37⋯H38
–
1.97
–
–
–
(3b)
N1—H26⋯N9
1.01
2.45
3.31
141.89
2.75
(3c)
N1—H20⋯N9
1.01
2.44
3.30
142.12
2.75
ωB97X/6-31G(d, p)
(3a)
N1—H27⋯N9
1.01
2.44
3.29
141.73
2.75
O26—H42⋯O19
0.98
1.69
2.57
147.03
2.72
H37⋯H38
–
2.00
–
–
–
(3b)
N1—H26⋯N9
1.01
2.44
3.29
141.58
2.75
(3c)
N1—H20⋯N9
1.01
2.45
3.31
142.02
2.75
4.7 Chemical reactivity
The chemical reactivity of molecule is described in three ways as: (i) Molar refractivity (MR) and (ii) Global and local electronic reactivity descriptors.
4.7.1 Molar refractivity (MR)
Molar refractivity (MR) is an important property used in quantitative structure property relationship (QSPR). It is directly related to the refractive index, molecular weight and density of steric bulk and responsible for the lipophilicity and binding property of investigated system. It can be calculated by the Lorentz–Lorentz equation (Padrón et al., 2002; Verma and Hansch, 2005) and defined as where n is the refractive index, MW is the molecular weight, ρ is the density, (MW/ρ) is the molar volume, N is the Avogadro Number, and α0 is the polarizability of molecular system. This equation holds for both liquid state and solid state of system. Using this equation, the molar refractivity (MR) for (3a–c) has been calculated as 109.28, 102.96, 119.14 esu, respectively. Molar refractivity is found to be related to the London dispersive forces that act in the drug–receptor interaction (Padrón et al., 2002). The molar refractivity has been reported as an important parameter in modulating the antimalarial activity (Uniyal et al., 2010), through charge transfer between drug and globin protein, indicating that the studied compounds may have activity against malarial disease. Molar refractivity based binding property of 4-Quinolinyl and 9-Acrydinyl hydrazones has been found to be potent antimalarial agents and active against C–Q resistant clone K1 Plasmodium falciparum strain (Uniyal et al., 2010).
4.7.2 Electronic reactivity descriptors
4.7.2.1 Global and Local reactivity descriptors
The chemical reactivity and site selectivity of the molecular systems have been determined on the basis of Koopman’s theorem (Parr and Yang, 1989). Global reactivity descriptors as electronegativity (χ) = −1/2(εLUMO + εHOMO), chemical potential (μ) = 1/2 (εLUMO + εHOMO), global hardness (η) = 1/2 (εLUMO − εHOMO), global softness (S) = 1/2η and electrophilicity index (ω) = μ2/2η are highly successful in predicting global reactivity trends (Rawat and Singh, 2014a, 2014b, 2019; Singh et al., 2013a, 2013b, 2013c, 2013d, 2013e; Singh and Rawat, 2013). According to Parr et al., electrophilicity index (ω) is a global reactivity index similar to the chemical hardness and chemical potential. This is positive and definite quantity and measures the stabilization in energy when the system acquires an additional electronic charge (ΔN) from the environment. The energies of frontier molecular orbitals (εHOMO, εLUMO), energy gap (εLUMO − εHOMO), electronegativity (χ), chemical potential (μ), global hardness (η), global softness (S), global electrophilicity index (ω) for (1), (2a–c), (3a–c) and ECT for reactant systems as [1 ↔ 2a], [1 ↔ 2b], [1 ↔ 2c] are listed in Table 6. The global electrophilicity index (ω = 5.41, 5.50, 8.11 eV) for (3a–c) shows that they behave as strong electrophiles and their electrophilic power is in the following order as (3a) < (3b) < (3c). εH, εL, εH − εL, χ, μ, η, ω (in eV) and S (in eV−1).
εH
εL
εL–εH
χ = −μ
η
S
ω
ECT
(1)
−6.5133
−2.8547
3.6585
4.6840
1.8293
0.2733
5.9969
(2a)
−6.1057
−1.2585
4.8472
3.6821
2.4236
0.2063
2.7971
(1.0412)[1↔2a]
(3a)
−5.8276
−2.5710
3.2561
4.1995
1.6283
0.3071
5.4163
(2b)
−7.121
−1.633
5.487
4.377
2.743
0.182
3.492
(0.965)[1↔2b]
(3b)
−5.876
−2.606
3.269
4.241
1.634
0.305
5.501
(2c)
−7.6391
−3.3064
4.3326
5.4727
2.1664
0.2308
6.9130
(0.0342)[1↔2c]
(3c)
−6.0934
−3.3483
2.7451
4.7209
1.3726
0.3642
8.1189
Electrophilic charge transfer (ECT) = (ΔNmax)A − (ΔNmax)B is defined as the difference between the ΔNmax values of interacting molecules A and B. If we consider two molecules A and B approach to each other (i) if ECT > 0, charge flow from B to A (ii) if ECT < 0, charge flow from A to B. ECT is calculated as 1.04, 0.96, 0.03 for reactant systems [1 ↔ 2a], [1 ↔ 2b], [1 ↔ 2c], respectively i.e. ECT > 0, which indicates that charge flows from (2a–c) to (1). Therefore, (1) acts as global electrophile and (2a–c) as global nucleophile.
Selected electrophilic reactivity descriptors (fk+, sk+, ωk+) for reactant (1) and nucleophilic reactivity descriptors (fk−, sk−, ωk−) for reactants (2a–c), using Hirshfeld charges are given in Table 7. The maximum values of local electrophilic reactivity descriptors (fk+, sk+, ωk+) at aldehyde carbon C6 of reactant (1) indicate that this is the most electrophilic site. The nucleophilic reactivity descriptors (fk−, sk−, ωk−) of reactant (2a–c) show that N atom of NH2 (N10) is the most nucleophilic site. Therefore, the nucleophilic attack of N10 of reactant (2a–c) at the most electrophilic site C6 of reactant (1) confirms the formation of product molecules or Schiff base linkage (C15⚌N16) in aroylhydrazones (3a–c). Selected reactivity descriptors as Fukui functions (fk+, fk−), local softnesses (sk+, sk−), local electrophilicity indices (ωk+, ωk−) for (3a–c), using Mulliken atomic charges are given in Table 8. The maximum values of local electrophilic reactivity descriptors (fk+, sk+, ωk+) at vinyl carbon (C6) of (3a–c) indicate that this site is more prone to nucleophilic attack and favors formation of the new unsymmetrical dipyrromethanes by attack of 2-unsubstituted pyrrole nucleophile at (C6). In the same way, for (3a–c) the maximum values of the nucleophilic reactivity descriptors (fk−, sk−, ωk−) at N17 nitrogen atom of —CH⚌N—NH— indicate that this site is more prone to electrophilic attack. fk+, fk– (in e); sk+, sk– (in eV−1) and ωk+, ωk– (in eV). fk+, fk– (in e); sk+, sk– (in eV−1) and ωk+, ωk– (in eV).
Sites
fk+
sk+
ωk+
Sites
fk–
sk–
ωk–
(1)
C6
0.1170
0.0320
0.7021
(2a)
N9
0.0459
0.0095
0.1281
C12
0.0384
0.0105
0.2307
N10
0.0672
0.0140
0.1873
(2b)
N9
0.1579
0.0287
0.5515
N10
0.2914
0.0530
1.0176
(2c)
N9
0.1066
0.0246
0.7374
N10
0.1312
0.0302
0.9071
Sites
fk+
sk+
ωk+
Sites
fk–
sk–
ωk–
(3a)
C6
0.0556
0.0170
0.3011
(3a)
N1
0.0005
0.0001
0.0031
C10
0.0434
0.0133
0.2350
N17
0.0277
0.0085
0.1504
C15
0.0411
0.0126
0.2230
(3b)
N1
0.0005
0.0002
0.0032
C18
0.0354
0.0108
0.1921
N17
0.039
0.012
0.216
(3b)
C6
0.052
0.016
0.289
(3c)
N1
0.00047
0.00017
0.00384
C10
0.041
0.012
0.227
N17
0.03379
0.01231
0.27436
C15
0.040
0.012
0.222
C18
0.037
0.011
0.203
(3c)
C6
0.03070
0.01118
0.24926
C10
0.02457
0.00895
0.19951
C15
0.02466
0.00898
0.20021
C18
0.02351
0.00856
0.19091
4.8 Static dipole moment (μ0), mean polarizability (|α0|), anisotropy of polarizability (Δα) and first hyperpolarizability (β0)
As hyperpolarizability is difficult task to measure directly, computational calculation is an alternate choice and provides another method to investigate extensive properties of materials. Polarizabilities and hyperpolarizabilities are described as response of a system in the presence of an applied electric field (Kleinmann, 1962). In order to investigate the relationship between molecular structure and NLO response, first hyperpolarizability (β0) of this novel molecular system, and related properties (|α0| and Δα) are calculated using B3LYP/6-31G(d, p), based on the finite-field approach and their calculated values are given in Table 9. Total static dipole moment (μ0), mean polarizability (|α0|), anisotropy of polarizability (Δα) and first hyperpolarizability (β0), using x, y, z, components are defined as
μ0 = (μ2x + μ2y + μ2z)1/2
|α0| = 1/3(αxx + αyy + αzz)
Δα = 2–1/2[(αxx − αyy)2 + [(αyy − αzz)2 + (αzz − αxx)2 + 6α2xx]1/2
β0 = [(βxxx + βxyy + βxzz)2 + (βyyy + βxxy + βyzz)2 + (βzzz + βxxz + βyyz)2]1/2
| (3a) | (3b) | (3c) | (3a) | (3b) | (3c) | ||
|---|---|---|---|---|---|---|---|
| μx | −1.942 | −0.733 | −2.707 | βxxx | 3117.61 | 578.17 | 2965.32 |
| μy | 5.625 | 5.057 | −5.255 | βxxy | −1200.40 | −1768.19 | 2380.40 |
| μz | 0.236 | −0.329 | −1.187 | βxyy | 22.32 | −170.65 | 402.86 |
| (μ0) | 5.955 | 5.120 | 6.030 | βyyy | 230.2 | 138.96 | −23.53 |
| αxx | 490.42 | 453.52 | 519.90 | βxxz | 26.76 | 30.69 | 487.92 |
| αyy | 291.82 | 271.67 | 319.86 | βyyz | −8.39 | −24.50 | −37.79 |
| αzz | 94.68 | 100.93 | 116.25 | βxzz | 11.81 | 18.18 | 7.99 |
| (|α0|) | 43.31 | 40.81 | 47.22 | βyzz | 1.94 | −8.92 | −6.13 |
| (Δα) | 135.74 | 124.90 | 143.15 | βzzz | 8.75 | 6.02 | 9.06 |
| (β0) | 28.48 | 14.62 | 35.76 |
Large value of particular component of the polarizability and hyperpolarizability indicates a substantial delocalization of charge in these directions. Since the value of the polarizability (|α0|) and first hyperpolarizability (β0) of Gaussian 09 output is reported in atomic unit (a.u.) these values are converted into electrostatic unit (esu) using converting factors as for α0: 1 a.u. = 0.1482 × 10−24 esu; for β0: 1 a.u = 0.008639 × 10−30 esu. The first hyperpolarizabilities (β0) of the title molecules (3a–c) are calculated as 28.48, 14.62, 35.76 × 10−30 esu, respectively with respect to p-Nitroaniline (p-NA) as a reference. The first hyperpolarizabilities of (3a–c) have been found to increase with electron withdrawing substituents and might be used as non-linear optical (NLO) material.
5 Conclusions
In this study we have presented a combined spectroscopic and quantum chemical studies on newly synthesized (3a–c) aryol hydrazones. The calculated 1H and 13C NMR chemical shifts are in agreement with the observed chemical shifts, experimentally. A combined experimental and theoretical UV–Visible spectral analysis indicates that observed wavelength absorption maxima (λex.max) have blue shifts compared to the calculated λex.max values. The molecular orbital coefficients and molecular orbital plots analysis suggests that the nature of electronic excitations involved in (3a) as π → π∗, whereas for (3b), (3c) as n → π∗. The emission spectra of (3a–c) show that title compounds are good photoluminescent material due to intense emission in green region and (3a), (3b) also show intense emission in yellow, blue region, respectively. The 1H NMR, NBO as well as vibrational analysis of (3a–c) are responsible for hydrogen bonding N1—H27/26/20⋯N9 due to downfield chemical shift, π1(C8—C9) → σ∗(N1—H27/H26/H20) interaction and vibrational red shift in the wave number of both proton donor pyrrole N–H and proton acceptor C≡N, respectively. AIM calculation also confirms the presence of these hydrogen bonds due to existence of the bond critical point at H27/26/20⋯N9 contact. Topological criteria (∇2ρBCP > 0 and HBCP > 0) show that nature of these hydrogen bonds is weak. The global electrophilicity index (ω = 5.41, 5.50, 8.11 eV) for (3a–c) indicates that these molecules behave as strong electrophiles. The electrophilic reactivity descriptors (fk+, sk+, ωk+) of (3a–c) indicate that the investigated molecules might be used as precursor for the target synthesis of new unsymmetrical dipyrromethane derivatives. These compounds exhibit strong effective intramolecular charge transfer (ICT) due to movement of π-electron cloud from donor to acceptor i.e. shows high polarity and responsible for the NLO properties of molecules. The first hyperpolarizabilities for (3a–c) (β0 = 28.48, 14.62, 35.76 × 10–30 esu) show that title molecules can be used as attractive material for non-linear optical (NLO) applications.
Acknowledgments
Authors are thankful to CSIR for financial support.
References
- Microwave assisted synthesis and antimicrobial activity of 2-quinoxalinone-3-hydrazone derivatives. Bioorg. Med. Chem.. 2010;18:214-221.
- [Google Scholar]
- Armbruster, F., Klingebiel, U., Noltemeyer, M.Z., 2006, Von Ketazinen zu 1,2-Diaza-3-phospha-cyclopent-5-enen,-penta-3,5-dienen,1,5-Diaza-2,6-diphospha-bicyclo[3.3.0]octa-3,7-dienund einem Cyclohexaphosphan. Naturforsch, 61b, 225–236.
- Atoms’ in Molecules. A Quantum Theory. Oxford: Oxford University Press; 1990.
- Synthesis and characterization of novel hydrazide-hydrazones and the study of their structure-antituberculosis activity. Eur. J. Med. Chem.. 2006;41:1253-1261.
- [Google Scholar]
- The Infrared Spectra of Complex Molecules (third ed.). London: Chapman and Hall; 1975.
- Standards for photoluminescence quantum yield measurements in solution. Pure Appl. Chem.. 2011;8:2213-2228.
- [Google Scholar]
- Res. J. Chem. Environ.. 2011;15:579-585.
- The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents III: the effect of the ligands on molecular targets involved in proliferation. Blood. 1999;94:781-792.
- [Google Scholar]
- Anticonvulsant properties of various acetylhydrazones, oxamoylhydrazones and semicarbazones derived from aromatic and unsaturated carbonyl compounds. Eur. J. Med. Chem.. 2000;35:241-248.
- [Google Scholar]
- Frisch, M.J., et al. Gaussian 09, Revision B.01, Gaussian Inc., Wallingford, CT, 2010.
- Vibrational spectra of palladium 5-nitrofuryl thiosemicarbazone complexes: experimental and theoretical study. Spectrochim. Acta Part A. 2007;68:341-348.
- [Google Scholar]
- Gauss-View 5.09 Program. Gaussian Inc., Wallingford, CT, 06492 USA.
- Genger, U.R., Pfeifer, D., Hoffmann, K., Flachenecker, G., Hoffman, A., Monte, C., 2008. In: Wolfbeis, O.S. (Ed.), Springer Series on Fluorescence, vol. 5. Springer, Berlin, pp. 65–99.
- Infrared spectra and photochemistry of matrix-isolated pyrrole-2-carbaldehyde. J. Phys. Chem. A. 2010;114:2506-2517.
- [Google Scholar]
- Gup, R. and Kırkan, B., 2005, Synthesis and spectroscopic studies of copper(II) and nickel(II) complexes containing hydrazonic ligands and heterocyclic coligand. 62, 1188–1195.
- Synthesis of some new hydrazide-hydrazones, thiosemicarbazides and thiazolidinones as possible antimicrobials. Eur. J. Med. Chem.. 1997;32:753-757.
- [Google Scholar]
- Porphodimethene-porphyrin interconversion: a tetrapyrrolic redox-switchable macrocycle. J. Am. Chem. Soc.. 2000;122:10476-10477.
- [Google Scholar]
- Ultrafast photoswitched charge transmission through the bridge molecule in a donor–bridge–acceptor system. J. Am. Chem. Soc.. 2000;122:5563-5567.
- [Google Scholar]
- Basis set and method dependence in atoms in molecules calculations. J. Phys. Chem. A. 2010;114:2240-2244.
- [Google Scholar]
- Kavlentis, E., 1988, Synthesis of phthalaldehyde bisguanylhydrazone (PABGH). Spectrophotometric analysis of Co(II)–Cu(II)–Ni(II) mixtures in the presence of several cations. Anal. Lett., 21, 689–697.
- Density functional theory/GIAO/CSGT studies of the 13 C NMR chemical shifts in 1-chlorosilatrane. Bull. Korean Chem. Soc.. 2000;21:148-150.
- [Google Scholar]
- Synthesis of pyrroles and a 1,2-Dihydropyrrolo[1,2-a]pyrazin-3(4H)-one identified as maillard reaction products. Acta Chem. Scand. B. 1979;33:125-132.
- [Google Scholar]
- Nonlinear dielectric polarization in optical media. Phys. Rev.. 1962;126:1977-1979.
- [Google Scholar]
- Characterization of C-H-0 Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem.. 1995;99:9747-9754.
- [Google Scholar]
- Principles of Fluorescence Spectroscopy (third ed.). Singapore: Springer Science & Business Media; 2006.
- An unusual type of H.H interaction: Ir–H–H–O and Ir–H–H–N hydrogen bonding and its involvement in σ-bond metathesis. J. Am. Chem. Soc.. 1994;116:11014-11019.
- [Google Scholar]
- Design, synthesis and in vitro antimalarial activity of an acylhydrazone library. Bioorg. Med. Chem. Lett.. 2006;16:31-35.
- [Google Scholar]
- Hydrazide-hydrazones in the synthesis of 1,3,4-oxadiazine, 1,2,4-triazine and pyrazole derivatives with anti-tumor activitiesOpen. Org. Chem. J.. 2010;4:8-14.
- [Google Scholar]
- Synthesis, characterization and antibacterial properties of some trivalent metal complexes with [(2-hydroxy-1-naphthaldehyde)-3-isatin]-bishydrazone. J. Enzyme Inhib. Med. Chem.. 2007;22:65-70.
- [Google Scholar]
- Molecular descriptor based on a molar refractivity partition using Randic-type graph-theoretical invariant. J. Pharm. Pharmaceut. Sci.. 2002;5:258-266.
- [Google Scholar]
- Density Functional Theory of Atoms and Molecules. Oxford, New York: Oxford University Press; 1989.
- Systematic ab initio gradient calculation of molecular geometries, force constants, and dipole moment derivatives. J. Am. Chem. Soc.. 1979;101:2550-2560.
- [Google Scholar]
- Synthesis and antimicrobial activity of some new hydrazones of 4-fluorobenzoic acid hydrazide and 3-acetyl-2,5-disubstituted-1,3,4-oxadiazolines. IL FARMACO. 2002;57:171-174.
- [Google Scholar]
- Spectral analysis, structural elucidation and evaluation of chemical reactivity of synthesized ethyl-4-[(2-cyano-acetyl)-hydrazonomethyl]-3,5-dimethyl-1H-pyrrole-2-carboxylate through experimental studies and quantum chemical calculations. J. Mol. Struct.. 2014;1074:201-212.
- [Google Scholar]
- Evaluation of molecular assembly, spectroscopic interpretation, intra-/inter molecular hydrogen bonding and chemical reactivity of two pyrrole precursors. J. Mol. Struct.. 2014;1075:462-470.
- [Google Scholar]
- Assessment of conformational, spectral, Antimicrobial activity, chemical reactivity and NLO application of Pyrrole-2, 5-dicarboxaldehyde bis(oxaloyldihydrazone) Spectrochim. Acta Part A. 2015;140:344-355.
- [Google Scholar]
- Synthesis, conformational, spectroscopic and chemical reactivity analysis of 2-cyano-3-(1H-pyrrol-2-yl)acrylohydrazide using experimental and quantum chemical approaches. J. Mol. Struc.. 2015;1082:118-130.
- [Google Scholar]
- Synthesis, spectral analysis and study of antimicrobial activity of 2,5-diformyl-1H-pyrrole bis(methan-1-yl-1-ylidene)dimalonohydrazone. Arab. J. Chem.. 2019;12:1219-1233. http://dx.doi.org/10.1016/j.arabjc.2014.10.050
- [Google Scholar]
- Crystal and molecular structure of 2- hydroxy-1-naphthaldehyde isonicotinoyl hydrazone (NIH) and its iron(III) complex: an iron chelator with antitumour. Biol. Inorg. Chem.. 1999;4:266-273.
- [Google Scholar]
- Biol. Activ. Hydraz. Derivat. Molec.. 2007;12:1910-1939.
- New α-(N)-heterocyclichydrazones: evaluation of anticancer, anti-HIV and antimicrobial activity. Eur. J. Med. Chem.. 2004;39:113-122.
- [Google Scholar]
- Charge carrier transporting molecular materials and their applications in devices. Chem. Rev.. 2007;107:953-1010.
- [Google Scholar]
- Spectrometric Identification of Organic Compounds (sixth ed.). New York: Jon Wiley Sons, Inc.; 1963.
- Spectral analysis, structural elucidation, and evaluation of both nonlinear optical properties and chemical reactivity of a newly synthesized ethyl-3,5-dimethyl-4-[(toluenesulfonyl)-hydrazonomethyl]-1H-pyrrole-2-carboxylate through experimental studies and quantum chemical calculations. J. Mol. Struct.. 2013;1054–1055:65-75.
- [Google Scholar]
- Synthesis, spectroscopic and structural evaluation of ethyl 2-cyano-3-{5-[(4-nitro-benzoyl)-hydrazonomethyl]-1H-pyrrol-2-yl}-acrylate using experimental and theoretical approaches. J. Mol. Struc.. 2013;1049:419-428.
- [Google Scholar]
- Investigation of spectroscopic, structural and non-linear optical properties of ethyl 3, 5-dimethyl-4-[(benzenesulfonyl)-hydrazonoethyl]-1H-pyrrol-2-carboxylate. J. Mol. Struc.. 2013;1054:123-133.
- [Google Scholar]
- Studies on molecular weaker interactions, spectroscopic analysis and chemical reactivity of synthesized ethyl 3,5-dimethyl-4-[3-(2-nitro-phenyl)-acryloyl]-1H-pyrrole-2-carboxylate through experimental and quantum chemical approaches. J. Mol. Struct.. 2013;1037:338-351.
- [Google Scholar]
- Synthesis, molecular structure, and spectral analyses of ethyl-4-[(2, 4-dinitrophenyl)-hydrazonomethyl]-3, 5-dimethyl-1H-pyrrole-2-carboxylate. Struct. Chem.. 2013;24:713-724.
- [Google Scholar]
- A combined experimental and quantum chemical (DFT and AIM) study on molecular structure, spectroscopic properties, NBO and multiple interaction analysis in a novel ethyl 4-[2-(carbamoyl)hydrazinylidene]-3,5-dimethyl-1H-pyrrole-2-carboxylate and its dimer. J. Mol. Struct.. 2013;1035:427-440.
- [Google Scholar]
- Synthesis, molecular structure, multiple interactions and chemical reactivity analysis of a novel ethyl 2-cyano-3-[5-(hydrazinooxalyl–hydrazonomethyl)-1H-pyrrol-2-yl]-acrylate and its dimer: A combined experimental and theoretical (DFT and QTAIM) approach. J. Mol. Struc.. 2014;1037:420-430.
- [Google Scholar]
- Synthesis, molecular structure, photoluminescence, multiple interaction, chemical reactivity and first hyperpolarizability analysis of Ethyl 2-cyano-3-{5-(4-methylbenzenesulfonyl)-hydrazonomethyl]-1H-pyrrol-2-yl}-acrylate: experimental and quantum chemical approaches. J. Mol. Struct.. 2014;1061:140-149.
- [Google Scholar]
- First evidence of a photoinduced spin change in an Fer (III) complex using visible light at room temperature. Eur. J. Inorg. Chem.. 1999;1999:2117-2119.
- [Google Scholar]
- Solid-to-solid reaction of 2,4Dinitrophenylhydrazine with several aromatic aldehydes bearing electron-donating groups. Supramol. Chem.. 2001;13:247-251.
- [Google Scholar]
- Synthesis of a novel host molecule of cross-linking-polymeric- -cyclodextrin-o-vanillin furfuralhydrazone and spectrofluorimetric analysis of its identifying cadmiumSpectrochim. Acta Part A. 2004;60:2425-2431.
- [Google Scholar]
- Quantifying the charge transfer phenomenon by molar refractivity in binding of 4-quinoinyl derivatives as antimalarials. Int. J. Chem. Tech. Res.. 2010;2:1468-1472.
- [Google Scholar]
- A comparison between two polarizability parameters in chemical–biological interactions. Bioorg. Med. Chem.. 2005;13:2355-2372.
- [Google Scholar]
- Third-order nonlinear optical properties in 4-[(E)-(2-phenylhydrazinylidene)methyl]tetrazolo[1,5-a]quinoline doped PMMA thin film using Z-scan technique. J. Mod. Optic. 2010;57:670-676.
- [Google Scholar]
- Study of third-order optical nonlinearities of substituted hydrazones in PMMA. J. Appl. Polym. Sci.. 2011;119:595-601.
- [Google Scholar]
- Textbook of Practical Organic Chemistry (third ed.). New York: Prentice Hall, John Wiley & Sons, Inc.; 1956.
- Vogt, R.F., Martin, G.E., Zenger, V., 2008. In: Wolfbeis, O.S. (Ed.), Springer Series on Fluorescence. Springer, Berlin, pp. 4-31.
- (E)-N′-(5-Bromo-2-hy-droxy-benzyl-idene)-4-(dimethyl-amino) benzohydrazide. Acta Cryst. E. 2012;68:o1304.
- [Google Scholar]
- Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc.. 1990;112:8251-8260.
- [Google Scholar]
- Synthesis and anion recognition of acetate ions using pyrrole-α-carboxaldhyde 2, 4-dinitrophenyl hydrazine. Ind. J. Chem. Sec A. 2007;46:1437-1439.
- [Google Scholar]
- Synthesis of novel substituted pyrazole-5-carbohydrazidehydrazone derivatives and discovery of a potent apoptosis inducer in A549 lung cancer cells. Bioorg. Med. Chem.. 2009;17:1957-1962.
- [Google Scholar]
- N′-(3-Fluorobenzylidene)-4-hydroxy-3-methoxybenzohydrazide methanol monosolvate. Acta Cryst. E. 2012;68:o1338.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.arabjc.2015.03.001.
Appendix A
Supplementary material
Supplementary data 1
Supplementary data 1
Supplementary data 2
Supplementary data 2




